

Background: Mammary gland branching morphogenesis is a highly regulated developmental process often disrupted in breast cancer.
Results: A chemical genetic screen in primary three-dimensional culture revealed that activation of the aryl hydrocarbon receptor promotes desmosomes to block branching.
Conclusion: Down-regulation of desmosomes is required for proper mammary branching morphogenesis.
Significance: Desmosomes are a novel mechanism through which exposure to environmental pollutants may affect mammary development.
Keywords: Aryl Hydrocarbon Receptor, Chemical Biology, Desmosome, Drug Screening, Mammary Gland
Abstract
During the process of branching morphogenesis, the mammary gland undergoes distinct phases of remodeling to form an elaborate ductal network that ultimately produces and delivers milk to newborn animals. These developmental events rely on tight regulation of critical cellular pathways, many of which are probably disrupted during initiation and progression of breast cancer. Transgenic mouse and in vitro organoid models previously identified growth factor signaling as a key regulator of mammary branching, but the functional downstream targets of these pathways remain unclear. Here, we used purified primary mammary epithelial cells stimulated with fibroblast growth factor-2 (FGF2) to model mammary branching morphogenesis in vitro. We employed a forward chemical genetic approach to identify modulators of this process and describe a potent compound, 1023, that blocks FGF2-induced branching. In primary mammary epithelial cells, we used lentivirus-mediated knockdown of the aryl hydrocarbon receptor (AHR) to demonstrate that 1023 acts through AHR to block branching. Using 1023 as a tool, we identified desmosomal adhesion as a novel target of AHR signaling and show that desmosomes are critical for AHR agonists to block branching. Our findings support a functional role for desmosomes during mammary morphogenesis and also in blocking FGF-induced invasion.
Introduction
Branching morphogenesis is a developmental process that maximizes surface area in order to enhance gas, nutrient, and waste exchange by tissue (1). Consequently, branching morphogenesis occurs during development of a variety of organs, including Drosophila trachea, mammalian lungs, kidneys, and salivary and mammary glands (2). Because this developmental event relies on coordination of major cellular processes, including proliferation, invasion, differentiation, and apoptosis (1), understanding the pathways that drive branching morphogenesis may lead to discoveries of mechanisms involved in human diseases, such as cancer, where these fundamental cellular pathways are aberrantly regulated.
In contrast to other organs, the mammary gland undergoes key stages of development postnatally. Due to its delayed development and capacity to branch in organotypic systems in vitro, the mammary gland is a uniquely accessible model organ of branching morphogenesis (3). At birth, the mammary gland consists of a small ductal network embedded in adipose tissue. Hormonal cues stimulate its development during puberty, which results in elongation of ducts that eventually fill the fat pad (4). Importantly, mammary ducts are composed of an epithelial bilayer, with a basal layer consisting of myoepithelial cells and inner layer of luminal cells. Each layer contains progenitor and differentiated cell populations that are poised to respond to developmental and hormonal changes. During pregnancy, alveolar buds branch from ducts and develop into lobuloalveolar acini, where functionally differentiated luminal cells produce milk during lactation. In addition, differentiated myoepithelial cells contract in response to oxytocin to facilitate milk delivery (5). Cumulatively, mammary branching morphogenesis functions to establish an extensive tubular gland capable of producing and delivering milk to newborn animals.
Studies using genetically modified mice and three-dimensional organotypic culture of mammary tissue have identified several molecular pathways important for mammary gland development. Among these pathways, epidermal growth factor (EGF) receptor and fibroblast growth factor (FGF) receptor have been shown to be critical drivers of proliferation and invasion, respectively (5). Although these studies support a pivotal role for EGF and FGF signaling in mammary development, the downstream cellular targets of these cascades remain unclear.
We sought to develop an in vitro model of mammary development well suited to define intracellular pathways targeted by growth factor signaling. In previously established organotypic models of mammary morphogenesis, fragments of murine mammary epithelium, called organoids, were embedded in extracellular matrix and stimulated with specific growth factors to induce branching (6–9). However, organoids are complex tissue structures consisting of both epithelial and stromal cells. As a result, one limitation of these studies is the inability to distinguish the effects of growth factor signaling directly on epithelial cells from the indirect effects exerted through stromal cells. We developed an in vitro three-dimensional branching model consisting of enriched, primary mammary epithelial cells (MECs)4 and employed a forward chemical genetic screen to identify modulators of branching morphogenesis. We describe a compound, 1023, which effectively blocks FGF2-induced branching while maintaining cells as normal mammary cysts. We demonstrate that 1023 acts through the aryl hydrocarbon receptor (AHR) to functionally inhibit branching by regulating desmosome adhesion complexes in mammary epithelium.
EXPERIMENTAL PROCEDURES
Isolation of Primary MECs
Organoids from the fourth inguinal mammary glands were isolated from 8–12-week-old female FVB/n mice as described previously (10). Protocol modifications and dissociation of organoids into single cells are described in the supplemental material.
Aggregation and Culture of Primary MECs in Matrigel
Frozen primary MECs were thawed, washed, and pelleted three times with 5 ml of DMEM/F-12 (HyClone) (150 × g, 3 min) to remove DMSO. To form aggregates, 2 × 106 primary MECs were resuspended in 1 ml of primary MEC growth medium (DMEM/F-12 supplemented with 5 μg/ml ITS-X (Invitrogen), 1 μg/ml hydrocortisone, 10 ng/ml murine EGF (BD Biosciences), 10% FBS (HyClone), and 1× penicillin/streptomycin/glutamine (Invitrogen)) and incubated overnight at 37 °C with 5% CO2 in a 24-well ultra-low attachment tissue culture plate (Costar). After 16 h, stromal contaminants were removed by differential centrifugation as described previously (6). To create a base layer, 20 μl of growth factor-reduced Matrigel (BD Biosciences) was plated in the center of each well of a 24-well glass SensoPlate (Greiner Bio-One) and allowed to solidify at 37 °C. Aggregates (250 per well) were plated on top of the hardened base layer in 40 μl of Matrigel per well and incubated at 37 °C with 5% CO2 to solidify Matrigel. Embedded aggregates were cultured in DMEM/F-12 supplemented with 5 μg/ml ITS-X, 1× penicillin/streptomycin/glutamine, and either murine EGF or human FGF2 (Invitrogen) (2.5 nm unless otherwise indicated). 1023, 1023-CF3, and 2,3,7,8-tetrachlorodibenzodioxin (TCDD) were added as indicated. Cells were incubated at 37 °C with 5% CO2 with medium changes every 48 h. When single cells were cultured in Matrigel, 2,500 single trypsinized primary MECs were embedded in 40 μl of Matrigel per well of a 24-well plate. For peptide experiments, aggregates were plated on top of Matrigel as described previously (11). Briefly, 50 μl of Matrigel was plated in each well of a 96-well plate and incubated at 37 °C to harden. Aggregates (250 per well) were resuspended in 100 μl of medium containing 2% Matrigel and plated on top of the base layer. We used this overlay technique to ensure that the peptides could access aggregates. To calculate the percentage of branching in all assays, samples were fixed in 4% paraformaldehyde and imaged with bright field microscopy at low magnification (×4). At least 100 aggregates/well were scored, with branching counted as three ducts or more, as established previously in organoid culture models (6). At least 3 independent samples/condition were averaged for each experiment.
Small Molecule Library Screen
Compound stocks were obtained from the University of Utah Department of Chemistry Collection and diluted to 10 μm in serum-free medium (DMEM/F-12 supplemented with 5 μg/ml ITS-X, 1× penicillin/streptomycin/glutamine, and 2.5 nm human FGF2). Aggregates were embedded as described above in 48-well tissue culture plates (BD Biosciences) with a 5-μl Matrigel base layer and 200 aggregates suspended in 10 μl of Matrigel on top. Medium was replaced with fresh compound-containing medium after 72 h in culture. After 144 h total, cells were fixed in 4% paraformaldehyde, and an Olympus CKX41 inverted microscope was used to score each compound based on the phenotype observed in greater than 50% of aggregates in each well.
Homology Modeling
Sequence alignment, homology modeling, and molecular docking of 1023 with human AHR is described in detail in the supplemental material.
Construction of shRNA Lentiviral Plasmids
RNA sequences (supplemental Table S2) for shRNA Ahr or Arnt (aryl hydrocarbon receptor nuclear translocator) knockdown were designed using the Dharmacon siDESIGN Center (available on the Thermo Scientific Web site). Oligonucleotides were annealed and ligated into HpaI and XhoI sites in pLentiLox5.0-GFP (12) following the molecular cloning protocol detailed in the supplemental material.
Virus Production and Transduction of Primary MECs
Lentivirus was produced and titrated as previously described (10, 13). High efficiency lentiviral transduction of primary MECs was conducted as described previously (14) with modifications detailed in the supplemental material.
Statistical Analysis
All values are shown as means ± S.E. or S.D., as indicated. p values were determined using Student's t test with two-tailed distribution and unequal variance.
Additional Experimental Procedures
Additional methodology is provided in the supplementary material.
RESULTS
Development of in Vitro Three-dimensional Branching Assay Using Primary MECs
To study the direct effect of growth factor signaling on epithelial cells, we developed a three-dimensional branching assay using isolated primary MECs devoid of stromal components. Primary MECs were prepared as single cells from the fourth inguinal mammary glands of 8–12-week-old FVB/n mice. Dissociated primary MECs were cultured in suspension to allow aggregation of epithelial cells, and after 16 h, the cell clusters were enriched from stromal cells by differential centrifugation as described previously (6). The MEC aggregates were embedded in Matrigel and grown in culture for 144 h in serum-free medium supplemented with either EGF or FGF2. In this assay, aggregates cultured with either growth factor underwent considerable reorganization, motility, and proliferation (Fig. 1, A and B). However, EGF and FGF2 induced distinct differences in morphology. Unlike in vitro organoid cultures (7), aggregated primary MECs in our assay did not branch efficiently in the presence of EGF and predominately formed hollow cysts with a fluid-filled lumen (Fig. 1A). The significantly reduced branching observed with EGF further supports its role as an indirect mediator of branching (8, 15) and is in agreement with the lack of stromal cells in our aggregate culture model (10) compared with the organoid model (16). In contrast to EGF, FGF2 stimulated extensive branching of MECs (Fig. 1B), which is consistent with the robust branching phenotype elicited by activation of the FGF receptor in mammary epithelial cells in vivo (17). To determine the optimal concentration of FGF2 required to stimulate branching in the aggregate model, we performed a dose-response analysis and found the half-maximal effective concentration (EC50) for FGF2-induced branching to be 0.41 ± 0.053 nm (Fig. 1C). Based on these results, we used 2.5 nm growth factor for all subsequent assays, which was the minimum concentration required to achieve maximal branching. At this concentration, 56.4% of primary MECs branched in the presence of FGF2 compared with only 8.9% with EGF (Fig. 1D).
FIGURE 1.

Primary MECs branch in the presence of FGF2. A, aggregated primary MECs grown in Matrigel with 2.5 nm EGF underwent extensive reorganization to form a fluid-filled cyst between 72 and 96 h in culture. Continued growth was observed through 144 h. B, 2.5 nm FGF2 stimulated robust branching. Aggregates initially reorganized to form a cyst, which collapsed near 72 h in culture. Extensive branching and increased growth was observed through 144 h in culture. Representative time course DIC images are shown. C, dose-response analysis for EGF- and FGF2-induced branching over 144 h. D, quantification of percentage of branching in primary MECs grown for 144 h with 2.5 nm EGF or 2.5 nm FGF2. Statistical analysis was performed using Student's t test. Results are shown as mean ± S.D. (error bars); n = 3; ****, p < 0.0001. E, mammary ducts are composed of an epithelial bilayer, as shown by immunofluorescence staining of a cross-section of a duct from a virgin FVB/n mouse with a luminal epithelial marker, K8 (green), and a myoepithelial marker, K14 (red). F, immunofluorescence staining for K8 and K14 in our three-dimensional primary MEC culture model with 2.5 nm EGF. G, immunofluorescence staining for K8 and K14 in our three-dimensional primary MEC culture model with 2.5 nm FGF2. E–G, interactions between luminal epithelial and myoepithelial cells were similar to those observed in vivo. Nuclei were stained with DAPI (in blue). The dashed white line defines the lumen. A, B, and E–G, scale bar, 40 μm.
We next used immunofluorescence to assess the cellular architecture of cyst and branched structures. Both EGF and FGF2 induced a cellular organization similar to that observed in vivo. Immunofluorescence staining showed that like in vivo mammary ducts (Fig. 1E), EGF-induced cysts were composed of an epithelial bilayer (Fig. 1F), with keratin-8 (K8)-positive luminal epithelial cells surrounded by keratin-14 (K14)-positive myoepithelial cells. In branched structures induced by FGF2, we found K8-positive luminal epithelial cells invading into the Matrigel at branch points and K14-positive myoepithelial cells retained at points of constriction at the base of branches (Fig. 1G), which is consistent with previous studies showing that branch points arise at breaks in the myoepithelial layer (16). Thus, our aggregate-based model recapitulated key aspects of in vivo mammary branching morphogenesis and allowed us to uncouple epithelial induced morphogenesis from stromal mediated events.
Chemical Genetic Screen to Identify Modulators of Branching Morphogenesis
With our three-dimensional assay, we next conducted a chemical genetic screen to identify strong modulators of branching, with the goal to use these compounds to elucidate signaling pathways in epithelial cells critical for this developmental process. We were specifically interested in compounds that completely blocked FGF2-induced invasion yet maintained the normal, bilayered morphology of the growing aggregate. Thus, we were interested in compounds that induced cyst morphology similar to EGF-treated aggregates. We hypothesized that compounds causing cyst arrest in FGF2-treated aggregates would more specifically target invasion rather than growth-stimulating pathways.
587 compounds from the University of Utah Chemistry Department Collection were screened in the FGF2 branching assay. Six dominant phenotypes were observed (Fig. 2, A and B): normal branching, hyperbranching, reduced branching, cyst arrest, filled, and cytotoxic. Additionally, 6% of compounds caused a mixed phenotype consisting of two or more dominant characteristics (Fig. 2B). The most potent compound that caused cyst arrest, a diphenyl oxadiazole named 1023 (Fig. 2C), exhibited an EC50 of 1.2 ± 0.050 μm in our branching assay (Fig. 2D) and had no significant toxicity or effect on growth compared with DMSO treatment (vehicle control) (Fig. 2E and supplemental Movies S1 and S2). The effect of impairing branching while simultaneously maintaining normal cyst growth (supplemental Fig. S1) suggested 1023 was a specific modulator of FGF2-induced branching. Thus, we reasoned that elucidating the target of 1023 would lead to better understanding of the molecular mechanisms downstream of FGF2 during mammary branching morphogenesis.
FIGURE 2.

Chemical library screen identifies 1023 as a potent inhibitor of in vitro branching morphogenesis. A, six dominant phenotypes were observed in a chemical library screen for alterations on FGF2-induced branching. Representative DIC images of the phenotypes observed are shown. B, quantification of the percentage of total compounds that induced each phenotype. C, structure of 1023, which was one of the most potent compounds to cause cyst arrest. D, dose-response analysis for FGF2-induced branching in the presence of 1023 over 144 h. E, representative time course DIC images of MECs grown in the FGF2 branching assay with DMSO (vehicle control) (top) or 10 μm 1023 (bottom). A and E, scale bar, 40 μm. Error bars, S.D.
Activation of the Aryl Hydrocarbon Receptor Blocks Branching Morphogenesis
For insight into the biological mechanism of action of 1023, we evaluated alterations in gene expression following compound treatment. We conducted microarray gene expression analysis on primary MECs grown in Matrigel for 72 h with FGF2 and either DMSO or 10 μm 1023. Compared with DMSO, 1023 highly up-regulated Cyp1a1, Ahrr, Gsta3, Aldh3a1, Nqo1, Cyp1b1, and Ugt-1a (Table 1, boldface type), an expression profile known to be induced by activation of AHR (18).
TABLE 1.
Genes of interest up-regulated by microarray in MECs treated for 72 h with 1023
Known AHR response genes are shown in boldface type.
| Gene identifier | Gene name | Gene symbol | Ratioa |
|---|---|---|---|
| NM_009992 | Cytochrome P450, family 1, subfamily a, polypeptide 1, transcript variant 1 | Cyp1a1 | 49.61 |
| NM_009644 | Aryl-hydrocarbon receptor repressor | Ahrr | 22.37 |
| XM_484705 | Desmoglein 1 α | Dsg1 | 10.51 |
| BY704100 | Desmocollin 1 | Dsc1 | 9.39 |
| NM_010356 | Glutathione S-transferase, α 3, transcript variant 2 | Gsta3 | 7.13 |
| NM_007436 | Aldehyde dehydrogenase family 3, subfamily A1, transcript variant 1 | Aldh3a1 | 5.37 |
| NM_008706 | NAD(P)H dehydrogenase, quinone 1 | Nqo1 | 4.09 |
| NM_009994 | Cytochrome P450, family 1, subfamily b, polypeptide 1 | Cyp1b1 | 3.49 |
| NM_030596 | Desmoglein 3 | Dsg3 | 3.38 |
| NM_201410 | UDP glucuronosyltransferase 1 family, polypeptide A6B | Ugt-1a | 2.91 |
a Log2 ratio for 1023-treated versus DMSO.
AHR is a ligand-activated transcription factor and member of the Per-Arnt-Sim family of basic helix-loop-helix proteins. Known for binding aromatic compounds (19, 20), AHR mediates response to chemicals by up-regulating expression of metabolic enzymes (21). However, a growing number of studies suggest that AHR has a wider physiologic role in regulating critical cellular processes, including proliferation, differentiation, and apoptosis (22, 23). To assess AHR as a potential target of 1023, we used a homology model structure of human AHR to predict binding modes of 1023 (24). Using the lowest energy conformer, 1023 was predicted to bind AHR with a free energy of −20.83 kcal/mol (Fig. 3A), which suggested a strong interaction between 1023 and AHR. In addition to van der Waals contacts, our modeling predicted Phe-287, His-291, Phe-324, Cys-300, His-337, and Gln-383 in the AHR binding cavity significantly contributed to the binding mode of 1023 through hydrogen bonding and other non-bonded contacts. Furthermore, the modeling data indicated that the addition of a trifluoromethyl group to the phenyl ring of 1023 would drastically reduce its ability to bind AHR due to steric clashes with His-291 and Phe-287. As a result, we synthesized the 1023-CF3 analog (Fig. 3B, top), which had a predicted binding energy of −11.21 kcal/mol, to use as a negative control in our assays.
FIGURE 3.

1023 activates the aryl hydrocarbon receptor. A, docking of 1023 (purple) with the homology model structure of human AHR (gray). Important interacting residues are noted in green. The binding free energy was estimated at −20.83 kcal/mol, suggesting a favorable interaction between 1023 and AHR. B, chemical structures of 1023-CF3 (top), a predicted inactive analog of 1023, and TCDD (bottom), a known activator of AHR. C, dose-response analysis for FGF2-induced branching in primary MECs treated with TCDD for 144 h. D, 1023-CF3 was less effective at blocking branching than 1023, as shown by dose-response analysis over 144 h for FGF2-induced branching. Error bars S.D. E, representative DIC images of the dominant phenotype observed in primary MECs grown in the FGF2 branching assay with DMSO (top left), 10 nm TCDD (top right), 10 μm 1023 (bottom left), or 10 μm 1023-CF3 (bottom right). Scale bar, 40 μm. F, in HEK-293T cells transiently expressing pACTAG2-HA-AHR, 10 nm TCDD and 10 μm 1023 increased nuclear localization of AHR compared with DMSO or 10 μm 1023-CF3 after 24 h of treatment. Quantification was based on immunofluorescence for HA. Results are shown as mean ± S.D.; n = 7; ****, p < 0.0001. G, relative gene expression of Cyp1a1, a known AHR response gene, was elevated in primary MECs treated for 48 h with 10 nm TCDD or 10 μm 1023 compared with DMSO or 10 μm 1023-CF3. Cyp1a1 gene expression was measured by RT-PCR and normalized to β-actin expression. Results are shown as mean ± S.E. (error bars); n = 3; ***, p < 0.001; **, p < 0.01. F and G, statistical analysis was performed using Student's t test.
With AHR as the putative cellular target of 1023, we hypothesized that TCDD (Fig. 3B, bottom), a widespread environmental pollutant and known activator of AHR (25, 26), would phenocopy 1023. In our three-dimensional branching assay, TCDD blocked branching of primary MECs (EC50 = 3.93 ± 0.17 pm) (Fig. 3C) and induced a strong cyst arrest phenotype (Fig. 3E, top right), similar to 1023 (Fig. 3E, bottom left). In contrast, 1023-CF3 did not efficiently block branching (EC50 = 71.1 ± 1.9 μm) compared with 1023 (EC50 = 1.2 ± 0.05 μm) (Fig. 3, D and E, bottom panels), as predicted by our modeling data. These results suggested that activation of AHR is sufficient to block mammary branching morphogenesis in vitro.
1023 Activates AHR to Disrupt Mammary Branching Morphogenesis
We next probed the AHR pathway to verify that 1023 was a strong activator of AHR. Following ligand binding, AHR undergoes nuclear translocation, dimerizes with ARNT, and activates transcription of downstream target genes (19, 27, 28). Considering this mechanism, we wanted to determine whether 1023 treatment led to an increased accumulation of AHR in the nucleus. To do so, we expressed an HA-tagged AHR construct, pACTAG2-HA-AHR, in HEK-293T cells. After 24 h of drug treatment, we performed immunofluorescence for HA and found that both 10 nm TCDD and 10 μm 1023 significantly increased the amount of nuclear AHR compared with DMSO or 10 μm 1023-CF3 (Fig. 3F and supplemental Fig. S2).
The increase in AHR nuclear translocation also correlated with induction of AHR response genes. Cyp1a1 (cytochrome P4501A1) is a direct AHR target and is used as a readout for AHR activation (29). In primary MECs treated for 48 h, 10 nm TCDD and 10 μm 1023 increased Cyp1a1 gene expression by 23.3- and 4.5-fold, respectively, compared with DMSO or 10 μm 1023-CF3 (Fig. 3G). Furthermore, we tested the specificity of Cyp1a1 induction and asked whether AHR and its transcriptional binding partner, ARNT, were required for 1023 to activate Cyp1a1 expression. We generated mouse mammary HC11 cell lines stably expressing either a nonspecific control shRNA, shRNA against Ahr (shAHR-1), or shRNA against Arnt (shARNT-1 or shARNT-2). In these cell lines, Ahr expression was reduced by 61% (shAHR-1) (supplemental Fig. S3A) and Arnt expression by 60% (shARNT-1) or 75% (shARNT-2) (supplemental Fig. S3B). Cells with Ahr or Arnt knockdown showed considerably reduced expression of Cyp1a1 than control cells after treatment with 10 nm TCDD (supplemental Fig. S3C) or 10 μm 1023 (Fig. 4A). Together, these results confirmed that 1023 activated AHR, leading to its nuclear translocation and induction of downstream gene targets.
FIGURE 4.

AHR is the biological target of 1023. A, induction of Cyp1a1 by 1023 was lower in HC11 cells stably expressing an shRNA against Ahr (shAHR-1) or Arnt (shARNT-1 or shARNT-2) than untransduced or control transduced cells. Cyp1a1 gene expression was measured after 6 days of treatment with DMSO or 10 μm 1023 and normalized to β-actin expression. Results are shown as mean ± S.E. (error bars). B, knockdown of Ahr in primary MECs rescued branching in the presence of AHR agonists. Shown is a quantification of the percentage of branching in transduced outgrowths from primary MECs infected with a lentiviral control shRNA (Control) or shRNA against Ahr (shAHR-1 or shAHR-2). Statistical analysis was performed using Student's t test. Results are shown as mean ± S.D. (error bars); n = 2; *, p < 0.05; **, p < 0.01. C, representative images of a clonal outgrowth from primary MECs transduced with a control shRNA. D, representative images of a clonal outgrowth from primary MECs transduced with shAHR-2. B–D, following transduction, cells were embedded in Matrigel as single cells and grown for 21 days in the presence of 2.5 nm FGF2 and DMSO, 10 nm TCDD, or 10 μm 1023. C and D, scale bar, 40 μm.
Although 1023 activated AHR, it was still unclear whether AHR was required for the observed branching defect. To address this, we used lentiviral shRNA constructs to knock down Ahr in primary MECs. Briefly, primary MECs were infected in monolayer with a lentiviral control hairpin or shRNA construct against Ahr (shAHR-1 or shAHR-2), and knockdown of Ahr was confirmed by RT-PCR (supplemental Fig. S3D). Following infection, primary MECs were embedded as single cells in Matrigel and grown for 21 days with FGF2. Within each well, single cells grew into clonal outgrowths that were either untransduced or transduced, as distinguished by GFP expression. As expected, control transduced outgrowths (Fig. 4, B and C) branched in the presence DMSO but generated cysts in the presence of 10 nm TCDD or 10 μm 1023. Similar results were observed with untransduced outgrowths that grew in the same well as transduced outgrowths (supplemental Fig. S3E). In contrast, transduction of MECs with lentiviruses containing either of two different shRNA hairpins against Ahr significantly rescued branching in the presence of 10 nm TCDD or 10 μm 1023 (Fig. 4, B and D and supplemental Fig. S3F). These results demonstrated that AHR was required for TCDD and 1023 to block FGF2-induced branching morphogenesis.
Desmosomal Adhesion Is Regulated by the AHR Pathway
After determining that 1023 targeted the AHR pathway, we sought to mechanistically understand how activation of AHR blocks mammary branching. Strikingly, our microarray analysis showed up-regulation of several genes involved in cellular adhesion, including desmosomal cadherins (Table 1 and supplemental Table S1). Because desmosomes are known to induce very tight intercellular contacts (30, 31) and are one of the dominant adhesion complexes found in the mammary gland (32), we hypothesized that desmosomal adhesion would be critical for maintenance of cysts caused by AHR activation.
First, we confirmed our microarray analysis by measuring gene expression of desmosomal cadherins in primary MECs treated for 96 h. RT-PCR results showed that both 10 nm TCDD and 10 μm 1023 significantly up-regulated gene expression of Dsc1 (desmocollin 1) (supplemental Fig. S4A), Dsg1 (desmoglein 1) (supplemental Fig. S4B), and Dsg3 (desmoglein 3) (Fig. 5A) compared with DMSO. Moreover, desmosomal cadherins were up-regulated in primary MECs grown in the presence of EGF compared with FGF2 (Fig. 5A and supplemental Fig. S4, A and B). To assess protein levels, we performed a Western blot for DSG3 in primary MECs extracted from Matrigel after 96 h of treatment. Consistent with our RT-PCR results, DSG3 protein levels were highly up-regulated in primary MECs grown with EGF, 10 nm TCDD, or 10 μm 1023 compared with FGF2 alone (Fig. 5B). Finally, knockdown of Ahr or Arnt significantly reduced induction of desmosomal cadherins (supplemental Fig. S4, C and D). Like Cyp1a1, Dsg1 gene expression was lower in HC11 cell lines with stable knockdown of Ahr or Arnt after treatment with 10 nm TCDD or 10 μm 1023. These experiments identified Dsc1, Dsg1, and Dsg3 as novel downstream targets of the AHR signaling pathway.
FIGURE 5.
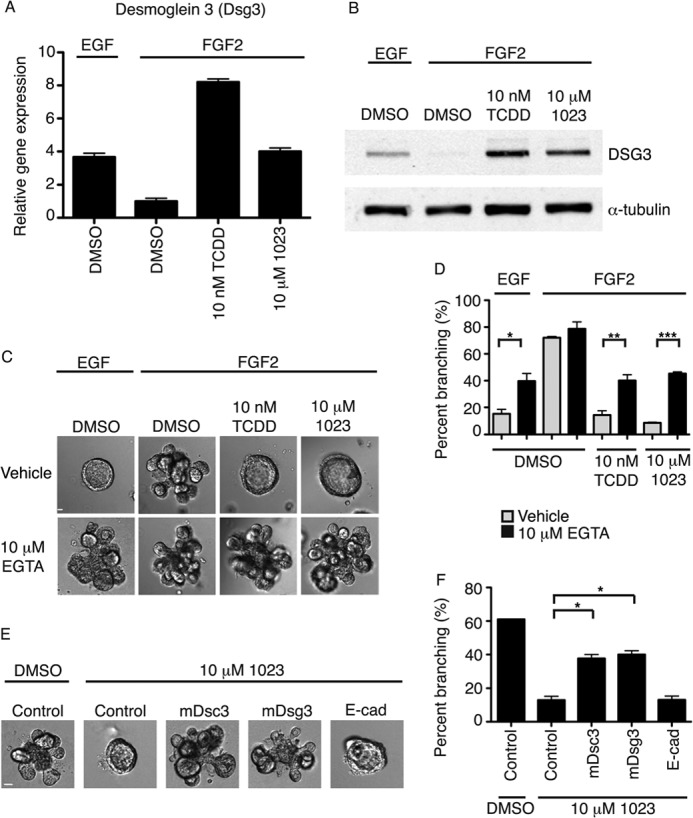
AHR signaling promotes desmosomal adhesion to block mammary branching morphogenesis. A, relative gene expression of Dsg3 (desmoglein 3) in aggregated primary MECs grown for 96 h in Matrigel with 2.5 nm EGF or 2.5 nm FGF2 and DMSO or 10 nm TCDD or 10 μm 1023. Gene expression was measured by RT-PCR and normalized to β-actin expression. Results are shown as mean ± S.E. (error bars). B, Western blot analysis of DSG3 in aggregated primary MECs grown for 96 h in Matrigel with 2.5 nm EGF or 2.5 nm FGF2 and DMSO or 10 nm TCDD or 10 μm 1023. C, the addition of 10 μm EGTA significantly increased branching of primary MECs grown in the presence of 2.5 nm EGF or in the presence of 2.5 nm FGF2 and 10 nm TCDD or 10 μm 1023 compared with vehicle control. Representative DIC images are shown. D, quantification of branching shown as mean ± S.D. (error bars); n = 3; *, p < 0.05; **, p < 0.01; ***, p < 0.001. C and D, EGTA was added after 48 h in culture, and branching was scored at 144 h. E, the addition of 2 mm blocking peptide against DSC3 or DSG3, but not E-cadherin or a nonspecific control peptide, significantly increased branching of primary MECs grown in the presence of FGF2 and 10 μm 1023. Representative DIC images are shown. F, quantification of branching shown as mean ± S.D. (error bars); n = 3; *, p < 0.05. E and F, peptide was added after 48 h in culture, and branching was scored at 144 h. C and E, scale bar, 40 μm. D and F, statistical analysis was performed using Student's t test.
Inhibition of Desmosomal Adhesion Promotes Branching of the Mammary Epithelium
To assess the functional importance of desmosomal adhesion in AHR-mediated disruption of branching morphogenesis, we sought to inhibit desmosome formation. Desmocollin and desmoglein are members of the cadherin family of proteins and require calcium to form functional desmosomes in vitro (33–36). We predicted that if cadherin junctions were required to maintain the cyst phenotype, then chelation of calcium would rescue the branching defect elicited by 1023. Thus, we added 10 μm EGTA (pH 7.4) to chelate calcium in our branching assay. After 144 h in culture, we found a significant increase in branching with EGTA in the presence 10 nm TCDD or 10 μm 1023 compared with vehicle control (Fig. 5, C and D). Furthermore, calcium chelation significantly increased branching in the presence of EGF, suggesting that desmosomes play a central role in the transition to a branched epithelial state.
Based on these results, we went on to selectively inhibit the formation of desmosome complexes using blocking peptides. These 10-mer peptides bind the cell adhesion recognition site (37) of a specific desmosome cadherin and functionally inhibit its ability to form adhesion complexes (38, 39). Accordingly, we added 2 mm peptide against DSC3 or DSG3 to primary MECs after 48 h in culture with 10 μm 1023 and observed a significant increase in branching at 144 h. In contrast, a nonspecific control peptide and a previously published peptide against E-cadherin (38) did not significantly affect branching in the presence of 10 μm 1023 (Fig. 5, E and F), suggesting a specific role for desmosomes. Together, these results demonstrate that desmosomes are a novel downstream target of ligand-induced AHR signaling and critical to block branching morphogenesis of the mammary gland.
DISCUSSION
Using a three-dimensional epithelial cell-based model of branching, we demonstrate that FGF2, but not EGF, is sufficient to drive morphogenesis of mammary epithelial cells, which is in agreement with the role of growth factors in mammary development (5, 40). In addition, we employed a novel chemical genetic approach to identify molecular pathways controlling the dynamics of mammary branching morphogenesis. The use of chemical genetics rather than a standard genetic approach allowed us to specifically activate the AHR signaling pathway and identify a new role for AHR in regulating adhesion complexes in the mammary gland.
Environmental exposure to chemical pollutants, including TCDD, is thought to contribute significantly to lactation deficiencies in women (41). These findings are supported by studies in rodents, which showed that TCDD exposure during gestation (42, 43) and pregnancy (44) decreased alveolar development and milk production, respectively. It remains controversial whether local or systemic mechanisms mediate the effect of TCDD on mammary differentiation. Although TCDD is known to modulate estrogen receptor signaling (45), studies have shown that it does not decrease circulating levels of prolactin, estradiol, or progesterone in compound-treated mice (44). Additionally, TCDD was shown to reduce alveolar development in ex vivo cultures of whole mammary glands (46). In agreement with these observations, our data show that AHR activation has direct effects on mammary cells.
The key mediators of the effects of TCDD have been difficult to study with in vitro organoid and in vivo transgenic models, due to the complex interactions between epithelial and stromal cells in these systems. Studies have shown that the DNA-binding domain of AHR is required for the deleterious effects of TCDD on the mammary gland (46), implicating AHR transcriptional regulation as a critical mechanism. Additionally, reciprocal transplant experiments with AHR knock-out mice suggest that both epithelial and stromal transcriptional targets of AHR contribute to the block in branching (46). Here, we demonstrate an epithelial mediated mechanism, in which AHR blocks alveolar development through desmosomal adhesion, which is a novel target of the AHR pathway.
There is growing evidence to suggest that desmosomes contribute significantly to mammary morphogenesis. In the 1970s, electron microscopy was used to assess the ultrastructure of the mouse mammary gland (47). These studies showed that desmosomes were a major adhesion component in the mammary gland, with large desmosome complexes connecting ductal epithelial cells in the virgin gland. Interestingly, desmosomal complexes were highly dynamic. At the onset of lactation, desmosomal adhesion was increased in major, nonsecretory ducts, whereas milk-producing alveoli lost all desmosome contacts during lactation. This stage-dependent shift in adhesion suggested that desmosomes may have a critical role maintaining ductal structure, and their down-regulation may functionally contribute to productive lactation in lobuloalveoli.
Since these early studies, a functional role for desmosomal adhesion in mammary epithelial cells has been slowly emerging. During branching morphogenesis, luminal epithelial cells migrate collectively through the outer myoepithelial layer of a cyst to form new branch points (16). EM studies on organoids grown in Matrigel showed that despite an overall loss of adhesion during this process, desmosomal adhesion is maintained between luminal epithelial cells (32). In another study, desmosome-blocking peptides disrupted the ability of a clonal cell line derived from mouse luminal epithelial cells to form clusters in an in vitro assay (38). Taken together, these results suggest that desmosomes are a critical form of adhesion between luminal epithelial cells.
Although our results show that down-regulation of desmosomal adhesion facilitates branching morphogenesis, the specific role of these adhesion complexes in myoepithelial and luminal cells is not clear. Our data suggest that the myoepithelial-specific desmosome cadherins, Dsc3 and Dsg3 (38), must be down-regulated for branching morphogenesis. Because branch points are known to initiate from breaks in the myoepithelial layer (16), the mechanical integrity of desmosomes may be selectively lost in myoepithelial cells to promote branching and differentiation. Loss of robust adhesion between myoepithelial cells would also facilitate contraction and milk delivery required during lactation.
Our data demonstrate that desmosomes form a critical and functional adhesion complex between mammary epithelial cells. Because desmosome cadherins are commonly mutated or silenced in human breast cancer (48, 49), desmosomes may be a critical adhesion complex that is lost in order to promote tumorigenesis. These findings are supported by previous work, which showed overexpression of desmosomal components inhibited invasion of nonadhesive cells in collagen (39). In our assay, desmosomal adhesion was sufficient to block the invasive mechanisms mediated by FGF2, consistent with an ability to promote tight epithelial cell interactions. Similarly, AHR agonists have been shown to inhibit invasion and increase differentiation of breast cancer cells (50). Thus, dissecting the role and regulation of desmosomes in vivo will be a critical next step toward understanding morphogenesis and transformation of the mammary gland.
Acknowledgments
We thank Dr. Oliver Hankinson (UCLA, Los Angeles, CA) for providing the AHR expression plasmid and Drs. James Bear (University of North Carolina, Chapel Hill, NC) and Thomas Marshall (University of Utah, Salt Lake City, UT) for providing the pLentiLox5.0-GFP vector.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-CA143815 and R01-CA140296. This work was also supported by Department of Defense Breast Cancer Research Program Grant W81XWH-09-1-04310. K. J. B. is supported by an NIH Developmental Biology Training Grant (5T32 HD07491).

This article contains supplemental Experimental Procedures, Tables S1 and S2, Figs. S1–S4, and Movies S1 and S2.
- MEC
- mammary epithelial cell
- AHR
- aryl hydrocarbon receptor
- K8
- keratin-8
- K14
- keratin-14
- TCDD
- 2,3,7,8-tetrachlorodibenzodioxin
- ARNT
- aryl hydrocarbon receptor nuclear translocator
- DIC
- differential interference contrast.
REFERENCES
- 1. Lu P., Werb Z. (2008) Patterning mechanisms of branched organs. Science 322, 1506–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu P., Sternlicht M. D., Werb Z. (2006) Comparative mechanisms of branching morphogenesis in diverse systems. J. Mammary Gland Biol. Neoplasia 11, 213–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smith B. A., Welm A. L., Welm B. E. (2012) On the shoulders of giants: A historical perspective of unique experimental methods in mammary gland research. Semin. Cell Dev. Biol. 23, 583–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sternlicht M. D. (2006) Key stages in mammary gland development. The cues that regulate ductal branching morphogenesis. Breast Cancer Res. 8, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sternlicht M. D., Kouros-Mehr H., Lu P., Werb Z. (2006) Hormonal and local control of mammary branching morphogenesis. Differentiation 74, 365–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fata J. E., Mori H., Ewald A. J., Zhang H., Yao E., Werb Z., Bissell M. J. (2007) The MAPK(ERK-1,2) pathway integrates distinct and antagonistic signals from TGFα and FGF7 in morphogenesis of mouse mammary epithelium. Dev. Biol. 306, 193–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Simian M., Hirai Y., Navre M., Werb Z., Lochter A., Bissell M. J. (2001) The interplay of matrix metalloproteinases, morphogens, and growth factors is necessary for branching of mammary epithelial cells. Development 128, 3117–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sternlicht M. D., Sunnarborg S. W., Kouros-Mehr H., Yu Y., Lee D. C., Werb Z. (2005) Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin. Development 132, 3923–3933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wiseman B. S., Sternlicht M. D., Lund L. R., Alexander C. M., Mott J., Bissell M. J., Soloway P., Itohara S., Werb Z. (2003) Site-specific inductive and inhibitory activities of MMP-2 and MMP-3 orchestrate mammary gland branching morphogenesis. J. Cell Biol. 162, 1123–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Welm B. E., Dijkgraaf G. J., Bledau A. S., Welm A. L., Werb Z. (2008) Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell 2, 90–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee G. Y., Kenny P. A., Lee E. H., Bissell M. J. (2007) Three-dimensional culture models of normal and malignant breast epithelial cells. Nat. Methods 4, 359–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cai L., Marshall T. W., Uetrecht A. C., Schafer D. A., Bear J. E. (2007) Coronin 1B coordinates Arp2/3 complex and cofilin activities at the leading edge. Cell 128, 915–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klages N., Zufferey R., Trono D. (2000) A stable system for the high-titer production of multiply attenuated lentiviral vectors. Mol. Ther. 2, 170–176 [DOI] [PubMed] [Google Scholar]
- 14. Vafaizadeh V., Klemmt P., Brendel C., Weber K., Doebele C., Britt K., Grez M., Fehse B., Desriviéres S., Groner B. (2010) Mammary epithelial reconstitution with gene-modified stem cells assigns roles to Stat5 in luminal alveolar cell fate decisions, differentiation, involution, and mammary tumor formation. Stem Cells 28, 928–938 [DOI] [PubMed] [Google Scholar]
- 15. Wiesen J. F., Young P., Werb Z., Cunha G. R. (1999) Signaling through the stromal epidermal growth factor receptor is necessary for mammary ductal development. Development 126, 335–344 [DOI] [PubMed] [Google Scholar]
- 16. Ewald A. J., Brenot A., Duong M., Chan B. S., Werb Z. (2008) Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev. Cell 14, 570–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Welm B. E., Freeman K. W., Chen M., Contreras A., Spencer D. M., Rosen J. M. (2002) Inducible dimerization of FGFR1. Development of a mouse model to analyze progressive transformation of the mammary gland. J. Cell Biol. 157, 703–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tijet N., Boutros P. C., Moffat I. D., Okey A. B., Tuomisto J., Pohjanvirta R. (2006) Aryl hydrocarbon receptor regulates distinct dioxin-dependent and dioxin-independent gene batteries. Mol. Pharmacol. 69, 140–153 [DOI] [PubMed] [Google Scholar]
- 19. Denison M. S., Nagy S. R. (2003) Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 43, 309–334 [DOI] [PubMed] [Google Scholar]
- 20. Denison M. S., Pandini A., Nagy S. R., Baldwin E. P., Bonati L. (2002) Ligand binding and activation of the Ah receptor. Chem. Biol. Interact. 141, 3–24 [DOI] [PubMed] [Google Scholar]
- 21. Nebert D. W., Dalton T. P., Okey A. B., Gonzalez F. J. (2004) Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 279, 23847–23850 [DOI] [PubMed] [Google Scholar]
- 22. Furness S. G., Whelan F. (2009) The pleiotropy of dioxin toxicity–xenobiotic misappropriation of the aryl hydrocarbon receptor's alternative physiological roles. Pharmacol. Ther. 124, 336–353 [DOI] [PubMed] [Google Scholar]
- 23. Lawrence B. P., Sherr D. H. (2012) You AhR what you eat? Nat. Immunol. 13, 117–119 [DOI] [PubMed] [Google Scholar]
- 24. Motto I., Bordogna A., Soshilov A. A., Denison M. S., Bonati L. (2011) New aryl hydrocarbon receptor homology model targeted to improve docking reliability. J. Chem. Inf. Model. 51, 2868–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fernandez-Salguero P. M., Hilbert D. M., Rudikoff S., Ward J. M., Gonzalez F. J. (1996) Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 140, 173–179 [DOI] [PubMed] [Google Scholar]
- 26. Mimura J., Fujii-Kuriyama Y. (2003) Functional role of AhR in the expression of toxic effects by TCDD. Biochim. Biophys. Acta 1619, 263–268 [DOI] [PubMed] [Google Scholar]
- 27. Heid S. E., Pollenz R. S., Swanson H. I. (2000) Role of heat shock protein 90 dissociation in mediating agonist-induced activation of the aryl hydrocarbon receptor. Mol. Pharmacol. 57, 82–92 [PubMed] [Google Scholar]
- 28. Pollenz R. S., Sattler C. A., Poland A. (1994) The aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator protein show distinct subcellular localizations in Hepa 1c1c7 cells by immunofluorescence microscopy. Mol. Pharmacol. 45, 428–438 [PubMed] [Google Scholar]
- 29. Whitlock J. P., Jr. (1999) Induction of cytochrome P4501A1. Annu. Rev. Pharmacol. Toxicol. 39, 103–125 [DOI] [PubMed] [Google Scholar]
- 30. Garrod D., Chidgey M. (2008) Desmosome structure, composition and function. Biochim. Biophys. Acta 1778, 572–587 [DOI] [PubMed] [Google Scholar]
- 31. Garrod D., Kimura T. E. (2008) Hyper-adhesion. A new concept in cell-cell adhesion. Biochem. Soc. Trans. 36, 195–201 [DOI] [PubMed] [Google Scholar]
- 32. Ewald A. J., Huebner R. J., Palsdottir H., Lee J. K., Perez M. J., Jorgens D. M., Tauscher A. N., Cheung K. J., Werb Z., Auer M. (2012) Mammary collective cell migration involves transient loss of epithelial features and individual cell migration within the epithelium. J. Cell Sci. 125, 2638–2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Demlehner M. P., Schäfer S., Grund C., Franke W. W. (1995) Continual assembly of half-desmosomal structures in the absence of cell contacts and their frustrated endocytosis. A coordinated Sisyphus cycle. J. Cell Biol. 131, 745–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garrod D. (2010) Desmosomes in vivo. Dermatol. Res. Pract. 2010, 212439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Penn E. J., Burdett I. D., Hobson C., Magee A. I., Rees D. A. (1987) Structure and assembly of desmosome junctions. Biosynthesis and turnover of the major desmosome components of Madin-Darby canine kidney cells in low calcium medium. J. Cell Biol. 105, 2327–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nagar B., Overduin M., Ikura M., Rini J. M. (1996) Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature 380, 360–364 [DOI] [PubMed] [Google Scholar]
- 37. Blaschuk O. W., Sullivan R., David S., Pouliot Y. (1990) Identification of a cadherin cell adhesion recognition sequence. Dev. Biol. 139, 227–229 [DOI] [PubMed] [Google Scholar]
- 38. Runswick S. K., O'Hare M. J., Jones L., Streuli C. H., Garrod D. R. (2001) Desmosomal adhesion regulates epithelial morphogenesis and cell positioning. Nat. Cell Biol. 3, 823–830 [DOI] [PubMed] [Google Scholar]
- 39. Tselepis C., Chidgey M., North A., Garrod D. (1998) Desmosomal adhesion inhibits invasive behavior. Proc. Natl. Acad. Sci. U.S.A. 95, 8064–8069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lu P., Ewald A. J., Martin G. R., Werb Z. (2008) Genetic mosaic analysis reveals FGF receptor 2 function in terminal end buds during mammary gland branching morphogenesis. Dev. Biol. 321, 77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Neville M. C., Walsh C. T. (1995) Effects of xenobiotics on milk secretion and composition. Am. J. Clin. Nutr. 61, 687S–694S [DOI] [PubMed] [Google Scholar]
- 42. Fenton S. E., Hamm J. T., Birnbaum L. S., Youngblood G. L. (2002) Persistent abnormalities in the rat mammary gland following gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. Sci. 67, 63–74 [DOI] [PubMed] [Google Scholar]
- 43. Lewis B. C., Hudgins S., Lewis A., Schorr K., Sommer R., Peterson R. E., Flaws J. A., Furth P. A. (2001) In utero and lactational treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs mammary gland differentiation but does not block the response to exogenous estrogen in the postpubertal female rat. Toxicol. Sci. 62, 46–53 [DOI] [PubMed] [Google Scholar]
- 44. Vorderstrasse B. A., Fenton S. E., Bohn A. A., Cundiff J. A., Lawrence B. P. (2004) A novel effect of dioxin. Exposure during pregnancy severely impairs mammary gland differentiation. Toxicol. Sci. 78, 248–257 [DOI] [PubMed] [Google Scholar]
- 45. Swedenborg E., Pongratz I. (2010) AhR and ARNT modulate ER signaling. Toxicology 268, 132–138 [DOI] [PubMed] [Google Scholar]
- 46. Lew B. J., Manickam R., Lawrence B. P. (2011) Activation of the aryl hydrocarbon receptor during pregnancy in the mouse alters mammary development through direct effects on stromal and epithelial tissues. Biol. Reprod. 84, 1094–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pitelka D. R., Hamamoto S. T., Duafala J. G., Nemanic M. K. (1973) Cell contacts in the mouse mammary gland. I. Normal gland in postnatal development and the secretory cycle. J. Cell Biol. 56, 797–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Klus G. T., Rokaeus N., Bittner M. L., Chen Y., Korz D. M., Sukumar S., Schick A., Szallasi Z. (2001) Down-regulation of the desmosomal cadherin desmocollin 3 in human breast cancer. Int. J. Oncol. 19, 169–174 [DOI] [PubMed] [Google Scholar]
- 49. Oshiro M. M., Kim C. J., Wozniak R. J., Junk D. J., Muñoz-Rodriguez J. L., Burr J. A., Fitzgerald M., Pawar S. C., Cress A. E., Domann F. E., Futscher B. W. (2005) Epigenetic silencing of DSC3 is a common event in human breast cancer. Breast Cancer Res. 7, R669–R680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Barhoover M. A., Hall J. M., Greenlee W. F., Thomas R. S. (2010) Aryl hydrocarbon receptor regulates cell cycle progression in human breast cancer cells via a functional interaction with cyclin-dependent kinase 4. Mol. Pharmacol. 77, 195–201 [DOI] [PubMed] [Google Scholar]