

Background: Many studies analyzing K-Ras function rely on overexpression.
Results: Knock-out and knock-in of endogenous mutant K-ras have modest effects on downstream signaling but strong effects on gene expression and transformation.
Conclusion: Genomic manipulation allows physiological determination of WT and mutant K-Ras consequences.
Significance: Gene expression patterns can be used to monitor inhibition of mutant K-Ras.
Keywords: Cell Signaling, Gene Knock-out, MAP Kinases (MAPKs), Oncogene, Ras, Transcription Target Genes, Isogenic
Abstract
To assess the consequences of endogenous mutant K-Ras, we analyzed the signaling and biological properties of a small panel of isogenic cell lines. These include the cancer cell lines DLD1, HCT116, and Hec1A, in which either the WT or mutant K-ras allele has been disrupted, and SW48 colorectal cancer cells and human mammary epithelial cells in which a single copy of mutant K-ras was introduced at its endogenous genomic locus. We find that single copy mutant K-Ras causes surprisingly modest activation of downstream signaling to ERK and Akt. In contrast, a negative feedback signaling loop to EGFR and N-Ras occurs in some, but not all, of these cell lines. Mutant K-Ras also had relatively minor effects on cell proliferation and cell migration but more dramatic effects on cell transformation as assessed by growth in soft agar. Surprisingly, knock-out of the wild type K-ras allele consistently increased growth in soft agar, suggesting tumor-suppressive properties of this gene under these conditions. Finally, we examined the effects of single copy mutant K-Ras on global gene expression. Although transcriptional programs triggered by mutant K-Ras were generally quite distinct in the different cell lines, there was a small number of genes that were consistently overexpressed, and these could be used to monitor K-Ras inhibition in a panel of human tumor cell lines. We conclude that there are conserved components of mutant K-Ras signaling and phenotypes but that many depend on cell context and environmental cues.
Introduction
Ras was one of the first human oncogenes to be discovered and remains one of the most frequently mutated genes across multiple human tumors. Ras is a small membrane-bound GTP-binding protein that recruits effector proteins such as Raf, phosphoinositide 3-kinase, and RalGDS when bound to GTP, resulting in activation of these proteins and their downstream signaling events. Activation of Ras causes a wide variety of biological consequences depending on the cell type and stimuli, including increased cell growth, proliferation, survival, differentiation, and morphogenesis. Ras is able to hydrolyze GTP to GDP, resulting in its self-inactivation, although this process is slow and is accelerated by a family of proteins termed GTPase-activating proteins (GAPs). Intrinsic exchange of GDP to GTP is also quite slow, and this process is accelerated by a family of proteins termed guanine nucleotide exchange factors (GEFs). Through the regulation of GAP and GEF activities, the GTP levels of Ras proteins are carefully adjusted depending on environmental conditions, resulting in the maintenance of appropriate cellular homeostasis. Mutations of Ras that occur in human tumors disrupt this process by preventing both intrinsic and GAP-mediated hydrolysis, causing constitutive GTP association and uncontrolled signaling and proliferation (for review, see Ref. 1).
Three ras genes are frequently mutated in human tumors, with characteristic frequencies in different tissues. H-ras is mutated in bladder cancer (∼10%) and salivary gland tumors (∼15%), N-ras is mutated in melanoma (∼20%) and hematopoietic tumors (∼15%), and K-ras is predominantly mutated in pancreatic ductal adenocarcinoma (∼60%), colorectal cancer (∼30%), non-small cell lung adenocarcinoma (∼20%), and endometrial cancer (∼15%) (frequencies as documented on the Sanger Center COSMIC website). ras mutations can be both prognostic for overall poor survival (2), as well as predictive for lack of response to chemotherapy and targeted therapies (3), showing the clinical importance of this gene. Genetically engineered mouse models have also confirmed that a single point mutation in K-ras or N-ras expressed in the relevant tissue is sufficient to develop disease that strongly resembles human tumors (4–6).
In addition to the well described pro-tumorigenic effects of mutant Ras alleles, there is some suggestive evidence that the wild type (WT) copy of Ras may possess tumor-suppressive effects. This was first noticed when mice expressing one copy of K-Ras generated more and larger lung tumors following chemical carcinogen treatment than mice expressing both alleles (7). A tumor-suppressive effect for N-Ras has also been noted in some mouse tumor models although this seems to be context-dependent (for review, see Ref. 8). Such a tumor-suppressive effect of WT Ras alleles is not as well characterized in human cancers.
The ability of mutant ras genes to activate downstream signaling pathways that contribute to cell transformation is mostly undisputed, although almost all of the studies drawing this conclusion have been performed using model systems that vastly overexpress the mutant ras gene, usually involving H-Ras. However, it has more recently become clear that overexpression of mutant ras has qualitatively different consequences compared with single copy gene mutations that presumably arise during the early stages of human tumorigenesis and that cell type also plays an important role in determining cellular responses. For example, overexpression of HRasV12 in immortalized mouse NIH3T3 cells causes transformation associated with activation of Raf and PI3K pathways (9), whereas overexpression of HRas in normal fibroblasts causes a senescent-like cell cycle arrest (10). In contrast, knock-in of a single copy of mutant K-ras into nontransformed mouse or human cells causes only very modest consequences on downstream signaling (11–14). The consequences on cell transformation (15) or tumor formation (11, 12) can also be surprisingly mild in the absence of additional genetic alterations. Moreover, the assumption of the strong transforming potential of mutant Ras needs to be reassessed in light of recent discoveries that certain developmental disorders such as Costello syndrome, cardiofacial cutaneous syndrome, and Noonans syndrome are caused by germ line-activating mutations in ras (for review, see Ref. 16).
In this study, we examined the consequences of single copy K-ras mutations in the context of human cancer cell lines, as well as in immortalized but nontransformed human mammary epithelial cells (HMECs).2 We utilized cell line derivatives in which the mutant or WT K-ras alleles had been deleted using targeted homologous recombination, or in which a single copy of mutant K-ras had been introduced using the same technology. We found that although mutant K-Ras has strong effects on cellular RasGTP levels, it has surprisingly mild consequences on downstream signaling through Raf and PI3K pathways. Mutant K-Ras is also able to initiate negative feedback signaling to the EGF receptor, which may have relevance in the response of K-Ras mutant tumors to EGFR inhibitors. Despite mild consequences on cellular signaling, flux through these pathways is likely altered as demonstrated by robust regulation of a small number of genes that seem to be consistently up-regulated by mutant K-Ras. We also demonstrate that the WT K-Ras allele may behave in a tumor-suppressive manner in the cancer cell lines analyzed.
EXPERIMENTAL PROCEDURES
Cell Culture and Lysate Preparation
Hec1A, HCT116, and SW48 cells were cultured in McCoy's 5A (Invitrogen) with 10% FBS and 2 mm l-glutamine. DLD1 cells were grown in RPMI 1640 medium with 10% FBS and 2 mm l-glutamine, and HMECs were cultured in 50:50 DMEM/F12 with 10% FBS, 20 ng/ml EGF, 0.01 mg/ml insulin, 500 ng/ml hydrocortisone, and 2 mm l-glutamine. Cells were cultured in a 37C/5% CO2 incubator. For standard Western blotting and immunoprecipitations, cells were harvested in radioimmuneprecipitation assay buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 2 mm EDTA, 1% Nonidet P-40, 0.1% SDS) with Complete Protease Inhibitor (Roche Applied Science) and Phosphatase Inhibitor Mixture II (Sigma). For Ras binding domain (RBD) pulldown experiments, cells were harvested in 1× MgCl buffer (Millipore) with Complete Protease Inhibitor.
siRNA Transfections
Cells were grown to 80% confluence in 10-cm dishes and transfected with nontargeting control or K-Ras OTP siRNAs (Dharmacon). Briefly, 900 pmol of siRNA and 20 μl of RNAiMax (Invitrogen) were diluted separately in 1.5 ml of OptiMEM each. The siRNA and lipid were then mixed together and incubated for 20 min at room temperature. The complexes (3 ml) were added to the cells and incubated for 72 h.
Immunoprecipitations/Immunoblots
To check for siRNA silencing of Ras, 2.5 mg of lysate was incubated with 10 μl of Pan Ras antibody EP1125Y (Millipore 04-1039) at 4 ºC overnight. 50 μl of protein A bead slurry was then added and incubated for 2 h at 4 ºC with rotation. Beads were washed three times with lysis buffer and resuspended in sample buffer. RBD pulldowns were performed using a Ras Activation Assay kit (Millipore 17-218) according to the manufacturer's protocol. Western blot samples were run on 4–20% Tris-glycine gels and transferred to PVDF membranes using the iBlot system (Invitrogen). Pan Ras immunoprecipitations were blotted for K-Ras (Santa Cruz sc-30). RBD immunoprecipitations were blotted for Pan Ras (Millipore 05-516), H-Ras, N-Ras, and K-Ras (Santa Cruz sc-29, sc-31, and sc-30, respectively). Cell lysates were also blotted for pERK (Cell Signaling 9101s), pAkt (Cell Signaling 9271s), pCbl (Cell Signaling 3555s), pEGFR-Y1125 (Abgent AP3376a), pEGFR-Y1069 (Upstate 07-715), total EGFR (Cell Signaling 4405s), total Cbl (Cell Signaling 2179s), β-actin (Sigma A2228), total Ras (Millipore 05-516), total ERK (Cell Signaling 9102s), and total Akt (Cell Signaling 4685s). In specified experiments, cells were treated with GDC0941 (Fisher RG007-25MG) or PD0325901 (Fisher RP02-10MG) diluted in dimethyl sulfoxide.
Soft Agar Assays
Base agar layer was prepared by mixing equal parts 2× medium + 20% FBS with 2.4% agarose. 1 ml of this base agar was then added to each well of a 12-well tissue culture plate (Grenier) and placed in a 4 ºC refrigerator to cool. Cells were trypsinized and counted, then diluted to 5000 cells/333 μl of 1× medium + 10% FBS. A 1:1 mixture of 2× medium + 20% FBS and 2.4% agarose was then prepared, and 667 μl was added to the cells, for a final agarose concentration of 0.8% in the cell layer. 1 ml of cell mixture was then pipetted on top of the base agar layer and placed at 4 ºC for 15 min to cool. The plates were placed in a 37 ºC/5% CO2 incubator overnight, and 1 ml of medium was added on top of the cell layer the next day. These assays were set up in triplicate.
Exome Alignment and Variant Calling
Illumina fastq reads were mapped to the UCSC human reference genome (GRCh37/hg19) using BWA (17) set to default parameters. Duplicate marking, local realignment, and variant calling was done as described previously (18). Variants included in dbSNP Build 131 (19) that were also absent in COSMIC v56 (20) were excluded.
Proliferation/Migration Assays
For proliferation assays, cells were trypsinized, counted, and plated at 2500 or 5000 cells in 150 μl/well, in a 96-well clear-bottomed black tissue culture plate (BD Falcon). Cells were left to proliferate over 7 days in an Incucyte instrument, which calculates cell density over specified intervals.
For migration assays, cells were plated the night before at 50,000 cells/well in a fibronectin- or collagen-coated ImageLock 96-well plate (Essen Bioscience). Cells were grown to confluence, then a wound was created using the WoundMaker apparatus (Essen Bioscience) according to the manufacturer's recommended protocol. The cells were then washed twice with PBS and medium replaced to 100 μl. Plates were imaged over 24 h at 2-h intervals.
Taqman Assays
RNA was isolated from cells using the RNeasy kit (Qiagen 74106), and cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems 4368814). Taqman assays were set up in multiplex, using GAPDH as the endogenous control. The target probes (DUSP5, DUSP6, ETV1, ETV5, NT5E, UPP1, K-ras, and IER3) were tagged with FAM dye. Assays were run in 384-well format, using 20 ng of input cDNA/reaction.
Additional procedures are described in the supplemental Materials and Methods.
RESULTS
To assess the contribution of single copy K-Ras mutations across multiple cell lines, we analyzed three previously published tumor cell lines in which the wild type or mutant K-ras allele was disrupted by homologous recombination. These cell lines are the colorectal cancer lines DLD1 and HCT116 (21, 22), which express heterozygous G13D mutations, and the endometrial cancer line Hec1A (23), which expresses a heterozygous G12D mutation. In addition, we also analyzed two cell lines in which a single copy of G13D mutant K-ras was introduced at its endogenous locus, a colorectal cancer cell line SW48 which evolved in the absence of mutant Ras, and nontransformed but immortalized HMECs (15). Whole genome copy number analysis shows that for the lines analyzed (DLD1 and Hec1A), although not truly “isogenic,” there are only very few and relatively minor differences at the chromosomal level between the parental lines and their isogenic derivatives (supplemental Fig. 1).
First we assessed the levels of RasGTP in these cell lines growing in 10% FBS, under starved conditions, and following short term stimulation with EGF. The RBD of cRaf was used as an affinity probe for RasGTP, as described previously (24). In all cases, knock-out and knock-in of mutant K-Ras decreased and increased KRasGTP levels, respectively (Fig. 1A). Interestingly, the presence or absence of mutant K-Ras affected NRasGTP levels; in HCT116 and SW48 cells, the presence of mutant K-Ras was associated with increased NRasGTP levels. This could be due to the previously described positive feedback loop caused by allosteric activation of the Ras exchange factor sos (25). In contrast, in Hec1A, HMECs, and to a lesser extent DLD1 cells, the presence of mutant K-Ras was associated with decreased NRasGTP levels, suggesting the presence of a negative feedback loop. We also analyzed the levels of HRasGTP using the same approach, but we did not detect any H-Ras present in the RBD pulldowns under the conditions tested (data not shown).
FIGURE 1.

Consequences of mutant K-ras knock-out and knock-in on RasGTP levels and downstream signaling. A, cell lines of the indicated genotypes were grown in 10% FBS, serum-starved for 18 h, or stimulated for 10 min with 100 ng/ml EGF. RasGTP was affinity-purified from 2.5 mg of cell lysate using GST-Raf-RBD and analyzed by Western blotting with Ras isoform-specific antibodies. In addition, 40 μg of lysate was used for Western blotting for total Ras protein. B, the indicated cell lines were treated as above, and 40 μg of protein lysates separated by SDS-PAGE and analyzed by Western blotting with anti-phospho-ERK, phospho-Akt, total ERK, total Akt, and β-actin antibodies.
The effects of mutant K-Ras on downstream signaling to ERK and Akt were examined next. In contrast to the dramatic effects on KRasGTP levels, the presence or absence of mutant K-Ras had very modest effects on ERK and Akt phosphorylation, under conditions of either basal proliferation in 10% FBS, serum starvation, or acute stimulation with EGF (Fig. 1B). Hec1A cells lacking mutant K-Ras actually showed slightly increased phosphorylation of these proteins following EGF stimulation, a paradoxical observation previously noted by Waldman and colleagues (23).
To better understand the impact of single copy mutant K-Ras on cell signaling, we performed a global phosphotyrosine analysis in the Hec1AG12D/− and Hec1A−/WT derivative cell lines using Epitome phosphotyrosine arrays. Similar to the effects seen by Western blot analysis, EGF-stimulated ERK1 and ERK2 phosphorylation was higher in the KRasG12D-deleted cells (supplemental Fig. 2A). The most dramatically altered proteins between these cell lines were EGFR and its downstream substrate Cbl (Fig. 2A). Both of these proteins showed increased phosphorylation in response to EGF in the mutant K-Ras knock-out clones compared with the WT K-Ras knock-out clone. We confirmed these results by Western blot analysis (Fig. 2B), which also analyzed the individual phosphorylation sites, Tyr-1125 (a Grb2 binding site) and Tyr-1069 (a Cbl binding site). To determine whether the inhibitory effect of mutant K-Ras on EGFR phosphorylation was a general phenomenon, we examined the additional cell lines for basal and stimulated EGFR and Cbl phosphorylation. DLD1 cells lacking mutant K-Ras also showed increased EGFR phosphorylation at Tyr-1125, even as total EGFR levels were decreased in this cell line derivative (Fig. 2B). In addition, Cbl also showed increased phosphorylation in the K-Ras knock-out DLD1 lines, although this was more apparent under basal or starved conditions (Fig. 2B). HMECs expressing mutant K-Ras also showed decreased EGFR phosphorylation (Fig. 2C). Increased EGFR phosphorylation was also seen in independently derived Hec1A WT and mutant K-Ras knock-out clones (supplemental Fig. 2B), showing that these effects are not due to clonal artifacts. However, neither HCT116 nor SW48 cell isogenic lines showed any obvious differences in EGFR or Cbl phosphorylation in the K-Ras knock-out or knock-in derivative cell lines (supplemental Fig. 2, C and D). Therefore, mutant K-Ras negatively regulates EGFR phosphorylation, as well as NRasGTP levels, in some cell lines.
FIGURE 2.

Mutant KRas negatively regulates EGFR signaling. A, Hec1A cells deleted for either WT or mutant KRas were serum-starved or starved and stimulated with 100 ng/ml EGF for 10 min. 1 mg of lysate was reduced, alkylated, digested with Lys-C, and used to probe antibody microarrays. Signal for EGFR and Cbl phosphorylation (mean ± S.D. (error bars, n = 3)) is shown. B, the indicated cell lines were cultured in 10% FBS, 0% FBS, or stimulated with 100 ng/ml EGF, and 40 μg of protein lysate was separated by SDS-PAGE and probed by Western blotting using the antibodies shown. C, results are the same as for B but using HMECs of the indicated genotypes.
To test the hypothesis that signaling downstream of mutant K-Ras might be responsible for suppressing EGF-induced EGFR phosphorylation, we utilized small molecule inhibitors of MEK and PI3K in the mutant K-Ras-expressing cells. Fig. 3A shows that PD0325901 was able to dramatically increase the response to EGF in the mut/WT and mut/− Hec1A and DLD1 cells. A smaller increase was also seen with GDC-0941. PD0325901 also increased EGFR phosphorylation in the Hec1A−/WT cell line, although this was not seen with GDC-0941 or by either compound in the DLD−/WT cells. Interestingly, NRasGTP levels were also increased with these inhibitors, suggesting a mechanistic link between EGFR phosphorylation and NRasGTP levels. To determine the nature of mutant K-Ras-induced EGFR inhibition, we asked whether conditioned media from mut/− cells could inhibit EGFR phosphorylation in −/WT cells. Supplemental Fig. 3 shows that there is a slight inhibition of EGFR phosphorylation in cells in Hec1A−/WT cells using conditioned media from Hec1Amut/− cells, but not in the similar DLD1 cell conditioned media swap. These results suggest that activation of ERK downstream of mutant K-Ras results in a signal that antagonizes response to EGF, which is partially (for Hec1A cells) as a result of a secreted factor. The use of a cytokine array did not help to clarify which secreted factor might be responsible for this (data not shown).
FIGURE 3.

Signaling through the MEK and PI3K pathways negatively regulates EGFR and NRas activity. Hec1A and DLD1 cells of the indicated genotypes were plated in 6-well dishes and at 70% confluence treated with the indicated concentration of the MEK inhibitor PD0325901 and the PI3K inhibitor GDC-0941. 18 h later, the cells were lysed, and 40 μg was subjected to SDS-PAGE and Western blotting using the antibodies shown.
We next examined the effects of endogenous mutant K-Ras on cellular phenotypes. Cell proliferation was not dramatically affected in any of the cell lines examined, although DLD1−/WT cells showed slightly decreased proliferation relative to parental and DLD1G12D/− clones, and SW48 cells showed slightly increased proliferation when mutant K-Ras was introduced (Fig. 4A). We then analyzed cell migration using a classic scratch wound-healing assay. DLD1 isogenic cell lines cells showed little consistent differences in cell migration, whereas Hec1A cells showed a slight decrease in migration upon loss of the mutant K-ras allele, but increased migration upon loss of the WT K-ras allele. Knock-in of mutant K-ras strongly decreased the migration of HMECs, which showed the fastest migration capacity of the cell types examined, with the wound being completely closed in 20 h (Fig. 4B). The migration of SW48 and HCT116 cells could not be assessed using this assay, as HCT116 cells did not remain attached to the plate during the wound-making process, and SW48 cells did not form a sufficiently confluent monolayer.
FIGURE 4.
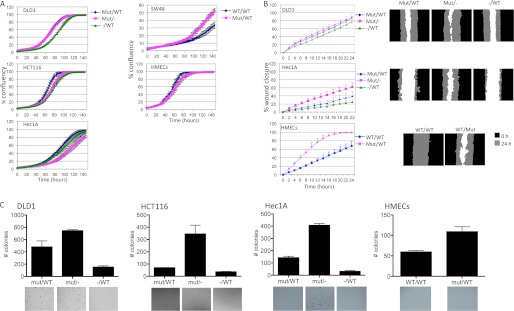
K-Ras status has modest effects on cell proliferation and migration but more dramatic effects on colony formation. A, the indicated isogenic cell lines were plated in 96-well plates at 5000 cells/well in 10% FBS, and confluence was tracked over 7 days using an Incucyte instrument. The means ± S.D. (error bars, n = 3) at 2-h intervals are shown. B, cell migration was analyzed by creating a scratch in the confluent cell monolayer and following wound closure over 24 h using an Incucyte instrument. The left panel shows the percentage wound closure measured at 2-h intervals (mean ± S.D., n = 3), and the right panels show images of the wound at 0 and 24 h. C, the isogenic cell lines were seeded in a 0.8% agar/growth medium layer, and colony formation was analyzed after 30 days. The graphs show the average ± S.D. of triplicate wells counted using a GelCount instrument, and the images show representative sections of one well.
We analyzed the transforming capacity of these cell lines using anchorage-independent growth in soft agar. As expected, and as previously reported, deletion of the mutant K-ras allele strongly decreased the number of colonies in soft agar in DLD1, HCT116, and Hec1A cells (Fig. 4C). There was also a modest increase in the number of colonies in the HMECs expressing mutant K-Ras, although the number of colonies in this line was lower than seen in the cancer cell lines. Interestingly, the number of colonies was significantly increased in all three cell lines lacking the wild type K-ras allele. SW48 cells did not form colonies under these conditions in our hands.
To attempt to understand why deletion of the WT K-ras allele could enhance soft agar growth, we compared the signaling pathways affected by this in Hec1A and HCT116 cells, the cells that were most dramatically affected in this assay. Supplemental Fig. 4 shows that of 45 phosphorylation sites on signaling proteins, only P-ERK (in Hec1A cells) and P-Akt (in HCT116 cells) showed any differences. To determine the frequency of WT K-ras allele loss in human tumor cell lines, we assessed the K-ras mutant allele frequency in a panel of 624 cell lines which had been subjected to exome-capture and deep sequencing.3 Of 130 cell lines with KRas variants, 35 showed KRas variant DNA read frequencies consistent with homozygous mutant K-ras status (Table 1). Moreover, even human tumor cell lines classified as heterozygous for mutant K-ras DNA frequently showed preferential expression of the mutant K-ras allele, as assessed by combined exome-seq and RNAseq analysis (Fig. 5). Combined, these data suggest that there is selective pressure to lose or reduce expression of the WT K-ras allele, at least in human cancer cell lines.
TABLE 1.
Frequency of heterozygous (Het) and homozygous (Hom) K-ras mutations in a panel of cell lines
| Gene | Mutation | Count | Het/Hom (count) | Tissue (counts) | Average variant allele frequency |
|---|---|---|---|---|---|
| K-ras | G12D | 40 | Het (37), Hom (3) | Stomach (1), lung (2), pancreas (19), uterus (10), ovary (2), colon (5), breast (1) | 0.52 |
| K-ras | G12V | 22 | Hom (9), Het (13) | Lung (4), colon (4), pancreas (13), breast (1) | 0.75 |
| K-ras | G12C | 16 | Hom (9), Het (7) | Lung (11), rectum (1), colon (2), ovary (1), urinary bladder (1) | 0.80 |
| K-ras | G13D | 15 | Het (14), Hom (1) | Lung (4), breast (1), colon (6), blood (1), ileum (1), lymph node (2) | 0.43 |
| K-ras | G12R | 7 | Het (6), Hom (1) | Pancreas (5), skin (1), breast (1) | 0.65 |
| K-ras | G12A | 6 | Hom (1), Het (5) | Blood (4), lung (1), colon (1) | 0.54 |
| K-ras | Q61H | 5 | Hom (5) | Lung (3), kidney (1), pancreas (1) | 1 |
| K-ras | G13C | 4 | Hom (1), Het (3) | lung (3), ovary (1) | 0.61 |
| K-ras | A146T | 3 | Het (2), Hom (1) | Blood (1), bone marrow (1), cecum (1) | 0.67 |
| K-ras | G12S | 2 | Hom (1), Het (1) | Lung (1), bone (1) | 0.69 |
| K-ras | Q61K | 2 | Hom (1), Het (1) | Stomach (1), lung (1) | 0.83 |
| K-ras | Q61L | 2 | Hom (1), Het (1) | Lung (1), colon (1) | 0.79 |
| K-ras | A146V | 1 | Het (1) | Blood (1) | 0.75 |
| K-ras | A59T | 1 | Het (1) | Bone (1) | 0.45 |
| K-ras | K117N | 1 | Hom (1) | Blood (1) | 1 |
| K-ras | L19F | 1 | Het (1) | Lung (1) | 0.51 |
| K-ras | P121H | 1 | Het (1) | Ovary (1) | 0.52 |
| K-ras | Y64C | 1 | Het (1) | Colon (1) | 0.48 |
FIGURE 5.

WT K-ras is either deleted or underexpressed in the majority of mutant KRas-expressing cell lines. The read counts of WT (y axis) and mutant (x axis) K-ras using RNA-seq data from 88 mutant K-Ras cell lines are plotted. Lines designated as homozygous mutant K-Ras are shown in blue; lines designated as heterozygous mutant K-Ras are shown in red.
To address whether the signaling and phenotypic consequences of chronic loss of mutant K-ras are contributed by either clonal anomalies or long term adaptation, we also analyzed the effects of short term K-Ras knockdown using siRNA. Knockdown of K-Ras (WT and mutant) in these cell lines showed similar modest effects on ERK and/or Akt phosphorylation in the basal and starved settings (Fig. 6, A–C). Interestingly, the increased EGFR phosphorylation seen in the K-Ras knock-out DLD1 and Hec1A cells was also seen following K-Ras knockdown, although the magnitude of this effect was diminished (Fig. 6, A and C). Transient knockdown of K-Ras in HCT116 cells also increased EGFR phosphorylation, despite incomplete knockdown efficiency in this cell line (Fig. 6B). The effects of K-Ras knockdown on proliferation and migration were also assessed. K-Ras knockdown had little effect on proliferation in any cell line (supplemental Fig. 5A), similar to the results with the mutant K-Ras knock-out derivatives. Knockdown of K-Ras in DLD1 cells had little effect on migration, whereas K-Ras knockdown decreased migration of Hec1A cells (supplemental Fig. 5B).
FIGURE 6.

K-Ras siRNA knockdown in parental cell lines shows similar effects as chronic mutant K-Ras deletion. A, 100 nm K-Ras siRNA pools were transfected into DLD1 cells and treated as shown. 72 h following transfection, 2.5 mg of protein lysates was subjected to pulldown with GST-RBD glutathione bead slurry, and total RasGTP and NRasGTP were analyzed by Western blotting with the indicated antibodies. Additionally, 40 μg of protein lysate was separated by SDS-PAGE and analyzed by Western blotting using the indicated antibodies. B and C, HCT116 and Hec1A cells were treated as described in A.
We then asked whether a combined analysis of K-Ras isogenic cell lines could be used to identify genes that are commonly regulated by mutant K-Ras expression. We profiled the Hec1A, DLD1, and SW48 isogenic cell lines on Affymetrix human genome U133 Plus microarrays. Knock-out or knock-in of K-ras did not have strong effects on global gene expression, cell lines clustered by parental cell line and not K-Ras genotype (Fig. 7A). Analysis of multiple individual clones of the same genotype (only performed for Hec1A cells) showed that there were only small differences that could be attributed to clone-specific alterations and that independent genotypically matched lines clustered together. Interestingly, WT K-ras-deleted lines clustered more closely to parental cell lines than mutant Kras-deleted lines, suggesting that in each cell line the mutant KRas allele has a stronger effect on global gene expression than the WT allele. The lists of genes affected by mutant K-Ras expression in these three cell lines are shown in supplemental Table 1. In general, the spectrum of genes regulated by the presence or absence of mutant K-Ras in the three different cell lines was quite distinct, as seen by overlaying the most significant up- and down-regulated genes from DLD1 cells onto Volcano plots of gene expression in the three cell lines (Fig. 7B). Similar disparate observations were also seen with the up- and down-regulated genes from Hec1A and SW48 cells (supplemental Fig. 6). Nevertheless, we searched for genes that displayed common regulation in response to mutant K-Ras and found two genes (NT5E and ETV1) that were commonly up-regulated by mutant K-Ras in all three cell lines (Fig. 7C). We also found several genes that were commonly up-regulated by mutant K-Ras in Hec1A and DLD1 cells, including ETV5, UPP1, IER3, DUSP5, and DUSP6.
FIGURE 7.
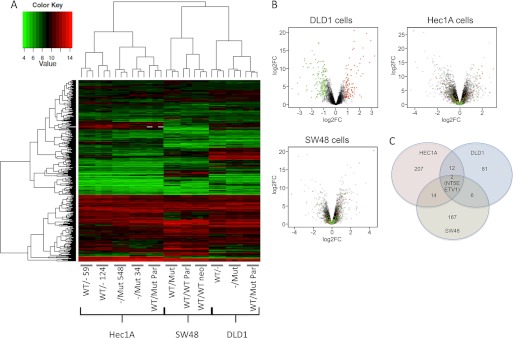
Mutant K-Ras initiates distinct gene transcription programs that are generally cell line-dependent. A, RNA from triplicate cultures of the indicated cell lines was hybridized onto Affymetrix U133 microarrays. The 403 genes that were differentially expressed in at least two cell lines were clustered using the complete linkage method as implemented in the function hclust. B, genes that were regulated by mutant K-Ras in DLD1 cells were overlaid onto all genes differentially regulated by mutant K-Ras in DLD1 cells (left plot), Hec1A cells (middle plot), and SW48 cells (right plot). C, genes up-regulated or down-regulated by mutant K-Ras expression >2-fold in different cell lines are shown.
We confirmed the mutant K-Ras-dependent regulation of these genes by Taqman analysis in these cell lines (Fig. 8A). All genes showed similar regulation by Taqman compared with the array; in addition although IER3, ETV5, and UPP1 were not identified as up-regulated by mutant K-Ras in SW48 cells by microarray, they did show up-regulation by Taqman. We next asked whether these genes could more generally be used as markers for K-Ras inhibition in a panel of K-Ras mutant cell lines and additionally asked whether they were also affected by K-Ras knockdown in WT K-Ras cell lines. K-Ras siRNA effectively decreased K-Ras expression in all of the lines examined (although knockdown was modest in HupT3 and HT55 cells) and decreased P-ERK in the mutant K-Ras-expressing cell lines (Fig. 8B). P-ERK was not affected in the K-Ras WT cell lines, nor was P-Akt decreased in any cell line. The basal expression of these genes in this panel of cell lines did not correlate with mutant K-Ras status (data not shown). However, all of the mutant K-Ras-specific genes identified from the isogenic cell lines were generally decreased following K-Ras knockdown in the mutant K-Ras-expressing cell lines. In contrast, their expression in the WT K-Ras cells was much less affected, confirming their usefulness as general markers for inhibition of mutant K-Ras signaling (supplemental Fig. 7 and summarized in Fig. 8C). This effect was statistically significant for all genes apart from ETV1, which had low/undetectable expression in this experiment in two cell lines.
FIGURE 8.

Expression of 7 genes are commonly regulated by mutant K-Ras. A, RNA was purified from the indicated cell lines and subjected to Taqman quantitative PCR analysis against the genes shown in each graph. Expression was normalized to the control gene GAPDH and expressed (mean ± S.D. (error bars), n = 3) relative to the levels seen in the parental cell line. B, a panel of 11 cell lines expressing WT or mutant K-Ras was treated with 100 nm NTC or K-Ras siRNA pools, and protein and RNA were harvested 72 h later from identically treated plates. K-Ras knockdown was assessed by immunoprecipitation-Western blotting using total and K-Ras-specific antibodies, and expression of P-ERK and P-Akt was assessed by Western blotting. C, RNA isolated from NTC or K-Ras siRNA-treated cells was subjected to Taqman quantitative PCR using the indicated probes, and the effects on gene expression in each cell line are indicated as a ratio of K-Ras:NTC siRNA treatment. The horizontal bar indicates the average expression of the gene across all cell lines. The open symbols represent WT K-Ras-expressing cells, and the filled symbols represent mutant K-Ras-expressing cells. p values are the results of an unpaired t test comparing the effects on mutant versus WT K-Ras-expressing cells.
To determine whether any of the genes induced by mutant K-Ras contributes to its transformation properties, we knocked down ETV1 and NT5E using siRNA pools in the same panel of 11 cell lines, and monitored proliferation in the second dimension on plastic and in the third dimension in hanging drop cultures (26). Knockdown relative to a nontargeting control is shown in supplemental Fig. 8, A–C. Whereas K-Ras knockdown inhibited proliferation in both the second and third dimensions selectively in the mutant K-Ras-expressing cell lines, there was little selective inhibition of proliferation following knockdown of either NT5E or ETV1 (supplemental Fig. 8, D and E).
DISCUSSION
K-Ras is widely recognized as an important human oncogene that is mutated in a large percentage of human epithelial tumors. However, most of the analyses of the cellular consequences of Ras expression have been performed using overexpression studies, often using H-Ras, and frequently using cell types that are either mesenchymal or murine. In this study, we have documented the effects of the endogenous mutated K-ras allele, using cell line derivatives generated by homologous recombination targeting. These studies have demonstrated that there are both common phenotypes to mutant K-Ras expression across multiple cell lines, as well as consequences that are unique to the cell line, and therefore likely also unique to the tumor tissue from which these were originally derived. The use of isogenic cell lines has several advantages over commonly utilized overexpression and knockdown approaches. Overexpression of mutant ras alleles results in a senescent-like phenotype that has been attributed to increased production of reactive oxygen species and associated stresses (27, 28) and is likely unrelated to the normal functions of single copy mutant ras, which results in tumor initiation without senescence (11). The use of RNAi against ras (similar to any gene knockdown) is problematic due to incomplete knockdown and the possibility of off-target effects associated with siRNA or shRNA sequences. Moreover, subtleties associated with activities of the remaining WT allele would also not be revealed using a conventional knockdown approach. The concerns of artifactual results due to cell line selection and clonal anomalies were partially alleviated in this study by the use of multiple clones (where available) and the use of transient knockdown to show similar effects on signaling and biological phenotypes.
One common consequence of mutant K-ras deletion was the detrimental effects on soft agar colony growth, an often used surrogate assay for tumorigenesis. This shows that, even with the multitude of genetic alterations present in these cell lines, deletion of a single allele has substantial effects on transformation. In contrast to the strong effect in transformation assays, deletion of mutant K-ras has mild or nonexistent effects on cell proliferation under the conditions assessed. This is unlikely to be a trait selected for during the generation of these lines, as knockdown of K-Ras in the parental lines also had only modest effects on cell proliferation. Recently, there has been an appreciation that K-Ras mutant cell lines vary in their response to K-Ras knockdown, with some cell lines showing a strong decrease in cell number due to enhanced apoptosis and/or growth arrest, and other cell lines showing little effect (29, 30). In these published studies, Hec1 (of which Hec1A is a derivative), HCT116, and DLD1 cells were all predicted and empirically shown to be insensitive to K-Ras knockdown in proliferation assays, consistent with the data shown here. It perhaps is not surprising that the isogenic K-Ras lines that exist are “insensitive,” as K-Ras-“sensitive” lines would likely be unable to expand following deletion of the mutant allele. Nevertheless, we would suggest that even though a cell line is classified as Ras-independent by proliferation criteria, it remains Ras-dependent by more stringent growth conditions that may more closely reflect the tumor environment.
One aspect of mutant K-Ras signaling that was common to some, but not all, of the cell lines studied here, was the cross-talk between K-Ras and N-Ras. This was most dramatically seen in the Hec1A cells and HMECs, but was also seen in DLD1 cells to a lesser extent. This also correlated with the ability of mutant K-Ras to down-regulate EGFR phosphorylation, an observation also recently made by in lung cancer cell lines (31) and in colorectal cancer cell lines (32). This could explain the relatively mild effects on downstream signaling in the different cell lines upon mutant K-ras deletion or expression, i.e. increased NRasGTP might compensate for the loss of KRasGTP and in the case of Hec1A cells, could explain the paradoxical increase in EGF-stimulated ERK phosphorylation upon mutant K-ras deletion. This observation may also contribute to the lack of effect of EGFR inhibitors in mutant K-Ras lung and colorectal tumor patients (33, 34). Although activated Ras could maintain downstream signaling in the presence of EGFR inhibition, it could also shut off EGFR signaling in these tumors, rendering them independent of EGF and therefore refractory to EGFR inhibition. Here we show that the decreased EGFR phosphorylation and NRasGTP levels in mutant K-Ras-expressing cell lines are largely due to negative feedback initiated by MEK signaling. This could be mediated, at least in part, by production of secreted factors that antagonize EGF-dependent signaling, although we were unable to define the exact factor that causes this.
Deletion or introduction of mutant K-ras caused surprisingly diverse effects on gene expression, showing that mutant K-Ras has specific functions depending on the tissue in which it is expressed. There have previously been several publications using “Ras gene signatures” to classify human tumors. Sweet-Cordero et al. (35) analyzed mutant K-Ras-driven lung tumors and compared them with normal mouse lung tissue and human K-Ras mutant lung cancers to derive 89 genes up-regulated in mutant K-Ras lung cancer. Bild et al. (36) overexpressed H-Ras in HMECs to derive 177 genes up-regulated by H-Ras. Arena et al. (37) used single copy K-Ras knock-in in mouse liver progenitor cells to derive 149 genes that were up-regulated upon this condition. Mutant K-Ras results in highly divergent consequences in different tissues and environments, as exemplified by the fact that only a single gene (DUSP6) is in common among these three sets of signatures. Although the approach used here to identify mutant K-Ras-regulated genes is likely no better or no worse, it confirms that only a small number of genes are consistently altered by mutant K-Ras. Notably, the seven genes identified and validated in this study have all been found associated with Ras expression or signaling in previous studies; DUSP6, DUSP5 (36, 38), NT5E, ETV1 (39), IER3 (36, 39), UPP1 (36, 40), and ETV5 (39, 41). The functions of these genes are quite diverse and represent mechanisms involved in negative feedback signaling (DUSP5 and 6), nucleoside metabolism (NT5E and UPP), transcriptional responses activated downstream of ERK signaling (ETV1 and ETV5), and protection from apoptosis (IER3). Knockdown of NT5E has previously been shown to decrease transformation properties of K-Ras mutant MDA-MB-231 cells (42). Although knockdown of NT5E did decrease proliferation in the second and third dimensions in K-Ras mutant A427 cells, there was no statistically significant correlation between the effects in mutant versus WT K-Ras cells (supplemental Fig. 6). It is likely that K-Ras uses multiple target genes to fully elicit transformation properties.
The increased transformation properties of Hec1A, HCT116, and DLD1 cells lacking the WT K-ras allele was unexpected but is reminiscent of studies in mice suggesting that the WT K-ras allele can display tumor-suppressive effects. This was first demonstrated in chemical carcinogen-induced tumors that result in K-ras mutation, that were more aggressive when one copy of K-ras was deleted in the germ line (7). Loss of the wild type K-ras allele was also recently observed in high grade, but not low grade lung tumors driven by conditional mutant K-Ras (43). A tumor-suppressive function for WT N-Ras has also been observed in mouse tumors driven by both chemical carcinogens, as well as by transgenic expression of mutant N-Ras (44). The potential for WT K-Ras to act as a tumor suppressor in human tumors has not been as well characterized, although our analysis of a large panel of human cancer cell lines shows that >one-quarter of mutant K-Ras-expressing cells have allelic imbalance/homozygous expression of the mutant allele. Even in cells that have heterozygous expression, a majority show enhanced expression of the mutant allele. These results clearly suggest a selective advantage for the tumor cells to lose the WT K-ras allele or to decrease its expression. The model systems used in this study, or other similar ones, should allow further dissection of this unexpected phenomenon.
Acknowledgments
We thank the members of the sequencing, bioinformatics, cell culture, and microarray core facilities at Genentech for expertise and timely analysis; Chris Klijn and Zemin Zhang for K-Ras RNA-seq information; Fred de Sauvage and Jeff Settleman for critical input during this project; and David Davis for many useful and thoughtful discussions.
S. Vartanian, C. Bentley, L. Li, M. J. Brauer, P. Haverty, E. Stawiski, Z. Modrusan, and D. Stokoe are paid employees of Genentech.

This article contains supplemental Table 1, Figs. 1–8, and Materials and Methods.
C. Klijn, E. Stawiski, Z. Zhang, S. Seshagiri, and members of the Genentech Cell Line Genomics Group, unpublished findings.
- HMEC
- human mammary epithelial cell
- EGFR
- EGF receptor
- RBD
- Ras binding domain.
REFERENCES
- 1. Karnoub A. E., Weinberg R. A. (2008) Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 9, 517–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mascaux C., Iannino N., Martin B., Paesmans M., Berghmans T., Dusart M., Haller A., Lothaire P., Meert A. P., Noel S., Lafitte J. J., Sculier J. P. (2005) The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br. J. Cancer 92, 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Linardou H., Dahabreh I. J., Kanaloupiti D., Siannis F., Bafaloukos D., Kosmidis P., Papadimitriou C. A., Murray S. (2008) Assessment of somatic K-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 9, 962–972 [DOI] [PubMed] [Google Scholar]
- 4. Jackson E. L., Willis N., Mercer K., Bronson R. T., Crowley D., Montoya R., Jacks T., Tuveson D. A. (2001) Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15, 3243–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hingorani S. R., Petricoin E. F., Maitra A., Rajapakse V., King C., Jacobetz M. A., Ross S., Conrads T. P., Veenstra T. D., Hitt B. A., Kawaguchi Y., Johann D., Liotta L. A., Crawford H. C., Putt M. E., Jacks T., Wright C. V., Hruban R. H., Lowy A. M., Tuveson D. A. (2003) Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 [DOI] [PubMed] [Google Scholar]
- 6. Li Q., Haigis K. M., McDaniel A., Harding-Theobald E., Kogan S. C., Akagi K., Wong J. C., Braun B. S., Wolff L., Jacks T., Shannon K. (2011) Hematopoiesis and leukemogenesis in mice expressing oncogenic NRasG12D from the endogenous locus. Blood 117, 2022–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang Z., Wang Y., Vikis H. G., Johnson L., Liu G., Li J., Anderson M. W., Sills R. C., Hong H. L., Devereux T. R., Jacks T., Guan K. L., You M. (2001) Wildtype K-ras2 can inhibit lung carcinogenesis in mice. Nat. Genet. 29, 25–33 [DOI] [PubMed] [Google Scholar]
- 8. Diaz R., Lopez-Barcons L., Ahn D., Garcia-Espana A., Yoon A., Matthews J., Mangues R., Perez-Soler R., Pellicer A. (2004) Complex effects of ras proto-oncogenes in tumorigenesis. Carcinogenesis 25, 535–539 [DOI] [PubMed] [Google Scholar]
- 9. White M. A., Nicolette C., Minden A., Polverino A., Van Aelst L., Karin M., Wigler M. H. (1995) Multiple Ras functions can contribute to mammalian cell transformation. Cell 80, 533–541 [DOI] [PubMed] [Google Scholar]
- 10. Serrano M., Lin A. W., McCurrach M. E., Beach D., Lowe S. W. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 [DOI] [PubMed] [Google Scholar]
- 11. Tuveson D. A., Shaw A. T., Willis N. A., Silver D. P., Jackson E. L., Chang S., Mercer K. L., Grochow R., Hock H., Crowley D., Hingorani S. R., Zaks T., King C., Jacobetz M. A., Wang L., Bronson R. T., Orkin S. H., DePinho R. A., Jacks T. (2004) Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5, 375–387 [DOI] [PubMed] [Google Scholar]
- 12. Guerra C., Mijimolle N., Dhawahir A., Dubus P., Barradas M., Serrano M., Campuzano V., Barbacid M. (2003) Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell 4, 111–120 [DOI] [PubMed] [Google Scholar]
- 13. Konishi H., Karakas B., Abukhdeir A. M., Lauring J., Gustin J. P., Garay J. P., Konishi Y., Gallmeier E., Bachman K. E., Park B. H. (2007) Knock-in of mutant K-ras in nontumorigenic human epithelial cells as a new model for studying K-ras-mediated transformation. Cancer Res. 67, 8460–8467 [DOI] [PubMed] [Google Scholar]
- 14. Braun B. S., Tuveson D. A., Kong N., Le D. T., Kogan S. C., Rozmus J., Le Beau M. M., Jacks T. E., Shannon K. M. (2004) Somatic activation of oncogenic K-ras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc. Natl. Acad. Sci. U.S.A. 101, 597–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Di Nicolantonio F., Arena S., Gallicchio M., Zecchin D., Martini M., Flonta S. E., Stella G. M., Lamba S., Cancelliere C., Russo M., Geuna M., Appendino G., Fantozzi R., Medico E., Bardelli A. (2008) Replacement of normal with mutant alleles in the genome of normal human cells unveils mutation-specific drug responses. Proc. Natl. Acad. Sci. U.S.A. 105, 20864–20869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schubbert S., Shannon K., Bollag G. (2007) Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 7, 295–308 [DOI] [PubMed] [Google Scholar]
- 17. Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DePristo M. A., Banks E., Poplin R., Garimella K. V., Maguire J. R., Hartl C., Philippakis A. A., del Angel G., Rivas M. A., Hanna M., McKenna A., Fennell T. J., Kernytsky A. M., Sivachenko A. Y., Cibulskis K., Gabriel S. B., Altshuler D., Daly M. J. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sherry S. T., Ward M. H., Kholodov M., Baker J., Phan L., Smigielski E. M., Sirotkin K. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Forbes S. A., Bindal N., Bamford S., Cole C., Kok C. Y., Beare D., Jia M., Shepherd R., Leung K., Menzies A., Teague J. W., Campbell P. J., Stratton M. R., Futreal P. A. (2011) COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 39, D945–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shirasawa S., Furuse M., Yokoyama N., Sasazuki T. (1993) Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science 260, 85–88 [DOI] [PubMed] [Google Scholar]
- 22. Yun J., Rago C., Cheong I., Pagliarini R., Angenendt P., Rajagopalan H., Schmidt K., Willson J. K., Markowitz S., Zhou S., Diaz L. A., Jr., Velculescu V. E., Lengauer C., Kinzler K. W., Vogelstein B., Papadopoulos N. (2009) Glucose deprivation contributes to the development of K-ras pathway mutations in tumor cells. Science 325, 1555–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim J. S., Lee C., Foxworth A., Waldman T. (2004) B-Raf is dispensable for K-ras-mediated oncogenesis in human cancer cells. Cancer Res. 64, 1932–1937 [DOI] [PubMed] [Google Scholar]
- 24. Taylor S. J., Shalloway D. (1996) Cell cycle-dependent activation of Ras. Curr. Biol. 6, 1621–1627 [DOI] [PubMed] [Google Scholar]
- 25. Boykevisch S., Zhao C., Sondermann H., Philippidou P., Halegoua S., Kuriyan J., Bar-Sagi D. (2006) Regulation of ras signaling dynamics by Sos-mediated positive feedback. Curr. Biol. 16, 2173–2179 [DOI] [PubMed] [Google Scholar]
- 26. Del Duca D., Werbowetski T., Del Maestro R. F. (2004) Spheroid preparation from hanging drops: characterization of a model of brain tumor invasion. J. Neurooncol. 67, 295–303 [DOI] [PubMed] [Google Scholar]
- 27. Lee A. C., Fenster B. E., Ito H., Takeda K., Bae N. S., Hirai T., Yu Z. X., Ferrans V. J., Howard B. H., Finkel T. (1999) Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 274, 7936–7940 [DOI] [PubMed] [Google Scholar]
- 28. DeNicola G. M., Karreth F. A., Humpton T. J., Gopinathan A., Wei C., Frese K., Mangal D., Yu K. H., Yeo C. J., Calhoun E. S., Scrimieri F., Winter J. M., Hruban R. H., Iacobuzio-Donahue C., Kern S. E., Blair I. A., Tuveson D. A. (2011) Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Singh A., Greninger P., Rhodes D., Koopman L., Violette S., Bardeesy N., Settleman J. (2009) A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 15, 489–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Singh A., Sweeney M. F., Yu M., Burger A., Greninger P., Benes C., Haber D. A., Settleman J. (2012) TAK1 inhibition promotes apoptosis in K-Ras-dependent colon cancers. Cell 148, 639–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sunaga N., Shames D. S., Girard L., Peyton M., Larsen J. E., Imai H., Soh J., Sato M., Yanagitani N., Kaira K., Xie Y., Gazdar A. F., Mori M., Minna J. D. (2011) Knockdown of oncogenic K-Ras in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol. Cancer Ther. 10, 336–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Houdt W. J., Hoogwater F. J., de Bruijn M. T., Emmink B. L., Nijkamp M. W., Raats D. A., van der Groep P., van Diest P., Borel Rinkes I. H., Kranenburg O. (2010) Oncogenic K-ras desensitizes colorectal tumor cells to epidermal growth factor receptor inhibition and activation. Neoplasia 12, 443–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. De Roock W., Jonker D. J., Di Nicolantonio F., Sartore-Bianchi A., Tu D., Siena S., Lamba S., Arena S., Frattini M., Piessevaux H., Van Cutsem E., O'Callaghan C. J., Khambata-Ford S., Zalcberg J. R., Simes J., Karapetis C. S., Bardelli A., Tejpar S. (2010) Association of K-Ras p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 304, 1812–1820 [DOI] [PubMed] [Google Scholar]
- 34. Mao C., Qiu L. X., Liao R. Y., Du F. B., Ding H., Yang W. C., Li J., Chen Q. (2010) K-Ras mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies. Lung Cancer 69, 272–278 [DOI] [PubMed] [Google Scholar]
- 35. Sweet-Cordero A., Mukherjee S., Subramanian A., You H., Roix J. J., Ladd-Acosta C., Mesirov J., Golub T. R., Jacks T. (2005) An oncogenic K-ras2 expression signature identified by cross-species gene-expression analysis. Nat. Genet. 37, 48–55 [DOI] [PubMed] [Google Scholar]
- 36. Bild A. H., Yao G., Chang J. T., Wang Q., Potti A., Chasse D., Joshi M. B., Harpole D., Lancaster J. M., Berchuck A., Olson J. A., Jr., Marks J. R., Dressman H. K., West M., Nevins J. R. (2006) Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 439, 353–357 [DOI] [PubMed] [Google Scholar]
- 37. Arena S., Isella C., Martini M., de Marco A., Medico E., Bardelli A. (2007) Knock-in of oncogenic K-ras does not transform mouse somatic cells but triggers a transcriptional response that classifies human cancers. Cancer Res. 67, 8468–8476 [DOI] [PubMed] [Google Scholar]
- 38. Tchernitsa O. I., Sers C., Zuber J., Hinzmann B., Grips M., Schramme A., Lund P., Schwendel A., Rosenthal A., Schäfer R. (2004) Transcriptional basis of K-ras oncogene-mediated cellular transformation in ovarian epithelial cells. Oncogene 23, 4536–4555 [DOI] [PubMed] [Google Scholar]
- 39. Packer L. M., East P., Reis-Filho J. S., Marais R. (2009) Identification of direct transcriptional targets of (V600E)BRAF/MEK signalling in melanoma. Pigment Cell Melanoma Res. 22, 785–798 [DOI] [PubMed] [Google Scholar]
- 40. Guerrero I., Villasante A., Corces V., Pellicer A. (1985) Loss of the normal N-ras allele in a mouse thymic lymphoma induced by a chemical carcinogen. Proc. Natl. Acad. Sci. U.S.A. 82, 7810–7814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pratilas C. A., Taylor B. S., Ye Q., Viale A., Sander C., Solit D. B., Rosen N. (2009) (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc. Natl. Acad. Sci. U.S.A. 106, 4519–4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhi X., Wang Y., Zhou X., Yu J., Jian R., Tang S., Yin L., Zhou P. (2010) RNAi-mediated CD73 suppression induces apoptosis and cell-cycle arrest in human breast cancer cells. Cancer Sci. 101, 2561–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Junttila M. R., Karnezis A. N., Garcia D., Madriles F., Kortlever R. M., Rostker F., Brown Swigart L., Pham D. M., Seo Y., Evan G. I., Martins C. P. (2010) Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 468, 567–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Diaz R., Ahn D., Lopez-Barcons L., Malumbres M., Perez de Castro I., Lue J., Ferrer-Miralles N., Mangues R., Tsong J., Garcia R., Perez-Soler R., Pellicer A. (2002) The N-ras proto-oncogene can suppress the malignant phenotype in the presence or absence of its oncogene. Cancer Res. 62, 4514–4518 [PubMed] [Google Scholar]