

Background: Plant arylalkylamine and aldehyde synthesizing aromatic amino acid decarboxylases (AAAD) are effectively indistinguishable.
Results: Mutagenesis of a single AAAD residue enables the interconversion of activities.
Conclusion: A single residue is primarily responsible for differentiating plant AAAD decarboxylase and aldehyde synthase activities.
Significance: AAAD activity differentiation enables primary sequence activity identification, generation of unusual AAAD enzyme products, and AAAD mechanistic insights.
Keywords: Amino Acid, Arabidopsis, Auxin, Decarboxylase, Tryptophan, Aromatic Acetaldehyde Synthese, Aromatic Amino Acid Decarboxylase, Indole-3-acetaldehyde, Phenylacetaldehyde Synthase, Tryptophan Decarboxylase
Abstract
Plant aromatic amino acid decarboxylase (AAAD) enzymes are capable of catalyzing either decarboxylation or decarboxylation-deamination on various combinations of aromatic amino acid substrates. These two different activities result in the production of arylalkylamines and the formation of aromatic acetaldehydes, respectively. Variations in product formation enable individual enzymes to play different physiological functions. Despite these catalytic variations, arylalkylamine and aldehyde synthesizing AAADs are indistinguishable without protein expression and characterization. In this study, extensive biochemical characterization of plant AAADs was performed to identify residues responsible for differentiating decarboxylation AAADs from aldehyde synthase AAADs. Results demonstrated that a tyrosine residue located on a catalytic loop proximal to the active site of plant AAADs is primarily responsible for dictating typical decarboxylase activity, whereas a phenylalanine at the same position is primarily liable for aldehyde synthase activity. Mutagenesis of the active site phenylalanine to tyrosine in Arabidopsis thaliana and Petroselinum crispum aromatic acetaldehyde synthases primarily converts the enzymes activity from decarboxylation-deamination to decarboxylation. The mutation of the active site tyrosine to phenylalanine in the Catharanthus roseus and Papaver somniferum aromatic amino acid decarboxylases changes the enzymes decarboxylation activity to a primarily decarboxylation-deamination activity. Generation of these mutant enzymes enables the production of unusual AAAD enzyme products including indole-3-acetaldehyde, 4-hydroxyphenylacetaldehyde, and phenylethylamine. Our data indicates that the tyrosine and phenylalanine in the catalytic loop region could serve as a signature residue to reliably distinguish plant arylalkylamine and aldehyde synthesizing AAADs. Additionally, the resulting data enables further insights into the mechanistic roles of active site residues.
Introduction
In many species, including humans, there is only one decarboxylase that selectively catalyzes the decarboxylation of aromatic amino acids. This enzyme is commonly named aromatic amino acid decarboxylase (AAAD).2 Within plants and insects, however, the similar enzyme has undergone extensive evolutionary divergence, resulting in multiple paralogs with divergent functions. The divergent functions of plant and insect AAADs are closely related to their corresponding substrate selectivity and catalytic reactions. For example, tyramine derived from tyrosine is the essential precursor for the biosynthesis of many plant secondary metabolites (including N-hydroxycinnamic acid amides and benzylisoquinoline alkaloids) (1–5). Tryptamine and 5-hydroxytryptamine (5-HT), produced by specific plant AAADs, are precursors for the synthesis of thousands of indole alkaloid compounds (1, 6, 7). Because their substrate specificity provides functional relevant information, some AAADs have been further annotated on their principle substrate as tyrosine decarboxylases (TyDC) and tryptophan decarboxylases (TDC) (8, 9).
Recent studies of AAAD proteins demonstrated that in addition to the typical decarboxylation activity established in TyDCs and TDCs, some annotated plant and insect AAAD proteins are actually aromatic acetaldehyde synthases (AASs) (10–12). These AASs catalyze a rather complicated decarboxylation-oxidative deamination process of aromatic amino acids, leading to the production of aromatic acetaldehydes, CO2, ammonia, and hydrogen peroxide rather than the AAAD-derived arylalkylamines and CO2 (Fig. 1). Research has implicated plant AAS enzymes in the production of volatile flower scents, floral attractants, and defensive phenolic acetaldehyde secondary metabolites (10–12) and insect AAS in soft cuticle hardening (13). Although the physiological functions of true AAADs vary with their substrate selection, AAS proteins perform additional distinct physiological roles as compared with true AAADs. Consequently, it is essential to be able to distinguish AAS proteins from true AAAD enzymes. Despite variations in substrate specificity and catalytic reactions, plant AAS proteins share great sequence similarity to true AAADs. Such high homology is specifically emphasized through the comparison of a characterized Arabidopsis thaliana AAS and a Capsicum annuum TDC (11, 14). Although the C. annuum TDC only catalyzes decarboxylation of indolic substrates and the A. thaliana AAS only catalyzes aldehyde synthesis on phenolic substrates, these two enzymes retain 74% identity and 84% homology. This extensive homology has lead to a major problem in distinguishing AAS enzymes from typical AAAD proteins. For example, all currently verified plant AAS proteins were initially annotated as TyDCs. Additionally, our recent study showed that even experimentally investigated AAAD enzymes are capable of inaccurate characterization. In the aforementioned study we demonstrate that the previously characterized Petroselinum crispum TyDC (Q06086) (15) is actually an AAS that principally catalyzes the conversion of tyrosine to p-hydroxyphenylacetaldehyde (12).
FIGURE 1.
Relative activities of AAAD and AAS.
To narrow the location of activity dictating AAAD and AAS residues we initiated this study by generating a chimeric enzyme, containing the N-terminal half of the Thalictrum flavum TyDC and the C-terminal half of the P. crispum AAS. The 70% identity between the P. crispum AAS and T. flavum TyDC (AAG60665) (12) suggests that their respective AAS and TyDC activities are likely dictated by a very limited number of active site residues. Activity analysis of the chimeric enzyme determined that the T. flavum-P. crispum hybrid has typical AAS activity. The resulting aldehyde synthase activity indicates that residues located in the C-terminal half of the mutant are predominantly responsible for activity differentiation. Next, analyses of published crystal structures were preformed to identify putative activity differentiating residues within plant AAAD and AAS enzymes. Several structures were analyzed, but due to the strong plant AAAD homology, the conservation of active site residues, and substantial electron density of an active site proximal catalytic loop (absent from other AAAD structures), the Human histidine decarboxylase (huHDC) structure was chosen as the primary model (16). Investigation of the Human HDC (PDB code 4E1O) crystal structure identified residues located within 6 Å of the pyridoxal 5′-phosphate inhibitor adduct. Comparison of these active site residues to their homologous residues in characterized plant AAAD and AAS sequences enabled the identification of active site residues potentially responsible for activity differentiation. Analysis of residues conserved in arylalkylamines synthesizing AAADs and absent in aromatic acetaldehyde synthesizing AASs enabled the exclusion of all but one putative activity dictating residue. The resulting analyses enabled the identification of a putative residue located in a catalytic loop proximal to the active site. The candidate residue is represented by a tyrosine 347 in the T. flavum enzyme and as a phenylalanine 346 in the P. crispum enzyme. Mutation of this candidate residue from phenylalanine to tyrosine within the A. thaliana (NP_849999) and the P. crispum (Q06086) aldehyde synthases converts the enzymes activities from decarboxylation-oxidative deamination to decarboxylation. Mutation of the homologous tyrosine residue in Catharanthus roseus TDC (P17770) and Papaver somniferum TYDC 9 (AAC61842) converted the enzymes activities from decarboxylation to decarboxylation-oxidative deamination. Our data demonstrated that the active site tyrosine and phenylalanine in the flexible loop of plant AAADs plays a primary role for true AAAD activity and AAS activity, respectively. Our progress represents a major step forward toward achieving a comprehensive understanding of substrate specificity and catalytic reactions in plant AAAD proteins.
EXPERIMENTAL PROCEDURES
Reagents
Acetonitrile, 2-mercaptoethanol, formic acid, tryptophan, tryptamine, indole-3-acetaldehyde·sodium bisulfite complex, indole-3-ethanol, phenylalanine, phenylethylamine, phthaldialdehyde, pyridoxal 5-phosphate (PLP), sodium bisulfite, and sodium borohydride were purchased from Sigma. The IMPACT-CN protein expression system was obtained from New England Biolabs (Ipswich, MA).
Plant Material and Growth Conditions
P. crispum seeds, T. flavum seeds, P. somniferum seeds, and C. roseus seeds were obtained from burpee.com, dianeseeds.com, onestoppoppyshoppe.com, and horizonherbs.com, respectively. A. thaliana seeds (wild type (CS60000), Columbia ecotype) were from The Arabidopsis Biological Resource Center (Columbus, OH). Seeds from these plants were germinated in Sunshine Pro Premium potting soil and grown under a 16-h photoperiod at 23 ºC at 100 microeinsteins.
RNA Isolation, cDNA Amplification, and Mutagenesis
Total RNA samples were isolated from 12-week-old A. thaliana, C. roseus, P. somniferum, P. crispum, and T. flavum plants using an Ambion® mirVanaTM miRNA Isolation Kit. These RNA samples were treated with DNase (Ambion TURBO DNA-freeTM Kit). First strand cDNAs were synthesized by RT-PCR of total RNA samples using Invitrogen SuperScriptTM III and a poly T17-primer. Specific primer pairs (supplemental Table S1), design based on coding sequences (CDS) of A. thaliana AAS (NP 849999), P. crispum AAS (Q06086), T. flavum (AAG60665), P. somniferum TyDC (AAC61842), and C. roseus TDC (P17770), were synthesized and used for the amplification of the respective coding sequences. Amplified full-length CDS cDNA were subsequently ligated into IMPACT-CN expression vector for protein expression. Completed expression vectors were used as templates to produce the AAS to AAAD and AAAD to AAS mutants.
To generate T. flavum-P. crispum chimeric enzyme, a specific reverse primer based on codons for T. flavum TyDC (AAG60665) residues 294–300 and a forward primer based on P. crispum AAS (Q06086) residues 300–306 (with both primers containing a SapI restriction site) were synthesized (supplemental Table S1). The new T. flavum reverse primer was paired with the original T. flavum forward primer and the new P. crispum forward primer was paired with the original P. crispum AAS reverse primer to amplify (using the respective full-length T. flavum TyDC and full-length P. crispum AAS CDS sequences as template) a DNA fragment encoding for the first 300 amino acids of T. flavum TyDC and a DNA fragment encoding for P. crispum residues 300–514, respectively. The two cDNA fragments were digested with SapI to generate a cohesive 3-end for the T. flavum cDNA fragment and cohesive 5-end for the P. crispum cDNA fragment. The two digested fragments were ligated together with the aid of T4 DNA ligase to produce a chimeric CDS coding for the 300 N-terminal side residues of the T. flavum TyDC and 214 C-terminal site of the P. crispum AAS (17).
Full-length CDS cDNA containing the F338Y mutation in A. thaliana AAS, the F346Y mutation in P. crispum AAS, the Y348F mutation in C. roseus TDC, and the Y350F mutation in P. somniferum TyDC were generated using a SapI mutagenesis procedure similar to that described for the production of the chimeric T. flavum-P. crispum enzyme. The variable region of the SapI restriction site was used to generate codon mutagenesis (supplemental Table S1).
Protein Expression and Purification
All cDNA products encoding for wild type TyDCs, AASs, TDCs, and their mutants were individually ligated into IMPACT-CN bacterial expression plasmids. DNA sequencing was utilized to verify the sequence and frame of each cDNA insert. Recombinant IMPACT-CN plasmids were utilized to transform Escherichia coli DE3. Transformed bacterial colonies, expressing the target proteins, were selected and used for large scale expression (16–20 liters of E. coli cells) of individual recombinant proteins. The detailed conditions for recombinant protein expression and purification were essentially the same as previously described (18).
Activity Assays
Typical reaction mixtures of 100 or 200 μl, containing 20–30 μg of recombinant TyDC, TDC, AAS, hybrid enzyme, or mutant protein and 5 mm substrate (tyrosine, DOPA, tryptophan, or phenylalanine) were prepared in 50 mm potassium phosphate buffer (pH, 7.5) and incubated at 25 °C in a water bath. The reactions were stopped (at a time point between 5 and 60 min) through the addition of an equal volume of 0.8 m formic acid. Supernatants of the reaction mixtures, obtained by centrifugation, were analyzed with a (Aqueous) Pierce Quantitative Peroxide Assay Kit to determine AAS activity. The supernatants were also analyzed by high performance liquid chromatography (HPLC) with electrochemical detection (HPLC-EC) or HPLC with ultraviolet detection (HPLC-UV). Details of HPLC-EC or HPLC-UV conditions were described in the figure captions. Product production was verified though the comparison of various aromatic amine standards under identical chromatography conditions.
AAAD Mutant Product Verification by Liquid Chromatography/Tandem Mass Spectrometry (HPLC/MS/MS)
To further verify the identity of the mutant enzymatic products, reaction mixtures containing physiological substrates and mutant enzymes were analyzed by LC/MS/MS. Reaction mixtures of 100 μl, containing 30 μg of C. roseus TDC Y348F and 10 mm tryptophan, or 30 μg of A. thaliana AAS F338Y and 10 mm phenylalanine were prepared in 50 mm phosphate buffer (pH 7.5) and incubated at 25 °C in a water bath for 30 min. Reactions were stopped by mixing an equal volume of 0.8 m formic acid. Prior to injection, indole-3-acetaldehyde, generated in the C. roseus TDC Y348F mutant and 10 mm tryptophan reaction mixture, was derivatized to the indole-3-acetaldehyde·sodium bisulfite complex using sodium bisulfite. Samples were analyzed by a LC-3200 Q Trap MS/MS system (AB Sciex) in either positive and negative ion mode, as appropriate. Identification of phenylethylamine from the A. thaliana AAS F338Y mutant reaction mixture and indole-3-acetaldehyde·sodium bisulfite complex from the derivatized C. roseus TDC Y348F mutant reaction mixture was based on their retention time and MS/MS spectra in comparison with those produced from authentic phenylethylamine and indole-3-acetaldehyde·sodium bisulfite complex standards at identical analytic conditions.
Kinetic Analysis
After the substrate specificity and catalytic reaction of enzymes were verified, their kinetic parameters were evaluated. The A. thaliana AAS (NP_849999) wild type and F338Y mutant enzymes were evaluated using phenylalanine as a substrate. The C. roseus TDC (P17770) wild type and Y348Y mutant enzymes were evaluated using tryptophan as a substrate. Kinetic data points were performed in triplicate and kinetic values were evaluated by hyperbolic regression. Reaction mixtures of 100 μl containing 5 μg of enzyme recombinant protein and varying concentrations of substrate (0.1–40 mm phenylalanine, 0.005–16 mm tryptophan) were prepared in 50 mm phosphate buffer (pH 7.5) and incubated at 25 °C. To detect phenylethylamine generated by A. thaliana F338Y, two volumes of 100% ethanol were added to the reaction mixtures (to stop the reaction) at 5 min after incubation and supernatants, obtained by centrifugation (14,000 × g for 5 min at 4 °C), were derivatized using OPT reagent and then analyzed by HPLC-EC and quantitated based on a standard curve generated using authentic OPT-derivatized phenylethylamine (19). Tryptamine generated by wild type C. roseus TDC was measured by stopping the reaction mixture using an equal volume of 0.8 m formic acid, obtaining the supernatant via centrifugation, and analyzing by HPLC-EC against a standard curve of tryptamine. To determine the AAS activity for the C. roseus TDC mutant and the wild type A. thaliana enzyme, an equal volume of 0.8 m formic acid was added to the reaction mixtures and supernatants (obtained by centrifugation) were analyzed with (Aqueous) Pierce Quantitative Peroxide Assay Kit to measure peroxide production. The amounts of H2O2 product in each reaction mixture was quantitated based on a standard curve generated using authentic peroxide.
RESULTS
AAAD/AAS Chimeric Mutant Analysis
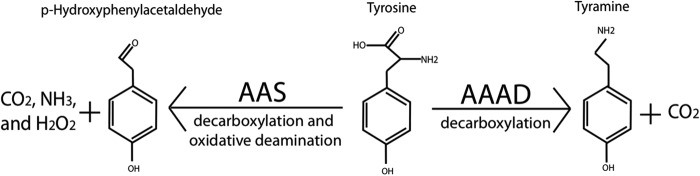
To narrow the location of the AAAD versus AAS activity differentiating residue/residues, a chimeric (hybrid) protein composed of T. flavum TyDC (AAG60665) residues 1–300 and P. crispum AAS (Q06086) residues 300–514 was generated and its catalytic reaction was assessed with tyrosine as a substrate. Incubation of the T. flavum-P. crispum chimeric enzyme in the presence of tyrosine resulted in the detection of a major broad peak overlapping with a minor sharp peak during HPLC-EC analysis (Fig. 2A). The broad peak was considered to be p-hydroxyphenylacetaldehyde (HPAA) because it displayed the same chromatographic behavior as the HPAA formed in the wild type P. crispum AAS and tyrosine reaction mixture (Fig. 2C). When both the hybrid enzyme and the wild type P. crispum enzyme reaction mixtures were treated with borohydride prior to HPLC-EC analysis, the broad product peaks (Fig. 2, A and C, respectively) were converted to sharp product peaks (Fig. 2, B and D, respectively) that coeluted with authentic 4-hydroxyphenylethanol (tyrosol) at different chromatographic conditions. Unlike the wild type AAS and tyrosine reaction mixture, a small amount of tyramine was formed in the mutant enzyme and tyrosine reaction mixture (Fig. 2, A and B). Despite this minor activity, the hybrid enzyme behaved primarily as a wild type P. crispum AAS. These results suggested that residues in the C-terminal half of plant AAADs are likely responsible for constraining decarboxylation and aldehyde synthase reactions.
FIGURE 2.
HPLC-EC analysis of T. flavum-P. crispum chimeric enzyme and P. crispum AAS activity with tyrosine as a substrate. y axis represents the output in microamps, the x axis represents retention time. Reaction mixtures of 100 μl containing either 30 μg of T. flavum-P. crispum chimeric enzyme or 20 μg of wild type AAS and 5 mm tyrosine were incubated at 25 °C and their reaction was stopped at 40 min after incubation by adding an equal volume of 0.8 m formic acid into the reaction mixture. The mixtures were centrifuged for 5 min at 14,000 × g and supernatants were injected for HPLC-EC analysis. To reduce HPAA to HPEA, some reaction mixtures were treated with an equal volume of 100% ethanol saturated with borohydride after a 40-min incubation. The borohydride-treated mixtures were incubated for 10 min on ice, treated with 0.8 m formic acid to decompose remaining borohydride, and centrifuged to obtain supernatants. Supernatants were injected for HPLC-EC analysis. Chromatograms (A and B) illustrate the accumulation of HPAA (the major broad peak) and tyramine (the minor sharp peak) in a T. flavum-P. crispum chimeric enzyme and tyrosine reaction mixture at 40 min after incubation and the reduction of 4-HPAA to 4-HPEA (tyrosol) in a borohydride-treated reaction mixture. Chromatograms C and D show the production of 4-HPAA in a wild type P. crispum AAS and tyrosine reaction mixture at 40 min after incubation and the reduction of HPAA to HPEA (tyrosol) in a borohydride-treated reaction mixture.
C-terminal Active Site Structural Analysis and Sequence Comparison
To impact the catalytic reaction, the activity dictating C-terminal residues should be in close proximity to the active site internal aldimine bond (that subsequently interacts with incoming substrate, leading to the formation of the external aldimine intermediate during catalysis). To elucidate active site proximal residues, the recently released mammalian histidine decarboxylase (huHDC) (NP_002103) (16) was investigated in PyMOL (20) to identify C-terminal residues located within 6 Å of the external aldimine inhibitor complex. The catalytic domain of HDC is homologous to that of plant AAADs. Additionally, most of the substrate interacting residues in AAADs are conserved in HDCs. Moreover, the active site loop of a ligand bound HDC complex displayed substantial electron density (absent from other AAAD structures). Results identified 13 active site proximal residues from the C-terminal portion of the huHDC structure. A multiple sequence alignment of the huHDC sequence, 4 characterized plant AAAD sequences, and 3 characterized plant AAS sequences was performed to identify homologous active site proximal residues from plant AAAD and AAS sequences (supplemental Fig. S1). The majority of these putative activity dictating residues were subsequently dismissed as activity dictating residues due to interactivity conservation or lack of intra-activity conservation. Ultimately, it became apparent that T. flavum TyDC Tyr-347 was stringently conserved in all identified TyDC and TDC (as well as many other predicted AAADs), whereas the similar position was occupied by phenylalanine in identified AAS proteins (one verified Rosa AAS contained a valine at the corresponding position) (10). The correlation between these hypothetical activity differentiating residues and their corresponding catalytic functions can be illustrated by viewing the multiple alignments of five characterized AAADs and 3 characterized AASs (Fig. 3). These analyses suggested that this tyrosine and/or phenylalanine might have an impact on AAAD and AAS catalytic reactions.
FIGURE 3.

Sequence alignment of A. thaliana AAS (NP_849999), Rosa hybrid cultivar AAS (ABB04522), P. crispum AAS (Q06086), P. somniferum TYDC 9 (AAC61842), T. flavum TyDC (AAG60665), A. thaliana TYDC (NP_001078461), C. roseus TDC (P17770), and Oryza sativa TDC (AK069031). The aforementioned plant sequences maintain an average 56% identity. The decarboxylation dictating tyrosine and the aldehyde synthase dictating phenylalanine are highlighted in yellow.
Conversion of A. thaliana and P. crispum AASs into AAADs through Mutation
To compare with P. crispum AAS, we previously expressed A. thaliana AAS (NP_849999) and verified its activity (12) (the activity of the A. thaliana AAS enzyme has previously been investigated) (11). The A. thaliana AAS has a Phe-338 occupying the same position as the conserved tyrosine in the flexible loop regions of the TyDC and TDC enzymes. To test the role this positional residue plays in catalysis, A. thaliana AAS Phe-338 was mutated to Tyr-338 and its mutant protein was expressed, purified, and compared with the wild type AAS in a catalytic reaction with phenylalanine as substrate. After the F338Y mutant was incubated for different time periods with phenylalanine and then assessed for the production of H2O2 in the reaction mixtures, no apparent H2O2 accumulation was observed in the reaction mixtures (Fig. 4, mutant curve). In contrast, production of H2O2 was observed in the wild type A. thaliana AAS and phenylalanine reaction mixtures and the relative amounts of H2O2 were approximately proportional to the incubation periods (Fig. 4, wild type curve). Inability to produce H2O2 in the A. thaliana AAS F338Y mutant phenylalanine reaction mixtures suggests that this particularly phenylalanine residue is important for the AAS activity of the A. thaliana enzyme.
FIGURE 4.
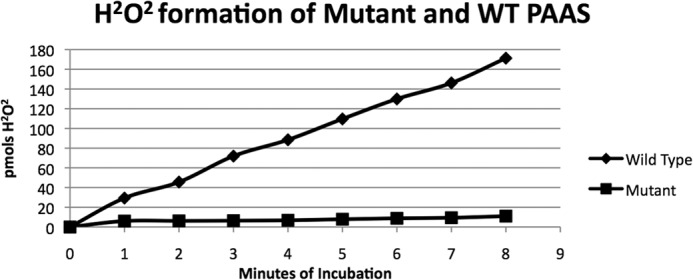
Analysis of hydrogen peroxide production in reaction mixtures containing phenylalanine and wild type A. thaliana AAS or its F338Y mutant. Reaction mixtures of 0.2 ml containing 5 mm phenylalanine and 20 μg of the wild type A. thaliana AAS or 20 μg of its F338Y mutant were prepared in 50 mm phosphate buffer, pH 7.5. The reaction mixtures were incubated at 25 °C. At each 1-min interval, 20 μl of reaction mixture was withdrawn and mixed into 150 μl of Pierce peroxide assay reagents solution. The wild type curve and mutant curve illustrate the amount of H2O2 accumulated in 20 μl of reaction mixture at a 1–8-min incubation periods.
Based on high conservation of the active loop tyrosine in TyDC and TDC at the corresponding position, the generated A. thaliana F338Y mutation might result in mutant decarboxylase activity. When the phenylalanine and A. thaliana AAS F338Y mutant reaction mixtures were analyzed by HPLC with UV detection at 265 nm at the described HPLC-EC separation conditions, a product with a retention time of 4.41 min was observed. The relative amounts of the product formed were approximately proportional to the incubation periods of the reaction mixtures (Fig. 5, A–C). The product has an identical retention time to the authentic phenylethylamine at the same HPLC separation conditions. HPLC/MS/MS analysis in the positive ion mode of an A. thaliana AAS F338Y mutant and phenylalanine reaction mixture revealed that the products of the MH+ ion m/z 122.1 and the relative intensities of its tandem spectrum (MS/MS spectrum) were identical to those generated using authentic phenylethylamine at identical conditions (Fig. 5, D and E). In addition, the product reacted with OPA-thio reagent producing an electrochemically active compound as other primary amines, further verifying the presence of the free primary amine of the product. To further verify the decarboxylation activity-dictating role of this active site tyrosine, we performed an additional phenylalanine to tyrosine mutation on the phenylalanine 346 residue of P. crispum AAS (Q06086). HPLC-EC comparison of the wild type enzyme versus the F346Y mutant confirmed an alteration in the primary activity from decarboxylation-deamination to decarboxylation (supplemental Fig. S2). In addition to the primary decarboxylation activity, the P. crispum F346Y mutant retained a small percentage of decarboxylation-deamination activity. The change from AAS activity to true AAAD activity in the A. thaliana AAS F338Y and the P. crispum F350Y mutants, in conjunction with the strict conservation of Tyr-338 equivalent within the identified TyDCs and TDCs (Fig. 2), indicates that this loop region tyrosine residue plays primarily a role in the typical decarboxylation reaction in plant AAADs.
FIGURE 5.

Detection/identification of phenylethylamine produced in A. thaliana AAS F338Y mutant and phenylalanine reaction mixtures by HPLC-UV and LC/MS/MS. Reaction mixtures of 0.1 ml containing 0.1 mg of Arabidopsis AAS F338Y mutant and 20 mm phenylalanine were prepared in 100 mm phosphate buffer (pH 7.5) and the reaction was stopped at different times after incubation by addition of an equal volume of 0.8 m formic acid. The mixtures were centrifuged for 5 min at 14,000 × g to obtain supernatants that were then injected for HPLC-UV analysis. Chromatograms (A-C) illustrate the relative amount of phenylethylamine (4.41-min peak) formed in the reaction mixtures at 5, 10, and 15 min after incubation, respectively. Chromatograms and MS/MS spectrum (D) illustrate the MH+ ion (122) and daughter ions of the product formed in A. thaliana AAS F338Y mutant and phenylalanine reaction mixture in comparison with those (E) produced by phenylethylamine standard under identical conditions of LC/MS/MS analysis.
Mutagenic Conversion of C. roseus TDC and P. somniferum TyDC into an Indole-3-acetaldehyde Synthase and a 4-Hydroxyphenylacetaldehyde Synthase, Respectively
We have previously expressed a C. roseus TDC (P17770) and verified its substrate specificity to primarily tryptophan and also 5-HTP at a reduced rate (12) (the activity of the C. roseus TDC enzyme has previously been investigated) (9). This TDC, like other verified TDC and TyDC, contains the conserved Tyr-348 at the position equivalent to Phe-338 of the A. thaliana AAS. To determine its role in the catalytic reaction, a Y348F mutant of the C. roseus TDC was produced and its catalytic reaction was assessed with tryptophan as a substrate. Similar to those obtained during analysis of the P. crispum AAS and tyrosine reaction mixtures (Fig. 2C), HPLC-EC analysis of the C. roseus TDC Y348F mutant and tryptophan reaction mixtures resulted in detection of a very broad peak (Fig. 6A) and treatment of the reaction mixture with borohydride converted the broad peak to a sharp peak (Fig. 6B), which contrasts to the typical tryptophan decarboxylation reaction by wild type C. roseus TDC (Fig. 6C). The sharp peak, detected in the borohydride-treated reaction mixture, had identical retention times as authentic indole-3-ethanol under the same conditions of HPLC-EC analysis and coeluted with the standard at different mobile phase conditions during HPLC-EC analysis (not shown). Indole-3-ethanol is not charged easily by electrospray ionization, but the indole-3-acetaldehyde·bisulfite adduct (formed easily by reacting indole-3-acetaldehyde with bisulfite in aqueous solution) is negatively charged in aqueous solution even at relatively acidic pH. Analysis of a sodium bisulfite-treated C. roseus TDC Y348F mutant and tryptophan reaction mixture by HPLC/MS/MS in negative mode verified that the MS-MS spectrum of [M-H]− precursor ion of the adduct m/z 240.1 was identical to those of authentic indole-3-acetaldehyde·bisulfite under identical analysis conditions (Fig. 6, D and E). The detection of a broad peak during HPLC-EC analysis of the TDC Y348F mutant and tryptophan reaction mixture, the conversion of the broad peak to a sharp peak by borohydride reduction, the coelution of the sharp peak with indole-3-ethanol, the reaction of the enzymatic product with bisulfite (indicative for the presence of aldehyde group), and the identical MS and MS/MS spectra of the its bisulfite adduct with those of indole-3-acetaldehyde·disulfite standards provide convincing evidences for the TDC Y348F enzymatic production of indole-3-acetaldehyde. Accordingly, mutation of Tyr-348 to Phe-348 changed C. roseus TDC into a tryptophan to indole-3-acetaldehyde catalyzing AAS. In addition to indole-3-acetaldehyde, a small amount of tryptamine was also detected in TDC Y348F mutant and tryptophan reaction mixtures. To substantiate the claim that this active site phenylalanine is responsible for decarboxylation-deamination activity, an additional tyrosine to phenylalanine mutation was performed upon the P. somniferum tyrosine 350 residue. Evaluation of the P. somniferum wild type and Y350F mutant enzyme activities demonstrates a conversion of activity from decarboxylation to decarboxylation-deamination (supplemental Fig. S3). In addition to the primary decarboxylation-deamination activity, the P. somniferum Y350F mutant retained a small percentage of its original decarboxylation activity. Changes of the catalytic reactions in the C. roseus and P. somniferum tyrosine to phenylalanine mutants, in conjunction with that of the P. crispum and A. thaliana phenylalanine to tyrosine mutants, further support the role the active site tyrosine and phenylalanine play in decarboxylation and decarboxylation-deamination activity, respectively.
FIGURE 6.

HPLC-EC and LC/MS/MS detection of indole-3-acetaldehyde generated from C. roseus TDC Y348F mutant and tryptophan reaction mixtures. Chromatograms A-C, y axis represents the output in microamps, the x axis represents retention time. Reaction mixtures of 0.1 ml containing 5 mm tryptophan and 20 μg of periwinkle TDC Y438F mutant or 20 μg of wild type periwinkle TDC were prepared in 100 mm phosphate buffer (pH 7.5). The reaction mixtures were incubated at 25 °C and the reaction was stopped after a 15-min incubation by adding an equal volume of 0.8 m formic acid into the reaction mixtures. The mixtures were centrifuged for 5 min at 14,000 × g to obtain supernatants that were then injected for HPLC-UV analysis. Chromatograms (A and B) illustrate the indole-3-acetaldehyde (the major broad peak) formed in a TDC Y348F mutant and tryptophan reaction mixtures after a 15-min incubation and the detection of indole-3-ethanol (tryptophanol) in another 15-min incubated reaction mixture that was treated with borohydride prior to HPLC-EC analysis. Chromatogram (C) shows the detection of tryptamine in a wild type TDC and tryptophan reaction mixture 15 min after incubation. Chromatogram and MS/MS spectrum (D) illustrate the [M-H]− ion (240) and daughter ions of the bisulfite-derivatized product formed in bisulfite-treated A. thaliana AAS F338Y mutant and phenylalanine reaction mixture in comparison with those (E) produced by a indole-3-acetaldehyde·bisulfite standard under identical conditions of HPLC/MS/MS analysis.
Substrate Specificity and Kinetic Analysis
The typical substrate of wild type enzymes was used for activity assays of the mutant enzymes. After the change of catalytic reaction for the mutant enzymes was established, they were screened for activity against other aromatic amino acids. Similar to wild type A. thaliana AAS, its F338Y mutant displayed a very low activity to tyrosine (∼5% of its activity to phenylalanine) and strong activity to DOPA (Fig. 7), but showed no detectable activity to tryptophan and 5-HTP; whereas the C. roseus TDC Y348F mutant showed activity to tryptophan and 5-HTP with no detectable activity to phenylalanine, tyrosine, and DOPA (not shown). These analyses established that although the catalytic reaction of these mutants was changed, their substrate specificity were the same as their wild type enzymes (9, 11).
FIGURE 7.

HPLC-EC detection of dopamine and 3,4-dihydrophenylethanol produced in A. thaliana AAS F338Y mutant and wild type reaction mixtures, respectively. y axis represents the output in microamps, the x axis represents retention time. Reaction mixtures of 0.1 ml containing 30 μg of A. thaliana AAS F338Y mutant or wild type and 5 mm DOPA were prepared in 100 mm phosphate buffer (pH 7.5). The reaction mixtures were incubated at 25 °C and the reaction was stopped at different times after incubation by adding an equal volume of 0.8 m formic acid. Chromatograms A–C illustrate the relative amount of dopamine formed in the reaction mixtures at 4, 12, and 24 min after incubation, respectively. Chromatogram D illustrates wild type enzyme and DOPA reaction mixture at 24 min after incubation and the reduction of 3,4-dihydroxylphenylacetaldehyde to 3,4-dihydrophenylethanol in a borohydride-treated reaction mixture. In the mutant enzyme reaction mixtures, the rate of product formation was decreased as dopamine accumulated in the reaction mixtures, the reaction could proceed basically to completion (CO2 is released, so no equilibrium is reached during reaction). At the applied conditions, essentially all DOPA was converted to dopamine, but no indication for further oxidative deamination of the dopamine product was observed.
Kinetic evaluations were conducted for both the wild type and mutant A. thaliana and C. roseus proteins using their respective physiological substrate. Under the applied conditions, the A. thaliana AAS F338Y and C. roseus TDC Y348F mutants displayed a similar affinity to phenylalanine and tryptophan, respectively, as compared with their respective wild type enzymes (Table 1). Similar Michaelis constants between wild type and mutant enzymes indicate that mutation of the active site tyrosine to phenylalanine or vise versa does not apparently alter the substrate binding affinity of the enzymes. In contrast, the specific activities of the mutant enzymes were quite different to those determined for the wild type enzymes (Table 1). The decarboxylase activity of the A. thaliana AAS F338Y mutant was ∼3-fold faster than the aldehyde synthase activity of its wild type enzyme to the same substrate, whereas the specific aldehyde synthase activity of the C. roseus Y348F mutant was ∼17-fold slower than the decarboxylase activity of the wild type TDC to tryptophan (Table 1). Such alterations in activity likely are reflected by the complexity of the reactions. The added chemistry necessary for decarboxylation-deamination as opposed to a simpler decarboxylation may result in the observed catalysis rate change.
TABLE 1.
Kinetic parameters of A. thaliana and C. roseus wild type and mutant enzymes
Values represent mean ± S.E. (n = 3).
| Organism | Enzyme | Vmax | Km | Substrate | Activity |
|---|---|---|---|---|---|
| nmol/min/mg protein | mm | ||||
| A. thaliana | PAAS (WT) | 112 ± 8 | 5.10 ± 0.70 | Phenylalanine | Aldehyde synthase |
| A. thaliana | PAAS (F338Y) | 341 ± 31 | 4.60 ± 0.55 | Phenylalanine | Decarboxylation |
| C. roseus | TDC (WT) | 2710 ± 110 | 0.12 ± 0.02 | Tryptophan | Decarboxylation |
| C. roseus | TDC (Y348F) | 160 ± 14 | 0.095 ± 0.02 | Tryptophan | Aldehyde Synthase |
DISCUSSION
Based on reactions they catalyze, plant AAADs can be classified as true AAADs (enzymes that catalyze the production of arylalkylamines from their respective aromatic amino acid substrates) or AASs (enzymes that catalyze the production of aromatic acetaldehydes from their respective aromatic amino acid substrates). Within each subclass, these enzymes can be further classified based on their substrate specificity, such as TyDCs and TDCs. Association of substrate selectivity and catalytic reaction with a given AAAD protein provides relevant functional linkages. Unfortunately, high sequence identity across plant AAADs makes it difficult to accurately assign substrate specificity and catalytic reaction based on a primary sequence. Our conversion of two AASs into AAADs through active site phenylalanine to tyrosine mutation and our conversion of two AAADs into AASs through active site tyrosine to phenylalanine mutation demonstrate that the conserved active site tyrosine and phenylalanine are predominantly responsible for their respective AAAD and AAS catalytic reactions. Although more experimental verification is necessary, our data, in conjunction with the strict conservation of the active site tyrosine in all verified TyDC and TDC, indicate that the presence of active site tyrosine is a genuine criterion to predict the decarboxylation activity of a given member of the AAAD family. We submit that the identification of a homologous active site tyrosine or phenylalanine will enable reliable differentiation of plant AAAD and AAS enzymes from a primary sequence basis. In addition, the identification of an activity implicative residue in AAADs and AASs provides some basis to expand on each of the activities respective reaction mechanisms.
PLP-dependent decarboxylases are believed to proceed by a single reaction mechanism (21). In this mechanism the external aldimine loses the α-carboxyl group to form the carbanion quinonoid intermediate (due to its interaction with catalytic residues). Subsequent α-carbon protonation by an active site residue generates the product monoamine. Previous investigations of AAAD enzymes suggest that the quinonoid intermediate may become protonated by the catalytic loop tyrosine to form the arylalkylamine product (22). The same report indicated that in the absence of α-carbon protonation, the molecular oxygen is capable of attacking the carbanion of the quinonoid intermediate to generate a peroxide. The peroxy-aldimine transition form will undergo heterolysis of the O–O bond allowing for the regeneration of PLP and formation of an imine complex that spontaneously decomposes to yield an aromatic aldehyde and ammonia. Based on the effective conversion of the two AASs into AAADs upon mutating the active site phenylalanine to tyrosine the proposed tyrosine α-carbon protonation mechanism seems plausible. However, the α-carbon protonation of the intermediate requires the ionization of the tyrosine phenolic hydroxyl group proton. With a pKa around 10, the phenolic hydroxyl group of tyrosine may not ionize easily under physiological conditions. Additionally, a lack of electron density for the loop region in the three available AAAD structures makes it difficult to accurately estimate the probable distance between the side chain hydroxyl group of tyrosine and the α-carbon of the external aldimine. Therefore, it is unknown whether the α-carbon and the catalytic loop tyrosine are within interacting distances.
Analysis of a recently released mammalian HDC (the only type II PLP decarboxylase with electron density for the catalytic loop) enables speculation on the interactions between the substrate α-carbon and the catalytic loop tyrosine (16). Careful examination of the HDC active site confirmation revealed that although its Tyr-344 from monomer B (the equivalent active site tyrosine in plant AAAD and mammalian DDC) is quite close to the α-carbon of the external aldimine complex (7.50 Å), its side chain hydroxyl group is actually much closer to the imidazole amine (ϵ-amine group) (4.42 Å) of monomer A His-194. Accordingly, within the HDC structure, the hydroxyl group of monomer B Tyr-344 should interact more easily with the imidazole of monomer A His-194 than it would with the α-carbon of the external aldimine complex. Such structural observations in conjunction with our experimental verifications of the catalytic loop tyrosine-mediated decarboxylation enable us to propose a new mechanism of α-carbon protonation. We believe that plant AAADs active site tyrosine does not directly protonate the intermediate α-carbon, but rather play a crucial role in stabilizing the proton transfer from His-194 to the α-carbon (in the HDC structure, Tyr-344 hydroxyl group is 4.39 Å from the histidine 194 N2). We propose that the phenolic hydroxyl group from the catalytic loop tyrosine forms a stabilizing hydrogen bond with the N2 histidine to enable the proton transfer from the histidine imidazole to the α-carbanion. The modeled active site conformation of C. roseus TDC and A. thaliana AAS (using PDB code 4E1O as template (16); generated through Modeler (23); visualized through PyMOL (20)) help illustrate the indirect role the active site tyrosine may play in stabilizing protonation of the α-carbon of the substrate-PLP complex through His-194 (Fig. 8). This proposed mechanism is further supported by the loss of decarboxylation activity in the tyrosine to phenylalanine AAAD mutants. Upon replacing the catalytic tyrosine with phenylalanine (Y348F mutation in C. roseus TDC, Y346F mutation in P. somniferum TyDC), there is no electronegative R group atom to stabilize the histidine proton transfer. Without such a stabilizing interaction, the substrate α-carbon is not protonated. This leaves the substrate carbanion susceptible to peroxidation and subsequent aromatic acetaldehyde formation.
FIGURE 8.
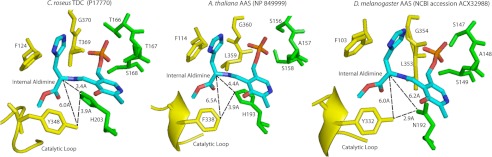
Models of active site residues and external aldimine interactions. The model on the left shows the relative locations of histidine 203, tyrosine 348, and the external aldimine from the C. roseus TDC model (generated using PDZB code 4E1O). The model in the middle shows the relative locations of histidine 193, phenylalanine 338, and the external aldimine from the A. thaliana AAS model (generated using PDB code 4E1O). The model on the right shows the relative locations of asparagine 192, tyrosine 332, and the external aldimine from the Drosophila melanogaster AAS model (generated using PDB code 4E1O). Residues shown in yellow correspond to the α chain, whereas residues in green correspond to the β chain.
The above hypothesis regarding the role of the active site tyrosine in α-carbon protonation (though reasonable) remains speculative in nature. However, the predicted active site conformation of insect AAS proteins provides a more compelling argument for our mechanistic proposal. Unlike plant AAS proteins that contain an active site phenylalanine, the equivalent position in insect AASs remains as a tyrosine (moreover, this tyrosine residue is stringently conserved in all available insect AAADs, regardless of their substrate specificity and catalytic reactions). This contrasts with what is observed in plant AAS proteins. However, analysis of the His-194 equivalent in insect AAAD and AAS determined that all typical insect AAADs contain the histidine in the same position, but the active site histidine was replaced by an asparagine in all verified insect AAS proteins. Asparagine has a greatly reduced likelihood of proton donation (nonionizable nature of its R group) as compared with histidine. Consequently, in the presence of an active site asparagine in insect AAS, the active site tyrosine does not seem to be able to play a stabilizing role in α-carbon proton transfer (Fig. 8). The presence of tyrosine and absence of histidine within insect AAS enzymes supports the role of tyrosine in promoting typical decarboxylation in plant AAADs through its interaction with the active site histidine. Through these analyses, it seems apparent that the active site tyrosine from insect AAAD and AAS proteins plays a different catalytic role from the tyrosine in plant AAADs. However, this by no means indicated that this active site tyrosine is not critical for insect AAAD and AAS substrate binding and catalysis. Proximity to the active site and the stringent conservation in all insect AAAD and AAS proteins indicates a probable role in substrate binding or catalysis.
Although our data suggested some apparent similarities in catalytic reactions of plant and non-plant AAADs, there are also considerable differences regarding the structural basis of substrate binding and catalysis. Mammalian DDC has been the most extensively studied AAAD in terms of substrate specificity and catalytic reactions. Based on literature, mammalian DDC can use both DOPA and 5-HTP as substrates (24, 25), but the plant AAADs or AASs use aromatic amino acids with either the indole ring or benzene ring, but never both (1, 8, 9). Other than its typical decarboxylation activity to DOPA and 5-HTP, mammalian DDC can also catalyze oxidative deamination of dopamine and 5-HT (26, 27). However, plant AAADs cannot catalyze any oxidative deamination of tyramine, dopamine, or tryptamine (analyzed with two available plant TyDC, one plant TDC, and three plant AASs in our laboratory). The same applies to the A. thaliana F338Y mutant as well. For example, at the applied assay conditions, essentially all DOPA substrate has been converted to dopamine after a 24-min incubation. HPLC-EC analyses showed no indication for any further oxidative deamination of dopamine product to its aldehyde derivative in the reaction mixture (Fig. 7C). This suggests that arylalkylamines, once released from the catalytic center of plant AAADs or AAS, cannot easily enter into their active site again, or that the external aldimine between the PLP cofactor and arylalkylamines cannot be easily formed at the active site of plant AAADs. Addition of tyramine at millimolar concentrations into the wild type P. crispum AAS and tyrosine reaction mixtures also did not noticeably slow down the AAS-catalyzed PHAA production from tyrosine. Additionally the tyramine levels remained at the same level during incubation (not shown), indicating the absence of tyramine oxidative-deamination. This reinforces the notion that the α-carboxyl group is essential for plant AAS substrate binding. Apparently, extensive studies about AAADs are necessary before being able to achieve a better overall understanding of the large AAAD family proteins across species.
The interconversion of AAAD and AAS activities enabled the generation of several unusual plant AAAD and AAS enzyme products. These products include 4-hydroxyphenylacetaldehyde, phenylethylamine, indole-3-acetaldehyde, and 5-hydroxy indole-3-acetaldehyde. 4-Hydroxyphenylacetaldehyde has only been documented as a product from a single AAS enzyme (12). Thus far the physiological function of this AAS product remains unknown. Phenylethylamine has only been observed as plant type II PLP decarboxylase product from a selection of Solanum lycopersicum enzymes (28). Plant AAAD enzymes typically retain ∼40% identity to one another, whereas these S. lycopersicum enzymes only demonstrate ∼11% identity to canonical AAADs. Further S. lycopersicum lycopersicum type II PLP decarboxylase sequence analysis demonstrates strong homology (∼55% identity) to the characterized A. thaliana (AF389349) and Brassica napus (BAA78331) serine decarboxylases (29). Therefore these S. lycopersicum enzymes cannot be viewed as canonical plant AAAD enzymes making phenylethylamine a truly unique plant AAAD product. Finally, the enzymatic conversion of tryptophan to indole-3-acetaldehyde and 5-hydroxytryptophan to 5-hydroxyindole-3-acetaldehyde by the C. roseus TDC mutant truly represents novel AAAD enzyme products. Among these unusual products, the C. roseus TDC mutant-generated indole-3-acetaldehyde is likely the most interesting. Indole-3-acetaldehyde is a proposed intermediate in the original tryptophan-dependent indole-3-pyruvic acid (IPA) auxin biosynthetic pathway (30, 31). Although recent biochemical and genetic data has demonstrated a clear alternate IPA pathway (tryptophan to IPA via tryptophan aminotransferases (32), and IPA to indole-3-acetic acid through YUCCA-catalyzed oxidative decarboxylation (33–35) (supplemental Fig. S4)), no experimental data has invalidated the role of indole-3-acetaldehydes as an alternate IPA pathway intermediate. Despite references regarding this putative auxin (indole-3-acetic acid) intermediate, no biosynthetic route has been identified. Although there have been verified tryptophan aminotransferases (32) capable of catalyzing the conversion of tryptophan to IPA, there have been no experimental data showing the presence of an IPA decarboxylase (IPA to indole-3-acetaldehyde) in plants. Interestingly, our mutant is capable of performing the function of a tryptophan aminotransferase and the putative IPA decarboxylase (converting tryptophan to indole-3-acetaldehyde) in one step (supplemental Fig. S4). The final auxin biosynthetic step in this proposed pathway involves the conversion of indole-3-acetaldehyde to indole-3-acetic acid. This reaction has been demonstrated by aldehyde oxidases (36, 37). Therefore, our TDC Y348F mutation could conceivably convert the standard TDC reaction into one capable of producing a putative intermediate for auxin biosynthesis. The introduction of a stable and active TDC Y348F mutant into wild type and auxin-deficient Arabidopsis plants should demonstrate the presence/absence of an IPA decarboxylase, demonstrate/disprove in vivo aldehyde oxidases activity, and produce insights into the putative role of indole-3-acetaldehyde as an auxin intermediate. Additionally, conversion of a regular TDC enzyme to a tryptophan aldehyde synthase only requires a single nucleotide substitution. One might think that somewhere and at some point throughout plant evolution this might have occurred and granted some level of reproductive fitness. Such an enzyme might have evolved in plants and may be producing an alternate auxin biosynthetic route (assuming the original IPA pathway is functional).
In summary, the AAAD family is a great example of protein functional expansion. Based on its ubiquitous distribution, it seems reasonable to consider that the typical DDC or TyDC, present from bacteria to humans, is the prototype of other more specific AAADs. Such divergent AAADs have likely evolved unique substrate specificities and activities to provide unique physiological requirements. The functions of AAADs vary considerably, depending upon their substrate specificity and catalytic reaction, but their high sequence similarity makes it extremely difficult to tell them apart based on primary sequences. Our data indicated that the presence of the conserved tyrosine or phenylalanine in the active site loop region could serve as signature residues for reliably predicting typical plant AAAD and AAS, respectively. This should help to more precisely distinguish between true AAAD and AAS sequences available in the database and to aid the proper annotation of numerous incoming plant AAAD sequences currently undergoing genome-sequencing projects. Our comparative analyses of active site conformations between different AAAD and AAS proteins lead to a new hypothesis concerning the roles the active site tyrosine and phenylalanine play in their typical decarboxylation reaction and decarboxylation-oxidative deamination reaction, respectively. Although the hypothesis remains to be further substantiated, the proposed mechanism gives some reasonable explanation and insight for AAAD- and AAS-catalyzed reactions, respectively, which should stimulate research in this area. In addition, the ability to catalyze tryptophan to indole-3-acetaldehyde suggests a useful research tool in the investigation of auxin biosynthesis. Overall, our progress in differentiating plant AAAD and AAS proteins is a major step forward toward proper classification/annotation of hundreds of individual plant AAADs in the database. However, much still remains to be established regarding the structural components and chemical mechanisms dictating the substrate specificity and variable activities of AAAD- and AAS-mediated reactions.
This work was supported by the Virginia Tech Department of Biochemistry, College of Agriculture and Life Sciences.

This article contains supplemental Table S1 and Figs. S1–S4.
- AAAD
- aromatic amino acid decarboxylase
- 5-HT
- 5-hydroxytryptamine
- TyDC
- tyrosine decarboxylase
- TDC
- tryptophan decarboxylase
- AAS
- aromatic acetaldehyde synthase
- PLP
- pyridoxal 5-phosphate
- HPAA
- p-hydroxyphenylacetaldehyde
- IPA
- indole-3-pyruvic acid
- DOPA
- 3,4-dihydroxyphenylalanine
- 5-HTP
- 5-hydroxytryptophan.
REFERENCES
- 1. Facchini P. J., Huber-Allanach K. L., Tari L. W. (2000) Plant aromatic l-amino acid decarboxylases. Evolution, biochemistry, regulation, and metabolic engineering applications. Phytochemistry 54, 121–138 [DOI] [PubMed] [Google Scholar]
- 2. Ellis B. E. (1983) Production of hydroxyphenylethanol glycosides in suspension-cultures of Syringa vulgaris. Phytochemistry 22, 1941–1943 [Google Scholar]
- 3. Leete E., Kirkwood S., Marion L. (1952) The biogenesis of alkaloids. VI. The formation of hordenine and N-methyltyramine from tyrosine in barley. Can. J. Chem. 30, 749–760 [Google Scholar]
- 4. Marques I. A., Brodelius P. E. (1988) Elicitor-induced l-tyrosine decarboxylase from plant cell suspension cultures. Plant Physiol. 88, 46–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Trezzini G. F., Horrichs A., Somssich I. E. (1993) Isolation of putative defense-related genes from Arabidopsis thaliana and expression in fungal elicitor-treated cells. Plant Mol. Biol. 21, 385–389 [DOI] [PubMed] [Google Scholar]
- 6. Meijer A. H., Verpoorte R., Hoge J. H .C. (1993) Regulation of enzymes and genes involved in terpenoid indole alkaloid biosynthesis in Catharanthus roseus. J. Plant Res. 3, 145–164 [Google Scholar]
- 7. Berlin J., Ruegenhagen C., Kuzovkina I. N., Fecker L. F., Sasse F. (1994) Are tissue cultures of Peganum harmala a useful model system for studying how to manipulate the formation of secondary metabolites? Plant Cell Tissue Organ Cult. 38, 289–297 [Google Scholar]
- 8. Lehmann T., Pollmann S. (2009) Gene expression and characterization of a stress-induced tyrosine decarboxylase from Arabidopsis thaliana. FEBS Lett. 583, 1895–1900 [DOI] [PubMed] [Google Scholar]
- 9. Noé W., Mollenschott C., Berlin J. (1984) Tryptophan decarboxylase from Catharanthus roseus cell suspension cultures. Purification, molecular and kinetic data of the homogenous protein. Plant Mol. Biol. 3, 281–288 [DOI] [PubMed] [Google Scholar]
- 10. Kaminaga Y., Schnepp J., Peel G., Kish C. M., Ben-Nissan G., Weiss D., Orlova I., Lavie O., Rhodes D., Wood K., Porterfield D. M., Cooper A. J., Schloss J. V., Pichersky E., Vainstein A., Dudareva N. (2006) Plant phenylacetaldehyde synthase is a bifunctional homotetrameric enzyme that catalyzes phenylalanine decarboxylation and oxidation. J. Biol. Chem. 281, 23357–23366 [DOI] [PubMed] [Google Scholar]
- 11. Gutensohn M., Klempien A., Kaminaga Y., Nagegowda D. A., Negre-Zakharov F., Huh J. H., Luo H., Weizbauer R., Mengiste T., Tholl D., Dudareva N. (2011) Role of aromatic aldehyde synthase in wounding/herbivory response and flower scent production in different Arabidopsis ecotypes. Plant J. 66, 591–602 [DOI] [PubMed] [Google Scholar]
- 12. Torrens-Spence M. P., Gillaspy G., Zhao B., Harich K., White R. H., Li J. (2012) Biochemical evaluation of a parsley tyrosine decarboxylase results in a novel 4-hydroxyphenylacetaldehyde synthase enzyme. Biochem. Biophys. Res. Commun. 418, 211–216 [DOI] [PubMed] [Google Scholar]
- 13. Vavricka C., Han Q., Huang Y., Erickson S. M., Harich K., Christensen B.M., Li J. (2011) From Dopa to dihydroxyphenylacetaldehyde. A toxic biochemical pathway plays a vital physiological function in insects. PLoS One 6, e16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park S., Kang K., Lee K., Choi D., Kim Y. S., Back K. (2009) Induction of serotonin biosynthesis is uncoupled from the coordinated induction of tryptophan biosynthesis in pepper fruits (Capsicum annuum) upon pathogen infection. Planta 230, 1197–1206 [DOI] [PubMed] [Google Scholar]
- 15. Kawalleck P., Keller H., Hahlbrock K., Scheel D., Somssich I. E. (1993) A pathogen-responsive gene of parsley encodes tyrosine decarboxylase. J. Biol. Chem. 268, 2189–2194 [PubMed] [Google Scholar]
- 16. Komori H., Nitta Y., Ueno H., Higuchi Y. (2012) Structural study reveals Ser-345 determines substrate specificity on human histidine decarboxylase. J. Biol. Chem. 287, 29175–29183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ko J. K., Ma J. (2005) A rapid and efficient PCR-based mutagenesis method applicable to cell physiology study. Am. J. Physiol. Cell Physiol. 288, C1273–1278 [DOI] [PubMed] [Google Scholar]
- 18. Han Q., Ding H., Robinson H., Christensen B. M., Li J. (2010) Crystal structure and substrate specificity of Drosophila 3,4-dihydroxyphenylalanine decarboxylase. PLOS One 5, e8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buteau C., Duitschaever C. L., Ashton G. C. (1984) High-performance liquid chromatographic detection and quantitation of amines in must and wine. J. Chromatogr. 284, 201–210 [DOI] [PubMed] [Google Scholar]
- 20. DeLano W. L. (2002) The PyMOL Molecular Graphics System, 0.99 Ed., DeLano Scientific, LLC, Palo Alto, CA [Google Scholar]
- 21. Eliot A. C., Kirsch J. F. (2004) Pyridoxal phosphate enzymes. Mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 73, 383–415 [DOI] [PubMed] [Google Scholar]
- 22. Bertoldi M., Gonsalvi M., Contestabile R., Voltattorni C. B. (2002) Mutation of tyrosine 332 to phenylalanine converts Dopa decarboxylase into a decarboxylation-dependent oxidative deaminase. J. Biol. Chem. 277, 36357–36362 [DOI] [PubMed] [Google Scholar]
- 23. Šali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 24. Udenfriend S., Clark C., Titus E. (1953) 5-Hydroxytryptophan decarboxylase. A new route of metabolism of tryptophan. J. Am. Chem. Soc. 75, 501–502 [Google Scholar]
- 25. Srinivasan K., Awapara J. (1978) Substrate specificity and other properties of DOPA decarboxylase from guinea pig kidneys. Biochim. Biophys. Acta 526, 597–604 [DOI] [PubMed] [Google Scholar]
- 26. Bertoldi M., Moore P. S., Maras B., Dominici P., Voltattorni C. B. (1996) Mechanism-based inactivation of Dopa decarboxylase by serotonin. J. Biol. Chem. 271, 23954–23959 [DOI] [PubMed] [Google Scholar]
- 27. Bertoldi M., Dominici P., Moore P. S., Maras B., Voltattorni C. B. (1998) Reaction of Dopa decarboxylase with α-methyldopa leads to an oxidative deamination producing 3,4-dihydroxyphenylacetone, an active site-directed affinity label. Biochemistry 37, 6552–6561 [DOI] [PubMed] [Google Scholar]
- 28. Tieman D., Taylor M., Schauer N., Fernie A. R., Hanson A. D., Klee H. J. (2006) Tomato aromatic amino acid decarboxylases participate in synthesis of the flavor volatiles 2-phenylethanol and 2-phenylacetaldehyde. Proc. Natl. Acad. Sci. U.S.A. 103, 8287–8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rontein D., Nishida I., Tashiro G., Yoshioka K., Wu W. I., Voelker D. R., Basset G., Hanson A. D. (2001) Plants synthesize ethanolamine by direct decarboxylation of serine using a pyridoxal phosphate enzyme. J. Biol. Chem. 276, 35523–35529 [DOI] [PubMed] [Google Scholar]
- 30. Woodward A. W., Bartel B. (2005) Auxin. Regulation, action, and interaction. Ann. Bot. 95, 707–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Koga J., Adachi T., Hidaka H. (1992) Purification and characterization of indolepyruvate decarboxylase. A novel enzyme for indole-3-acetic acid biosynthesis in Enterobacter cloacae. J. Biol. Chem. 267, 15823–15828 [PubMed] [Google Scholar]
- 32. Stepanova A. N., Robertson-Hoyt J., Yun J., Benavente L. M., Xie D. Y., Dolezal K., Schlereth A., Jürgens G., Alonso J.M. (2008) TAA1-mediated auxin biosynthesis is essential for hormone crosstalk and plant development. Cell 133, 177–191 [DOI] [PubMed] [Google Scholar]
- 33. Stepanova A. N., Yun J., Robles L. M., Novak O., He W., Guo H., Ljung K., Alonso J. M. (2011) The Arabidopsis YUCCA1 flavin monooxygenase functions in the indole-3-pyruvic acid branch of Auxin biosynthesis. Plant Cell 23, 3961–3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mashiguchi K., Tanaka K., Sakai T., Sugawara S., Kawaide H., Natsume M., Hanada A., Yaeno T., Shirasu K., Yao H., McSteen P., Zhao Y., Hayashi K., Kamiya Y., Kasahara H. (2011) The main auxin biosynthesis pathway in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 108, 18512–18517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dia X., Mashiguchi K., Chen Q., Kasahara H., Kamiya Y., Ojha S., DuBois J., Ballou D., Zhao Y. (November 27, 2012) The biochemical mechanism of auxin biosynthesis by an Arabidopsis YUCCA flavin-containing monooxygenase. J. Biol. Chem. 287, 10.1074/jbc.M112.424077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seo M., Akaba S., Oritani T., Delarue M., Bellini C., Caboche M., Koshiba T. (1998) Higher activity of an aldehyde oxidase in the auxin-overproducing superroot1 mutant of Arabidopsis thaliana. Plant Physiol. 116, 687–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sekimoto H., Seo M., Kawakami N., Komano T., Desloire S., Liotenberg S., Marion-Poll A., Caboche M., Kamiya Y., Koshiba T. (1998) Molecular cloning and characterization of aldehyde oxidases in Arabidopsis thaliana. Plant Cell Physiol. 39, 433–442 [DOI] [PubMed] [Google Scholar]