

Background: γ-Secretase activating protein, GSAP, was identified as a novel regulator of γ-secretase and amyloid-β production.
Results: Reducing GSAP expression in cells decreased amyloid-β generation. However, overexpression of GSAP in cells and recombinant GSAP in vitro did not modulate amyloid-β generation.
Conclusion: The relationship between GSAP and γ-secretase is unclear.
Significance: GSAP is not a validated therapeutic target for Alzheimer disease.
Keywords: Alzheimers Disease, Amyloid, Amyloid Precursor Protein, Secretases, siRNA, GSAP, Imatinib
Abstract
γ-Secretase is a large enzyme complex comprising presenilin, nicastrin, presenilin enhancer 2, and anterior pharynx-defective 1 that mediates the intramembrane proteolysis of a large number of proteins including amyloid precursor protein and Notch. Recently, a novel γ-secretase activating protein (GSAP) was identified that interacts with γ-secretase and the C-terminal fragment of amyloid precursor protein to selectively increase amyloid-β production. In this study we have further characterized the role of endogenous and exogenous GSAP in the regulation of γ-secretase activity and amyloid-β production in vitro. Knockdown of GSAP expression in N2a cells decreased amyloid-β levels. In contrast, overexpression of GSAP in HEK cells expressing amyloid precursor protein or in N2a cells had no overt effect on amyloid-β generation. Likewise, purified recombinant GSAP had no effect on amyloid-β generation in two distinct in vitro γ-secretase assays. In subsequent cellular studies with imatinib, a kinase inhibitor that reportedly prevents the interaction of GSAP with the C-terminal fragment of amyloid precursor protein, a concentration-dependent decrease in amyloid-β levels was observed. However, no interaction between GSAP and the C-terminal fragment of amyloid precursor protein was evident in co-immunoprecipitation studies. In addition, subchronic administration of imatinib to rats had no effect on brain amyloid-β levels. In summary, these findings suggest the roles of GSAP and imatinib in the regulation of γ-secretase activity and amyloid-β generation are uncertain.
Introduction
Alzheimer disease (AD)3 is a devastating neurodegenerative disorder affecting more than 30 million people worldwide (1). The disease is characterized pathologically by the presence of extracellular senile plaques and intracellular neurofibrillary tangles in the brain (2, 3). The core constituents of senile plaques are insoluble aggregates of the 4-kDa amyloid-β (Aβ) peptides, Aβ40 and Aβ42, which are generated following sequential proteolytic processing of the amyloid precursor protein (APP) by β-site APP cleaving enzyme 1 (BACE1) and γ-secretase (4). Although Aβ40 is the most abundantly produced Aβ peptide, the slightly longer and less abundant Aβ42 peptide has been implicated as the key pathogenic species in AD brain (5).
γ-Secretase is a large protein complex composed of four components, presenilin (PS), nicastrin, presenilin enhancer 2, and anterior pharynx-defective 1 (Aph-1) that are required for γ-secretase activity (6). Additional ancillary proteins such as CD147 and TPM21 that can associate with the γ-secretase complex and regulate Aβ generation have also been identified (7, 8). γ-Secretase cleaves APP at γ-sites within the transmembrane domain to generate Aβ peptides of varying length and also at the ϵ-site near the cytoplasmic membrane border of APP to generate the APP intracellular domain (AICD) (9). In analogy to the other membrane proteins that are subject to γ-secretase cleavage, including the Notch receptor (10), it has been proposed that AICD can translocate to the nucleus and regulate gene transcription (11).
Due to its pivotal role in the generation of Aβ peptides, γ-secretase is an attractive therapeutic target for AD. Early drug discovery efforts focused on the development of γ-secretase inhibitors. However, due to the fundamental role γ-secretase plays in the intramembrane proteolysis of a number of other proteins (10), the development of these compounds was hindered by mechanism-based toxicities associated with inhibition of Notch signaling (12–14). Consequently, drug discovery efforts have shifted to the development of γ-secretase modulators that selectively lower Aβ42 production, in the absence of any effect on Notch processing (15, 16) and therefore, should be safer and better tolerated than γ-secretase inhibitors.
Alternative approaches to regulate γ-secretase activity and Aβ generation are of immense interest for the treatment of AD. Recently, a novel 16-kDa γ-secretase activating protein (GSAP-16K) was identified that forms a ternary complex with γ-secretase and the juxtamembrane region of the APP C-terminal fragment (CTF) and thereby, regulates cleavage at the γ- and ϵ-sites and hence, generation of Aβ and AICD (17). Decreasing GSAP expression in cells significantly reduced Aβ levels but had no effect on Notch cleavage. In vitro, recombinant GSAP-16K was shown to elevate γ-secretase activity. In addition, chronic depletion of GSAP in vivo was found to reduce brain Aβ levels and Aβ plaque development. Intriguingly, imatinib (also known as STI571 or Gleevec®), an Abl kinase inhibitor and anti-cancer drug that was previously shown to selectively inhibit Aβ production in cells (18), was shown to mediate its Aβ lowering activity by binding GSAP and preventing its interaction with APP CTF (17). These findings highlighted GSAP as a potentially novel therapeutic target for the treatment of AD. The aim of this study was to further characterize the role of GSAP and imatinib in the regulation of γ-secretase activity.
EXPERIMENTAL PROCEDURES
Cell Lines
Mouse neuroblastoma Neuro2a (N2a) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with l-glutamine (Invitrogen) supplemented with 5% heat-inactivated fetal bovine serum (FBS) (Lonza, Basel, Switzerland) and 100 units/ml of penicillin and streptomycin. Human embryonic kidney cells overexpressing APP Swedish variantK595N/M596L (HEK-APPsw) or APP wild type (HEK-APPwt) were cultured in DMEM with l-glutamine supplemented with 10% heat-inactivated FBS, 100 units/ml of penicillin and streptomycin, and 100 or 200 μg/ml of hygromycin B (Invitrogen), respectively. Human T-lymphoma SUP-T1 cells were cultured in Roswell Park Memorial Institute Medium (RPMI 1640) (Invitrogen) supplemented with 10% heat-inactivated FBS. Human neuroblastoma SH-SY5Y cells overexpressing the γ-secretase substrate precursor (SHSY5Y-SPA4CT) were cultured in 1:1 minimal essential medium with Earle's salt and glutamine and F12 medium (Invitrogen), 1× nonessential amino acids, 10% FBS, 100 units/ml of penicillin and streptomycin, and 300 μg/ml of hygromycin B. All cell lines were cultured at 37 °C in a humidified atmosphere of 5% CO2, 95% air.
siRNA Knockdown Studies
For cellular knockdown studies, siRNA to GSAP of the following sequence were designed: sense sequence, 5′-CUUUGCUGGUAGAAAUACATT-3′ and antisense sequence, 5′-UGUAUUUCUACCAGCAAAGTT-3′ (Microsynth, Balgach, Switzerland). A nontargeting siRNA pool (Dharmacon Inc., Lafayette, CO) was used as a negative control. Mouse neuroblastoma N2a cells were reverse transfected with 50 nm siRNA using DharmFECT2 transfection reagent (Thermo Fischer Scientific Inc., Waltham, MA) according to the manufacturer's instruction. 24 h post-transfection, medium was removed and fresh medium was added. In the inhibitor studies, the fresh medium contained either 0.5% dimethyl sulfoxide (DMSO) or 10 μm imatinib (AK Scientific Inc., Union City, CA). 48 h post-transfection, medium was removed for Aβ analysis and cells were lysed for extraction of total RNA.
Quantitative RT-PCR
Total RNA was extracted from cells using TRIzol (Invitrogen) as described by the manufacturer. RNA was quantified and then reverse transcribed using the iScript cDNA Synthesis Kit (Bio-Rad). Quantification of mouse or human GSAP or GAPDH (housekeeping gene) mRNA transcripts were performed using the quantitative PCR using FastStart Universal SYBR Green Master mix and the LightCycler® 480 Real-Time PCR System (both from Roche Applied Science). Primer sequences used for mouse GSAP were 5′-TCCAGATCACCAGAGAAG-3′ (forward sense) and 5′-ATCCCACTGAGCCCAAAC-3′ (reverse sense), and for human GSAP were 5′-AATTCTGGCCATCTCCCAAG-3′ (forward sense) and 5′-ACTGAGCCCAAACGAAATCC-3′ (reverse sense) (Thermo Fischer Scientific Inc.). A mouse or human GAPDH primer mixture was used as an internal control (Qiagen, Hilden, Germany). Quantitative PCR were run in 12 μl in triplicate with 3 μl of cDNA solution and 9 μl of PCR target-specific reaction mixture. Reactions were performed with 1200 nm mouse GSAP primers and 1× FastStart Universal SYBR Green Master (ROX) reagent (Roche Applied Science). PCR conditions were as follows: an initial cycle of 95 °C for 5 min followed by 50 cycles of 95 °C for 10 s, 60 °C for 30 s, and a final melting cycle at 97 °C. Raw Ct values were used to calculate % GSAP expression relative to the housekeeping gene GAPDH.
Transient Overexpression Studies
A plasmid encoding full-length human GSAP with a C-terminal HA tag, pReceiver-M07-GSAP-HA (GeneCopoeia, Rockville, MD) was used for overexpression of full-length GSAP-HA in cells. The 16-kDa C-terminal fragment of GSAP, GSAP-16K (amino acids 733–854 of full-length GSAP) was amplified from pReceiver-M07-GSAP-HA and subcloned into pRevM07 vector with a C-terminal HA tag. HEK-APPsw cells, HEK-APPwt, or N2a cells were transiently transfected with GSAP using FuGENE HD transfection reagent (Roche Applied Science) according to the manufacturer's instructions. 24 h post-transfection, medium was removed and fresh medium was added. 48 h post-transfection, medium was removed for Aβ analysis and cells were lysed in 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40 containing an EDTA-free protease inhibitor mixture (Roche Applied Science).
Aβ Analysis
Medium from cells was assayed for Aβ40 or Aβ42 using a sensitive immunoassay employing a biotinylated mid-region Aβ antibody, 4G8 (Covance, Princeton, NJ), and a ruthenylated C-terminal Aβ40 antibody G2–10 (Millipore, Billerica, MA) or Aβ42-specific antibody A387 (licensed from Torrey Pines Therapeutics). In addition to detecting Aβ peptides, this assay also detected the minor p3 species generated via the nonamyloidogenic/α-secretase pathway. Plates were analyzed on a SECTOR® Imager 6000 (Meso Scale Discovery, Gaithersburg, MD).
Rat brain quadrisects were homogenized in 0.2% diethylamine (Thermo Fischer Scientific Inc.) in 50 mm NaCl, pH 10 (10% v/w), and centrifuged for 35 min at 355,000 × g. The supernatant was removed and neutralized with 0.5 m Tris-HCl, pH 6.8 (10% v/v). DEA brain extracts were assayed in triplicate for Aβ40 as described above. Plasma and CSF samples were diluted 2-fold in PBS containing 2% bovine serum albumin (BSA), 0.2% Tween 20, and a protease inhibitor mixture (Roche Applied Science) and assayed in triplicate for Aβ40 as described above. For quantification of Aβ, standard curves were constructed using human Aβ40 (Anaspec, Fremont, CA).
SDS-PAGE and Western Blot Analysis
Purified recombinant GSAP-16K (8 μg) was resolved on a 4–12% BisTris NuPAGE gel (Invitrogen) under reducing or nonreducing conditions as described by the manufacturer and protein bands were visualized with Coomassie Blue. For Western blot analysis, cell lysates were resolved by SDS-PAGE and transferred to nitrocellulose (Invitrogen). Membranes were probed with an anti-HA antibody (Abcam, Cambridge, UK), anti-APP CTF antibody CT695 (Invitrogen), anti-PS-1 antibody (Calbiochem, Darmstadt, Germany), anti-cleaved Notch 1 (Val-744) antibody (Cell Signaling, Boston, MA), or anti-actin antibody (Millipore) for detection of GSAP-HA, APP CTF, PS-1 CTF, NICD, and actin, respectively. Aβ and AICD-FLAG generated in the in vitro γ-secretase assay were detected by Western blot analysis with Aβ-specific antibody 6E10 (Covance) and FLAG-specific M2 antibody (Sigma), respectively.
Co-immunoprecipitation Studies
HEK-APPsw cells were transiently transfected with GSAP-16KHA. 24 h post-transfection, cells were incubated with the γ-secretase inhibitor, DAPT (1 μm; Calbiochem). 48 h post-transfection cells were lysed in 50 mm HEPES, 150 mm NaCl, 5 mm MgCl2, 5 mm CaCl2, 1% CHAPSO containing a protease inhibitor mixture. Prior to immunoprecipitation, cell lysates were diluted in lysis buffer lacking CHAPSO to give 0.25% final CHAPSO concentration. Cell lysates were incubated for 3 h at room temperature with 5 μg of anti-HA antibody or 5 μg of anti-APP C-terminal antibody CT695. Dynabeads® M-280 sheep anti-rabbit IgG (50 μl; Invitrogen) were added and samples were incubated overnight at 4 °C. A control incubation of cell lysates with Dynabeads alone was also conducted. Dynabeads were collected and washed 5 times with lysis buffer containing 0.25% CHAPSO. Bound proteins were eluted with SDS sample buffer containing reducing agent and subject to Western blot analysis as described above.
Expression and Purification of GSAP-16K
Human GSAP-16K was amplified from pReceiver-M07-GSAP-HA (GeneCopoeia) and subcloned into pET30a vector (Novagen, Darmstadt, Germany) with a C-terminal His6 tag for expression in BL21DE3 Escherichia coli. The E. coli cell paste was resuspended in 5 volumes (ml/g) of 0.1 m Tris-HCl, pH 7.5, 10 mm MgCl2 and homogenized with a Polytron. Benzonase (50 units/g of bacteria) was added and the sample was incubated at room temperature on a magnetic stirrer for 15 min. The bacteria were disrupted by 2 passages through a French press at 1600 bar and the sample was centrifuged at 25,000 × g for 2 h. The pellet corresponding to the inclusion bodies was solubilized in 0.1 m Tris-HCl, pH 8.0, 6 m guanidine HCl, 20 mm DTT, homogenized as above, and heated for 30 min at 60 °C prior to centrifugation at 100,000 × g for 1 h. The supernatant was filtered through a 0.22-μm filter and loaded on a 280-ml Source 30 RPC column (GE Healthcare) equilibrated in 0.1% trifluoroacetic acid in water and bound proteins were eluted with a gradient of elution buffer (0.1% trifluoroacetic acid, 80% acetonitrile in water). Fractions were collected, analyzed by SDS-PAGE, and the fractions containing the GSAP protein were pooled and lyophilized. The lyophilized protein was solubilized in 0.1 m Tris-HCl, pH 8.0, 6 m guanidine HCl, 2 mm DTT and refolded through an overnight 20-fold dilution with 0.1 m Tris-HCl, pH 8.5, 0.5 m arginine at 4 °C. The refolded sample was adjusted to pH 8.5, 0.05% Brij-35 was added and the sample was filtered before loading on a 20-ml nickel-nitrilotriacetic acid-agarose column (Qiagen) equilibrated in 50 mm Tris-HCl, pH 8.5, 0.05% Brij-35. Bound proteins were eluted with 50 mm Tris-HCl, pH 8.5, 300 mm imidazole, 0.05% Brij 35. Fractions containing the GSAP protein were identified by SDS-PAGE, pooled, and dialyzed against PBS, 0.05% Brij-35.
In Vitro γ-Secretase Assays
Membranes from SHSY5Y-SPACT cells were prepared according to Beher et al. (19). For in vitro generation of Aβ peptides, membranes (equivalent to 50 μg of proteins) were incubated for 4 h at 37 °C in the presence of recombinant GSAP-16K (0.2, 2, and 7 μg), BSA (0.2, 2, and 7 μg), 0.05% DMSO, 10 μm imatinib, or 1 μm γ-secretase inhibitor XIX (Calbiochem). GSAP-16K was pre-treated with 5 mm DTT for 30 min at 37 °C for assays under reducing conditions. De novo production of Aβ40 and Aβ42 was quantified using the immunoassay described above.
In vitro γ-secretase assays using the recombinant APP-based C100-FLAG substrate and purified γ-secretase were performed as previously reported (20). γ-Secretase and GSAP were solubilized in 0.2% (w/v) CHAPSO, HEPES, pH 7.0, 150 mm NaCl, 5 mm CaCl2, 5 mm MgCl2 and preincubated for 30 min with 0.1% phosphatidylcholine and 0.025% (w/v) phosphatidyethanolamine before addition of 1 μm APP-based C100-FLAG. Assays were performed with 0, 2.3, or 23 μg/ml of GSAP, either under reducing (5 mm DTT) or nonreducing (0 mm DTT) conditions. Prior to assays performed under reducing conditions, GSAP was pre-treated for 30 min with 5 mm DTT at 37 °C. Consistent with previous results, the presence of DTT alone did not affect γ-secretase activity (21). All assay reactions were incubated 4 h at 37 °C and the resulting products, Aβ and AICD-FLAG, were detected by Western blot analysis as described above or analyzed by immunoprecipitation and mass spectrometry as previously described (21).
Compound Treatment
For 16 h treatment with inhibitor, cells were cultured to 70–90% confluence in 96-well plates. Imatinib was dissolved in DMSO and added to cells at the appropriate test concentration, ensuring that the final DMSO concentration was <0.5%. A DMSO solvent control was included in each experiment. The effect of compounds on cell viability was assessed using alamarBlue® (Invitrogen). Medium was collected for Aβ analysis as described above. To determine the effect of inhibitors on Notch processing, SUP-T1 cells were lysed in 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.5% Nonidet P-40, 1% Triton X-100, 0.2% SDS containing a mixture of protease inhibitors (Roche Applied Science) for 30 min at 4 °C.
For the in vivo study, male Sprague-Dawley rats (Janvier Laboratory, La Mayenne, France) weighing between 150 and 200 g were housed for 4 days on a 12-h light-dark schedule with water and food available ad libitum before the initiation of the study. Animals were dosed by intraperitoneal injection with saline (NaCl 0.9%, n = 8) or imatinib mesylate (30 mg/kg, n = 8; Enzo Life Sciences GmbH, Lörrach, Germany) twice daily for 5 days. A second group of animals (n = 5–7/group) were subject to a single oral administration of vehicle (0.5% (w/v) methylcellulose, 0.25% (w/v) Tween 20) or the γ-secretase inhibitor (GSI), LY-411575 (10 mg/kg) (12). Two hours post-final dose on day 5 for the imatinib study or 4 h post-dose for the GSI study, rats were anesthetized under isofluorane (induction box, 3–5%) and then injected intraperitoneally with a combination of xylasine (10 mg/kg) and ketamine (75 mg/kg) diluted in saline to maintain the animals under deep anesthesia. The top of the head was sprayed with 70% ethanol and an incision with scissors was performed in the skin to expose the parietal region of the skull. Twenty to 30 μl of CSF was collected from the cisterna magna with a syringe MYJECTOR 29G (Terumo, Tokyo, Japan) and stored at −80 °C for Aβ analysis. An intracardiac puncture was performed to collect 0.6 ml of blood. 500 μl of blood was transferred into EDTA containing tubes for Aβ analysis and 100 μl of blood was transferred into heparinized tubes for determination of imatinib concentration. Blood samples were centrifuged at 6000 × g for 10 min at 4 °C and plasma was collected and stored at −20 or −80 °C for subsequent analysis of imatinib or Aβ, respectively. Brains were removed and divided into four equal parts. The right frontal quarter was collected in a precellys tube (VWR International, Radnor, PA) and stored at −20 °C for subsequent determination of imatinib concentration in the brain. The parietal quarter was collected in a 1.5-ml Falcon tube and stored at −80 °C prior to analysis for Aβ.
Determination of Drug Concentrations
The concentration of imatinib in plasma and brain was determined using a method based on protein precipitation followed by HPLC/MS/MS analysis. Plasma samples (5 μl) were diluted in methanol (150 μl) and an internal standard (15 μl) was added. All samples were centrifuged at 12,000 × g for 5 min. Supernatant (50 μl) was diluted in 400 μl of acetonitrile/H2O (10:90, v/v) and assayed for imatinib by HPLC using a Chromolith® fast gradient RP-18e, 50-3 mm column (Agilent Technologies, Santa Clara, CA). The mobile phase was composed of two eluents: eluent A consisted of water containing 0.1% formic acid and eluent B consisted of pure acetonitrile, with a flow rate of 0.8 ml/min and a run time of 3.5 min. Brains were homogenized in 1 ml of ethanol and incubated for 30 min prior to centrifugation at 12,000 × g for 5 min. Brain homogenates (10 μl) were diluted in acetonitrile/H2O (10:90, v/v) and 15 μl of an internal standard added. Samples were analyzed by HPLC as described above. Analytes were detected using an API400 Mass Spectrometer (Applied Biosystems, Carlsbad, CA). The lower limit of quantification of imatinib was 16 ng/ml (blood) and 165 ng/g (brain).
Statistical Analysis
Statistical analysis was conducted using an unpaired t test or a one-way ANOVA with post-hoc Dunnett's multiple comparison test in GraphPad PRISM 5.0 (GraphPad Software, Inc., La Jolla, CA).
RESULTS
Effect of GSAP siRNA on Aβ Generation
To investigate the level of endogenous GSAP expression in various cell lines and rat brain tissue, quantitative RT-PCR studies were first performed. GSAP mRNA expression was detectable in all cell lines, albeit at very low levels ranging from 0.01 to 0.05% of the housekeeping gene, GAPDH (Fig. 1a). The level of GSAP mRNA was slightly higher in rat brain tissue at 0.2% of the housekeeping gene, GAPDH (Fig. 1a). Next, to determine whether GSAP expression could modulate Aβ generation, siRNA knockdown studies were conducted. Mouse neuroblastoma N2a cells were selected for these studies as both GSAP mRNA expression and endogenous Aβ levels were detectable in these cells and also because this cell line was used by He et al. (17) in their studies on GSAP. 48 h post-siRNA treatment, GSAP mRNA expression was reduced by 74% in N2a cells (Fig. 1b). This was associated with a significant decrease in the levels of Aβ40 (36%, p < 0.0001) and Aβ42 (42%, p < 0.0001) in the medium of these cells (Fig. 1c).
FIGURE 1.

siRNA-mediated knockdown of GSAP in N2a cells. a, total RNA was extracted from different cell lines or rat brain tissue and the mRNA levels of GSAP and the housekeeping gene GAPDH were determined by quantitative RT-PCR. Data are mean ± S.D. GSAP expression is expressed as a % of the housekeeping gene GAPDH. b and c, N2a cells were transfected with GSAP siRNA or a scramble siRNA control (Ctrl). 48 h post-transfection, the effects on GSAP mRNA expression (b) and Aβ generation (c) were determined. Data are mean ± S.E. Statistical analysis was conducted using an unpaired t test or a one-way ANOVA with Dunnett's multiple comparison test (***, p < 0.0001).
Effect of GSAP Overexpression on Aβ Generation
Given that a knockdown in GSAP expression resulted in a decrease in Aβ levels, we hypothesized that increasing GSAP expression in cells would increase Aβ generation. Full-length human GSAP with a C-terminal HA tag (GSAP-FLHA) was overexpressed in HEK cells stably expressing APP Swedish variant (HEK-APPsw) or in HEK cells stably expressing APP wild type (APPwt) and the effect on Aβ levels in the medium was determined. GSAP-FLHA migrated on SDS-PAGE as a protein of ∼98 kDa (Fig. 2, a and c). In HEK-APPsw cells, expression of GSAP-FLHA resulted in a small yet significant decrease in the level of Aβ40 in the medium (21% p < 0.0001) but had no effect on the level of Aβ42 (Fig. 2b). In contrast, in HEK-APPwt cells, expression of GSAP-FLHA resulted in a small yet significant increase in the level of Aβ42 in the medium (18% p < 0.0001) but had no effect on the level of Aβ40 (Fig. 2d). These modest effects on Aβ levels were observed with transfection efficiencies in the range of 60–70 and 30–50% for GSAP-FLHA in HEK-APPsw and HEK-APPwt cells, respectively. Unfortunately, attempts to overexpress full-length GSAP in N2a cells, the cell line used in the studies by He et al. (17), were not successful.
FIGURE 2.

Overexpression of GSAP in HEK-APPsw and HEK-APPwt cells. HEK-APPsw cells (a and b) or HEK-APPwt (c and d) were transiently transfected with empty vector (Ctrl) or cDNA encoding full-length human GSAP (GSAP-FLHA) or the C-terminal fragment GSAP-16KHA. 48 h post-transfection, medium was removed for Aβ analysis (b and d) and cell lysates were generated for Western blot analysis of GSAP expression with an anti-HA antibody (a and c). Aβ data are mean ± S.E. Statistical analysis was conducted using a one-way ANOVA with Dunnett's multiple comparison test (***, p < 0.0001).
He et al. (17) proposed that the predominant form of GSAP in N2a cells was not the full-length 98-kDa protein but instead a 16-kDa C-terminal fragment of human GSAP (GSAP-16K). Intriguingly, we did not observe any proteolytic processing of GSAP-FLHA to GSAP-16K following overexpression in HEK-APPsw or HEK-APPwt cells. To exclude the possibility that the lack of a profound effect of GSAP expression on Aβ generation in these cells was due to the absence of GSAP-16K, we also expressed GSAP-16K with a C-terminal HA tag (GSAP-16KHA) in HEK-APPsw and HEK-APPwt cells and determined its effect on Aβ generation. Like GSAP-FLHA, expression of GSAP-16KHA in HEK-APPsw cells caused a small yet significant decrease (29%, p < 0.0001) in the level of Aβ40 in the medium, but had no effect on the level of Aβ42 (Fig. 2b). In HEK-APPwt cells, expression of GSAP-16KHA resulted in a small yet significant decrease in both Aβ40 and Aβ42 levels (17%, p < 0.0001; Fig. 2d). Furthermore, transient expression of GSAP-16KHA in N2a cells was found to have no significant effect on the level of Aβ peptides (Fig. 3, a and b). The transfection efficiencies for GSAP-16KHA in all three cell lines were high and ranged from 50 to 75%.
FIGURE 3.

Overexpression of GSAP in N2a cells and GSAP co-immunoprecipitation studies. N2a cells were transiently transfected with cDNA encoding GSAP-16KHA. 48 h post-transfection, medium was removed for Aβ analysis (b) and cell lysates were generated for Western blot analysis of GSAP expression with an anti-HA antibody (a). Aβ data are mean ± S.E. Statistical analysis was conducted using a one-way ANOVA with Dunnett's multiple comparison test (#, not significant). c, HEK-APPsw cells were transiently transfected with human GSAP-16KHA. 48 h post-transfection, cell lysates were generated. Immunoprecipitation (IP) of GSAP-16KHA (HA) or APP CTF (CTF) was performed followed by Western blot analysis to detect the bound proteins. To control for nonspecific binding, incubation with Dynabeads (beads) alone was conducted.
GSAP and APP CTF Interaction Studies
To determine whether GSAP could directly interact with the APP CTF to modulate γ-secretase activity as proposed by He et al. (17), co-immunoprecipitation studies were conducted. GSAP-16KHA was overexpressed in HEK-APPsw cells and cell lysates were generated. Immunoprecipitation of GSAP-16KHA or APP CTF was subsequently performed followed by Western blot analysis to detect the bound proteins. GSAP-16KHA was efficiently captured with the anti-HA antibody (Fig. 3c, upper panel). No APP CTF was pulled down with GSAP-16KHA, suggesting these proteins do not interact with each other (Fig. 3c, middle panel). In addition, no interaction of GSAP-16KHA with the PS-1 CTF was evident (Fig. 3c, lower panel). To confirm these observations, the immunoprecipitation was also conducted in the opposite manner. APP CTF was efficiently captured with the anti-CTF antibody (Fig. 3c, middle panel). No interaction with GSAP-16KHA was observed (Fig. 3c, upper panel). However, we could confirm a specific interaction of the APP CTF with the PS-1 CTF (Fig. 3c, lower panel).
Effect of Recombinant GSAP on Aβ Generation in Vitro
To further investigate whether GSAP could modulate γ-secretase cleavage and Aβ production, the effect of purified GSAP was explored in two different and well characterized in vitro γ-secretase assays. Recombinant GSAP-16K with a C-terminal His6 tag (GSAP-16KHis6) was expressed in E. coli and purified as described under “Experimental Procedures.” On SDS-PAGE under nonreducing conditions, purified GSAP-16KHis6 migrated as a dimer with a predicted molecular mass of 32 kDa that was dissociated to monomers with a predicted molecular mass of 16 kDa under reducing conditions (Fig. 4a). Recombinant GSAP-16KHis6 was subsequently incubated with membranes from SHSY5Y-SPA4CT cells that contain both endogenous γ-secretase and overexpressed APP CTF substrate (22) and the effect on Aβ generation was determined. Consistent with our overexpression studies, no increase in the generation of Aβ40 was observed in the presence of various concentrations of recombinant GSAP-16KHis6 (Fig. 4b). In contrast, a potent GSI reduced Aβ generation in this assay (Fig. 4b). To determine whether the dimeric nature of GSAP may influence its activity in vitro, studies were also conducted with GSAP-16KHis6 that was pre-treated with DTT to generate monomeric GSAP-16KHis6 prior to its incubation with membranes from SHSY5Y-SPA4CT cells. Neither DTT alone nor monomeric GSAP-16KHis6 had any effect γ-secretase activity and Aβ generation in vitro (Fig. 4b).
FIGURE 4.

Effect of recombinant GSAP-16K in a membrane-based γ-secretase assay. a, recombinant GSAP-16K with a C-terminal His6 tag was expressed in E. coli and purified on a nickel-nitrilotriacetic acid column. The protein was analyzed by SDS-PAGE under nonreducing (lane 1) and reducing (lane 2) conditions. b, membranes from SHSY5Y-SPA4CT cells were incubated for 4 h at 37 °C in the presence of BSA, GSAP-16KHis6, 0.5% DMSO, or 1 μm GSI and the level of Aβ40 generated was quantified by immunoassay. GSAP-16KHis6 was either untreated or pre-treated with 5 mm DTT for 30 min at 37 °C prior to the assay. Data are mean ± S.E. Statistical analysis was conducted using a one-way ANOVA with Dunnett's multiple comparison test (**, p < 0.001).
To extend these findings, the effect of recombinant GSAP-16KHis6 on the generation of Aβ peptides and the AICD was determined in an alternative assay utilizing purified γ-secretase (20). In this assay, purified γ-secretase was incubated with exogenous APP CTF (C100-FLAG) substrate in the absence or presence of GSAP-16KHis6 that was either untreated or pre-treated with DTT. Assays were performed with γ-secretase:GSAP-16KHis6 molar ratios of 1:1 (2.3 μg/ml of GSAP-16KHis6) and 1:10 (23 μg/ml of GSAP-16KHis6). The presence of GSAP-16KHis6 had no effect on the levels of Aβ peptides or the AICD generated in vitro (Fig. 5a). In addition, no evident change in the Aβ peptide profile was observed by mass spectrometric analysis of the peptides generated in this assay (Fig. 5b).
FIGURE 5.

Effect of recombinant GSAP-16K on purified γ-secretase. a, recombinant C100-FLAG was incubated with purified γ-secretase for 4 h at 37 °C in the presence of buffer (Ctrl) or GSAP-16KHis6 (2.3 or 23 μg/ml). GSAP-16KHis6 was untreated or pre-treated with 5 mm DTT for 30 min at 37 °C prior to the assay. The reactions were analyzed by Western blot for C100-FLAG and the cleavage products Aβ and AICD-FLAG. b, samples from the C100-FLAG assay were also analyzed for Aβ peptides by immunoprecipitation and MALDI-TOF mass spectrometry. Representative spectra are shown.
Effect of Imatinib on Aβ Generation and Notch Processing
GSAP was identified as the molecular target responsible for the Aβ lowering activity of imatinib (17). We therefore, investigated the effect of imatinib on Aβ generation in N2a cells. Treatment of N2a cells with imatinib resulted in a concentration-dependent decrease in Aβ40 and Aβ42 levels with an IC50 of ∼3–6 μm (Fig. 6a). Imatinib was also profiled in SHSY5Y cells overexpressing the C-terminal fragment of APP (SHSY5Y-SPA4CT). As shown in Fig. 6b, a concentration-dependent decrease in Aβ40 and Aβ42 levels was observed in these cells resulting in ∼40% inhibition at 10 μm. No detrimental effect on cell viability was observed in either cell lines. The direct effect of imatinib on γ-secretase activity was also investigated in vitro. Imatinib was found to have no effect on Aβ40 generation in our membrane-based in vitro γ-secretase assay, whereas treatment with GSI markedly reduced Aβ production (Fig. 6c).
FIGURE 6.

Effect of imatinib on Aβ generation and Notch processing in cells. a, N2a cells, or b, SHSY5Y-SPA4CT cells were grown in the absence or presence of increasing concentrations of imatinib. 24 h later, medium was removed for Aβ analysis and the cells subject to a cellular viability assay (alamarBlue®). c, membranes from SHSY5Y-SPA4CT cells were incubated for 4 h at 37 °C in the presence of 0.5% DMSO, 10 μm imatinib, or 1 μm GSI and the level of Aβ40 generated was quantified by immunoassay. Data are mean ± S.E. d, SUP-T1 cells were grown in the presence of 0.5% DMSO, 10 μm imatinib, or 1 μm GSI. 24 h later, cells were lysed and subject to Western blot analysis for the Notch-intracellular domain (NICD) or actin. The graph depicts the % change in the level of NICD normalized to the actin control. Data are mean ± S.E. Statistical analysis was conducted using a one-way ANOVA with Dunnett's multiple comparison test (***, p < 0.0001).
Imatinib reportedly inhibits γ-secretase cleavage of APP with no effect on Notch processing (18). To confirm these findings, human T-lymphoma SUP-T1 cells, which constitutively express a truncated Notch receptor (23) were grown in the presence of 10 μm imatinib or 1 μm GSI. Treatment of cells with imatinib had no effect on NICD generation (Fig. 6d). In contrast, the γ-secretase inhibitor abolished Notch cleavage and NICD generation (Fig. 6d).
To determine whether the effects of imatinib on Aβ generation in cells were mediated via inhibition of GSAP, the effect of imatinib in cells treated with GSAP siRNA was investigated. N2a cells were treated with either control siRNA or GSAP siRNA in the absence of presence of imatinib (10 μm). As observed previously, knockdown of GSAP expression in cells (75%, p < 0.0001) resulted in a significant decrease in the levels of Aβ40 (31%, p < 0.0001) and Aβ42 (30%, p < 0.0001) in the medium (Fig. 7). Interestingly, co-treatment with imatinib resulted in an additional reduction in Aβ levels to 54 (p < 0.0001) and 46% (p < 0.0001) of control levels for Aβ40 and Aβ42, respectively (Fig. 7b). These reductions were similar to those observed following imatinib treatment alone (Fig. 7b).
FIGURE 7.
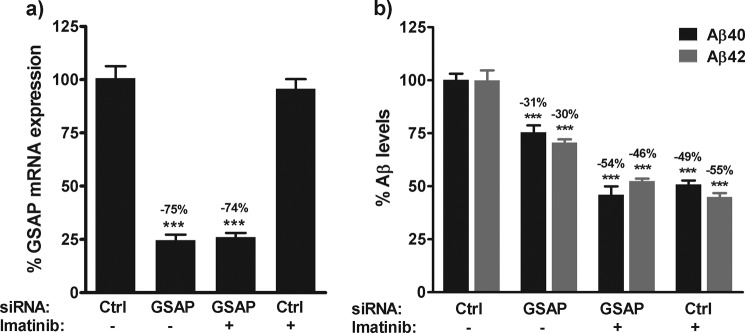
Effect of imatinib in GSAP siRNA-treated cells. N2a cells were transfected with GSAP siRNA or a scramble siRNA control (Ctrl). 24 h post-transfection medium was replaced with medium containing 0.5% DMSO or 10 μm imatinib. 48 h post-transfection, the effect on GSAP mRNA expression (a) and Aβ generation (b) was determined. Data are mean ± S.E. Statistical analysis was conducted using a one-way ANOVA with Dunnett's multiple comparison test (***, p < 0.0001).
Finally, to determine whether imatinib could modulate Aβ levels in vivo, a subchronic study was conducted. Rats were subject to intraperitoneal administration of saline or imatinib (30 mg/kg) twice daily for 5 days and the effect on the level of Aβ40 in plasma, CSF, and brain was determined. This dosing paradigm resulted in plasma and brain exposure of imatinib of 13.3 ± 4.9 and 1.56 ± 0.72 μm, respectively. However, no significant decrease in the level of Aβ40 was observed in plasma or brain following imatinib treatment (Fig. 8, a and b). Imatinib treatment also had no effect on the level of Aβ40 in CSF (data not shown). To demonstrate that effective Aβ lowering could be achieved in this model, a separate cohort of animals was administered a single oral dose of GSI, LY-411575 (10 mg/kg) (12). This resulted in a significant decrease in the level of Aβ40 in plasma (98%, p < 0.0001) and brain (96%, p < 0.0001) (Fig. 8, c and d).
FIGURE 8.
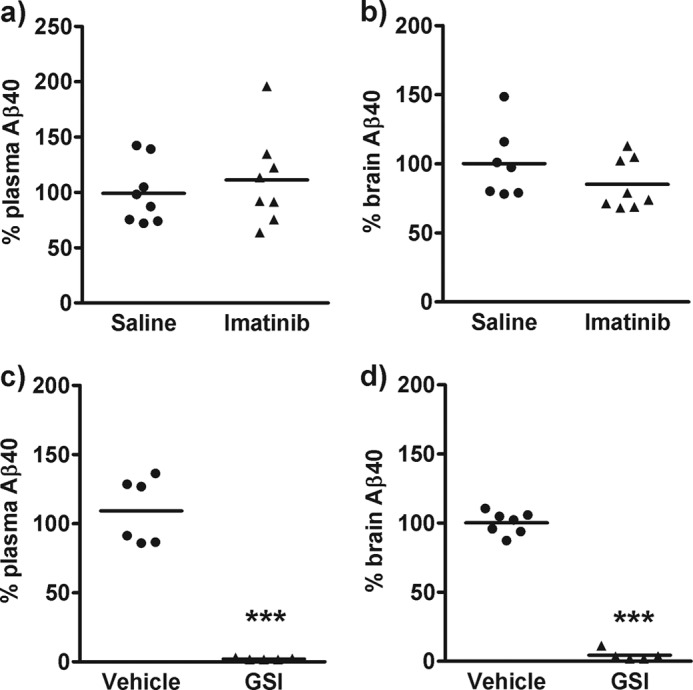
Effect of imatinib in vivo. Rats were administered with saline or imatinib (30 mg/kg) twice daily for 5 days or vehicle and a GSI (10 mg/kg) for 4 h. Plasma (a and c) or brain extracts (b and d) were assayed for Aβ40 using a sensitive immunoassay. Statistical analysis was conducted using a one-way ANOVA with Dunnett's multiple comparison test (***, p < 0.0001).
DISCUSSION
The modulation of γ-secretase activity to selectively decrease Aβ levels without inhibiting cleavage of other substrates such as Notch is an attractive therapeutic approach for AD. The identification of the novel protein, GSAP as a regulator of γ-secretase activity highlighted this protein as a potential new target for AD. In this study we have further investigated the role of GSAP in the regulation of γ-secretase activity.
In siRNA knockdown studies we demonstrated that a reduction of endogenous GSAP expression significantly reduced Aβ levels in N2a cells. The degree of GSAP mRNA lowering and the decrease in Aβ levels achieved were consistent with published findings (17) and suggested that endogenous GSAP can modulate Aβ generation. Based on these observations we postulated that overexpression of GSAP in cells would have the opposite effect and increase Aβ levels. However, overexpression of GSAP in HEK-APPsw or HEK-APPwt cells had no overt effect on Aβ levels regardless of whether full-length GSAP or the 16-kDa CTF of GSAP (GSAP-16K) was expressed. Similarly, overexpression of GSAP-16K in N2a cells had no effect on Aβ generation. Moreover, the modest decreases in Aβ levels observed following overexpression of GSAP in cells were inconsistent with the siRNA results. Importantly, these findings also contrasted with the published study where transfection of GSAP in HEK cells overexpressing APP-CTF resulted in an increase in Aβ production when monitored by pulse-chase labeling (17). Unfortunately, the level of GSAP expression required to observe the increase in Aβ was unclear as the expression of GSAP achieved in these experiments was not shown. One plausible explanation for why we failed to see an effect on Aβ levels in our cell system could be that the level of endogenous GSAP in these cells is sufficient to modulate γ-secretase activity and the presence of excess GSAP protein cannot further enhance γ-secretase cleavage of APP. Unfortunately, in the absence of a well validated commercially available antibody to GSAP we were unable to determine the level of endogenous GSAP protein in these cell lines. However, at the mRNA level, the expression of GSAP was very low suggesting GSAP is not an abundantly expressed protein. He et al. (17) also proposed that the predominant form of GSAP in N2a cells at steady state was the C-terminal 16-kDa fragment of GSAP, GSAP-16K. In our hands, following overexpression of full-length GSAP in HEK-APPsw or HEK-APPwt cells, no proteolytic processing to generate GSAP-16K was evident.
The contradictory findings of the GSAP knockdown and overexpression studies raise queries over the potential role of GSAP in the modulation of γ-secretase activity and Aβ generation. It is conceivable that the decrease in Aβ levels observed in the siRNA knockdown studies is not due to a direct effect of GSAP on γ-secretase activity but instead due to an indirect effect on the trafficking or assembly of the γ-secretase complex or other components of the APP processing pathway. In support of this hypothesis, no specific interaction between GSAP-16K and APP CTF or PS-1 CTF was observed in co-immunoprecipitation studies suggesting these proteins do not form a complex. Once again, these observations were inconsistent with the findings of He et al. (17).
To further explore the potential role of GSAP in the regulation of γ-secretase activity, we generated recombinant GSAP-16K and assessed its activity in two distinct and well characterized in vitro γ-secretase assays. Purified recombinant GSAP-16K was predominantly a dimer that could be converted to monomer in the presence of the reducing agent, DTT. Both forms of exogenous GSAP-16K were tested in the in vitro γ-secretase assays. First, we investigated the effect of recombinant GSAP-16K in a membrane-based γ-secretase assay that was similar to the one utilized by He et al. (17). Surprisingly and in contrast to published findings (17), we observed no increase in de novo Aβ40 generation in the presence of dimeric or monomeric GSAP-16K. We therefore investigated the effect of recombinant GSAP-16K in an alternative assay using purified γ-secretase and exogenous C100-FLAG substrate. One of the advantages of this assay was that we could regulate the ratio of GSAP and γ-secretase enzyme. However, again we found that exogenous GSAP-16K had no effect on the levels of Aβ or AICD generated in vitro, even when present in excess. These findings were consistent with our overexpression studies and suggested GSAP-16K does not regulate γ-secretase activity and Aβ generation in vitro. As no experimental details were provided on the purification of recombinant GSAP-16K utilized by He et al. (17), we were unable to compare the nature of our recombinant GSAP-16K to their GSAP-16K. Hence, we cannot exclude the possibility that under certain specific conditions an active form of GSAP-16K can be generated that can form a ternary complex with γ-secretase and APP CTF to regulate cleavage at the γ- and ϵ-sites. However, a recent publication on the purification and characterization of human GSAP found that recombinant GSAP did not associate with imatinib or APP CTF in vitro (24), further supporting our findings.
To extend these findings, we conducted studies with imatinib, an Abl tyrosine kinase inhibitor that reportedly reduces Aβ levels by preventing the interaction of GSAP with APP CTF, by a mechanism that does not involve kinase inhibition (17, 18). Treatment of N2a cells and SHSY5Y-SPA4CT cells with imatinib resulted in a concentration-dependent decrease in Aβ levels that was consistent with an action on the γ-secretase pathway. Moreover, imatinib had no effect on γ-secretase processing of Notch and NICD production in SUP-T1 cells confirming it is a specific modulator of γ-secretase activity in cells. In contrast to the findings in cells, imatinib had no effect on Aβ generation in our membrane-based γ-secretase assay suggesting it does not target γ-secretase directly. These observations were consistent with previously published results (18, 25) and suggest that imatinib can selectively modulate γ-secretase cleavage of APP via an indirect cellular mechanism. This would be consistent with the notion that imatinib binds an alternative target such as GSAP to modulate Aβ production in cells. To investigate this further, we determined whether imatinib had the potential to further reduce Aβ generation in cells treated with GSAP siRNA. In contrast to He et al. (17) who reported that imatinib had little or no additional effect on Aβ lowering in N2a cells treated with GSAP siRNA, we found that imatinib could further reduce Aβ levels in N2a cells with reduced GSAP expression. We cannot exclude the possibility that some endogenous GSAP protein may still be present in the GSAP siRNA-treated N2a cells, which could be inhibited by imatinib and give rise to this further reduction in Aβ levels. However, our findings do give rise to the possibility that imatinib can decrease Aβ levels by some alternative mechanism independent of GSAP. For instance, it has been shown that imatinib can regulate Aβ and AICD levels in cells by a mechanism involving increased neprilysin expression (26, 27).
Recently it was reported that subchronic administration of imatinib in mice can reduce Aβ levels in plasma and that this decrease in peripheral Aβ was sufficient to reduce Aβ levels in the brain (28). Contrary to these findings, we found subchronic administration of imatinib in rats had no effect on the level of Aβ40 in plasma despite achieving a plasma exposure in excess of the cellular IC50. Consistent with published findings (29), administration of imatinib resulted in a low exposure in the brain that was far below the cellular IC50 to have a pharmacodynamic effect on brain Aβ. Our findings clearly demonstrate that imatinib has no effect on Aβ levels in vivo and these findings coupled with its poor brain penetration demonstrate that imatinib is of limited therapeutic value for the treatment of AD.
In summary, our studies highlight that the relationship between GSAP, imatinib, and γ-secretase is unclear. A number of findings initially reported by He et al. (17) were not reproducible in our studies, thereby raising doubts on the potential validity of GSAP as novel target for the treatment of AD.
Acknowledgments
We thank Aurelie Baguet, Arman Saric, Martine Bertrand, Catherine Cavegn-Battier, and Virginie Zimmer for technical support. We also thank Bruno Antonsson, François Fossiez, and Antoine Attinger for technical advice and support.
This work was supported by Swiss National Science Foundation Grant 31003A_134938/1 (to P. C. F.) and an Ecole Polytechnique Fédérale de Lausanne (EPFL)-Merck Serono grant (to J. R. A. and M. D.).
- AD
- Alzheimer disease
- Aβ
- amyloid-β peptide
- APP
- amyloid precursor protein
- AICD
- amyloid precursor protein intracellular domain
- PS
- presenilin
- CTF
- carboxyl-terminal fragment
- DMSO
- dimethyl sulfoxide
- GSAP
- γ-secretase activating protein
- GSI
- γ-secretase inhibitor
- NICD
- Notch intracellular domain
- ANOVA
- analysis of variance
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- CHAPSO
- 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonic acid.
REFERENCES
- 1. Barnes D. E., Yaffe K. (2011) The projected effect of risk factor reduction on Alzheimer's disease prevalence. Lancet Neurol. 10, 819–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Glenner G. G., Wong C. W. (1984) Alzheimer's disease. Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890 [DOI] [PubMed] [Google Scholar]
- 3. Goedert M., Wischik C. M., Crowther R. A., Walker J. E., Klug A. (1988) Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease. Identification as the microtubule-associated protein Tau. Proc. Natl. Acad. Sci. U.S.A. 85, 4051–4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Selkoe D. J. (1994) Cell biology of the amyloid β-protein precursor and the mechanism of Alzheimer's disease. Annu. Rev. Cell Biol. 10, 373–403 [DOI] [PubMed] [Google Scholar]
- 5. Hardy J. A., Higgins G. A. (1992) Alzheimer's disease. The amyloid cascade hypothesis. Science 256, 184–185 [DOI] [PubMed] [Google Scholar]
- 6. De Strooper B. (2003) Aph-1, Pen-2, and nicastrin with presenilin generate an active-secretase complex. Neuron 38, 9–12 [DOI] [PubMed] [Google Scholar]
- 7. Zhou S., Zhou H., Walian P. J., Jap B. K. (2005) CD147 is a regulatory subunit of the γ-secretase complex in Alzheimer's disease amyloid β-peptide production. Proc. Natl. Acad. Sci. U.S.A. 102, 7499–7504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pardossi-Piquard R., Böhm C., Chen F., Kanemoto S., Checler F., Schmitt-Ulms G., St George-Hyslop P., Fraser P. E. (2009) TMP21 transmembrane domain regulates γ-secretase cleavage. J. Biol. Chem. 284, 28634–28641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu X. (2009) γ-Secretase catalyzes sequential cleavages of the AβPP transmembrane domain. J. Alzheimers Dis. 16, 211–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haapasalo A., Kovacs D. M. (2011) The many substrates of presenilin/γ-secretase. J. Alzheimers Dis. 25, 3–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beckett C., Nalivaeva N. N., Belyaev N. D., Turner A. J. (2012) Nuclear signalling by membrane protein intracellular domains. The AICD enigma. Cell Signal. 24, 402–409 [DOI] [PubMed] [Google Scholar]
- 12. Wong G. T., Manfra D., Poulet F. M., Zhang Q., Josien H., Bara T., Engstrom L., Pinzon-Ortiz M., Fine J. S., Lee H. J., Zhang L., Higgins G. A., Parker E. M. (2004) Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits β-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem. 279, 12876–12882 [DOI] [PubMed] [Google Scholar]
- 13. Imbimbo B. P., Panza F., Frisardi V., Solfrizzi V., D'Onofrio G., Logroscino G., Seripa D., Pilotto A. (2011) Therapeutic intervention for Alzheimer's disease with γ-secretase inhibitors. Still a viable option? Expert Opin. Investig. Drugs 20, 325–341 [DOI] [PubMed] [Google Scholar]
- 14. Schor N. F. (2011) What the halted phase III γ-secretase inhibitor trial may (or may not) be telling us. Ann. Neurol. 69, 237–239 [DOI] [PubMed] [Google Scholar]
- 15. Wolfe M. S. (2007) γ-Secretase modulators. Curr. Alzheimer Res. 4, 571–573 [DOI] [PubMed] [Google Scholar]
- 16. Beher D. (2008) γ-Secretase modulation and its promise for Alzheimer's disease. A rationale for drug discovery. Curr. Top. Med. Chem. 8, 34–37 [DOI] [PubMed] [Google Scholar]
- 17. He G., Luo W., Li P., Remmers C., Netzer W. J., Hendrick J., Bettayeb K., Flajolet M., Gorelick F., Wennogle L. P., Greengard P. (2010) γ-Secretase activating protein is a therapeutic target for Alzheimer's disease. Nature 467, 95–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Netzer W. J., Dou F., Cai D., Veach D., Jean S., Li Y., Bornmann W. G., Clarkson B., Xu H., Greengard P. (2003) Gleevec inhibits β-amyloid production but not Notch cleavage. Proc. Natl. Acad. Sci. U.S.A. 100, 12444–12449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beher D., Fricker M., Nadin A., Clarke E. E., Wrigley J. D., Li Y. M., Culvenor J. G., Masters C. L., Harrison T., Shearman M. S. (2003) In vitro characterization of the presenilin-dependent γ-secretase complex using a novel affinity ligand. Biochemistry 42, 8133–8142 [DOI] [PubMed] [Google Scholar]
- 20. Wu F., Schweizer C., Rudinskiy N., Taylor D. M., Kazantsev A., Luthi-Carter R., Fraering P. C. (2010) Novel γ-secretase inhibitors uncover a common nucleotide-binding site in JAK3, SIRT2, and PS1. FASEB J. 24, 2464–2474 [DOI] [PubMed] [Google Scholar]
- 21. Alattia J. R., Kuraishi T., Dimitrov M., Chang I., Lemaitre B., Fraering P. C. (2011) Mercury is a direct and potent γ-secretase inhibitor affecting Notch processing and development in Drosophila. FASEB J. 25, 2287–2295 [DOI] [PubMed] [Google Scholar]
- 22. Shearman M. S., Beher D., Clarke E. E., Lewis H. D., Harrison T., Hunt P., Nadin A., Smith A. L., Stevenson G., Castro J. L. (2000) L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid beta-protein precursor γ-secretase activity. Biochemistry 39, 8698–8704 [DOI] [PubMed] [Google Scholar]
- 23. Ellisen L. W., Bird J., West D. C., Soreng A. L., Reynolds T. C., Smith S. D., Sklar J. (1991) TAN-1, the human homolog of the Drosophila Notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 66, 649–661 [DOI] [PubMed] [Google Scholar]
- 24. Deatherage C. L., Hadziselimovic A., Sanders C. R. (2012) Purification and characterization of the human γ-secretase activating protein. Biochemistry 51, 5153–5159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fraering P. C., Ye W., LaVoie M. J., Ostaszewski B. L., Selkoe D. J., Wolfe M. S. (2005) γ-Secretase substrate selectivity can be modulated directly via interaction with a nucleotide-binding site. J. Biol. Chem. 280, 41987–41996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eisele Y. S., Baumann M., Klebl B., Nordhammer C., Jucker M., Kilger E. (2007) Gleevec increases levels of the amyloid precursor protein intracellular domain and of the amyloid-β degrading enzyme neprilysin. Mol. Biol. Cell. 18, 3591–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bauer C., Pardossi-Piquard R., Dunys J., Roy M., Checler F. (2011) γ-Secretase-mediated regulation of neprilysin. Influence of cell density and aging and modulation by imatinib. J. Alzheimers Dis. 27, 511–520 [DOI] [PubMed] [Google Scholar]
- 28. Sutcliffe J. G., Hedlund P. B., Thomas E. A., Bloom F. E., Hilbush B. S. (2011) Peripheral reduction of β-amyloid is sufficient to reduce brain β-amyloid. Implications for Alzheimer's disease. J. Neurosci. Res. 89, 808–814 [DOI] [PubMed] [Google Scholar]
- 29. Takayama N., Sato N., O'Brien S. G., Ikeda Y., Okamoto S. (2002) Imatinib mesylate has limited activity against the central nervous system involvement of Philadelphia chromosome-positive acute lymphoblastic leukaemia due to poor penetration into cerebrospinal fluid. Br. J. Haematol. 119, 106–108 [DOI] [PubMed] [Google Scholar]