

Background: All known ligands of EGF receptor (EGFR) are characterized by the EGF motif and generated from transmembrane precursors.
Results: Prolidase, a cytosolic dipeptidase devoid of EGF motif, binds and activates EGFR independent of its dipeptidase activity when present outside of cell.
Conclusion: Prolidase is a novel EGFR ligand.
Significance: This shows a new function of prolidase and new mechanism of EGFR activation.
Keywords: Cell Surface Receptor, Epidermal Growth Factor Receptor (EGFR), Multifunctional Protein, Receptor Tyrosine Kinase, Signal Transduction, EGFR Ligand, Cyclooxygenase-2, Prolidase
Abstract
Prolidase, also known as Xaa-Pro dipeptidase or peptidase D (PEPD), is a ubiquitously expressed cytosolic enzyme that hydrolyzes dipeptides with proline or hydroxyproline at the carboxyl terminus. In this article, however, we demonstrate that PEPD directly binds to and activates epidermal growth factor receptor (EGFR), leading to stimulation of signaling proteins downstream of EGFR, and that such activity is neither cell-specific nor dependent on the enzymatic activity of PEPD. In line with the pro-survival and pro-proliferation activities of EGFR, PEPD stimulates DNA synthesis. We further show that PEPD activates EGFR only when it is present in the extracellular space, but that PEPD is released from injured cells and tissues and that such release appears to result in EGFR activation. PEPD differs from all known EGFR ligands in that it does not possess an epidermal growth factor (EGF) motif and is not synthesized as a transmembrane precursor, but PEPD binding to EGFR can be blocked by EGF. In conclusion, PEPD is a ligand of EGFR and presents a novel mechanism of EGFR activation.
Introduction
Epidermal growth factor receptor (EGFR)2 is a cell surface receptor that plays a major role in a variety of cellular responses (1, 2). It is a member of four closely related receptor tyrosine kinases: EGFR (ErbB-1), Her2/c-neu (ErbB-2), Her3 (ErbB-3), and Her4 (ErbB-4). Ligand binding to the extracellular domain of these receptors leads to homo- or heterodimerization, followed by receptor tyrosine phosphorylation and phosphorylation/activation of many signaling proteins involved in an array of cellular events (3). Key signaling pathways downstream of ErbB receptors include the PI3K/AKT/mTOR pathway, the Ras/Raf/ERK pathway, and the JAK/STAT pathway. ErbB activation results in an increase in DNA synthesis, cell growth, cell proliferation, cell differentiation, and cell migration, which perhaps are most clearly demonstrated in cancer cells (4, 5). Indeed, the ErbB receptors have become major drug targets in cancer therapy (6). Many peptide ligands of ErbB receptors have been identified, including epidermal growth factor (EGF), heparin-binding EGF-like growth factor (HB-EGF), TGF-α, amphiregulin, epiregulin, β-cellulin, and neuregulin 1–4. All of these ligands contain one or more of the conserved EGF motif (CX7CX4–5CX10–13CXCX8GXRC, where X represents any amino acid) (7, 8), but they differ in receptor specificity (9). Moreover, all ligands are synthesized as transmembrane precursors and are released by ectodomain shedding via proteolysis (10).
Prolidase, also known as peptidase D (PEPD) and Xaa-Pro dipeptidase, is ubiquitously expressed and hydrolyzes dipeptides with proline or hydroxyproline at the carboxyl terminus. PEPD is a homodimeric protein; each subunit of human PEPD is 54 kDa; the human PEPD gene is located on the short arm of chromosome 19 (11). PEPD is believed to play an important role in collagen turnover and matrix modeling, as proline and hydroxyproline are very abundant in collagen. In this article, however, we demonstrate that PEPD, which we initially identified as an inducer of cyclooxygenase-2 (Cox-2), is a ligand of EGFR and that EGFR activation by PEPD leads to stimulation of signaling proteins downstream of EGFR and induction of Cox-2. Data are also presented to show that its dipeptidase activity is not required for PEPD to activate EGFR and that PEPD activates EGFR only when it is present in the extracellular space. Although PEPD is known to be a cytosolic protein, our study indicates that PEPD is released from injured cells and tissues and such a release appears to activate EGFR. PEPD differs from all other EGFR ligands in that it does not possess an EGF motif, that it is not pre-synthesized as a transmembrane protein, and also that there is no evidence that PEPD undergoes proteolytic maturation. Thus, PEPD not only is a new EGFR ligand but also presents a novel mechanism by which EGFR is activated.
EXPERIMENTAL PROCEDURES
Reagents
CCl4, glycyl-proline, proline, and hydroxyproline were from Sigma. The EGFR inhibitor (324674) and cetuximab (a monoclonal antibody that binds to the ectodomain of EGFR) were from Calbiochem and ImClone, respectively. Recombinant human IgG1 Fc (110-HG-100) and recombinant EGFR-Fc (a chimera of the extracellular domain (Met1-Ser645) of human EGFR and the Fc fragment of human IgG1 (344-ER-050), recombinant human EGF (236-EG-200), and a biotinylated anti-human EGF antibody (BAF236) were from R&D Systems. Other antibodies used in the study include: anti-Cox-2 (Santa Cruz, sc-1745), anti-PEPD (Abcam, ab86507), anti-EGFR (Santa Cruz, sc-365829), anti-p-EGFR (Santa Cruz, sc-81488), anti-β-cellulin (Santa Cruz, sc-5802), anti-amphiregulin (Santa Cruz, sc25436), anti-epiregulin (Santa Cruz, sc-30215), anti-EGF (Santa Cruz, sc-1342), anti-HB-EGF (Santa Cruz, sc-28908), anti-TGF-α (Santa Cruz, sc-9043), anti-p-EGFR (Cell Signaling, 4407S), anti-STAT3 (Cell Signaling, 4904P), anti-p-STAT3 (Cell Signaling, 9145P), anti-AKT (Cell Signaling, 4691S), anti-p-AKT (Cell Signaling, 4060S), anti-ERK (Cell Signaling, 9102), anti-p-ERK (Cell Signaling, 9101S), goat anti-human IgG-Fcγ fragment specific (Jackson, 109-005-008), anti-GAPDH (Millipore, MAB374), anti-human IgG1 for detection of Fc (Santa Cruz, sc-2453), FITC-conjugated anti-polyhistidines (His, Abcam, ab1206), biotin-conjugated anti-His (Bethyl, A190–113B), and phycoerythrin-conjugated goat anti-mouse IgG1 (Santa Cruz, sc-3764). HRP-conjugated streptavidin was from Thermo Scientific (N100).
Cell Culture
RT-4 cells were from ATCC. Hepa1c1c7 cells were originally generated by Oliver Hankinson (12). 32D cells and 32D cells with stable expression of wild-type human EGFR and its kinase-inactive K721M mutant were kindly provided by Gibbes R. Johnson, US Food and Drug Administration (13). RT-4 cells were cultured in McCoy's 5A medium with 10% FBS. Hepa1c1c7 cells were cultured in minimal essential medium α supplemented with glutamine and 10% FBS. 32D cells and their derivatives were cultured in RPMI 1640 medium supplemented with 10% FBS, 5% WEHI-3B cell-conditioned medium, and 0.1% 2-mecaptoethanol. All cells were cultured in a humidified incubator at 37 °C with 5% CO2.
Western Blot Analysis
Cells were harvested from culture by trypsin treatment and centrifugation. After washing twice with PBS, cells were lysed in 1× cell lysis buffer (Cell Signaling) supplemented with 2 mm phenylmethanesulfonyl fluoride and a proteinase inhibitor mixture (Roche Applied Science). Cell lysis was enhanced by sonication. The samples were cleared by centrifugation at 13,000 × g for 10 min at 4 °C. Liver tissues were mixed with RIPA buffer (25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with the proteinase inhibitor mixture from Roche Applied Science and stroked in a Dounce homogenizer, and the homogenate was cleared by centrifugation at 13,000 × g for 15 min at 4 °C. Protein concentrations of all specimens were measured by the BCA assay kit (Pierce). The samples were mixed with 4× loading dye, heated for 5 min at 95 °C, and resolved by SDS-PAGE (8–10%). In each experiment, an equal amount of proteins was loaded across lanes. The proteins were transferred to polyvinylidene fluoride membrane and probed with specific antibodies, and protein bands were detected using the ECL Plus kit (Amersham Biosciences) or the SuperSignal West Pico kit (Thermo Scientific).
Preparation of Recombinant Human PEPD and Its Mutant
The bacterial pBAD/TOPO ThioFusion expression system (Invitrogen) was used to produce and purify PEPD and a PEPD mutant. In brief, pCMV6-XL5-PEPD (Origene) was used as a template to amplify the full-length human PEPD by PCR using primers, forward 5′-AATACGACTCACTATAGGGCG-3′ and reverse 5′-CTTGGGGCCAGAGAAGG-3′, which were designed to express PEPD as a native protein with His6 tagged to the carboxyl terminus (but without the N-terminal Thio). The resulting PCR fragments were subcloned into the pBAD/Thio-TOPO expression vector by TA cloning. The construct was sequenced to ensure the integrity of the entire coding sequence. Expression and purification were performed as indicated by the manufacturer. The PEPD G278D mutant was generated by site-directed mutagenesis using the QuikChange Lightning Multi Site-directed Mutagenesis kit (Agilent Technologies) and using the above described PEPD expression vector as the template. The construct was sequenced to ensure the correct mutation. Both the wild-type PEPD and mutant PEPD were purified via nickel-nitrilotriacetic acid-agarose chromatography (Qiagen). The proteins were further purified using Ultracel YM-30 Centricon (Millipore). SDS-PAGE was performed in 8–10% acrylamide gels under denaturing and reducing conditions, and the gels were stained with Coomassie Blue to examine protein purity. The preparations were also checked for potential contamination of lipopolysaccharides, using the E-TOXATE Kit (Sigma), following the manufacturer's instruction, but no lipopolysaccharides were detected (detection limit: 0.005 endotoxin unit per 0.1-ml sample).
Aspartate Aminotransferase (AST) Assay and Prolidase Activity Assay
AST activity was measured using the InfinityTM AST (GOT) Reagent kit (Thermo, TR70021). Prolidase activity was measured using glycyl-proline as a substrate as previously described (14).
Transient Gene Transfection and siRNA Transfection
Cells were grown in 6-well plates. Transfection of pCMV6-XL5 plasmid (empty vector) or pCMV6-XL5-PEPD (expressing human PEPD) was performed using FuGENE 6 (Roche Applied Science) at 1–2 μg of DNA per well for 24 h. Transfection of siRNA was performed using Lipofectamine RNAiMAX (Invitrogen) at 50 nm siRNA per well. A nonspecific scrambled siRNA (Hi-GC siRNA) and a siRNA targeting EGFR (primer 1: GGA GGA GAG GAG AAC UGC CAG AAA U; primer 2: AUU UCU GGC AGU UCU CCU CUC CUC C) were from Invitrogen.
Immunofluorescence Staining
Cells were grown on glass cover slides and incubated with recombinant human PEPD at 0.27 μm for 4 h at 37 °C. Following wash with ice-cold PBS, the cells were fixed in 4% paraformaldehyde. The cells were then treated with 1% BSA and then an FITC-conjugated His6 tag antibody. In the case of Hepa1c1c7 cells, DNA was stained with propidium iodide at 0.5 μg/ml for 15 min at room temperature. The cells were thoroughly washed with wash buffer and examined by fluorescence microscopy (Axiovert 40 CFL, Carl Zeiss) at room temperature. Images were taken with A-Plan 40 × 0.50 objective lenses (Carl Zeiss) and a Flex camera (Spot) using the Spot advanced acquisition software. ImageJ (National Institutes of Health) was used for image analysis. For confocal analysis, cells were grown in chamber slides and subjected to the same treatment and fixation as mentioned above. For co-treatment with PEPD and EGF, cells were treated simultaneously with both agents. After blocking with 1% BSA, the cells were incubated with anti-EGFR and then incubated with FITC-conjugated anti-His6 tag and a goat anti-mouse IgG1-phycoerythrin (for EGFR detection). The cells were then examined by confocal microscopy (LSM 510 META, Carl Zeiss) equipped with a microscope (Axiovert 200 M, Carl Zeiss) at room temperature. Images were taken with Plan Apochromat ×63 1.40 NA oil lens (Carl Zeiss). LSM 5 software (Carl Zeiss) was used for image acquisition and analysis.
Immunoprecipitation
Cells were lysed in M-PER Buffer (Thermo Scientific) supplemented with a proteinase inhibitor mixture (Roche Applied Science). After measurement of protein concentration, cell lysates (0.5 mg of protein) were incubated with recombinant human PEPD (20 μg) in a 0.5-ml volume for 1 h at 37 °C, followed by incubation with anti-EGFR or an isotype-matched IgG overnight at 4 °C. As a control, cells lysates were incubated with anti-EGFR without prior incubation with PEPD. The mixture was then incubated with protein G-Sepharose beads for 1 h at room temperature. The beads were washed with immunoprecipitation washing buffer and then subjected to Western blot analysis. For detection of direct and specific binding of PEPD to EGFR, recombinant human PEPD (0.4 μm) was incubated in M-PER buffer with recombinant human EGFR-Fc (0.04 μm), EGFR-Fc (0.04 μm) plus BSA (19 μm), or recombinant human Fc (0.04 μm) for 1 h at 37 °C and then overnight at 4 °C, followed by incubation with protein A-Sepharose beads for 1 h at room temperature. The beads were washed with immunoprecipitation washing buffer and then subjected to Western blot analysis. In another experiment, recombinant human PEPD (1 μm) was incubated with EGFR-Fc (0.04 μm) in the presence or absence of recombinant human EGF (5 or 50 nm) overnight at 4 °C, followed by incubation with protein A-Sepharose beads for 1 h at room temperature. The beads were washed with immunoprecipitation washing buffer and then subjected to Western blot analysis.
ELISA for Detection of Serum PEPD Concentration and Detection of PEPD/EGF Binding to EGFR
For detection of serum levels of PEPD, 96-well ELISA plates were coated with 100 μl/well of diluted anti-PEPD mouse monoclonal antibody (2.5 μg/ml) overnight at 4 °C. The plates were then washed three times with phosphate-buffered saline with Tween 20 (PBST) and blocked with 200 μl/well of blocking buffer (incubation for at least 2 h at room temperature). The plates were washed with PBST and then incubated with 100 μl/well of PEPD standard or samples, which were appropriately diluted, for 2 h at room temperature. The plates were washed three times with PBST, and each well was then incubated with 100 μl of a detection antibody (an anti-PEPD rabbit polyclonal antibody) for 2 h at room temperature. After washing the plates three times with PBST, 100 μl of secondary reagent (goat anti-rabbit IgG-HRP, 1:2500 dilution) was added to each well, followed by a 1-h incubation at room temperature. The plates were washed again with PBST three times, and each well was then incubated with 100 μl of a HRP substrate solution (3,3′,5,5″-tetramethylbenzidine from Cell Signaling, number 7004). After adequate color development, 100 μl of stop solution (Cell Signaling, number 7002) was added to each well, and absorbance at 450 nm was recorded by a microtiter plate reader. Pure PEPD was used as a standard.
To measure PEPD or EGF binding to EGFR, 96-well ELISA plates were coated overnight at 4 °C with 100 μl/well of goat anti-human IgG Fc (10 μg/ml). After washing three times with PBST, residual protein binding sites in the wells were saturated by incubating for 1 h at room temperature with 300 μl/well of assay buffer (1% BSA/PBS). After removing the assay buffer, 60 μl of diluted human recombinant EGF or human recombinant PEPD were added to each well, followed by addition of 60 μl of EGFR-Fc (1 μg/ml) to each EGF- or PEPD-containing well and incubation at 4 °C overnight. The plates were then washed three times with PBST, and 100 μl of biotin-conjugated goat anti-human EGF (1 μg/ml) or biotin-conjugated goat anti-His (1:10000 dilution) were added to each well and incubated for 2 h at room temperature. After another round of washing with PBST, 100 μl of streptavidin-conjugated HRP (1:10,000 dilution) was added to each well and incubated for 45 min at room temperature. The wells were washed again with PBST, and the bound HRP was detected by addition of 100 μl/well of 3,3′,5,5″-tetramethylbenzidine as a peroxidase substrate. The reaction was terminated by addition of 100 μl/well of stop solution, and absorbance reading at 450 nm was recorded by a microtiter plate reader. To measure the effect of EGF on PEPD binding to EGFR, the concentration of PEPD was kept constant at 500 μg/ml (4.6 μm) in the binding assay mentioned above, and PEPD binding to EGFR was measured in the presence of increasing concentrations of EGF (0, 10, 100, and 500 ng/ml).
BrdU Assay
Cells were grown in 48-well plates and treated with recombinant human PEPD in serum-free medium for 24 h. BrdU incorporation into DNA was measured using the BrdU In-Situ Detection kit (BD Pharmingen) according to the manufacturer's instruction. Briefly, cells were incubated with BrdU at the final concentration of 10 μm in cell culture medium for 2 h. Following wash with ice-cold PBS, the cells were fixed in fixation buffer. The cells were then treated with diluent buffer and then 0.3% H2O2. After washing, cells were incubated with a biotin-conjugated anti-BrdU antibody and streptavidin-HRP for 1 h and 30 min at room temperature, respectively, followed by incubation with diaminobenzidine for staining. Cells were examined with an Axiovert 40 CFL microscope (Carl Zeiss). Images were taken with A-Plan 10 × 0.25 objective lenses (Carl Zeiss) and a Flex camera (Spot) using Spot advanced acquisition software. For each well, 2000 cells over 5 randomly chosen microscopic fields were counted using ImageJ. Staining intensity was divided into none (density score at <1), weak (density score at 1–1.5), moderate (density score at 1.6–2.5), and strong (density score >2.5).
Mice
C57BL/6N male mice (6 weeks of age) were purchased from Taconic and were randomly assigned to receive a single dose of CCl4 (0.5 mg/g body weight) or vehicle (corn oil, ∼25 μl/mouse) by intraperitoneal injection, 4 mice per group; 24 h after dosing, the animals were sacrificed; blood, kidney, and liver were collected. The blood specimens were processed to prepare serum samples, which were stored at −80 °C before use. Half of each liver was snap frozen and stored at −80 °C before use, and the other half was fixed in 10% buffered formalin. All kidney specimens were snap frozen and stored at −80 °C. The animal protocol was approved by the Institutional Animal Care and Use Committee at Roswell Park Cancer Institute.
RESULTS
PEPD Induces Cox-2, and It Does So without Requiring Its Dipeptidase Activity but Only When Present in the Extracellular Space
Cox-2 plays a key role in the biosynthesis of prostaglandins, which act as autocrine and paracrine signaling molecules that are important for a variety of biological activities (15). Overexpression of Cox-2 and the ensuring oncogenesis have been well documented (16–18). During our recent study of COX-2 gene regulation, we noticed that treatment of human bladder cancer RT-4 cells with the rat liver S9 fraction resulted in significant induction of Cox-2 (supplemental “Materials and Methods” and supplemental Fig. S1A). Further studies indicated that PEPD was responsible at least in part for Cox-2 induction by S9 (supplemental “Materials and Methods” and supplemental Figs. S1, B-I, and S2). Subsequently, full-length human PEPD was cloned to an expression vector (pBAD/TOPO), and the recombinant PEPD with His6 tagged to its carboxyl terminus was generated in Escherichia coli, which was purified by nickel-nitrilotriacetic acid-agarose chromatography and further purified by Ultracel YM-30 Centricon. Its purity was confirmed by SDS-PAGE and Coomassie Blue staining (Fig. 1A). The preparation was free of lipopolysaccharide contamination. Treatment of RT-4 cells or murine hepatoma Hepa1c1c7 cells with 2.7, 27, and 270 nm recombinant human PEPD (based on its dimeric molecular weight) for 24 h resulted in dose-dependent induction of Cox-2 (Fig. 1C). PEPD induced Cox-2 in both cell lines but appeared to be more active in Hepa1c1c7 cells. Although other inducers of Cox-2 might be present in liver S9, e.g. hepatocyte growth factor (19), our results of the recombinant PEPD show unambiguously that PEPD is an inducer of Cox-2.
FIGURE 1.

PEPD induces Cox-2 and does so without requiring its enzymatic activity, but occurs only when it is present in the extracellular space. A, purity of recombinant human PEPD and its mutant, measured by SDS-PAGE and Coomassie Blue staining. B, the enzymatic activities of recombinant wild-type human PEPD and its G278D mutant (mean ± S.D.). One unit of PEPD activity is the amount of enzyme that releases 1 μmol of proline/h from glycyl-proline. C, cells were treated with PEPD, its G278D mutant or vehicle (same as in the other groups) for 24 h. Cell lysates were measured for Cox-2 by Western blotting. D, cells were treated with proline, hydroxyproline, or vehicle (same as in the other groups) for 24 h. Cell lysates were measured for Cox-2 by Western blotting. E, cells were transfected with a plasmid expressing human PEPD or the empty vector for 24 h. Cell lysates were measured for PEPD and Cox-2 by Western blotting. F, cells were incubated with PEPD at 0.27 μm or vehicle (same as in the PEPD group) for 4 h, followed by immunostaining with FITC-conjugated anti-His and fluorescence microscopy. DNA in Hepa1c1c7 cells, but not RT-4 cells, was stained with propidium iodide. Bar, 5 μm.
Next, we asked if the enzymatic activity of PEPD was needed for Cox-2 induction. We introduced a point mutation (833G → A) to the human PEPD sequence, generating the mutant with G278D change. The recombinant PEPD mutant protein was generated and purified by the same procedure as described above for the wild-type protein (Fig. 1A). The mutant PEPD showed only 0.6% of the enzymatic activity of the wild-type PEPD (Fig. 1B), as measured using glycyl-proline as a substrate, which is similar to a previously published result (20). Incubation of both Hepa1c1c7 and RT-4 cells with the mutant PEPD resulted in Cox-2 induction, which was comparable with that caused by the wild-type protein (Fig. 1C). Furthermore, because the dipeptidase activity of PEPD generates proline and hydroxyproline, we also examined the effects of these compounds on Cox-2 expression. Thus, Hepa1c1c7 and RT-4 cells were treated with proline or hydroxyproline up to 2.5 mm for 24 h; but no significant induction of Cox-2 was detected (Fig. 1D). High concentrations of these compounds were used, in view of their abundance in collagen (21). Based on these results, we concluded that the enzymatic activity of PEPD was not involved in its induction of Cox-2.
However, forced expression of human PEPD in both RT-4 and Hepa1c1c7 cells, via transient gene transfection, failed to induce Cox-2 (24 h after transfection) (Fig. 1E). Thus, PEPD was able to induce Cox-2 only when it is present outside of the cells. Consistent with the above observation, incubation of cells with PEPD (270 nm, 4 h) followed by incubation with FITC-conjugated anti-His showed that the exogenous PEPD bound to the cell surface membrane (Fig. 1F).
PEPD Directly Binds to EGFR
Because PEPD could induce Cox-2 only when it was present in the extracellular space and PEPD could bind to the cell surface membrane, as described above, we hypothesized that PEPD induced Cox-2 by interacting with a membrane protein. Interestingly, several previous studies have indicated that EGFR activation leads to transcriptional activation of Cox-2 (22–24). To find out if PEPD could bind to EGFR, cell lysates were prepared from Hepa1c1c7 and RT-4 cells, and each lysate (0.5 mg of protein) was incubated with the recombinant PEPD (20 μg of protein) in a 0.5-ml volume and the mixture was then subjected to immunoprecipitation with anti-EGFR or an isotype-matched IgG, followed by Western blotting with anti-His (for detection of the recombinant PEPD). Cell lysates were also incubated with anti-EGFR without prior incubation with PEPD, to serve as a control. Our result showed that PEPD bound to EGFR (Fig. 2A).
FIGURE 2.

PEPD binds to EGFR. A, cell lysates (1 mg of protein/ml) were incubated with PEPD (40 μg/ml) for 1 h at 37 °C and then incubated with anti-EGFR or an isotype-matched IgG overnight at 4 °C. As a control, an equal amount of cell lysates were incubated with anti-EGFR without prior incubation with PEPD. All samples were then subjected to pull-down by protein G-Sepharose beads and Western blotting for PEPD. B, PEPD (0.4 nmol/ml) was incubated with EGFR-Fc (0.04 nmol/ml), EGFR-Fc (0.04 nmol/ml) plus BSA (19 nmol/ml), or Fc (0.04 nmol/ml) for 1 h at 37 °C and then overnight at 4 °C. In a separate experiment, PEPD (1 nmol/ml) was incubated with EGFR-Fc (0.04 nmol/ml) in the absence or presence of EGF (+, 5 pmol/ml; ++, 50 pmol/ml) for 1 h at 37 °C and then overnight at 4 °C. All samples were then subjected to pull-down by protein A-Sepharose beads and Western blotting for PEPD, Fc, and/or EGFR-Fc. C–E, binding of PEPD or EGF to EGFR or inhibition of PEPD-EGFR binding by EGF, measured by an ELISA as described under “Experimental Procedures.” Each value is mean ± S.D. The Kd values were estimated by nonlinear regression (GraphPad Prim 4 Software, r2 > 0.99). F, cells were incubated with 0.27 μm PEPD (with or without 0.027 μm EGF) or vehicle (same as in the PEPD/EGF groups) for 4 h, and after paraformaldehyde fixing and BSA blocking, were incubated with anti-EGFR and then a phycoerythrin-conjugated secondary antibody and FITC-conjugated anti-His, followed by confocal microscopy. Bar, 10 μm.
To confirm direct and specific binding of PEPD to EGFR, recombinant human PEPD (0.4 μm) was incubated with the ectodomain of human EGFR (0.04 μm EGFR-Fc, a recombinant chimera of the extracellular domain of EGFR and the Fc fragment of human IgG1) with or without the presence of 47.5-fold excess of BSA (19 μm)), or incubated with the Fc (0.04 μm) of human IgG1 as a control, followed by pull-down with protein A-Sepharose beads and Western blotting. As shown in Fig. 2B, PEPD bound to EGFR but not Fc, and the binding was not affected by BSA. In another experiment, the recombinant PEPD (1 μm) was incubated with EGFR-Fc (0.04 μm) with or without recombinant human EGF (5 or 50 nm). Whereas EGF at the low concentration did not appear to interfere with PEPD binding to EGFR, at the high concentration it significantly blocked PEPD binding to EGFR (Fig. 2B). In an ELISA using EGFR-Fc, we showed that PEPD bound to EGFR in a dose-dependent manner and showed a typical receptor-ligand binding mode, with an estimated Kd value of 5.3 μm (Fig. 2C). Under the same assay condition, the estimated Kd value of EGF binding to EGFR was 14.6 nm (Fig. 2D). Thus, the binding affinity of PEPD toward EGFR is ∼360-fold lower than that of EGF. Using the same ELISA and keeping PEPD constant at 4.5 μm, we also showed that EGF blocked PEPD binding to EGFR in a dose-dependent manner; EGF at 1.7 nm was virtually ineffective, but EGF at 17 and 83 nm blocked PEPD binding to EGFR by 87 and 96%, respectively (Fig. 2E).
To further document binding of PEPD to EGFR on the cell surface membrane, Hepa1c1c7 cells were incubated with vehicle or PEPD at 0.27 μm for 4 h and then subjected to immunofluorescence staining of EGFR and PEPD, followed by confocal microscopy. Our result showed that PEPD and EGFR co-localized to the cell surface membrane (Fig. 2F). The spotty distribution of the proteins is consistent with known distribution of EGFR in lipid rafts. Moreover, as expected, in the presence of 27 nm EGF, PEPD binding to EGFR was blocked (Fig. 2F).
PEPD Activates EGFR, Which Does Not Require the Dipeptidase Activity of PEPD but Requires Its Presence in the Extracellular Space
PEPD, when added to cell culture medium, caused EGFR phosphorylation in both Hepa1c1c7 cells and RT-4 cells; two key phosphorylation sites (Tyr1068 and Tyr1173) were examined, and both were stimulated by PEPD (Fig. 3, A and B). Although increased phosphorylation of EGFR could be detected after 30 min of PEPD treatment, the peak level of p-EGFR was not reached until ∼4 h after PEPD treatment was initiated. PEPD is a potent activator of EGFR, as treatment of Hepa1c1c7 cells with 2.7 nm PEPD resulted in significant phosphorylation of EGFR and phosphorylation of key signaling molecules downstream of EGFR, including AKT, STAT3 (both STAT3α/p86 and STAT3β/p79), and ERK (both ERK1/p44 and ERK2/p42) (Fig. 3, C and D). In the experiment shown in Fig. 3C, vehicle control was included for each time point, but neither EGFR nor phosphorylated EGFR showed time-related changes in the control samples. The extent to which PEPD caused phosphorylation of EGFR, AKT, STAT3, and ERK was similar to that caused by EGF at the same concentration (Fig. 3, C and E), although the time course of such changes caused by the two ligands differed significantly as discussed below. Moreover, the enzymatically inactive PEPD mutant (G278D mutant) was also similarly effective in stimulating EGFR phosphorylation and the phosphorylation of AKT, STAT3, and ERK (Fig. 3F), indicating that the enzymatic activity of PEPD is not required for EGFR activation, which is similar to its effect on Cox-2 induction as described above. However, several differences between EGF and PEPD in our results are worth noting. First, EGF activated EGFR much more rapidly than did PEPD, as the peak level of p-EGFR was detected within 0.5 h of EGF treatment but at 4 h in PEPD-treated cells. Second, whereas EGFR phosphorylation appeared to be closely coupled temporally to the phosphorylation of AKT, STAT3, and ERK in response to PEPD, signal transmission from EGFR to these downstream proteins in response to EGF was much slower, as maximal levels of phosphorylation of these downstream proteins were not detected until 5 h after EGF treatment (Fig. 3, C–F). Finally, the EGFR level began to decrease after 2 h of EGF treatment and was virtually undetectable at 5 h (Fig. 3E), which likely resulted from endocytosis and degradation in the lysosome of activated EGFR (25). In contrast, the EGFR level remained largely unchanged during a 5-h PEPD treatment (Fig. 3, C, D, and F), although the EGFR level was clearly lower in cells treated with PEPD for 24 h (Fig. 3, A and B). Thus, PEPD-induced EGFR endocytosis and degradation may be much slower than that induced by EGF.
FIGURE 3.

PEPD activates EGFR. A–C, cells were treated with recombinant human PEPD, recombinant human EGF, or vehicle (same as in the other groups). Cell lysates were measured for EGFR and p-EGFR by Western blotting. D–F, cells were treated with recombinant human PEPD, recombinant human EGF, the G278D mutant of human PEPD, or vehicle (same as in the other groups). Cell lysates were measured for changes of various proteins by Western blotting. G, cells were transfected with a plasmid expressing human PEPD or the empty plasmid for 24 h. Cell lysates were measured for changes of various proteins by Western blotting.
As in the case of Cox-2 induction, forced expression of PEPD in Hepa1c1c7 cells by transient gene transfection had no effect on EGFR phosphorylation and the phosphorylation of AKT, STAT3, and ERK (Fig. 3G). These results provide further evidence that PEPD binds to and activates EGFR when it is present in the extracellular space. Moreover, in line with the growth-promoting activity of EGFR, treatment of Hepa1c1c7 and RT-4 cells with PEPD in the medium strongly and dose-dependently stimulated DNA synthesis in both cell lines, as measured by BrdU labeling (Fig. 4). For example, in cells treated with 27 nm PEPD for 24 h, the overall number of BrdU-labeled nuclei increased 3.9-fold (Hepa1c1c7) and 7.5-fold (RT-4), respectively, and the combined number of nuclei with moderate and strong BrdU labeling increased 11.1-fold (Hepa1c1c7) and 23.9-fold (RT-4), respectively (Fig. 4).
FIGURE 4.

PEPD stimulates DNA synthesis. Cells were treated with recombinant human PEPD or vehicle (same as in the PEPD group) in serum-free medium for 24 h before measurement of BrdU incorporation. A total of 2000 cells were counted per sample using ImageJ. Each value is a mean ± S.D. (n = 3). Staining intensity was divided into none (density score at <1), weak (density score at 1–1.5), moderate (density score at 1.6–2.5), and strong (density score >2.5). Bar, 5 μm.
Cox-2 Induction and ERK Activation by PEPD Reply on EGFR Activation
Having shown that PEPD binds and activates EGFR, it was important to find out whether Cox-2 induction by PEPD required EGFR activation. We also measured ERK as a representative of PEPD-induced phosphorylation of signaling proteins, to further document the relationship between PEPD and EGFR. First, EGFR in Hepa1c1c7 cells was silenced by an EGFR siRNA or treated by a control siRNA (48 h treatment), followed by treatment with PEPD (2.7 nm) for 5 h for measurement of ERK phosphorylation or for 24 h for measurement of Cox-2 induction. For comparison purpose, cells were also treated with EGF (2.7 nm) for 5 and 24 h, respectively. The EGFR siRNA was highly effective, as the EGFR level in the treated cells was undetectable by Western blotting (Fig. 5A). PEPD and EGF showed similar inductive effects on ERK phosphorylation (both ERK1/p44 and ERK2/p42) and Cox-2 expression in cells treated by the control siRNA. The inductive effects of both PEPD and EGF on ERK and Cox-2 were significantly attenuated or almost totally abolished in EGFR-depleted cells (Fig. 5A). It is unlikely that such a blockage as described above was an off-target effect, because in cells depleted of the EGFR by the EGFR siRNA Cox-2 could still be significantly induced by dihydrotestosterone (supplemental “Materials and Methods” and supplemental Fig. S3), a known Cox-2 inducer via androgen receptor (26).
FIGURE 5.
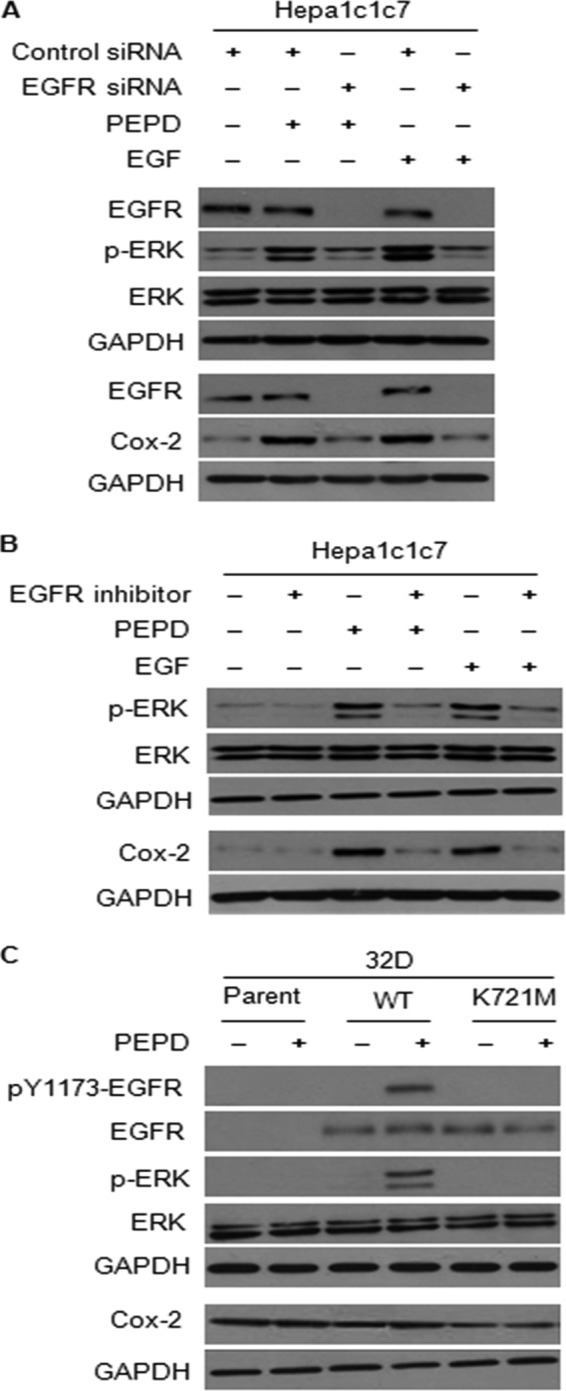
EGFR is required for ERK phosphorylation and Cox-2 induction by PEPD. A, cells were treated with a control siRNA or an EGFR siRNA for 48 h, followed by treatment with recombinant human PEPD (2.7 nm), recombinant human EGF (2.7 nm), or vehicle (same as in the other groups) for 5 h for measurement of ERK phosphorylation or 24 h for measurement of Cox-2 induction. B, cells were treated with or without recombinant human PEPD (2.7 nm) or recombinant human EGF (2.7 nm) for 5 h for measurement of ERK phosphorylation or 24 h for measurement of Cox-2 induction, whereas another group of cells were pretreated with the EGFR tyrosine kinase inhibitor 324674 (5 μm) for 1 h and then co-treated with both the inhibitor (5 μm) and PEPD (2.7 nm) or EGF (2.7 nm) for 5 or 24 h; the same amount of vehicle was used in the control groups. C, parent 32D cells and their derivatives expressing wild-type human EGFR or its K721M mutant were treated with or without recombinant human PEPD (2.7 nm) for 6 h for measurement of phosphorylation of EGFR and ERK, and for 24 h for measurement of Cox-2 levels; the same amount of vehicle was used in the control groups. All cell lysates were analyzed by Western blotting.
Next, cells were treated with solvent, PEPD (2.7 nm) or EGF (2.7 nm) in the absence or presence of a specific EGFR tyrosine kinase inhibitor (cyclopropanecarboxylic acid-(3-(6-(3-trifluoromethyl-phenylamino)-pyrimidin-4-ylamino)-amide, also known as 324674) at 5 μm for 5 h, followed by measurement of ERK phosphorylation, or for 24 h, followed by measurement of Cox-2 induction. The activity of this inhibitor against EGFR has been previously shown (27). The inhibitor appears to almost completely prevent PEPD and EGF from causing ERK phosphorylation and Cox-2 induction (Fig. 5B).
Murine myeloid 32D cells are known to express no EGFR or its family members. Johnson and co-workers (13) previously generated 32D cells with stable expression of human EGFR or its kinase-inactive K721M mutant and showed that EGF could only stimulate ERK phosphorylation in cells expressing the wild-type EGFR. We obtained these cells from Dr. Johnson. After treatment with 2.7 nm PEPD for 6 h, significant phosphorylation of EGFR at tyrosine 1173 was detected in cells expressing wild-type EGFR, but not in cells expressing the K721M mutant (Fig. 5C). This is consistent with a previous finding that the K721M mutation abolished EGF-induced EGFR autophosphorylation at tyrosine 1173 (13). PEPD (2.7 nm, 6 h) was also able to stimulate ERK phosphorylation only in cells expressing wild-type EGFR (Fig. 5C). Interestingly, Cox-2 was readily detected in parent 32D cells, but EGFR does not appear to up-regulate Cox-2 in these cells, and the EGFR/K721M mutant seems to inhibit Cox-2 expression somewhat (Fig. 5C). Also, PEPD (2.7 nm, 24 h) showed little effect on Cox-2 expression in all three cell lines (Fig. 5C). Likewise, EGF could not induce Cox-2 in any of the three cell lines (result not shown).
PEPD Directly Activates EGFR
Besides showing that the K721M mutation abolishes PEPD-induced EGFR phosphorylation at tyrosine 1173, as described above, we also asked if blocking PEPD binding to EGFR would lead to inhibition of EGFR phosphorylation. Thus, Hepa1c1c7 cells were pretreated for 1 h with cetuximab (20 μg/ml) (28), a monoclonal antibody that binds to the ectodomain of EGFR and blocks ligand binding to this receptor, followed by co-treatment with cetuximab (20 μg/ml) and PEPD (2.7 nm) for 5 h. Phosphorylation at both tyrosine 1068 and tyrosine 1173 of EGFR were measured, and cetuximab completely blocked PEPD-induced phosphorylation at both sites (Fig. 6A). Previous studies have shown that EGFR tyrosine phosphorylation may be modulated by protein-tyrosine phosphatases (29). However, the result of the cetuximab experiment described above makes it highly unlikely that PEPD-induced EGFR phosphorylation may be mediated by down-regulation or inhibition of a tyrosine phosphatase. The EGF family ligands are synthesized as transmembrane precursors, and ectodomain shedding via proteolysis is a recognized process to yield soluble and active ligands from cells as mentioned before. Thus, it was of interest to determine whether shedding of any known EGFR ligands plays a role in EGFR activation by PEPD. Hepa1c1c7 cells were treated with 2.7 nm PEPD for 5 h, a condition that was shown to activate EGFR (Fig. 3, C and D). Cells were then harvested for measurement of the precursor levels of all 6 known EGFR ligands, including EGF, TGF-α, HB-EGF, amphiregulin, epiregulin, and β-cellulin (3). All precursors could be detected in these cells, although the expression levels of pro-TGF-α, pro-epiregulin, and pro-β-cellulin may be relatively higher than that of the other precursors. However, PEPD showed no effect on the expression level of any of these proteins (Fig. 6B), indicating that PEPD does not cause release of the EGFR ligands from the cells. It is of note that multiple bands were detected for some of the proteins, pro-epiregulin in particular, but only a single band for other proteins, which could be due to a combination of protein glycosylation, which is known to occur in these proteins, and the specificity/sensitivity of the particular antibody used.
FIGURE 6.
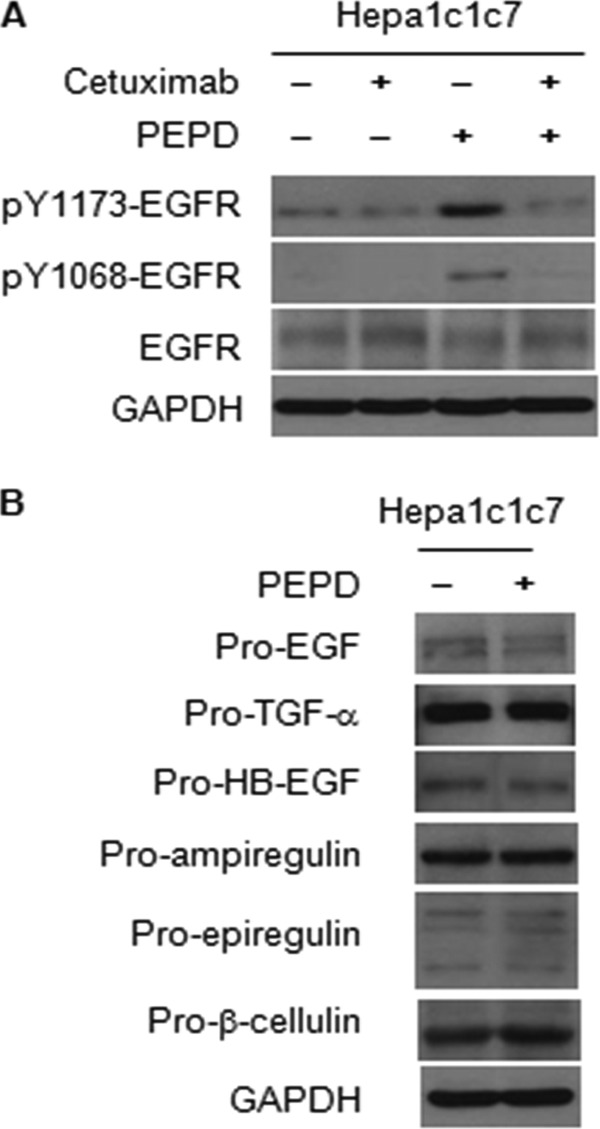
Inhibition of PEPD-induced EGFR phosphorylation by cetuximab, and the effect of PEPD on the levels of EGFR ligand precursors. A, cells were pretreated with or without cetuximab (20 μg/ml) for 1 h and then co-treated with cetuximab (20 μg/ml) and recombinant human PEPD (2.7 nm) for 5 h. B, cells were treated with or without recombinant human PEPD (2.7 nm) for 5 h. The same amount of vehicle was used in the control groups. Cells lysates were measured by Western blotting for various proteins.
PEPD Is Released from Damaged Cells and Tissues, and Such Release Appears to Cause EGFR Activation
Given that PEPD activated EGFR only when it was present outside of cells, but PEPD was known to be a cytosolic protein, we sought to determine whether PEPD was released from cells and tissues. PEPD could be detected in the blood of healthy mice (Fig. 7A). Carbon tetrachloride (CCl4) is known to cause liver damage (30). C57BL/6 mice were given a single intraperitoneal dose of vehicle or CCl4 (0.5 mg/g body weight); 24 h later, blood, kidney, and liver were removed from these animals for analysis. CCl4 caused significant liver damage, as the serum level of AST increased 44.7-fold after CCl4 treatment (Fig. 7B). Histological analysis also showed that CCl4 caused significant tissue necrosis (supplemental “Materials and Methods” and supplemental Fig. S4). The serum level of PEPD increased 3.8-fold to 3.3 nm after CCl4 treatment (Fig. 7A), which was measured by ELISA, suggesting that PEPD was released to blood from damaged liver cells and tissues. Indeed, increase in the blood PEPD level was associated with a decrease in liver tissue PEPD levels (Fig. 7C). The livers of CCl4-treated mice also showed a decrease in the EGFR level but increased EGFR phosphorylation and induction of Cox-2 (Fig. 7C), indicating activation of EGFR signaling. As mentioned before, studies have shown that activated EGFR may undergo endocytosis and degradation in the lysosome, and the EGFR level in cultured cells decreased after PEPD treatment for 24 h (Fig. 3, A and B). Given that these changes were associated with PEPD release from liver tissues, it seems likely that PEPD may cause EGFR activation and Cox-2 induction by acting as an autocrine or paracrine ligand to EGFR. Indeed, we showed that in cultured cells, PEPD at 2.7 nm, which is lower than the serum concentration of PEPD detected in CCl4-treated mice, activated EGFR and EGFR signaling (Fig. 3, C and D) and induced Cox-2 (Fig. 1C). Also, in the kidneys of CCl4-treated mice, tissue levels of PEPD did not decrease, yet there was increased phosphorylation of EGFR and induction of Cox-2, but a decrease in total EGFR level (Fig. 7D).
FIGURE 7.
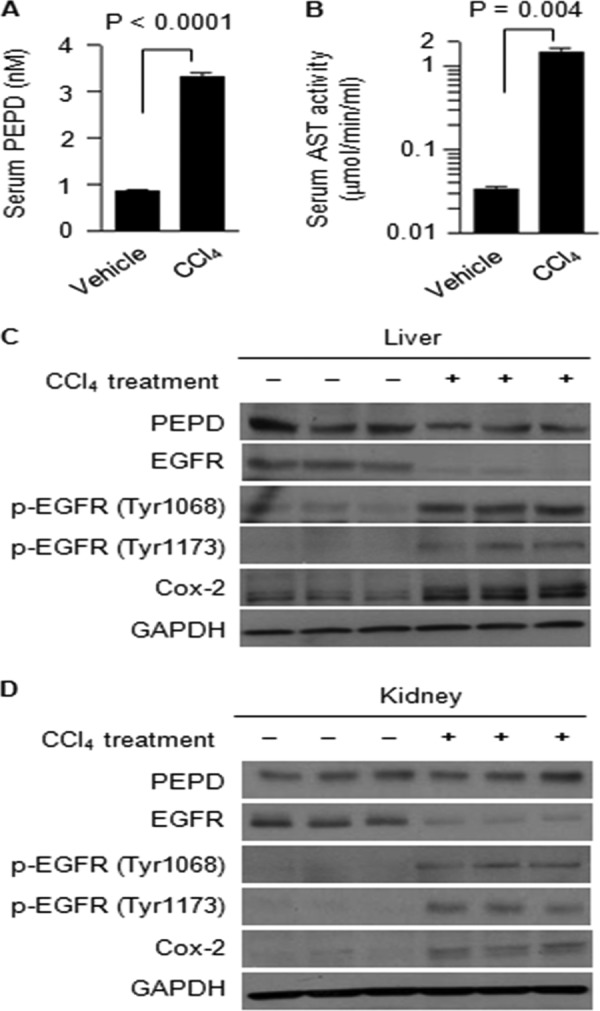
Release of PEPD from damaged cells and tissues is associated with EGFR activation. Groups of four C57BL/6N male mice were given a single intraperitoneal dose of CCl4 or vehicle; 24 h later, the mice were sacrificed, from which blood, kidney, and liver were collected. Serum levels of PEPD (A) and AST (B) were measured by an ELISA and enzymatic activity, respectively (mean ± S.D.). Two-tail Student's t test was used for statistical analysis. C and D, liver and kidney homogenates from 3 control mice and 3 CCl4-treated mice were used for Western blotting.
DISCUSSION
Our recent incidental observation that the liver S9 fraction strongly induced Cox-2 in cultured cells and our subsequent attempt to identify the inducer led us to discover that PEPD not only mediates Cox-2 induction by S9 but also is a ligand of EGFR. Many lines of evidence are presented in this article to show that PEPD directly binds and activates EGFR, e.g. specific binding of PEPD to the EGFR ectodomain and blockage of such binding by EGF in the immunoprecipitation/immunoblot assay, ELISA, and immunofluorescence analysis (Fig. 2, B–F), inhibition of PEPD-induced EGFR activation by cetuximab (Fig. 6A), the inability of PEPD to induce phosphorylation of kinase-dead EGFR (Fig. 5C), and the finding that PEPD does not modulate ectodomain shedding of other EGFR ligands (Fig. 6B). We also showed that the dipeptidase activity of PEPD is not required for this protein to bind and activate EGFR but that PEPD can activate EGFR only when present in the extracellular space. Thus, EGFR activation by PEPD contrasts with other activities of PEPD that have been previously reported, including stimulation of hypoxia inducible factor 1α and its target gene products (vascular endothelial growth factor and glucose transporter-1) (31) as well as stimulation of transforming growth factor β1 (21), as these activities of PEPD were shown to be related to the metabolism of imidodipeptides by PEPD and to require the intracellular presence of PEPD. EGFR activation by PEPD is further documented by the ensuing phosphorylation of all tested downstream signaling proteins, including AKT, ERK, and STAT3, in PEPD-treated cells and the dependence of such activation on EGFR activation, which was demonstrated using siRNA-mediated EGFR depletion (Fig. 5A), pharmacologic inhibition of EGFR kinase activity (Fig. 5B), and cells that express kinase-inactive EGFR (Fig. 5C). It is also gratifying that we were able to show that not only could PEPD induce Cox-2 but this induction also depended on EGFR activation. Although the detailed mechanism by which PEPD induces Cox-2 remains to be elucidated, Xu and Shu (22) reported that EGFR activation in human glioma cells transcriptionally activated Cox-2 via p38 MAPK and Sp1/Sp3, and Lo et al. (32) reported that nuclear EGFR bound together with STAT3 to the Cox-2 gene promoter and stimulated Cox-2 gene transcription in human glioma cells.
PEPD differs in many ways from other EGFR ligands. As mentioned before, all known peptide ligands of EGFR family receptors possess one or more EGF domains, which are thought to be critical for ligand-receptor binding. Yet, examination of the amino acid sequence of human PEPD (NCBI Reference Sequence NM_000285.3) revealed no EGF domain. The action of PEPD, therefore, shows that an EGF motif is not essential for a ligand to bind and activate EGFR. Given that PEPD does not carry an EGF motif, the PEPD binding site on EGFR may differ from that of other ligands. Interestingly, EGF was able to block PEPD binding to EGFR (Fig. 2, B–F). Does this suggest that the conformation change of EGFR induced by EGF binding (33) may prevent PEPD binding or in fact PEPD and EGF may share the same binding site? Further study is needed to address this intriguing question. All other peptide ligands of EGFR family receptors are synthesized as transmembrane precursors and are released by ectodomain shedding via proteolysis (10). In contrast, PEPD is a cytosolic protein (34, 35), and there is no evidence that PEPD undergoes proteolytic maturation. The exact PEPD sequence to bind and activate EGFR remains to be elucidated. Although PEPD exists as a homodimer (36, 37), thus raising the question of whether PEPD binds to EGFR as a dimer, all other ligands are known to exist and to bind to EGFR as a monomer. In our ELISA experiments, the binding affinity of human PEPD to the ectododomain of human EGFR (estimated Kd of 5.3 μm) was ∼360-fold lower than that of EGF (estimated Kd of 14.6 nm) (Fig. 2, C and D). Yet, PEPD and EGF, when each tested at 2.7 nm, showed similar efficacy in activating EGFR and its downstream signaling molecules (AKT, ERK and STAT3) and in inducing Cox-2 in cultured cells (Fig. 3, C–F, and 5, A and B). This may suggest that PEPD is much more efficient than EGF in inducing autophosphorylation of EGFR or that the interaction between EGFR and PEPD in an ELISA setting does not fully reflect their interaction on the cell surface. Further study is needed to better understand the interaction between them. Interestingly, our results suggest that PEPD-induced EGFR endocytosis and degradation may be much slower than that induced by EGF. It is therefore conceivable that the slower ligand-induced internalization rate of EGFR may translate into higher and more sustained EGFR activation. There is also a difference between EGF and PEPD in the time course of EGFR activation and the activation of its downstream signals. EGF caused rapid EGFR phosphorylation (reaching peak level within 30 min), whereas PEPD was a slower EGFR activator (reaching peak level at ∼4 h) (Fig. 3, C–F). Yet, signal transmission from EGFR to the downstream proteins (AKT, ERK, and STAT3) in response to EGF appears to be much slower than that in response to PEPD, as little phosphorylation of AKT, ERK, and STAT3 was detected within 2 h of EGF treatment, but phosphorylation of these proteins was closely linked to the phosphorylation of EGFR during PEPD treatment (Fig. 3, D–F). Taken together, although the exact mechanism by which PEPD binds and activates EGFR remains to be worked out, it is clear that PEPD represents a novel mechanism of EGFR activation. Our findings also raise the question as to whether PEPD may bind and activate other ErbB family receptors.
In view of the critical importance of EGFR in a variety of cellular processes, the implication of our discovery that PEPD binds and activates EGFR may be far-reaching. Our results suggest that PEPD may mediate tissue injury, inflammation, and repair by stimulating autocrine or paracrine EGFR signaling. The serum PEPD level increased 3.8-fold in mice 24 h after a single treatment of CCl4, which damaged the liver (Fig. 7A). The increase in the serum level of PEPD in CCl4-treated mice was associated with PEPD loss, EGFR activation, and Cox-2 induction in the liver tissues of these mice (Fig. 7C). Circulating PEPD levels in CCl4-treated mice (3.3 nm in serum, Fig. 7A) was higher than what was shown to activate EGFR and induce Cox-2 in cultured cells (2.7 nm; Figs. 1C, 3, C, D, and F, and 5). Indeed, EGFR was also activated in the kidneys of CCl4-treated mice, where the tissue PEPD level did not change (Fig. 7D). It is also tempting to speculate that PEPD present in the blood of control mice (0.9 nm in serum) may play a role in eliciting a basal level of EGFR activation in vivo systemically. In our preliminary experiments, 1.4 nm PEPD was still able to activate EGFR and induce Cox-2 in Hepa1c1c7 cells (result not shown). The serum level of EGF in healthy mice was reported to be ∼30–80 pm (38), which is similar to the normal human plasma concentration of EGF (∼60 pm) (39). Thus, the normal circulating PEPD level is markedly higher than that of EGF.
Serum PEPD activity was found to be significantly higher in patients with endometrial cancer than in controls (40). PEPD activity was also shown to be significantly higher in human breast cancer tissues (41) and human lung cancer tissues (42) than in their corresponding control tissues. These results, in light of the well established role of EGFR in cancer (43), suggest that PEPD may influence cancer growth and progression via EGFR stimulation. In the present study, we have clearly shown that PEPD induces Cox-2, a major oncogene, via EGFR activation.
PEPD deficiency is an autosomal recessive disease caused by mutation or partial deletion in the gene. PEPD-deficient patients present massive urinary elimination of iminodipeptides (44, 45) and show a wide range of clinical outcome, such as intractable skin ulcer, recurrent infection, and mental retardation (46–48). The disease mechanism of PEPD deficiency remains poorly understood. Previous studies of PEPD deficiency have been focused exclusively on the loss of its enzymatic activity, but therapies aimed at correcting the enzymatic loss have been largely ineffective (49–51). Our present findings raise the question of whether EGFR signaling is involved in disease development of PEPD deficiency and whether stimulating EGFR signaling is a therapeutic option for this disease.
Acknowledgments
We thank our colleagues Drs. Xiaoyin Chen and Arup Bhattacharya for technical support, and Dr. Hans Minderman for discussion on image cytometry. We also thank Dr. Daniel J. Leahy of Johns Hopkins University for valuable advice. The 32D cells and their derivatives expressing EGFR and its K721M mutants were generous gifts of Dr. Gibbes R. Johnson of the United States Food and Drug Administration.
This work was supported, in whole or in part, by National Institutes of Health Grants R01CA120533, R01CA124627, and P30CA016056 from the NCI.

This article contains supplemental “Materials and Methods” and Figs. S1–S4.
- EGFR
- epidermal growth factor receptor
- AST
- aspartate aminotransferase
- Cox-2
- cyclooxygenase-2
- HB-EGF
- heparin-binding EGF-like growth factor
- PBST
- phosphate-buffered saline with Tween 20
- PEPD
- prolidase or peptidase D.
REFERENCES
- 1. Sibilia M., Kroismayr R., Lichtenberger B. M., Natarajan A., Hecking M., Holcmann M. (2007) The epidermal growth factor receptor. From development to tumorigenesis. Differentiation 75, 770–787 [DOI] [PubMed] [Google Scholar]
- 2. Lurje G., Lenz H. J. (2009) EGFR signaling and drug discovery. Oncology 77, 400–410 [DOI] [PubMed] [Google Scholar]
- 3. Yarden Y., Sliwkowski M. X. (2001) Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2, 127–137 [DOI] [PubMed] [Google Scholar]
- 4. Hynes N. E., Lane H. A. (2005) ERBB receptors and cancer. The complexity of targeted inhibitors. Nat. Rev. Cancer 5, 341–354 [DOI] [PubMed] [Google Scholar]
- 5. Baselga J. (2002) Why the epidermal growth factor receptor? The rationale for cancer therapy. Oncologist 7, 2–8 [DOI] [PubMed] [Google Scholar]
- 6. Ciardiello F., Tortora G. (2008) EGFR antagonists in cancer treatment. N. Engl. J. Med. 358, 1160–1174 [DOI] [PubMed] [Google Scholar]
- 7. Jones J. T., Akita R. W., Sliwkowski M. X. (1999) Binding specificities and affinities of EGF domains for ErbB receptors. FEBS Lett. 447, 227–231 [DOI] [PubMed] [Google Scholar]
- 8. Dunbar A. J., Goddard C. (2000) Structure-function and biological role of betacellulin. Int. J. Biochem. Cell Biol. 32, 805–815 [DOI] [PubMed] [Google Scholar]
- 9. Wong R. W. (2003) Transgenic and knock-out mice for deciphering the roles of EGFR ligands. Cell Mol. Life Sci. 60, 113–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murthy A., Defamie V., Smookler D. S., Di Grappa M. A., Horiuchi K., Federici M., Sibilia M., Blobel C. P., Khokha R. (2010) Ectodomain shedding of EGFR ligands and TNFR1 dictates hepatocyte apoptosis during fulminant hepatitis in mice. J. Clin. Invest. 120, 2731–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Endo F., Tanoue A., Nakai H., Hata A., Indo Y., Titani K., Matsuda I. (1989) Primary structure and gene localization of human prolidase. J. Biol. Chem. 264, 4476–4481 [PubMed] [Google Scholar]
- 12. Hankinson O. (1979) Single-step selection of clones of a mouse hepatoma line deficient in aryl hydrocarbon hydroxylase. Proc. Natl. Acad. Sci. U.S.A. 76, 373–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fan Y. X., Wong L., Deb T. B., Johnson G. R. (2004) Ligand regulates epidermal growth factor receptor kinase specificity. Activation increases preference for GAB1 and SHC versus autophosphorylation sites. J. Biol. Chem. 279, 38143–38150 [DOI] [PubMed] [Google Scholar]
- 14. Zbucka M., Miltyk W., Bielawski T., Surazynski A., Palka J., Wolczynski S. (2007) Mechanism of collagen biosynthesis up-regulation in cultured leiomyoma cells. Folia Histochem. Cytobiol. 45, S181–185 [PubMed] [Google Scholar]
- 15. Turini M. E., DuBois R. N. (2002) Cyclooxygenase-2. A therapeutic target. Annu. Rev. Med. 53, 35–57 [DOI] [PubMed] [Google Scholar]
- 16. Koki A. T., Khan N. K., Woerner B. M., Seibert K., Harmon J. L., Dannenberg A. J., Soslow R. A., Masferrer J. L. (2002) Characterization of cyclooxygenase-2 (COX-2) during tumorigenesis in human epithelial cancers. Evidence for potential clinical utility of COX-2 inhibitors in epithelial cancers. Prostaglandins Leukot. Essent. Fatty Acids 66, 13–18 [DOI] [PubMed] [Google Scholar]
- 17. Oshima M., Dinchuk J. E., Kargman S. L., Oshima H., Hancock B., Kwong E., Trzaskos J. M., Evans J. F., Taketo M. M. (1996) Suppression of intestinal polyposis in Apc δ716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 87, 803–809 [DOI] [PubMed] [Google Scholar]
- 18. Klein R. D., Van Pelt C. S., Sabichi A. L., Dela Cerda J., Fischer S. M., Fürstenberger G., Müller-Decker K. (2005) Transitional cell hyperplasia and carcinomas in urinary bladders of transgenic mice with keratin 5 promoter-driven cyclooxygenase-2 overexpression. Cancer Res. 65, 1808–1813 [DOI] [PubMed] [Google Scholar]
- 19. Zeng Q., McCauley L. K., Wang C. Y. (2002) Hepatocyte growth factor inhibits anoikis by induction of activator protein 1-dependent cyclooxygenase-2. Implication in head and neck squamous cell carcinoma progression. J. Biol. Chem. 277, 50137–50142 [DOI] [PubMed] [Google Scholar]
- 20. Ledoux P., Scriver C. R., Hechtman P. (1996) Expression and molecular analysis of mutations in prolidase deficiency. Am. J. Hum. Genet. 59, 1035–1039 [PMC free article] [PubMed] [Google Scholar]
- 21. Surazynski A., Miltyk W., Prokop I., Palka J. (2010) Prolidase-dependent regulation of TGF β (corrected) and TGFβ receptor expressions in human skin fibroblasts. Eur. J. Pharmacol. 649, 115–119 [DOI] [PubMed] [Google Scholar]
- 22. Xu K., Shu H. K. (2007) EGFR activation results in enhanced cyclooxygenase-2 expression through p38 mitogen-activated protein kinase-dependent activation of the Sp1/Sp3 transcription factors in human gliomas. Cancer Res. 67, 6121–6129 [DOI] [PubMed] [Google Scholar]
- 23. Kajanne R., Leppä S., Luukkainen P., Ustinov J., Thiel A., Ristimäki A., Miettinen P. J. (2007) Hydrocortisone and indomethacin negatively modulate EGF-R signaling in human fetal intestine. Pediatr. Res. 62, 570–575 [DOI] [PubMed] [Google Scholar]
- 24. Yoon J. H., Gwak G. Y., Lee H. S., Bronk S. F., Werneburg N. W., Gores G. J. (2004) Enhanced epidermal growth factor receptor activation in human cholangiocarcinoma cells. J. Hepatol. 41, 808–814 [DOI] [PubMed] [Google Scholar]
- 25. Dikic I. (2003) Mechanisms controlling EGF receptor endocytosis and degradation. Biochem. Soc. Trans. 31, 1178–1181 [DOI] [PubMed] [Google Scholar]
- 26. Osterlund K. L., Handa R. J., Gonzales R. J. (2010) Dihydrotestosterone alters cyclooxygenase-2 levels in human coronary artery smooth muscle cells. Am. J. Physiol. Endocrinol. Metab. 298, E838–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu Z., Cui B., Jin Y., Chen H., Wang X. (2011) Novel irreversible EGFR tyrosine kinase inhibitor 324674 sensitizes human colon carcinoma HT29 and SW480 cells to apoptosis by blocking the EGFR pathway. Biochem. Biophys. Res. Commun. 411, 751–756 [DOI] [PubMed] [Google Scholar]
- 28. Perez-Torres M., Guix M., Gonzalez A., Arteaga C. L. (2006) Epidermal growth factor receptor (EGFR) antibody down-regulates mutant receptors and inhibits tumors expressing EGFR mutations. J. Biol. Chem. 281, 40183–40192 [DOI] [PubMed] [Google Scholar]
- 29. Xu Y., Tan L. J., Grachtchouk V., Voorhees J. J., Fisher G. J. (2005) Receptor-type protein-tyrosine phosphatase-κ regulates epidermal growth factor receptor function. J. Biol. Chem. 280, 42694–42700 [DOI] [PubMed] [Google Scholar]
- 30. Roomi M., Kalinovsky T., Roomi N. W., Ivanov V., Rath M., Niedzwiecki A. (2008) A nutrient mixture suppresses carbon tetrachloride-induced acute hepatic toxicity in ICR mice. Hum. Exp. Toxicol. 27, 559–566 [DOI] [PubMed] [Google Scholar]
- 31. Surazynski A., Donald S. P., Cooper S. K., Whiteside M. A., Salnikow K., Liu Y., Phang J. M. (2008) Extracellular matrix and HIF-1 signaling. The role of prolidase. Int. J. Cancer 122, 1435–1440 [DOI] [PubMed] [Google Scholar]
- 32. Lo H. W., Cao X., Zhu H., Ali-Osman F. (2010) Cyclooxygenase-2 is a novel transcriptional target of the nuclear EGFR-STAT3 and EGFRvIII-STAT3 signaling axes. Mol. Cancer Res. 8, 232–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leahy D. J. (2004) Structure and function of the epidermal growth factor (EGF/ErbB) family of receptors. Adv. Protein Chem. 68, 1–27 [DOI] [PubMed] [Google Scholar]
- 34. Lupi A., Della Torre S., Campari E., Tenni R., Cetta G., Rossi A., Forlino A. (2006) Human recombinant prolidase from eukaryotic and prokaryotic sources. Expression, purification, characterization and long-term stability studies. FEBS J. 273, 5466–5478 [DOI] [PubMed] [Google Scholar]
- 35. Liu G., Nakayama K., Sagara Y., Awata S., Yamashita K., Manabe M., Kodama H. (2005) Characterization of prolidase activity in erythrocytes from a patient with prolidase deficiency. Comparison with prolidase I and II purified from normal human erythrocytes. Clin. Biochem. 38, 625–631 [DOI] [PubMed] [Google Scholar]
- 36. Besio R., Alleva S., Forlino A., Lupi A., Meneghini C., Minicozzi V., Profumo A., Stellato F., Tenni R., Morante S. (2010) Identifying the structure of the active sites of human recombinant prolidase. Eur. Biophys. J. 39, 935–945 [DOI] [PubMed] [Google Scholar]
- 37. Maher M. J., Ghosh M., Grunden A. M., Menon A. L., Adams M. W., Freeman H. C., Guss J. M. (2004) Structure of the prolidase from Pyrococcus furiosus. Biochemistry 43, 2771–2783 [DOI] [PubMed] [Google Scholar]
- 38. Perheentupa J., Lakshmanan J., Hoath S. B., Beri U., Kim H., Macaso T., Fisher D. A. (1985) Epidermal growth factor measurements in mouse plasma. Method, ontogeny, and sex difference. Am. J. Physiol. 248, E391–396 [DOI] [PubMed] [Google Scholar]
- 39. Brzeziński J., Lewiński A. (1998) Increased plasma concentration of epidermal growth factor in female patients with non-toxic nodular goitre. Eur. J. Endocrinol. 138, 388–393 [DOI] [PubMed] [Google Scholar]
- 40. Arioz D. T., Camuzcuoglu H., Toy H., Kurt S., Celik H., Aksoy N. (2009) Serum prolidase activity and oxidative status in patients with stage I endometrial cancer. Int. J. Gynecol. Cancer 19, 1244–1247 [DOI] [PubMed] [Google Scholar]
- 41. Cechowska-Pasko M., Pałka J., Wojtukiewicz M. Z. (2006) Enhanced prolidase activity and decreased collagen content in breast cancer tissue. Int. J. Exp. Pathol. 87, 289–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Karna E., Surazynski A., Palka J. (2000) Collagen metabolism disturbances are accompanied by an increase in prolidase activity in lung carcinoma planoepitheliale. Int. J. Exp. Pathol. 81, 341–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Woodburn J. R. (1999) The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol. Ther. 82, 241–250 [DOI] [PubMed] [Google Scholar]
- 44. Arata J., Umemura S., Yamamoto Y., Hagiyama M., Nohara N. (1979) Prolidase deficiency. Its dermatological manifestations and some additional biochemical studies. Arch. Dermatol. 115, 62–67 [DOI] [PubMed] [Google Scholar]
- 45. Aytug A. F., Ergun T., Ratip S., Elcioglu N., Gultepe M., Mercan E., Gurbuz O. (2006) Prolidase deficiency associated with hemoglobin O trait and microcytic anemia. Int. J. Dermatol. 45, 867–868 [DOI] [PubMed] [Google Scholar]
- 46. Lupi A., Rossi A., Campari E., Pecora F., Lund A. M., Elcioglu N. H., Gultepe M., Di Rocco M., Cetta G., Forlino A. (2006) Molecular characterization of six patients with prolidase deficiency. Identification of the first small duplication in the prolidase gene and of a mutation generating symptomatic and asymptomatic outcomes within the same family. J. Med. Genet. 43, e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Falik-Zaccai T. C., Khayat M., Luder A., Frenkel P., Magen D., Brik R., Gershoni-Baruch R., Mandel H. (2010) A broad spectrum of developmental delay in a large cohort of prolidase deficiency patients demonstrates marked interfamilial and intrafamilial phenotypic variability. Am. J. Med. Genet. B Neuropsychiatr. Genet. 1538, 46–56 [DOI] [PubMed] [Google Scholar]
- 48. Isemura M., Hanyu T., Gejyo F., Nakazawa R., Igarashi R., Matsuo S., Ikeda K., Sato Y. (1979) Prolidase deficiency with imidodipeptiduria. A familial case with and without clinical symptoms. Clin. Chim. Acta 93, 401–407 [DOI] [PubMed] [Google Scholar]
- 49. Charpentier C., Dagbovie K., Lemonnier A., Larregue M., Johnstone R. A. (1981) Prolidase deficiency with iminodipeptiduria. Biochemical investigations and first results of attempted therapy. J. Inherit. Metab. Dis. 4, 77–78 [DOI] [PubMed] [Google Scholar]
- 50. Leoni A., Cetta G., Tenni R., Pasquali-Ronchetti I., Bertolini F., Guerra D., Dyne K., Castellani A. (1987) Prolidase deficiency in two siblings with chronic leg ulcerations. Clinical, biochemical, and morphologic aspects. Arch. Dermatol. 123, 493–499 [PubMed] [Google Scholar]
- 51. Haywood M. T. (2011) Prolidase deficiency. A child with persistent lower extremity ulcerations. The Foot and Ankle Online Journal 4, 4 [Google Scholar]