

Background: Viruses must interact with membranes to access host cell machinery for replication.
Results: SV40 VP2 and VP3 are shown to form pores in membranes and pore formation is required for viral propagation.
Conclusion: VP2 and VP3 act as viral-encoded membrane pore forming proteins or viroporins.
Significance: These versatile proteins have evolved to function as both soluble and membrane proteins to support viral propagation.
Keywords: Membrane, Membrane Proteins, Membrane Trafficking, Membrane Transport, Virus, Membrane Pores, Viroporins
Abstract
For nonenveloped viruses such as Simian Virus 40, the mechanism used to translocate viral components across membranes is poorly understood. Previous results indicated that the minor structural proteins, VP2 and VP3, might act as membrane proteins during infection. Here, purified VP2 and VP3 were found to form pores in host cell membranes. To identify possible membrane domains, individual hydrophobic domains from VP2 and VP3 were expressed in a model protein and tested for their ability to integrate into membranes. Several domains from the late proteins supported endoplasmic reticulum membrane insertion as transmembrane domains. Mutations in VP2 and VP3 were engineered that inhibited membrane insertion and pore formation. When these mutations were introduced into the viral genome, viral propagation was inhibited. This comprehensive approach revealed that the viroporin activity of VP2 and VP3 was inhibited by targeted disruptions of individual hydrophobic domains and the loss of membrane disruption activity impaired viral infection.
Introduction
Viruses exploit and overcome membrane barriers to infect cells. Nonenveloped viruses lack a membrane and must translocate viral components across a host cell membrane. The model nonenveloped virus Simian Virus 40 (SV40)2 infects host cells via binding at the cell surface to the ganglioside receptor GM1, endocytosis into caveolae-coated vesicles and trafficking to the endoplasmic reticulum (ER) (1–3). In the ER lumen, molecular chaperones and protein-disulfide isomerases are thought to induce structural rearrangements that uncoat or prime the virus for penetration (4). The mechanism used to subvert the ER membrane for eventual delivery of the viral minichromosome to the nucleus has not been characterized.
The SV40 genome is composed of circular dsDNA that encodes early gene products including large T (LT) and small T antigen, which dictate viral genome replication and control synthesis of the late mRNA transcripts (5, 6). The major structural protein VP1 forms pentamers that possess a central hydrophobic binding pocket (7). The minor structural proteins VP2 and VP3 are synthesized from successive in-frame start Met residues so their C termini are equivalent. A single copy of VP2 or VP3 is bound to each VP1 pentamer using a C-terminal region of the minor structural proteins (8). Assembly of 72 pentamers bound to VP2 and VP3 oligomerize around the viral genome to produce virus particles.
As entry and delivery of the viral genome to the nucleus is a central question for many nonenveloped viruses, several studies have investigated the early binding and entry events of SV40 (9–11). Virus-like particles (VLP) lacking VP2 and VP3 have some defects in virion assembly and viral penetration (12, 13). Recent results demonstrate that SV40 VP2 and VP3 serve critical roles in the viral life cycle, and these roles involve their ability to interact with host cell membranes (12, 14). However, the mechanism of action of VP2 and VP3 remains elusive. VP4 was recently shown to possess lytic activity thereby acting as a viroporin (15, 16). As the VP4 sequence is contained within VP2 and VP3, this suggested that VP2 and VP3 might also have membrane disruption activity.
To investigate the interaction of the minor structural proteins with membranes, VP2 and VP3 were purified from Escherichia coli and shown to disrupt cell membranes by forming pores. A model protein derived from E. coli Leader Peptidase (Lep) was used to identify domains in VP2 and VP3 that support membrane integration. Membrane integration of these domains was inhibited by the introduction of charged residues and membrane insertion inhibition diminished the perforation activity of the purified proteins. Finally when the same mutations were introduced into the virus, viral propagation was perturbed. Further analysis suggested that viral infection was reduced in the absence of viroporin activity. This supports the conclusion that the pore formation or viroporin activity of VP2 and VP3 is involved in viral infection.
EXPERIMENTAL PROCEDURES
Reagents
The SP6 RNA polymerase was purchased from New England BioLabs (Ipswich, MA). The RNasin and components of the rabbit reticulocyte cell-free translation system were purchased from Promega (Madison, WI). Rough ER-derived microsomes were purchased from tRNA Probes (College Station, TX). [35S]Met/Cys was acquired from PerkinElmer (Waltham, MA). The bacterial propagation competent pSV40 plasmid, which encodes WT SV40 (strain 776), was a generous gift from Dr. H. Kasamatsu (Los Angeles, CA). BS-C-1 and COS7 cells were obtained from ATCC (Manassas, VA). The following antibodies were acquired as indicated: Large T (LT) (Merck KGaA, Darmstadt, Germany); VP1 and VP2/3 (Abcam, Cambridge, MA). CytoTox 96 cytotoxicity assay kit for quantification of lactate dehydrogenase (LDH) release was purchased from Promega. All tissue culture reagents and Lipofectamine 2000 were purchased from Invitrogen (Carlsbad, CA). All other reagents were purchased from Sigma-Aldrich.
Construction of Plasmids
GST-VP2 and GST-VP3 were created by subcloning into the pGEX-6P-1-TEV-His as previously described (16). Lep constructs used to test NCyto (Lep H2, Fig. 2), NLumen (Lep H3, data not shown) and single domain (Lep H1, Fig. 3) membrane insertion were generous gifts from Drs. G. von Heijne and I. Nilsson (17, 18). Individual segments of VP2 were PCR amplified, digested, and ligated into each Lep reporter construct using standard techniques. Phusion site-directed mutagenesis was used according to the manufacturer's recommendations to generate all HD mutants in the GST constructs, Lep constructs and the pSV40 plasmid. Wild type revertants were created for all SV40 mutants in the present study and shown to restore viral propagation to rule out unintended mutation of the plasmid backbone. All cloning and mutagenesis was verified by sequencing (Genewiz, South Plainfield, NJ).
FIGURE 2.

Transmembrane domain predictions for VP2 and HD1, HD2, and HD6 insert into ER microsomes with NCyto topology. A, schematic representation of the SV40 protein VP2 with TMD predicted by a variety of algorithms (supplemental Fig. S2) denoted by black rectangles. HD are numbered from the N-to-C termini. Start sites for VP2, VP3, and VP4 are indicated on the composite. B, diagram of the model protein Lep H2 used to determine the membrane insertion of individual domains of late viral proteins (17). Glycosylation sites are indicated as circles, with filled circles indicating the glycosylation site is occupied, and open circles that the site is not modified. Vector (a bona fide TMD) or individual hydrophobic domains (HD1–6) of VP2 were introduced into Lep H2 (gray box). The star denotes a possible site for cleavage by the signal peptidase complex. C–F, [35S]Met/Cys-labeled proteins were synthesized in reticulocyte lysate for 1 h at 27 °C in the presence of ER rough microsomes (MS) as indicated. Wild type (WT) and mutants of the individual HD were tested. The translations were either untreated (Lys) or centrifuged through a sucrose cushion (Pellet). Half of the lysates were loaded compared with pellet samples. One-half of each sample was treated with Endo H as indicated to remove glycans. Proteins were resolved by reducing SDS-PAGE, followed by autoradiography. Unglycosylated proteins are indicated with an open circle whereas singly and doubly glycosylated proteins are denoted with filled circles. Recognition as a signal sequence by signal peptidase produced an internal cleavage product that was glycosylated or deglycosylated as indicated with black or white stars, respectively. Quantification of the percent TMD insertion is noted below the corresponding sample. The percentage of TMD insertion for each HD was quantified as the fraction inserted divided by total fraction targeted to ER rough microsomes. For Lep H2, %TM = (f2 + fc)/(f2 + fc + f1) × 100 where the fraction of TM targeting was the doubly glycosylated (f2) and cleaved product (fc) divided by the sum of the total targeted including the doubly glycosylated, cleaved product, and targeted but not inserted singly glycosylated (f1). The species of unglycosylated protein, which was not targeted to microsomes, was ignored. Standard deviations are based on at least three independent experiments.
FIGURE 3.

HD6 inserts into RER-derived microsomes in multiple orientations. A, diagram of model Lep H1 used to determine autonomous membrane insertion of individual domains. B and C, HD1–6 domains were analyzed as described in Fig. 2. D, proteins were translated in the absence (−) or presence of MS (Co) as indicated. The reaction lacking MS was treated with cycloheximide to inhibit translation and the reaction was split and microsomes were added post-translationally where indicated (Po) and incubated for 1 h at 27 °C. Half of each reaction was treated with Endo H as indicated to remove glycans. The lysate samples correspond to half of the pellet samples. Wild type (WT) and mutant forms of the various HD were tested. Quantification of the percent TMD insertion is noted below the corresponding sample. The fraction of each translocated (Trans) species including NLumen (f1), NCyto (f2), and N&CLumen (f3) was divided by the sum of all species including the unglycosylated (f0). Hence the %Trans = (f1 + f2 + f3)/(f1 + f2 + f3 + f0) × 100 or individual targeting e.g. %NLumen = f1/(f1 + f2 + f3 + f0) × 100. Standard deviations are based on at least three independent experiments.
Bacterial Expression and Purification of GST-tagged Proteins
Bacterial expression and purification of GST-VP2, GST-VP3, and their mutants were performed as described previously with minor modifications (15). For expression of GST-VP2 protein expression was induced with 1 mm IPTG in the presence of 400 mm NaCl and 20 mm proline for 3 h at 30 °C. Expression of GST-VP3 was carried out at 16 °C for 12 h after inducing with 1 mm IPTG. All further affinity purification with GST Sepharose 4B (Amersham Biosciences) and Ni-NTA His Bind resin (Novagen) were carried out as previously described (15).
LDH Release Assay
COS7 cells were maintained in DMEM supplemented with 10% FBS and Pen-Strep in a humidified CO2 incubator at 37 °C. COS7 cells (10,000 cells) were incubated with purified protein (232 nm) for 30 min at 37 °C. The CytoTox 96 cytotoxicity assay kit was used according to the manufacturer's instructions to quantify the release of LDH from the cells. The percent release was normalized to detergent lysis (2% Triton X-100) and buffer control. Osmoprotection was determined as previously described (15).
In Vitro Translations
The [35S]Met/Cys radiolabeled in vitro translation reactions were carried out in rabbit reticulocyte lysate as per the manufacturer's instructions (Promega). Proteins were synthesized by in vitro translation in the absence or presence of rough ER (RER)-derived microsomes. Microsome samples were recovered by ultracentrifugation through a sucrose cushion (500 mm sucrose, 50 mm TEA (pH 7.5) and 1 mm DTT) for 10 min at 72,000 × g at 4 °C. Samples were analyzed by reducing SDS-PAGE and developed by autoradiography.
Viral Genome Manipulation, Transfection, and Immunoblotting
BS-C-1 cells were maintained in DMEM supplemented with 5% FBS and Pen-Strep in humidified 5% CO2 incubator at 37 °C. All mutant versions of SV40 were created by site-directed mutagenesis as described above and confirmed by sequencing. To rule out off-target mutagenesis of the genome backbone wild type revertants were created for all mutants used in this study and displayed wild type levels of viral propagation based on quantification of LT by flow cytometry (see below). Wild type or mutant SV40 genome preparations were performed as previously described (14, 19). Briefly, bacterial replication competent viral genomes (pSV40) were digested with BamHI-HF (New England BioLabs) to remove the bacterial replication elements. Subsequently, viral genomes were produced by ligating the genome under dilute conditions to favor re-circularization. BS-C-1 cells were transfected with viral genomes using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Non-adherent and adherent cells were collected and lysed in Hepes (pH 7.4) and 200 mm NaCl with 1% Nonidet P-40 and protease inhibitors (50 μm calpain inhibitor I, 1.5 μm aprotinin, 20 μm leupeptin, 1 μm pepstatin, 400 μm phenylmethylsulfonyl fluoride, and 20 mm N-ethylmaleimide) as previously described (14). The insoluble nuclear fractions and media were recovered and analyzed by immunoblotting.
Flow Cytometry
The percentage of transfected or infected cells was quantified based on expression of LT antigen using flow cytometry as previously described (12, 19). Nonadherent cells were collected by centrifugation at 1,000 × g for 5 min at 4 °C. Adherent cells were trypsinized and pooled with the nonadherent cells by centrifugation at 1,000 × g for 5 min at 4 °C. Cells were fixed with 4% formaldehyde and permeabilized with 0.05% saponin. Samples were incubated in flow cytometry buffer (20 mm EDTA, 0.02% sodium azide, 2% BSA, 0.1% saponin, and PBS at pH 7.4) with anti-LT antibody followed by anti-mouse Alexa fluor 488 antibodies. A total of 30,000 cells were counted and analyzed using FACS Diva software. Cells were gated, and the percentage of LT-positive cells was determined relative to untransfected or mock-infected cells used as negative controls.
Virus Purification, Genome Encapsidation, and Infection
BS-C-1 cells were transfected with wild type or HD mutant SV40. Viral supernatants were recovered ∼12 days P.T. by freeze thawing three times as previously described (14). Cell lysates were centrifuged at 14,000 × g for 10 min to remove cell debris. Supernatants were passed through a 0.45 μm filter and centrifuged at 180,000 × g for 30 min at 4 °C to purify viral particles. Viral supernatants were subjected to DNase I digestion for 1 h to degrade non-encapsidated viral genome. As a control, 3 μg of viral plasmid was digested. DNase I was heat inactivated and the capsids were disassembled by boiling in SDS buffer. DNA was purified by phenol chloroform extraction and ethanol precipitation. Purified DNA was PCR amplified with VP1-specific primers. All infections were normalized based on VP1, the major structural protein. Confluent monolayers of BS-C-1 cells were infected for 6 h in a humidified 5% CO2 incubator at 37 °C. Unbound virus was removed and replaced with fresh DMEM, 5% FBS, and Pen-Strep.
Transmission Electron Microscopy
Cells were harvested 3 days post transfection and fixed with 2% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.2, for 1 h and then rinsed three times with the same buffer over 30 min. Cells were post-fixed with 1% osmium tetroxide in the same buffer on ice for 4 h, followed by rinsing three times with water. The washed pellet was resuspended in 25 ml of melted 2% agarose Type IX at 25 °C, pelleted at 1500 × g and chilled on ice to set. The enrobed pellet was en bloc stained with 1% aqueous uranyl acetate for 4 h, dehydrated by a graded ethanol series (30%, 50%, 70%, 80%, 95%, 100%), changed to 100% acetone, infiltrated with Epon-Araldite epoxy resin (25%, 50%, 75% in acetone) for 1 h each, then 100% epoxy, and polymerized 24 h at 65 °C. Sections (60 nm) were mounted on copper grids and stained with 2% aqueous uranyl acetate for 30 min, 0.025% alkaline lead citrate for 10 min. Images were taken on Kodak 4489 Electron Image Film in a JEOL JEM-100S electron microscope. Negatives were digitized at 1200 dpi on a Microtek Scanmaker i900 flatbed scanner. Images were analyzed using ImageJ and prepared for figures using Adobe Photoshop.
RESULTS
VP2 and VP3 Form Pores in Host Cell Membranes
To determine if VP2 or VP3 possess membrane disruption activity that contributes to their role in the viral life cycle, each protein was expressed and purified from E. coli as an N-terminal GST fusion protein with a C-terminal His tag (supplemental Fig. S1). Size-exclusion chromatography of purified VP2 and VP3 found that they were predominantly monomeric (supplemental Fig. S1C). The isolated proteins were probed for their ability to disrupt membranes with the SV40 permissive host COS7 cells. Membrane disruption was analyzed by monitoring the release of cytoplasmic lactate dehydrogenase (LDH) from the cells after exposure to the purified proteins (16).
Incubation of COS7 cells (10,000 cells) with purified GST-VP2 (232 nm) resulted in release of more than 67% of LDH while GST-VP3 displayed enhanced activity with the release of 81% of the cellular LDH (Fig. 1A). The GST tag alone did not have membrane disruption activity indicating that VP2 and VP3 possess intrinsic membrane disruption activity.
FIGURE 1.
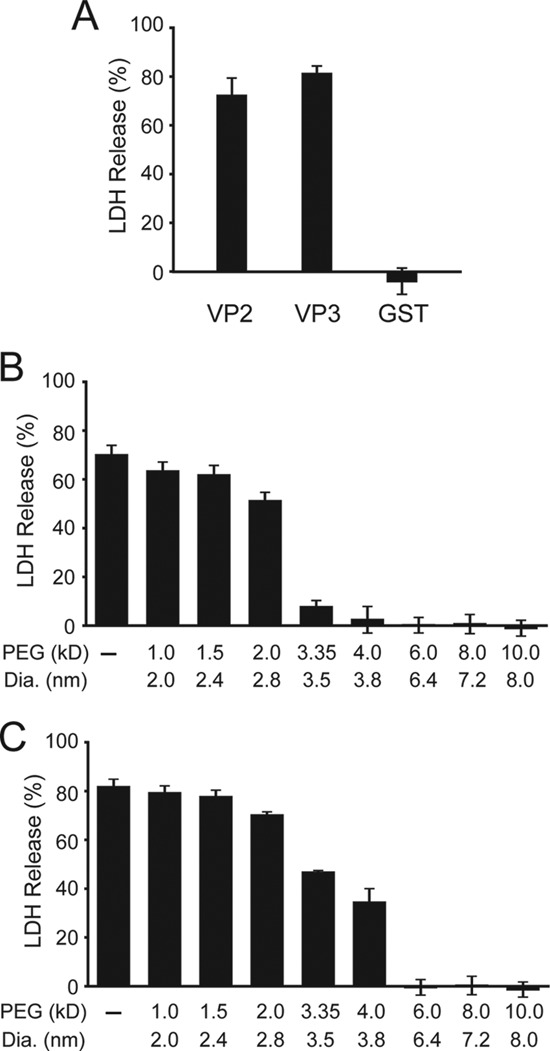
VP2 and VP3 form pores in host cell membranes. A, equimolar concentrations (232 nm) of bacterial expressed and purified GST-VP2-His (VP2), GST-VP3-His (VP3), or GST-His (GST) was incubated with COS7 cells (10,000 cells) for 30 min at 37 °C. The release of LDH in the medium was determined and normalized to detergent lysis and buffer control. B and C, determination of pore size by osmoprotection of COS7 cells by different molecular weight PEG for VP2 (B) and VP3 (C). Error bars represent the standard deviation of three independent experiments.
Proteins can disrupt membranes by forming membrane pores or by the solubilization of membranes using a detergent-like mechanism (20). To differentiate between these two mechanisms of membrane disruption, the ability of VP2 and VP3 to form discrete pores in COS7 membranes was characterized. LDH release from COS7 cells was monitored in the presence of different diameter polyethylene glycols (PEG) (Fig. 1, B and C). If the PEG is larger in diameter than the pore formed, it will increase the osmolality of the media thereby conferring protection from lysis (15, 21). Conversely if the polymer is smaller than the pore formed, it will equilibrate across the membrane and cell lysis will be observed. This experimental approach provides evidence of both the presence of a pore and an estimate of its inner diameter.
In the absence of PEG, GST-VP2 disrupted COS7 cell membranes resulting in release of ∼70% of LDH (Fig. 1B). A modest reduction in lysis was observed in the presence of PEG polymers with a diameter up to 2.8 nm. In contrast, PEG larger than 3.5 nm in diameter inhibited cell lysis by osmoprotection. This demonstrated that GST-VP2 formed pores in COS7 cell membranes with an inner diameter of ∼3 nm. Pore formation activity was also determined for GST-VP3 under the same conditions. Purified GST-VP3 induced cell lysis in the presence of PEG up to 3.8 nm in diameter (Fig. 1C). PEG of 6.4 nm in diameter and larger provided osmoprotection against GST-VP3 treatment. Based on the osmoprotection assay, VP2 and VP3 formed distinct pores in COS7 cell membranes with inner diameters of ∼3 nm and 4–6 nm, respectively.
VP2 and VP3 Possess Multiple Hydrophobic Domains
Transmembrane domains (TMD) are generally comprised of α-helical hydrophobic domains (HD) (22). A number of algorithms are available that identify HD that might act as TMD from the amino acid sequence of a protein (23–25). To gain insight into critical domains for the membrane disruption activities of VP2 and VP3, a bioinformatics analysis was performed using a variety of the established algorithms that predict α-helical TMD (Fig. 2A and supplemental Fig. S2).
The late viral proteins VP3 and VP4 are produced from successive Met residues within the VP2 transcript so they share a common C terminus. VP2 contains a unique N terminus (amino acids 1–118). The unique N terminus of VP2 is predicted to contain at least one TMD. The extreme N terminus (AA 1–27, HD1) was recently shown to be critical for SV40 infection supporting the importance of investigating the activity of VP2 during infection (12). All of the algorithms identified a HD designated as HD2, which extends from approximately amino acid 70 to 100 in VP2 (supplemental Fig. S2). Only Membrane Protein Explorer (MPEx) indicated that the overlapping VP2/3 region contained several HD (HD3–5), while half of the algorithms identified a HD in the overlapping VP2/3/4 region (HD6). A composite that contains all the predicted HD using all the algorithms shows that VP2 is comprised of 6 total HD (Fig. 2A and supplemental Fig. S2).
HD1, HD2, and HD6 Each Support Membrane Insertion with NCyto Topology
To determine if the individual HD of VP2 support membrane insertion, a reporter system was employed. Two variations of Lep, an E. coli secretory membrane protein, have been engineered to measure integration of potential membrane spanning domains in either the NCyto-CLumen or NLumen-CCyto orientation (17, 18). Individual segments of amino acids that might support membrane integration are incorporated into an acceptor site in Lep. To quantify the insertion of α-helical TMD, glycosylation sites have been introduced within Lep since N-linked glycan addition is restricted to the lumen of the ER. This experimental system has allowed the von Heijne group to develop a biological prediction of the free energy of TMD helix insertion (18, 26). To determine if the individual HD predicted within the late viral proteins could support membrane integration each domain was empirically tested for membrane insertion using the Lep constructs.
To quantify membrane insertion, an in vitro assay using RER-derived microsomes was utilized. Lep contains two TMD such that the N and C termini face the lumen of the ER (27). A modified version of Lep (Lep H2, Fig. 2B) contains an N-terminal extension with a glycosylation sequence and a second glycosylation site after the second TMD (17). The first TMD (TM1) is engaged by the signal recognition particle (SRP) and the protein is targeted to the Sec61 translocon for translocation into the ER lumen. Consequently, all proteins targeted to RER-derived microsomes will contain at least one glycan. To evaluate membrane integration, each HD was swapped for the second TMD (shaded gray). Thus if the HD supports membrane insertion, the protein is doubly glycosylated (Fig. 2B, left); whereas RER-derived microsome targeted but non-inserted domains will be singly glycosylated (Fig. 2B, right). The intensity of each of the glycosylated species can be readily quantified to determine the insertion efficiency of each HD.
The endogenous Lep TMD (TM2) was assayed in the acceptor site to determine the robustness of RER-derived microsome targeting and TMD insertion (Fig. 2C, lanes 1–4). In the absence of RER-derived microsomes, untranslocated and unglycosylated protein was observed. When the protein was translated in the presence of RER-derived microsomes doubly and singly glycosylated proteins were detected, which were deglycosylated by the addition of endoglycosidase H (Endo H) (Fig. 2C, compare lanes 3 and 4). The insertion efficiency of the native membrane domain was almost 80%, which demonstrates that the domain was efficiently recognized and that the Lep reporter effectively reflects membrane insertion propensity.
When Lep H2 HD1 was translated in the presence of RER-derived microsomes, a fragment was unexpectedly detected (filled star) that increased in mobility upon Endo H treatment (open star) indicating that it was glycosylated (Fig. 2C, compare lanes 7 to 8). This glycosylated fragment was likely caused by the internal cleavage of Lep H2 by the signal peptidase complex (Fig. 2B, star) (17). Since the active site of the signal peptidase complex is located in the lumen of the ER, HD1 inserted into the ER membrane and was subsequently recognized and cleaved. As cleavage only occurs if the domain is integrated into the lipid bilayer, HD1 was efficiently recognized and targeted for membrane integration. Therefore, membrane insertion of HD1 was significant as ∼25% was cleaved to the smaller fragment.
HD2 and HD6 also possessed internal cleavage sites that produced similar glycosylated fragments observed with HD1 but with greater efficiencies of 67 and 76%, respectively (Fig. 2C, lanes 11 and 27). Of the six potential TMD predicted, HD3, HD4, and HD5 did not support membrane insertion as less than 11% TMD insertion was observed for each (Fig. 2C, lanes 15, 19, and 23). The insertion efficiency in an NCyto topology was HD6>HD2≫HD1 and each of the three HD was recognized by the signal peptidase complex. Altogether, this indicated that the unique N terminus of VP2 contained two domains that supported membrane insertion and the C-terminal HD6 shared by VP2, VP3, and VP4 was inserted into the membrane with the highest efficiency.
Next, mutations were designed in the HD that would potentially disrupt membrane insertion so that the necessity of this activity could be tested in membrane disruption and viral propagation assays. Mutations in HD1, HD2, and HD6 domains were tested for membrane integration as these domains efficiently inserted into ER membranes. Membrane domains contain hydrophobic amino acids so charged residues were substituted for nonpolar residues in an attempt to inhibit membrane integration (22). Initially, charge substitutions were screened in silico for their effect on membrane integration using the algorithms that accurately predicted each respective membrane domain. Single amino acid mutations were not predicted to influence the membrane insertion properties of the HD. As double and triple substitutions within the HD were anticipated to abolish their membrane insertion properties, these mutations were assessed using the Lep H2 construct (Fig. 2, D–F).
Substitution of two or three hydrophobic residues with charged residues abolished the ability of HD1 to insert into the RER-derived membranes (Fig. 2D, lanes 11 and 15). In contrast, a conserved Glu in HD1 of VP2 was shown to be important for SV40 viral infection because this residue mediates interaction with a membrane-spanning domain in BAP31, an ER-resident membrane protein (12). The E17A mutation previously shown to inhibit viral infection was assessed in this in vitro targeting system to determine if it influenced membrane integration. Introduction of the hydrophobic residue within HD1 enhanced membrane integration over the wild type sequence almost 2-fold (Fig. 2D, compare lane 7 and 3).
Generally introduction of charged residues significantly inhibited membrane incorporation of HD2 and HD6 (Fig. 2, E and F). Mutations reduced membrane incorporation from half of the wild type HD to background levels. However, in some cases inhibition was position dependent. For example, Lep H2 HD2 A75K/V87E insertion was 25% compared with the same net mutation F80K/L84E, with only 10% insertion (Fig. 2E). Likewise charged residues at positions M295/S305 reduced membrane targeting from ∼76% to 32%, whereas charged residues at L298/L302 completely inhibited membrane incorporation to less than 10% (Fig. 2F). This supports the conclusion that some regions within each HD do not tolerate charged residues for membrane insertion.
To establish if the HD within the late viral proteins favored insertion in the alternative NLumen-CCyto direction, a modified version of Lep was used termed Lep H3 (18). Using this construct, HD1, HD4, and HD6 displayed only slight levels of membrane insertion with NLumen topology ranging from 5–12% above background (data not shown). This indicated that while these HD were recognized for membrane incorporation, the efficiency was low indicating that the NCyto-CLumen topology for HD1, HD2, and HD6 was favored.
HD6 Supports Autonomous Membrane Insertion
Membrane insertion using Lep H2 and Lep H3 is SRP-dependent because the N-terminal TMD of Lep (TM1) is recognized for RER-derived microsome insertion. This means that TM1 effectively tethers a test HD in the vicinity of the membrane, which might enhance membrane incorporation. To determine if any of the HD in VP2 could support membrane targeting on their own, an additional Lep construct was employed (Lep H1, Fig. 3A). The HD acceptor site (gray) is flanked by a single glycosylation site on the N terminus and two glycosylation sites on the C terminus. Thus if the protein were singly, doubly, or triply glycosylated it adopted an NLumen-CCyto, NCyto-CLumen, or N&CLumen orientation in the membrane, respectively. Proteins that are not targeted to the RER membranes will be unglycosylated.
To assess the background levels of targeting, a sequence that is hydrophobic but not targeted to RER-derived microsomes (1 Leu flanked by 9 Ala on either side; 9Ala-1Leu-9Ala) was tested for membrane targeting (Fig. 3B, lanes 1–4). As previously observed (17), the vector was not glycosylated and did not support RER-derived microsome targeting. Similarly, HD1-HD3 and HD5 did not exhibit detectable glycosylation so they were unable to directly integrate into RER membranes (Fig. 3B, lanes 7, 11, 15, and 23). Since HD1 and HD2 were efficiently ER targeted in Lep H2 (Fig. 2C), this suggested that these domains were reliant on other regions in the protein for membrane integration.
For HD4 a low level (∼10%) of singly glycosylated species was observed, suggesting that HD4 supported weak insertion with the NLumen-CCyto topology. This orientation was detected with Lep H3 at similar levels suggesting that this domain prefers an NLumen-CCyto orientation but incorporation was inefficient (Fig. 3B, lane 19 and data not shown). The targeting of HD4 was noteworthy as only one prediction algorithm (MPEx) suggested that this region could support membrane incorporation (supplemental Fig. S2).
Strikingly, HD6 produced a ladder of three glycosylated species in the presence of RER-derived microsomes (Fig. 3B, lane 27). This indicated that HD6 adopted three topologies in microsomes: NLumen-CCyto; NCyto-CLumen; and N&CLumen. Furthermore, each glycosylated species was observed in almost equal abundance suggesting that the orientations were favored equally (Fig. 3B, lane 27 and Fig. 3D, lane 3). HD6 displayed the most effective autonomous membrane targeting and insertion of all the HD tested yet it was variable in its topology. HD6 insertion required the microsomes to be present during translation, as the glycosylation ladder was not observed if the microsomes were added post-translationally (Fig. 3D, lane 5). Altogether, these results suggested that HD6, the domain shared by VP2, VP3, and VP4, displayed the highest inherent ability to target ER membranes.
To determine if the changes in membrane incorporation observed with Lep H2 HD6 mutations would be recapitulated with independent targeting of Lep H1 HD6, the charge substitutions were tested in Lep H1 HD6 (Fig. 3C). All charge substitutions tested abolished targeting of Lep H1 HD6. Overall, the self-directed targeting of Lep H1 HD6 was found to be more sensitive to charge substitutions than in the context of Lep H2.
HD Mutations Inhibit Membrane Disruption Activity of the Purified Late Proteins
To determine if the HD mutations associated with membrane insertion defects would affect the membrane disruption activity of the late proteins, the mutations were introduced into VP2, VP3, and VP4 for expression of the proteins in E. coli and the mutants were purified for characterization of their membrane disruption abilities. First, the addition of purified GST-VP2, GST-VP3 or GST-VP4 to mammalian cells resulted in release of ∼70, 80, and 97% of LDH, respectively (Fig. 4). However, when charged residues were incorporated into an individual HD, cell lysis was significantly inhibited for VP2, VP3, and VP4. In general, the effects on membrane incorporation studied with the Lep constructs as models for HD insertion, correlate with the membrane disruption activity of the late proteins. The targeted disruption of membrane insertion of an individual HD resulted in loss of membrane disruption activity. GST-VP2 E17A had 3-fold diminished membrane disruption activity compared with GST-VP2. Since the E17A mutation enhanced the membrane incorporation of Lep H2 HD1, likely due to increasing the hydrophobicity of the domain, this suggested that despite having enhanced membrane insertion abilities, the E17A mutation decreased the perforation activity of VP2.
FIGURE 4.
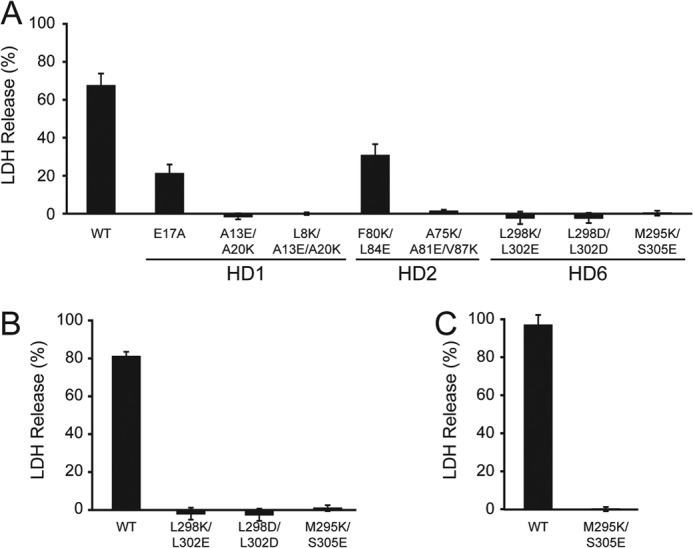
Purified VP2, VP3, and VP4 require HD for activity. A, bacterial expressed and purified wild type and mutant forms of VP2 (232 nm) were assayed as in Fig. 1 for their ability to disrupt the membranes of COS7 cells (10,000 cells). B and C, overlapping HD6 mutations were monitored for membrane disruption activity as in Fig. 1 with VP3 (B) and VP4 (C) as indicated. Error bars represent the standard deviation of three independent experiments.
Lastly, VP4 was recently shown to require L298 and L302 for pore formation because substitution of charged residues inhibited membrane binding (16). Since the same mutations inhibit the membrane disruption activity of GST-VP2 and GST-VP3 (Fig. 4), this supports that the same features are critical for activity of the purified proteins. Altogether these results demonstrated that VP2, VP3, and VP4 all require HD6 for their membrane disruption activity.
Introduction of Charged Residues into Individual HD Inhibits Viral Propagation
Several mutations were identified that inhibited the viroporin activity of VP2, VP3, and VP4. To determine if pore formation activity is critical in the viral life cycle, HD mutations were introduced into the viral genome and tested for propagation. To initiate viral propagation, BS-C-1 cells were transfected with the viral genome to trigger virus production and virus propagation was monitored. Initial samples were analyzed 2 days post-transfection (P.T.) after protein synthesis but before cell lysis and virion release (14). Propagation was determined 7 days P.T. to measure the ability of the viruses to infect the remaining population of cells. To score propagation levels, synthesis of the early gene product LT was quantified by flow cytometry (12, 14, 28). To uncover if the expression of the viral proteins was affected by the mutations that were introduced, Western blot analysis was used to monitor production of LT, VP1, VP2, and VP3. Nuclear fractions were inspected since all of these proteins are nuclear localized. Transfection bypassed the infection process and introduced the viral genome directly to the nucleus for virus production.
The percentage of LT-positive cells observed in wild type and HD mutant viral genome transfected cells was similar after 2 days (Fig. 5A, ∼1.5% each). Likewise immunoblot analysis of nuclear lysates 2 days P.T. indicated that LT was produced at similar levels (Fig. 5B, lanes 2–10). This demonstrated that the initial transfection level of cells was similar and early gene synthesis was not affected by the HD mutations. The HD1 and HD2 mutants did not change the expression levels of VP1 or VP2 and VP3 compared with wild type SV40 (Fig. 5B, lanes 2–7). In addition, the M295K/S305E mutation did not have an impact on the level of VP1 but slightly enhanced the production of VP2 and VP3 (Fig. 5B, lane 10). Protein synthesis was not perturbed for many of the HD mutations.
FIGURE 5.
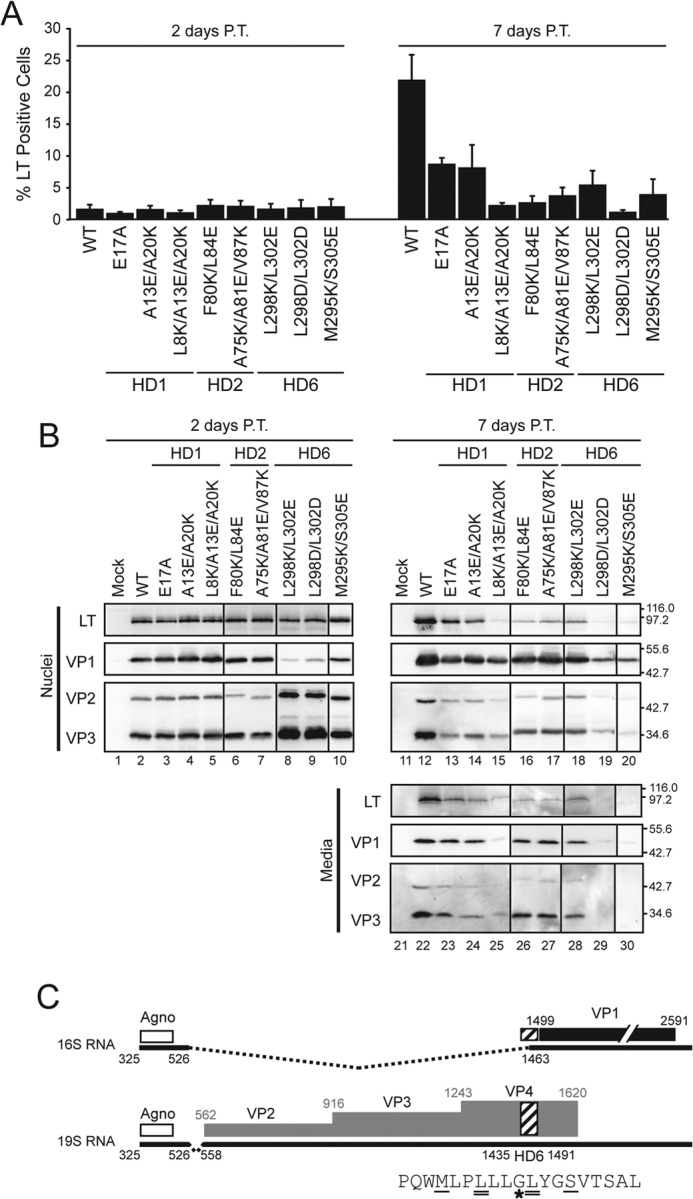
Introducing charged residues into individual hydrophobic domains inhibit viral propagation. A, BS-C-1 cells were transfected with wild type or mutant SV40 genomes as indicated. The percentage of infected cells was determined based on LT expression using flow cytometry after 2 days (left plot) and 7 days (right plot). Error bars represent the standard deviation of at least three independent experiments. B, BS-C-1 cells were transfected as in A, and the nuclei and media were immunoblotted and probed with the indicated antibodies after 2 days (left panel) or 7 days (right panel) post-transfection (P.T.). Molecular weight markers are indicated in kDa on the right. C, splicing of SV40 mRNA. The white, black, and gray regions denote the coding region for agnoprotein (Agno), VP1 and VP2/3/4, respectively. The 16S and 19S mRNA species are indicated with black lines where dashed pieces are removed by splicing to the mature forms. HD6 is highlighted by the hatched box and indicated within the splice site utilized to produce the 16S mRNA. The HD6 amino acid sequence (shown below) denotes the mutated L298/L302 residues as designated by double underlines and the M295K/S305E mutations is denoted by single underline. The star denotes the Gly codon that is the major splice site used to produce the 16S mRNA. This figure was adapted from Ref. 31.
In contrast, HD6 L298/L302 charge mutants displayed drastically reduced levels of VP1 and enhanced production of VP2 and VP3 compared with wild type SV40 and the other HD6 mutant (Fig. 5B, compare lane 2 to lanes 8 and 9). The HD6 Leu mutations inhibited production of VP1. Splicing of the late transcripts is known to control the temporal production of VP1 prior to VP2 and VP3 from 16S and 19S mRNA, respectively (29, 30). The main splice site utilized to produce the VP1-encoding 16S transcript lies in the HD6 coding region (31, 32) (Fig. 5C). This suggested that mutating the L298/L302 codons inhibited splicing of the 16S mRNA and led to premature expression of VP2 and VP3 from the 19S transcript. Consequently, both of the L298/L302 charge mutants drastically affected the expression of the late viral capsid proteins and were not characterized further.
As expected, wild type SV40 propagated in cell cultures as 22% of cells were LT-positive 7 days P.T. (Fig. 5A). Propagation was, however, inhibited for several of the HD mutants. Each mutant in HD1 displayed diminished propagation to various degrees compared with wild type SV40. For SV40 E17A and A13E/A20K propagation was less than half of wild type SV40 but for the triple mutant SV40 L8K/A13E/A20K propagation was completely inhibited as only 2% LT-positive cells were observed after 7 days. The E17A result was consistent with a recent study that demonstrated that it was defective for viral infection due to inhibition of interaction with the ER membrane protein BAP31 (12). Both charge mutations tested for HD2 completely inhibited viral propagation of SV40 (Fig. 5A). The HD6 mutations inhibited viral propagation as mutation of the central L298/L302 inhibited propagation along with M295K/S305E. This indicated that HD1, HD2, and HD6 each contain hydrophobic residues that were required for viral propagation.
Analysis of the expression level of the viral proteins 7 days after transfection was consistent with the propagation observed by flow cytometry (Fig. 5B, lanes 12–20). HD mutant viruses produced lower levels of LT, VP1, VP2, and VP3 compared with wild type SV40.
To determine if the mutant viruses were released from cells, the media was probed for viral proteins. The viral structural proteins were each detected in the media suggesting that cell lysis occurred for virus release (Fig. 5B, lanes 22–30). As with the flow cytometry results, viruses that propagate to a greater extent had increased levels of the viral proteins in the media. In addition, the nuclear-localized nonstructural viral protein LT was detected in the media at higher levels for viruses that supported low levels of propagation implying that cell lysis occurred. Markedly, LT was not detected in the media fraction for the M295K/S305E mutant. Overall, viral induced cell lysis was observed suggesting that the defects in propagation observed for the charge mutants in HD1, HD2, and HD6 could be attributed to some other step in the replication cycle.
HD Mutant Viruses Are Properly Assembled
To assess if viral encapsidation of the SV40 minichromosome was impaired in the HD mutants, a DNase protection assay was utilized. Viruses produced by transfection were subjected to DNase digestion and PCR of a genome fragment. Consequently if the capsid conferred nuclease protection, it suggested that the capsid was assembled around the viral genome. For wild type SV40 and the mutant viruses, an amplified PCR fragment was observed by agarose gel electrophoresis (Fig. 6A). However, this assay might be unable to differentiate between tight or loose-assembled viral particles as the genome from both may be inaccessible to DNase.
FIGURE 6.

SV40 mutants are properly assembled. A, integrity of the wild type and mutant viruses produced by transfection and purified by sedimentation was tested. PCR of the viral DNA was performed after treatment of the virus with DNase to determine if the viral capsid protected the DNA. PCR products were resolved by agarose gel electrophoresis. Viruses were also analyzed for VP1 content by performing an immunoblot. B, BS-C-1 cells were transfected for 3 days with wild type SV40 genome and harvested for transmission electron microscopy (TEM), the inset area is shown in C. Scale bar indicates 500 nm. C, representative images of virus particles observed in BS-C-1 cell nuclei after 3 days of transfection with mutant viral genomes as indicated. Scale bar corresponds to 100 nm. Diameter of viral particles observed by TEM as indicated. Standard deviations are from 100 particles measured.
To directly observe virus particle assembly, electron microscopy was utilized. BS-C-1 cells were transfected and subjected to thin section transmission electron microscopy (TEM). Virions in wild type transfected cells were observed throughout the nucleoplasm (Fig. 6B). Wild type virus was ∼45 nm in diameter as previously described (Fig. 6C) (7). In addition, virus particles were observed for all of the SV40 HD mutants characterized and these viruses were also ∼45 nm in diameter. Taken together, wild type and the HD mutant SV40 viruses appeared to be properly assembled indicating the propagation defects associated with HD mutants did not involve virus assembly.
Viroporin Activity Is Required for Viral Infection
To determine if infection was impaired for the SV40 HD mutants, BS-C-1 cells were infected with purified wild type or mutant viruses. A VP1 immunoblot of purified viral particles was used to normalize viral infections (Fig. 7A). Viral infectivity was quantified with flow cytometry based on LT expression 24 h postinfection.
FIGURE 7.
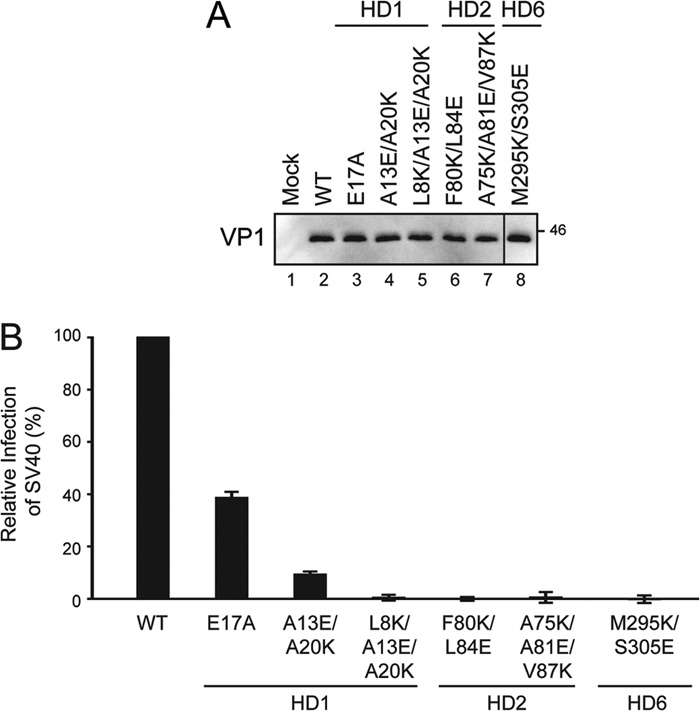
HD are required for viral infection. A, VP1 immunoblot of purified SV40 wild type and mutant viral particles to normalize infection levels used in B. Amino acids mutated in VP2/3/4 are indicated along with their location in the corresponding HD. B, percentage of infected cells was determined by flow cytometry 24 h postinfection and normalized to wild type SV40 using VP1 levels. Error bars represent the standard deviation of 30,000 cells measured in three independent experiments.
When charged residues were introduced into a HD, the infection level was reduced compared with wild type SV40 (Fig. 7B). The HD1 and HD2 charge mutants were infected at a level that was 10% or less of wild type. The substitution of charged residues in HD1 or HD2 was not tolerated for viral infection. In contrast, removal of the previously studied charged residue in HD1 (E17A) reduced the infectivity of the virus by 60% (12). SV40 M295K/S305E viral particles were defective for infection and propagation. Therefore, mutation of each HD-disrupted productive viral infection.
DISCUSSION
We have determined that the minor structural proteins from SV40 possess viroporin activity that is critical for viral propagation. Using an in vitro translation and membrane insertion assay, three HD in VP2 were identified that supported membrane integration. Purified VP2 and VP3 formed size-selective pores in COS7 cells. Targeted disruption by introducing charged residues into the HD inhibited membrane targeting and the viroporin activity of the purified proteins. Mutations in the late proteins that affected their membrane perforation activity also inhibited the propagation of SV40 demonstrating that viroporin activities of the late proteins were important for virus proliferation.
Purified VP4 possess lytic properties that are deployed during later stages in the viral life cycle to aid in the release of SV40 from host cells (15, 16). VP2 and VP3 contain the complete VP4 sequence, and they also possess membrane disruption properties. These studies were performed with bacterial expressed and purified GST constructs. When GST was removed from the purified proteins by TEV cleavage, the late proteins aggregated, hindering the ability to directly study the untagged proteins. The large N-terminal tags helped to maintain their solubility and inhibit their lethality in bacteria, as VP2 and VP3 proteins were toxic without the N-terminal tag (19). The minor structural proteins were each able to disrupt the plasma membrane of host cells as monitored by LDH release. A cell-based osmoprotection assay indicated that VP2 and VP3 form pores with diameters of ∼3 nm and 4–6 nm, respectively, though it is possible that the GST moiety might affect the size of the pore formed through steric hindrance.
To determine if pore formation by the minor structural proteins was required for the progression of the viral life cycle, HD mutations that affected the viroporin activity of the purified proteins and membrane targeting of the Lep constructs were introduced into the viral genome and tested for virus propagation. Viruses with mutations in HD1, HD2, or HD6 did not propagate as efficiently as wild type SV40. Introducing charged residues into HD1 or HD2 reduced the infection to less than 10% of wild type. In addition, introduction of charged residues within HD6 at M295/S305 reduced viral infection to a negligible level. Mutation of these critical residues in HD1, HD2, or HD6: 1) impaired membrane insertion as measured with Lep; 2) inhibited the membrane disruption activity of the full-length purified proteins; 3) perturbed viral propagation; 4) did not affect the production of the viral proteins; and 5) did not inhibit viral assembly. Taken together, this supports the conclusion that the viroporin activity of VP2 and VP3 was required for efficient infection.
After internalization and trafficking to the ER, SV40 particles are acted upon by oxidoreductases that direct viral uncoating (4). This process may release VP2 and VP3 to disrupt the ER membrane to support penetration of the subviral particle. However, the poorly defined penetrating subviral particle is expected to be larger than the 3–6 nm pores created by the purified proteins. Alternatively, uncoating may liberate domains of VP2 and VP3 that disrupt the membrane in the context of being attached to the uncoated viral particle. VP2 has been recently found to interact through its N terminus with the ER membrane protein BAP31 (12). VP2 is also myristoylated at its N terminus (33). Together, the BAP31 interaction and lipid modification may help VP2 to engage the membrane and support local membrane disruption with the aid of HD such as HD6. Membrane disruption may also be aided by membrane accessibility of VP3. Further analysis is required to demonstrate that the viroporin activity of VP2 and VP3 was not required for earlier stages of infection including binding and intracellular trafficking before conclusions about penetration may be reached.
The sequences of VP2 and VP3 can vary widely in different polyomaviruses yet the C-terminal domain corresponding to HD6 is highly conserved (34). Introduction of charged residues into HD6 inhibited the membrane disruption activity of VP2 and VP3. The minor structural proteins bind to a hydrophobic pocket in VP1 pentamers to coordinate capsid assembly (35). The region of VP2 and VP3 that mediates binding to VP1 comprises part of HD6, one of the HD that supported membrane incorporation in Lep H2 and H1. As inhibition of membrane insertion of HD6 impaired the viroporin activity of VP2 and VP3, this supports the conclusion that HD6 was utilized for both membrane perforation and virus assembly. In addition, the nucleotide sequence corresponding to HD6 contains the mRNA splice site used to produce the 16S mRNA for synthesis of VP1. The region of the viral genome corresponding to HD6 is used for viroporin activity, assembly of virions and splicing of the late mRNA. The SV40 M295K/S305E mutant virus was properly assembled but unable to support viral propagation or infection. Altogether, this suggests that HD6 is highly conserved in polyomaviruses because selective pressure on the virus prevents mutations in this critical region of the viral genome.
Viruses have a limited protein coding capacity due to their size restrictions. Therefore, viral proteins often serve versatile functions in the viral life cycle. Viroporins are an understudied class of proteins that have been characterized most thoroughly for their role in virus release; however several have recently been shown to perform crucial functions for other stages of viral life cycle. The influenza virus viroporin M2 is critical for efficient uncoating of the subviral particle in the cytoplasm for infection, depolarizing the plasma membrane to aid in virus release and to facilitate membrane scission for release of virus particles from infected cells (36, 37). Likewise the p7 viroporin from hepatitis C virus functions in the secretory pathway to promote virus assembly and release of active viral particles (38, 39). Mutagenesis or chemical inhibition of M2 or p7 prevents viral propagation, providing direct evidence that their viroporin activity is required for productive viral replication. Based on findings presented here, the nonenveloped virus SV40 utilizes the viroporin activity of the minor structural proteins VP2 and VP3 for effective infection. Agnoprotein from JC polyomavirus has been recently shown to be a viroporin (40). If agnoprotein from SV40 also possesses viroporin activity, combined with our VP2, VP3, and VP4 results (15), this suggests that four of the seven total SV40 encoded proteins have viroporin activity. Future studies will be needed to investigate the specific stage(s) of viral infection that require viroporin activity.
Acknowledgments
We thank Dale Callaham from the University of Massschusetts Central Microscopy Facility for help with performing the transmission electron microscopy of viral particles. We would also like to acknowledge current and past members of the Hebert laboratory for helpful discussions including Dr. Robert Daniels (University of Stockholm). In addition we would like to thank Drs. Gunnar von Heijne and IngMarie Nilsson (University of Stockholm) for their assistance with the Lep constructs.
This work was supported by United States Public Health Grant AI078142 (to D. N. H.) and supported in part by National Science Foundation, Integrative Graduate Education and Research Traineeship (IGERT), Institute for Cellular Engineering (DGE-0654128), and by National Institutes of Health Chemistry-Biology Interface Training Grant T32GM00815 (to K. M. G.).

This article contains supplemental Figs. S1 and S2.
- SV40
- Simian Virus 40
- ER
- endoplasmic reticulum
- LT
- large T antigen
- VLP
- virus-like particles
- LDH
- lactate dehydrogenase
- PEG
- polyethylene glycol
- TMD
- transmembrane domain
- Endo H
- endoglycosidase H
- P.T.
- post-transfection
- HD
- hydrophobic domain
- Lep
- leader peptidase.
REFERENCES
- 1. Campanero-Rhodes M. A., Smith A., Chai W., Sonnino S., Mauri L., Childs R. A., Zhang Y., Ewers H., Helenius A., Imberty A., Feizi T. (2007) N-glycolyl GM1 ganglioside as a receptor for simian virus 40. J. Virol. 81, 12846–12858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tsai B., Gilbert J. M., Stehle T., Lencer W., Benjamin T. L., Rapoport T. A. (2003) Gangliosides are receptors for murine polyoma virus and SV40. EMBO J. 22, 4346–4355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderson H. A., Chen Y., Norkin L. C. (1996) Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell 7, 1825–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schelhaas M., Malmström J., Pelkmans L., Haugstetter J., Ellgaard L., Grünewald K., Helenius A. (2007) Simian Virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell 131, 516–529 [DOI] [PubMed] [Google Scholar]
- 5. Reddy V. B., Thimmappaya B., Dhar R., Subramanian K. N., Zain B. S., Pan J., Ghosh P. K., Celma M. L., Weissman S. M. (1978) The genome of simian virus 40. Science 200, 494–502 [DOI] [PubMed] [Google Scholar]
- 6. Fiers W., Contreras R., Haegemann G., Rogiers R., Van de Voorde A., Van Heuverswyn H., Van Herreweghe J., Volckaert G., Ysebaert M. (1978) Complete nucleotide sequence of SV40 DNA. Nature 273, 113–120 [DOI] [PubMed] [Google Scholar]
- 7. Stehle T., Gamblin S. J., Yan Y., Harrison S. C. (1996) The structure of simian virus 40 refined at 3.1 A resolution. Structure 4, 165–182 [DOI] [PubMed] [Google Scholar]
- 8. Chen X. S., Stehle T., Harrison S. C. (1998) Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. EMBO J. 17, 3233–3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pelkmans L., Püntener D., Helenius A. (2002) Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 296, 535–539 [DOI] [PubMed] [Google Scholar]
- 10. Damm E. M., Pelkmans L., Kartenbeck J., Mezzacasa A., Kurzchalia T., Helenius A. (2005) Clathrin- and caveolin-1-independent endocytosis: entry of simian virus 40 into cells devoid of caveolae. J. Cell Biol. 168, 477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Engel S., Heger T., Mancini R., Herzog F., Kartenbeck J., Hayer A., Helenius A. (2011) The role of endosomes in SV40 entry and infection. J. Virol. 85, 4198–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geiger R., Andritschke D., Friebe S., Herzog F., Luisoni S., Heger T., Helenius A. (2011) BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 13, 1305–1314 [DOI] [PubMed] [Google Scholar]
- 13. Kawano M. A., Inoue T., Tsukamoto H., Takaya T., Enomoto T., Takahashi R. U., Yokoyama N., Yamamoto N., Nakanishi A., Imai T., Wada T., Kataoka K., Handa H. (2006) The VP2/VP3 minor capsid protein of simian virus 40 promotes the in vitro assembly of the major capsid protein VP1 into particles. J. Biol. Chem. 281, 10164–10173 [DOI] [PubMed] [Google Scholar]
- 14. Daniels R., Rusan N. M., Wadsworth P., Hebert D. N. (2006) SV40 VP2 and VP3 Insertion into ER Membranes Is Controlled by the Capsid Protein VP1: Implications for DNA Translocation out of the ER. Mol. Cell 24, 955–966 [DOI] [PubMed] [Google Scholar]
- 15. Raghava S., Giorda K. M., Romano F. B., Heuck A. P., Hebert D. N. (2011) The SV40 Late Protein VP4 Is a Viroporin that Forms Pores to Disrupt Membranes for Viral Release. PLoS Pathog. 7, e1002116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giorda K. M., Raghava S., Hebert D. N. (2012) The Simian Virus 40 Late Viral Protein VP4 Disrupts the Nuclear Envelope for Viral Release. J. Virol. 86, 3180–3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lundin C., Kim H., Nilsson I., White S. H., von Heijne G. (2008) Molecular code for protein insertion in the endoplasmic reticulum membrane is similar for N(in)-C(out) and N(out)-C(in) transmembrane helices. Proc. Natl. Acad. Sci. U.S.A. 105, 15702–15707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hessa T., Kim H., Bihlmaier K., Lundin C., Boekel J., Andersson H., Nilsson I., White S. H., von Heijne G. (2005) Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433, 377–381 [DOI] [PubMed] [Google Scholar]
- 19. Daniels R., Rusan N. M., Wilbuer A. K., Norkin L. C., Wadsworth P., Hebert D. N. (2006) Simian virus 40 late proteins possess lytic properties that render them capable of permeabilizing cellular membranes. J. Virol. 80, 6575–6587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brogden K. A. (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250 [DOI] [PubMed] [Google Scholar]
- 21. Scherrer R., Cabrera Beaman T., Gerhardt P. (1971) Macromolecular sieving by the dormant spore of Bacillus cereus. J. Bacteriol. 108, 868–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. von Heijne G. (2006) Membrane-protein topology. Nat. Rev. Mol. Cell Biol. 7, 909–918 [DOI] [PubMed] [Google Scholar]
- 23. Claros M. G., von Heijne G. (1994) TopPred II: an improved software for membrane protein structure predictions. Comput. Appl. Biosci. 10, 685–686 [DOI] [PubMed] [Google Scholar]
- 24. Shen H., Chou J. J. (2008) MemBrain: improving the accuracy of predicting transmembrane helices. PLoS One 3, e2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones D. T. (2007) Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 23, 538–544 [DOI] [PubMed] [Google Scholar]
- 26. Hessa T., Meindl-Beinker N. M., Bernsel A., Kim H., Sato Y., Lerch-Bader M., Nilsson I., White S. H., von Heijne G. (2007) Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450, 1026–1030 [DOI] [PubMed] [Google Scholar]
- 27. Moore K. E., Miura S. (1987) A small hydrophobic domain anchors leader peptidase to the cytoplasmic membrane of Escherichia coli. J. Biol. Chem. 262, 8806–8813 [PubMed] [Google Scholar]
- 28. Drayman N., Kler S., Ben-nun-Shaul O., Oppenheim A. (2010) Rapid method for SV40 titration. J. Virol. Methods 164, 145–147 [DOI] [PubMed] [Google Scholar]
- 29. Sedman S. A., Good P. J., Mertz J. E. (1989) Leader-encoded open reading frames modulate both the absolute and relative rates of synthesis of the virion proteins of simian virus 40. J. Virol. 63, 3884–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Good P. J., Welch R. C., Barkan A., Somasekhar M. B., Mertz J. E. (1988) Both VP2 and VP3 are synthesized from each of the alternative spliced late 19S RNA species of simian virus 40. J. Virol. 62, 944–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu Y., Alwine J. C. (2006) 19S late mRNAs of simian virus 40 have an internal ribosome entry site upstream of the virion structural protein 3 coding sequence. J. Virol. 80, 6553–6558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lai C. J., Dhar R., Khoury G. (1978) Mapping the spliced and unspliced late lytic SV40 RNAs. Cell 14, 971–982 [DOI] [PubMed] [Google Scholar]
- 33. Streuli C. H., Griffin B. E. (1987) Myristic acid is coupled to a structural protein of polyoma virus and SV40. Nature 326, 619–622 [DOI] [PubMed] [Google Scholar]
- 34. Johne R., Buck C. B., Allander T., Atwood W. J., Garcea R. L., Imperiale M. J., Major E. O., Ramqvist T., Norkin L. C. (2011) Taxonomical developments in the family Polyomaviridae. Arch. Virol. 156, 1627–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liddington R. C., Yan Y., Moulai J., Sahli R., Benjamin T. L., Harrison S. C. (1991) Structure of Simian virus 40 at 3.8-Å resolution. Nature 354, 278–284 [DOI] [PubMed] [Google Scholar]
- 36. Martin K., Helenius A. (1991) Nuclear transport of influenza virus ribonucleoproteins: the viral matrix protein (M1) promotes export and inhibits import. Cell 67, 117–130 [DOI] [PubMed] [Google Scholar]
- 37. Rossman J. S., Jing X., Leser G. P., Lamb R. A. (2010) Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142, 902–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boson B., Granio O., Bartenschlager R., Cosset F. L. (2011) A concerted action of hepatitis C virus p7 and nonstructural protein 2 regulates core localization at the endoplasmic reticulum and virus assembly. PLoS Pathog. 7, e1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jones C. T., Murray C. L., Eastman D. K., Tassello J., Rice C. M. (2007) Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 81, 8374–8383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Suzuki T., Orba Y., Okada Y., Sunden Y., Kimura T., Tanaka S., Nagashima K., Hall W. W., Sawa H. (2010) The human polyoma JC virus agnoprotein acts as a viroporin. PLoS Pathog. 6, e1000801. [DOI] [PMC free article] [PubMed] [Google Scholar]