

Background: The N111G and N111W mutations make the AT1 receptor constitutively active and inactivable, respectively.
Results: The orientation and interactions of D742.50 are influenced by the residue at position 1113.35.
Conclusion: H-bond formation between D742.50 and N461.50 is critical for AT1 receptor activation.
Significance: This novel molecular switch could be involved in the GPCR activation mechanism as it involves highly conserved residues D2.50 and N1.50.
Keywords: 7-Helix Receptor, Angiotensin II, G Protein-coupled Receptors (GPCR), Molecular Dynamics, Molecular Modeling, AT1, Activation Mechanism
Abstract
G protein-coupled receptors contain selectively important residues that play central roles in the conformational changes that occur during receptor activation. Asparagine 111 (N1113.35) is such a residue within the angiotensin II type 1 (AT1) receptor. Substitution of N1113.35 for glycine leads to a constitutively active receptor, whereas substitution for tryptophan leads to an inactivable receptor. Here, we analyzed the AT1 receptor and two mutants (N111G and N111W) by molecular dynamics simulations, which revealed a novel molecular switch involving the strictly conserved residue D742.50. Indeed, D742.50 forms a stable hydrogen bond (H-bond) with the residue in position 1113.35 in the wild-type and the inactivable receptor. However, in the constitutively active mutant N111G-AT1 receptor, residue D74 is reoriented to form a new H-bond with another strictly conserved residue, N461.50. When expressed in HEK293 cells, the mutant N46G-AT1 receptor was poorly activable, although it retained a high binding affinity. Interestingly, the mutant N46G/N111G-AT1 receptor was also inactivable. Molecular dynamics simulations also revealed the presence of a cluster of hydrophobic residues from transmembrane domains 2, 3, and 7 that appears to stabilize the inactive form of the receptor. Whereas this hydrophobic cluster and the H-bond between D742.50 and W1113.35 are more stable in the inactivable N111W-AT1 receptor, the mutant N111W/F77A-AT1 receptor, designed to weaken the hydrophobic core, showed significant agonist-induced signaling. These results support the potential for the formation of an H-bond between residues D742.50 and N461.50 in the activation of the AT1 receptor.
Introduction
In recent years, a growing number of crystal structures of various family A G protein-coupled receptors (GPCRs)3 in complex with either agonists or antagonists have been resolved (1–5). Although the sequence similarity of these receptors is rather low (6), there are a few highly conserved residues in every transmembrane domain (TMD), and the three-dimensional arrangement of the TMDs is highly similar in all available crystal structures. These characteristics coupled with results from studies on conformational changes that occur during activation of different family A GPCRs suggest that they may share common activation mechanisms (7–9).
The angiotensin II type 1 (AT1) receptor and its cognate ligand, the octapeptide hormone angiotensin II (AngII), are part of the renin-angiotensin-aldosterone system responsible for controlling blood pressure and water retention via smooth muscle contraction and ion transport, respectively. The AT1 receptor signaling can also induce steroidogenesis in the adrenal gland, neurosecretion, neuronal activation, cell growth, and proliferation (10).
Structure-activity studies on the AT1 receptor have shown that substituting the asparagine residue in position 1113.35 (see “Experimental Procedures” for details on the residue numbering scheme) for glycine produces a constitutively active mutant receptor, whereas an inactivable mutant is produced when the asparagine is substituted for tryptophan (11). Asparagine 111 (N1113.35) is highly, but not strictly, conserved among the family A GPCRs. Homology models of the AT1 receptor that we generated in previous studies (12, 13) suggested that N1113.35 is in close proximity to D742.50, which is strictly conserved, as well as N2957.46. An interaction between the residues corresponding to N1113.35 and D742.50 is notably present in the crystal structures of the human CXC chemokine receptor type 4 (CXCR4) and the mouse μ- and δ- and human κ- and nociceptin/orphanin FQ opioid receptors (2–5, 14), whereas an interaction between the residues corresponding to D742.50 and N2957.46 is featured in the crystal structures of the four opioid receptors mentioned above. Key mutations have stressed the importance of some residues in AT1 receptor activation. Among them, mutation of D742.50 for asparagine or for glutamate abolished the activity (15), whereas mutation of N2957.46 for serine conferred pseudo-constitutive activity to the AT1 receptor (16). The constitutively active N111G-AT1 receptor has been used to study the mechanism of activation of the AT1 receptor using a variety of approaches (12, 13, 17–23, 25–35). These methods have highlighted differences between the AT1 receptor and the N111G-AT1 receptor regarding the solvent-accessible residues delimiting the binding pocket and the ligand contact points. Unlike the AT1 receptor, the constitutively active N111G-AT1 receptor maintains a high affinity conformation despite being uncoupled from its cognate G protein Gq/11α, whereas the non-activable N111W-AT1 receptor does not couple to Gq/11α (11). These results clearly suggest that residue 111 significantly influences the conformational states of the AT1 receptor, but its role at the molecular level is unclear. We hypothesized that residue N111 is part of an interaction network including residues D742.50 and N2952.49 that stabilizes an inactive state of the AT1 receptor. To investigate this hypothesis, we used molecular dynamics (MD) simulations of the AT1 receptor (WT, N111G, and N111W) in a solvated lipid bilayer. The simulations revealed H-bond networks that appear specific to the inactive and the active states of the receptor, and they suggest conformational changes required for receptor activation. These simulations have been validated by mutagenesis and in cellulo functional assays. The results highlight the importance of the H-bond formed between residues D742.50 and N461.50 in the activation process of the AT1 receptor.
EXPERIMENTAL PROCEDURES
Materials
The two computers used to run the simulations have an Intel Core-i7 quad core processor at 2.67 and 2.93 GHz with 12 and 8 GB of RAM, respectively. All reagents were from Sigma-Aldrich unless otherwise indicated. Culture media, trypsin, FBS, penicillin, and streptomycin were from Wisent (St-Bruno, Quebec, Canada). Opti-MEM was from Invitrogen. Polyethyleneimine was from Polysciences (Warrington, PA). The cDNA clone for the human AT1 receptor was kindly provided by Dr. Sylvain Meloche (University of Montréal). 125I-[Sar1,Ile8]AngII (specific radioactivity, ∼1000 Ci/mmol) was prepared with Iodo-GEN® (Perbio Science, Erembodegem, Belgium) as reported previously (36).
Residue Numbering Scheme
Residues of the AT1 receptor are given two numbering schemes. First, residues are numbered according to their positions in the AT1 receptor sequence. Second, residues are also indexed according to their position relative to the most conserved residue in the TMD in which they are located. By definition, the most conserved residue is assigned the position index “50”; e.g. in TMD2, D74 is the most conserved residue and was designated D742.50, whereas the upstream residue is designated A732.49, and the downstream residue is designated L752.51. This indexing simplifies the identification of aligned residues in different GPCRs (37).
Homology Modeling
We used the I-TASSER server to generate multiple template homology structures of the AT1 receptor. The primary structure of the AT1 receptor used for the modeling can be seen in Fig. 1. The resulting five best structures provided in the output had near identical orientations of the side chains of the H-bond network. We selected the only structure that featured both known disulfides bonds, which had a high confidence score of 0.99 (38, 39). The backbone of the model is very similar to the crystal structure of the CXCR4 receptor (Protein Data Bank code 3ODU) with a root-mean-square deviation distance of 0.900 Å between the positions of Cα atoms. Superposition of the two structures is shown in Fig. 2, and sequence alignment between AT1 and CXCR4 is shown in Table 1. The homology model was also analyzed with PROCHECK (40), and the Ramachandran plot indicated that over 97% of the residues were in the “most favored” and “additional allowed” regions. The rest of the stereochemistry was also of high quality. The unstructured N-terminal and C-terminal portions of the model were truncated by removing residues 1–14 and 319–359, respectively, to keep the simulation box as small as possible. This enables better performances for the MD simulations. Models of the N111G-AT1 receptor and N111W-AT1 receptor were generated by replacing residue N111 by the corresponding residue using the mutagenesis feature in PyMOL.
FIGURE 1.

Two-dimensional schematic representation of the primary amino acid structure of the AT1 receptor. The dashed rectangle represents the lipid bilayer where the seven TMD are located. Residues in red correspond to the most conserved residue in each TMD that are notably important for sequence alignment during homology modeling procedures. Labeled residues in red and residues in blue represent important residues discussed in this study.
FIGURE 2.
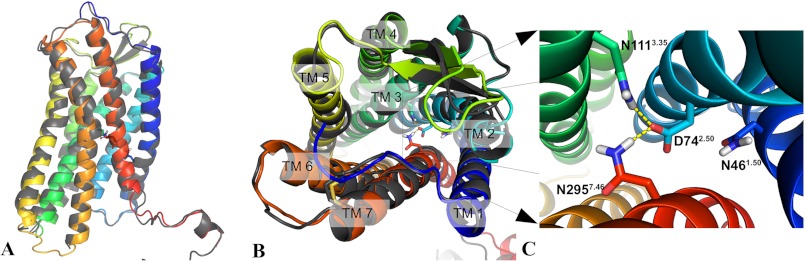
The I-TASSER server was used to produce a homology model of the AT1 receptor. A, the backbone of the AT1 receptor homology model (colored) closely matches the backbone of the crystal structure of the CXCR4 receptor (gray). The side chains of residues N461.50, D742.50, N1113.35, and N2957.46 of the AT1 receptor are shown as sticks visible through the semitransparent backbone representation. B, view from the top (from the extracellular side looking toward the intracellular side) of the superposition of the AT1 receptor homology model (colored) and the CXCR4 receptor (gray). C, zoomed-in view showing the H-bonds (yellow dashes) formed between N1113.35, D742.50, and N2957.46 in the AT1 receptor homology model. Hydrogen, nitrogen, and oxygen atoms are shown in white, blue, and red, respectively. Carbon atoms are colored according to the TMD in which they are located.
TABLE 1.
Sequence alignment of the AT1 receptor, CXCR4 receptor, κ-opioid receptor, and nociceptin/orphanin FQ receptor
Sequence alignment was performed by the I-TASSER server. Residues in red represent the most conserved residues within each transmembrane domain. Residues in blue represent other important residues discussed in the text. Numbers represent the Ballestcros numbering of the residue aligned under the last number. Sequences for the CXCR4, κ-opioid receptor (KOR), and nociceptin/orphanin FQ (N/OFQ) receptors are truncated and represent the actual sequences found in the crystal structures of those receptors.

Molecular Dynamics Simulations
The GROMACS software suite (41–44) was used to prepare and run the simulations. The AT1 receptor, N111G-AT1 receptor, and N111W-AT1 receptor models were inserted in a lipid bilayer consisting of 128 molecules of dioleoylphosphatidylcholine using the InflateGRO approach (45). Simulation parameters were based on previous work (46–48). The membrane-receptor system was solvated with the simple point charge water model (49). Counterions were added at random positions, replacing water molecules, to keep the net charge of the system at 0. The ffg53a6 force field, modified to use the Berger lipid parameter (50), was used for the calculations. Parameters for the dioleoylphosphatidylcholine molecules and the Protein Data Bank file of the bilayer, developed by Tieleman and co-workers (51–53), were obtained from Peter Tieleman. Equilibration of the system in conditions of constant number of atoms, volume and temperature was performed for 100 ps to reach the desired 310 K temperature. This was followed by equilibration in conditions of constant number of atoms, pressure and temperature for 15 ns with the pressure set at 1 bar. Such long equilibration is necessary for proper equilibration of the lipids after embedding a protein in the membrane (54). We monitored the size of the system in the x, y, and z axes to confirm the stabilization on the dioleoylphosphatidylcholine bilayer. The position of all heavy atoms of the receptor was restrained during equilibration. System size after equilibration in conditions of constant number of atoms, pressure and temperature was ∼72 × 71 × 79 Å, ensuring that the AT1 receptor molecule could not interact with its periodic image. Unrestrained MD simulations were run for 84 ns in 2-fs steps. The 84-ns simulation length was deemed long enough for our study, which aimed to analyze side chain reorientation in the core of the AT1 receptor and, to a certain extent, local backbone rearrangements caused by mutations. These motions occur on the nanosecond time scale (55). The simulations were run in periodic boundary conditions at constant temperature (310 K) and pressure (1 bar) using the Nose-Hoover thermostat (56, 57) with τT = 0.2 ps and the Parrinello-Rahman barostat with τP = 5 ps, respectively. Simulation data were saved every 2 ps for a total of 41,001 frames. Stability of the systems was assessed by calculating the root-mean-square deviation distance between the positions of Cα atoms of the TMDs during the simulations. In all three instances, the root-mean-square deviations converged to a similar and stable value, close to 2.5Å, indicating that equilibrium was reached before initiating the MD simulation of all three systems.
Trajectory Analysis
MD trajectories output from GROMACS were converted to Protein Data Bank files with 211 frames (one for every 200 saved) for visual inspection with PyMOL (58) and to compressed .xtc trajectory files with 2101 frames (one for every 20 saved) for other analyses. Evaluation of the presence of H-bonds during the simulations was performed with the g_hbond tool in GROMACS using the default cutoff angle value of 30° and a cutoff radius of 0.35 nm. Data regarding distances, dihedral angles, water occupancy, and water molecule count were performed with the g_dist, g_angle, g_spatial, and g_mindist tools, respectively, within GROMACS. Statistical analyses were performed using Student's t test. Results were considered statistically significant when p < 0.01 (*).
Oligodeoxynucleotide Site-directed Mutagenesis
Site-directed mutagenesis was performed on the AT1 receptor or relevant mutants using the QuikChange II XL mutagenesis kit (Stratagene, La Jolla, CA) as recommended by the manufacturer. Briefly, forward and reverse oligonucleotides were constructed to introduce the N46G mutation in the AT1 receptor or in the N111G-AT1 receptor background and the F77A mutation in the AT1 receptor or in the N111W-AT1 receptor background. Site-directed mutations were then confirmed by automated DNA sequencing by aligning the AT1 receptor sequence with multiAlin (59).
Cell Culture and Transfection
HEK293 cells were maintained in DMEM supplemented with 10% FBS, 100 IU/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a humidified 5% CO2 atmosphere. The day prior to transfection, cultured cells were washed with phosphate-buffered saline (PBS) at room temperature, trypsinized, and seeded at 150,000 cells/well in a 6-well plate. For transfection, 2 μg of the DNA construct containing the appropriate AT1 receptor construct was added to 100 μl of Opti-MEM containing 6 μg of polyethyleneimine, and the mixture was incubated for 20 min before being added to the cultured cells as described previously (60).
Inositol Phosphate Production
Inositol phosphate (IP1) production was determined using the IP-One assay (Cisbio Bioassays, Bedford, MA). Necessary dilutions of the agonist AngII were prepared in stimulation buffer (10 mm Hepes, 1 mm CaCl2, 0.5 mm MgCl2, 4.2 mm KCl, 146 mm NaCl, 5.5 mm glucose, 50 mm LiCl, pH 7.4). 48 h after transfection, the cells were washed with PBS at room temperature. The cells were trypsinized and distributed at 20,000 cells/well (7 μl) in a white 384-well plate in stimulation buffer. Cells were stimulated at 37 °C for 30 min with increasing concentrations of AngII. Cells were then lysed with 3 μl of IP1 coupled to the dye d2. After addition of 3 μl of anti-IP1 cryptate terbium conjugate, cells were incubated for 1 h at room temperature under agitation. FRET signal was measured with a Tecan M1000 plate reader.
Binding Experiments
HEK293 cells were grown for 36 h after transfection in 100-mm culture dishes and subjected to one freeze-thaw cycle. Broken cells were then gently scraped into washing buffer (25 mm Tris-HCl, pH 7.4, 100 mm NaCl, 5 mm MgCl2), centrifuged at 2500 × g for 15 min at 4 °C, and resuspended in binding buffer (25 mm Tris-HCl, pH 7.4, 100 mm NaCl, 5 mm MgCl2, 0.1% bovine serum albumin, 0.01% bacitracin, 0.01% soybean trypsin inhibitor). Saturation binding experiments were done by incubating broken cells (20–40 μg of protein) for 1 h at room temperature with increasing concentrations of 125I-[Sar1,Ile8]AngII in a final volume of 500 μl. Nonspecific binding was determined in the presence of 1 μm unlabeled [Sar1,Ile8]AngII. Bound radioactivity was separated from free ligand by filtration through GF/C filters presoaked for at least 3 h in binding buffer. Receptor-bound radioactivity was evaluated by γ counting. Results are presented as means ± S.D. Binding data (Bmax and Kd) were analyzed with Prism version 5.0 for Windows (GraphPad Software, San Diego, CA) using a one-site binding hyperbola nonlinear regression analysis. Dose displacement experiments were done by incubating broken cells (20–40 μg of protein) for 1 h at room temperature with 0.8 nm 125I-[Sar1,Ile8]AngII as tracer and increasing concentrations of AngII. The Kd values in the displacement studies were determined from the IC50 values using the Cheng-Prusoff equation (61).
RESULTS
Rearrangement of the N111-D74-N295 H-bond Network Caused by the N111G Mutation
An updated version of the homology model of the AT1 receptor (Fig. 1) that includes the latest structural knowledge has been generated using the I-TASSER server (38, 39) (Fig. 2, A and B). The backbone structure of the homology model closely matches that of the CXCR4 crystal structure (Protein Data Bank code 3ODU) with a root-mean-square deviation of 0.900 Å between the Cα atoms. In this model, a network of H-bonds involving residues D742.50, N1113.35, and N2957.46 is revealed (Fig. 2C). This network displayed a high stability throughout the 84-ns MD simulation of the AT1 receptor (Fig. 3A). More precisely, analysis of the MD simulation indicated that an H-bond was present between the side chain of N111 and those of D74 and N295 64.6 and 56.4% of the time, respectively (data not shown). In addition, an H-bond between the side chains of D74 and N295 was present 98.0% of the time. On the other hand, an H-bond formed 1.4% of the time between the side chains of D74 and N46. Interestingly, the MD simulation showed that within the constitutively active N111G-AT1 receptor the side chain of residue D742.50 reorients toward TMD1 within 1 ns to form an H-bond with the side chain of N461.50 (Fig. 3B). Indeed, analysis of the MD trajectory using g_hbond indicated that the H-bond between the side chains of N461.50 and D742.50 was present 34.1% of the time in the N111G-AT1 receptor. As to the H-bond between the side chains of D742.50 and N2957.46, the analysis revealed that it was present 88% of the time in the N111G-AT1 receptor. Analysis of the rotamers of residue D742.50 revealed that χ1 maintains a gauche (−) conformation 99.5% of the time throughout the simulation of the AT1 receptor, whereas in the N111G-AT1 receptor, it explored a trans conformation 35.6% of the time (Fig. 4A). These results suggest that the N111-D74-N295 H-bond network rearranges to form the new N46-D74-N295 H-bond network in the context of receptor constitutive activity.
FIGURE 3.
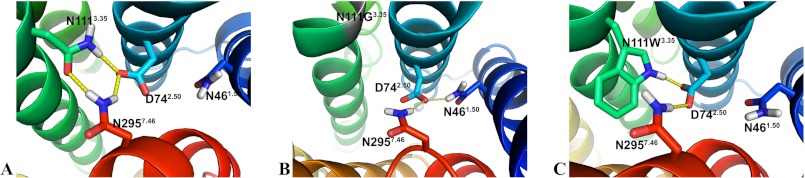
The orientation of D742.50 and the N111-D74-N295 H-bond network are affected by the residue at position 1113.35. A, snapshot of the 84-ns MD simulation of the AT1 receptor showing the N111-D74-N295 H-bond network. B and C, snapshots of the 84-ns MD simulations showing the N46-D74-N295 network in the N111G-AT1 receptor (B) and the W111-D74-N295 network in the N111W-AT1 receptor (C). H-bonds are shown as yellow dashed lines. Hydrogen, nitrogen, and oxygen atoms are shown in white, blue, and red, respectively. Carbon atoms are colored according to the TMD in which they are located.
FIGURE 4.

Analysis of the χ1 and χ2 angles (x and y axes, respectively) of the side chain of residue D742.50 (A) and N461.50 (B) from 2100 frames of the 84-ns MD simulation of the AT1 receptor (blue), N111G-AT1 receptor (red) and N111W-AT1 receptor (green). Colored squares represent the average χ1 and χ2 values in each simulation.
Structural Changes Induced by the N111G Mutation
The most important conformational change observed in the backbone of the N111G-AT1 receptor mutant was located in TMD7. Indeed, the α-helical geometry is distorted in the portion of TMD7 encompassing residues I2907.41 to F2937.44 during the MD simulation (Fig. 5A). The Ramachandran plots for these residues clearly show that the backbone explored conformations in the β-strand quadrant for A2917.42 and Y2927.43 (data not shown). The associated increase in pitch for this portion of TMD7 was assessed by measuring the distance between the α-carbon atoms of residues C2897.40 and N2947.45. On average, the distance between these two atoms was 9.1 ± 0.5 Å for the AT1 receptor and 13.2 ± 0.8 Å for the N111G-AT1 receptor (Fig. 5, B and C).
FIGURE 5.

The helicity of TMD7 is disturbed in the N111G-AT1 receptor. A, the final frame of the 84-ns MD simulations of the N111G-AT1 receptor (colored) shows that the helicity of TMD7 (red) is locally disturbed between residues I290 and F293 (pink section shown within circled area). The final frame of the 84-ns simulation of the AT1 receptor (gray) is superimposed for comparison. B, the zoomed-in view of the circled area of TMD7 shows the increased distance between the Cα atoms of C2897.40 and N2947.45 (shown as spheres). The blue and yellow dashed lines represent the measured distances between residues C2897.40 and N2947.45 in the AT1 receptor and N111G-AT1 receptor, respectively. C, distance between the α-carbon of residues C2897.40 and N2947.45. Data are the mean ± S.D. (error bars) of 2100 frames from each 84-ns MD trajectory. *, significantly different (p < 0.01).
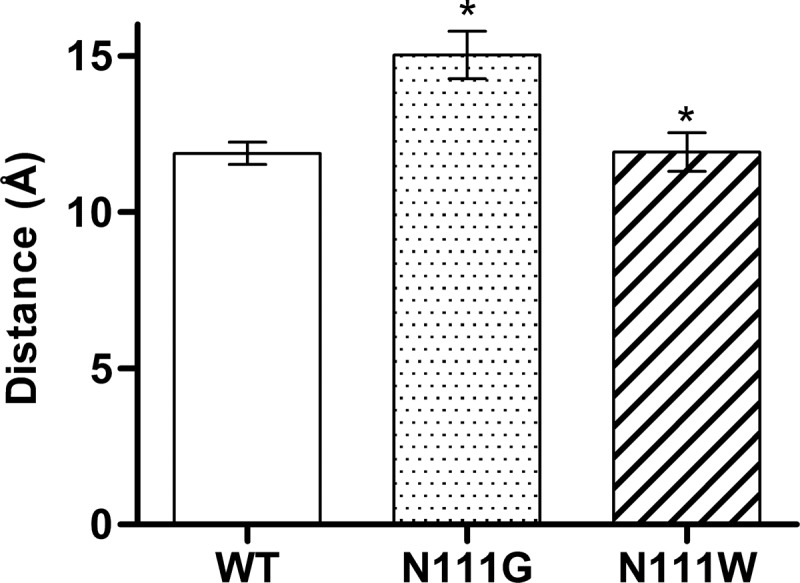
To identify additional structural differences between the AT1 receptor and the N111G-AT1 receptor, we measured the distance between Cα atoms of the residues involved in the H-bond networks described above. Over the course of the MD simulations, the average distance between the Cα atoms of residues 1113.35 and N2957.46 increased by 1.7 Å from 9.4 Å in the AT1 receptor to 11.1 Å in the N111G-AT1 receptor (Fig. 6). Interestingly, the average distance between the Cα atoms of residues N461.50 and N2957.46 decreased by 1.7 Å from 10.1 Å in the AT1 receptor to 8.4 Å in the N111G-AT1 receptor (Fig. 6). Although significant (due to the large number of data), a very minor decrease in the distance between the Cα atoms of D742.50 and N2957.46 was observed (6.9 Å in the AT1 receptor and 6.8 Å in the N111G-AT1 receptor) (Fig. 6).
FIGURE 6.

Conformational changes between the AT1 receptor and N111G-AT1 receptors revealed by the analysis of the MD trajectories. Histograms of the average distance between the Cα atoms of residues 1113.35 and N2957.46, N461.50 and N2957.46, and D742.50 and N2957.46 are shown. Data are the mean ± S.D. (error bars) of 2100 frames from each 84-ns MD trajectory. *, significantly different from AT1 receptor (p < 0.01).
These results suggest that during the process of activation the reorientation of D742.50 is accompanied with a concerted movement of N2957.46 away from TMD3 and toward TMD1 that causes a local conformational change of TMD7. We propose that this movement is facilitated by the apparent stability of the H-bond between the side chains of D742.50 and N2957.46.
To evaluate the impact of the side chain rearrangement and movement of TMD7 on the water accessibility within the receptor, we used the spatial density function (62) of GROMACS to calculate the water occupancy in the molecular dynamics trajectories of the AT1 receptor and the N111G-AT1 receptor (Fig. 7). A difference was observed in the upper portions of TMD3 and TMD7 (toward the extracellular side) roughly up to two helical turns above N2957.46. This area was much more accessible to water molecules within the constitutively active N111G-AT1 receptor (Fig. 7B) than within the AT1 receptor (Fig. 7A). Interestingly, this area includes the portion of TMD7 that experiences conformational changes in the N111G-AT1 receptor. This area contains a cluster of hydrophobic residues consisting of residues V1083.33, L1123.36, I2887.39, A2917.42, Y2927.43, and F772.53. The MD simulations suggested that, within the AT1 receptor, these residues form a tight hydrophobic core (Fig. 7A), which is disrupted within the N111G-AT1 receptor (Fig. 7B). To support the information obtained from the spatial density function, we evaluated the number of water molecules within a 5-Å distance of the cluster of hydrophobic residues. Analysis of the MD trajectories using the GROMACS tool g_mindist revealed that, on average, 16 water molecules were within 5 Å of the hydrophobic cluster in the AT1 receptor, whereas 26 water molecules were within 5 Å of the hydrophobic cluster in the N111G-AT1 receptor (Fig. 8). Interestingly MD trajectories indicate that in the N111G-AT1 receptor water molecules form H-bonds with the backbone carbonyl of I288 and A291, two residues located in the region of TMD7 that has lost its helicity (Fig. 9). This gain in water accessibility within the N111G-AT1 receptor was accompanied by a 3-Å increase in the average distance between TMD3 and TMD7 in the portion of helices containing residues V1083.33 to L1123.36 and I2887.39 to Y2927.43 (Fig. 10).
FIGURE 7.
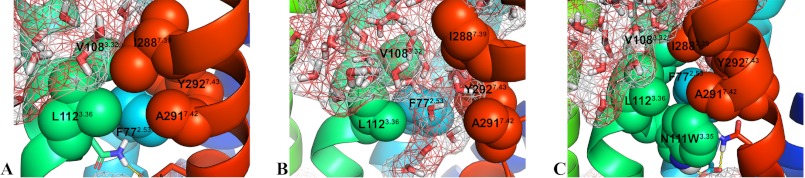
Snapshots of the 84-ns MD simulations showing the water molecules (red and white sticks; molecular surface in mesh) in the area near the hydrophobic cluster between TMD2 (light blue), TMD3 (green), and TMD7 (red). The hydrophobic residues F772.53 (light blue), V1083.33 (green), L1123.36 (green), I2887.39 (red), A2917.42 (red), and Y2927.43 (red) are displayed in spheres. Water molecules are shown in the AT1 receptor (A), the N111G-AT1 receptor (B), and the N111W-AT1 receptor.
FIGURE 8.
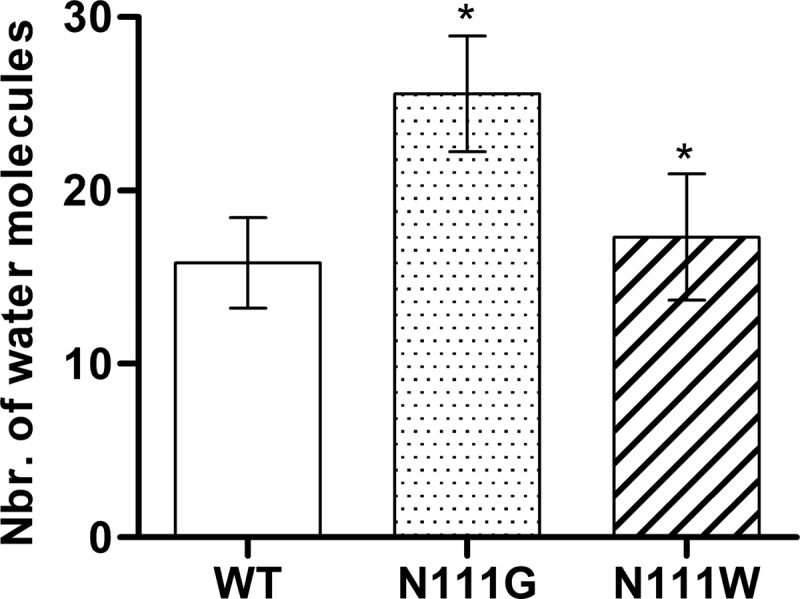
Number of water molecules in the area near the hydrophobic cluster of the AT1 receptor, the N111G-AT1 receptor, and the N111W-AT1 receptor. Analyses of the 84 ns MD trajectories shows the average number of water molecules within 5Å of residues V1083.33, L1123.36, I2887.39, A2917.42, Y2927.43 and F772.53. Data are the mean ± S.D. (error bars) of 2100 frames from each 84 ns MD trajectory. *, significantly different from AT1 receptor (p < 0.01).
FIGURE 9.

Snapshot of the 84-ns MD trajectory of the N111G-AT1 receptor showing water molecules interacting with backbone atoms of TMD7. Water molecules and backbone atoms are shown as sticks. Side chain atoms are shown as thin lines. Hydrogen, nitrogen, and oxygen atoms are shown in white, blue, and red, respectively. Carbon atoms are green or as specified. Yellow dashes represent intrabackbone H-bonds. Light blue dashes represent H-bonds involving water. One water molecule interacts with the backbone carbonyl of I2887.39 (magenta), and one water molecule interacts with the backbone carbonyl of A2917.42 (orange) and with the side chain of N2957.46.
FIGURE 10.
Histograms showing the average distance between the portions of helices (center of mass) containing residues V1083. 33 to L1123.36 on TMD3 and I2887.39 to Y2927.43 on TMD7. Data are the mean ± S.D. (error bars) of 2100 frames from each 84-ns MD trajectory.*, significantly different from AT1 receptor (p < 0.01).
Interestingly, the hydrophobic cluster is adjacent to W2536.48, a conserved tryptophan residue that is part of the CWXP motif in TMD6 and commonly known as the “rotamer toggle switch” (63, 64). The MD trajectories of the AT1 receptor suggested that W2536.48 maintains its hydrophobic indole moiety in an orientation allowing a direct contact with the hydrophobic core, more precisely with residues A2917.42 and L1123.36 (Fig. 11A). Conversely, the MD trajectories of the N111G-AT1 receptor revealed that the indole moiety of W2536.48 is oriented away from the hydrophobic cluster toward TMD5 (Fig. 11B). These results suggest that the N111G mutation disrupts the hydrophobic cluster and increases the solvent accessibility in that area of the receptor, apparently influencing the orientation of the side chain of W2536.48.
FIGURE 11.
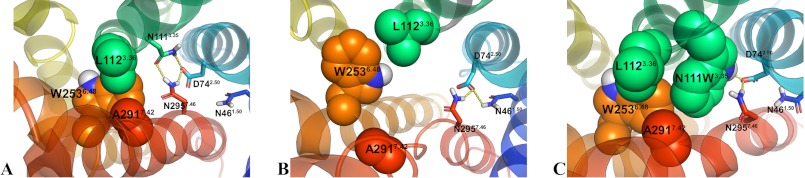
A stable hydrophobic core involving W2536.48 is observed in the inactivable N111W-AT1 receptor. Snapshots of the 84-ns MD simulations show the orientation of the highly conserved W2536.48 (orange spheres). A, in the AT1 receptor, the indole group of W2536.48 is oriented toward the hydrophobic core (L112 in green spheres; A291 in red spheres). B, in the N111G-AT1 receptor, the indole group of W2536.48 is oriented toward TMD5 (yellow helix). C, in the N111W-AT1 receptor, the indole group of W2536.48 is oriented toward the hydrophobic core (N111W3.35 and L1123.36 in green spheres; A2917.42 in red spheres).
Conserved Residue N461.50 Is Important for AT1 Receptor Activation
MD simulations suggested that a highly stable N111-D74-N295 H-bond network within the AT1 receptor rearranges to form a new N46-D74-N295 H-bond network within the constitutively active N111G-AT1 receptor. The H-bond between D742.50 and N461.50 thus appears to play an important role in receptor activation. To support this possibility, we evaluated the functional properties of mutant receptors in which N46 was substituted for a glycine (N46G-AT1 receptor and N111G/N46G-AT1 receptor) and compared them with the AT1 receptor and the N111G-AT1 receptor. Table 2 shows that all the constructs were efficiently expressed and exhibited high binding affinities. The functional properties of the wild-type and mutant AT1 receptors were evaluated by assessing the basal and AngII-induced production of IP1 in transiently transfected HEK293 cells (Table 3). Fig. 12A shows that the basal level of IP1 in cells expressing the constitutively active N111G-AT1 receptor was significantly higher (75.9 ± 4.4 nm) than the basal level of IP1 in cells expressing the AT1 receptor (27.2 ± 8.0 nm). Upon stimulation with increasing concentrations of AngII, the constitutively active receptor increased the IP1 to a maximal level (224.3 ± 5.6 nm) that was not significantly different from that obtained with the AT1 receptor (235.4 ± 7.3 nm). The basal level of IP1 in cells expressing the N111G/N46G-AT1 receptor (9.2 ± 0.7 nm) was not significantly different from the level of IP1 in mock-transfected cells (7.1 ± 0.7 nm). Furthermore, AngII-induced IP1 production in cells expressing the N111G/N46G-AT1 receptor was completely blunted. Fig. 12A also shows that the basal level of IP1 in cells expressing the N46G-AT1 receptor was very low (8.2 ± 1.1 nm) and that this level marginally increased (57.5 ± 1.6 nm) upon stimulation with a maximal concentration of AngII. These results indicate that residue N461.50 plays an important role in agonist-induced activation of the AT1 receptor, and it also stabilizes an active state of the AT1 receptor in the absence of any agonist. At the molecular level, the N111G mutation appears to disrupt the interaction between N1113.35 and D742.50 that reorients toward N461.50, thus inducing a conformational change responsible for the constitutive activity.
TABLE 2.
Binding properties of 125I-[Sar1,Ile8]AngII to AT1 receptor mutants
Cells expressing the indicated receptor were assayed for their binding properties as described under “Experimental Procedures.” Binding affinities (Kd) and maximal binding capacities (Bmax) were obtained in saturation binding experiments using 125I-[Sar1,Ile8]AngII. In dose displacement experiments, the tracer 125I-[Sar1,Ile8]AngII competed with increasing concentrations of AngII, and the IC50 values were converted to Kd values using the Cheng-Prusoff equation. All values are expressed as the means ± S.D. of values obtained in n independent experiments performed in duplicate.
| Receptor mutant |
Kd |
Bmax | n | |
|---|---|---|---|---|
| 125I-[Sar1,Ile8]AngII | AngII | |||
| nm | pmol/mg | |||
| AT1 | 1.9 ± 0.7 | 3.8 ± 0.3 | 1.5 ± 0.2 | 8 |
| N111G-AT1 | 2.7 ± 0.1 | 1.5 ± 0.4 | 0.9 ± 0.1 | 5 |
| N111W-AT1 | 7.7 ± 2.3 | 5.7 ± 1.5 | 0.7 ± 0.1 | 4 |
| N46G-AT1 | 6.4 ± 0.9 | 9.6 ± 2.8 | 1.1 ± 0.1 | 4 |
| N111G/N46G-AT1 | 6.9 ± 2.5 | 6.8 ± 1.5 | 0.7 ± 0.1 | 4 |
| D74N-AT1 | 4.1 ± 1.2 | 4.5 ± 1.3 | 0.4 ± 0.03 | 3 |
| N111G/D74N-AT1 | 4.4 ± 1.0 | 6.4 ± 1.1 | 0.2 ± 0.02 | 3 |
| F77A-AT1 | 4.3 ± 1.4 | 3.2 ± 1.1 | 1.7 ± 0.4 | 5 |
| N111W/F77A-AT1 | 3.1 ± 0.6 | 2.2 ± 0.8 | 1.5 ± 0.2 | 5 |
TABLE 3.
Functional properties of mutant AT1 receptors
HEK293 cells expressing mutant AT1 receptors were assayed for their IP1 production as described under “Experimental Procedures.” EC50 values are expressed as the means ± S.D. of values obtained in n independent experiments performed in duplicate. ND, no detectable response.
| EC50 | n | |
|---|---|---|
| nm | ||
| AT1 | 2.4 ± 1.5 | 12 |
| N111G-AT1 | 3.3 ± 1.1 | 6 |
| N111W-AT1 | ND | 7 |
| N46G-AT1 | 35.8 ± 13.5 | 6 |
| N111G/N46G-AT1 | ND | 6 |
| D74N-AT1 | ND | 5 |
| N111G/D74N-AT1 | ND | 5 |
| F77A-AT1 | 2.1 ± 1.1 | 7 |
| N111W/F77A-AT1 | 3.7 ± 1.8 | 7 |
FIGURE 12.

Dose-response curve of IP1 production for AT1 receptor mutants. HEK293 cells were transfected with the indicated receptor, and their IP1 production was assayed as described under “Experimental Procedures.” Each point represents the mean of duplicate determinations of a typical experiment, which is representative of at least three independent experiments. Error bars represent S.D.
We also verified whether the inactivable D74N-AT1 receptor could be rescued by the N111G mutation. Fig. 12B shows that the basal level of IP1 in cells expressing the D74N-AT1 receptor was low (27.8 ± 4.7 nm) and barely increased upon stimulation with AngII (41.6 ± 4.9 nm). In cells expressing the D74N/N111G-AT1 receptor, the basal level was slightly lower compared with cells expressing the N111G-AT1 receptor (45.1 ± 3.8 nm). Stimulation of this mutant receptor with AngII produced only a weak increase in IP1 level (85.8 ± 6.6 nm). The presence of an aspartate at position 742.50 appears to be crucial to the mechanism of activation as substitution of residue D742.50 for an asparagine significantly hampered constitutive and agonist-induced activity of the receptor.
The H-bond between Residues 1113.35 and D742.50 Is Stabilized by the N111W Mutation
The MD trajectories of the N111W-AT1 receptor showed that an H-bond network involving the side chains of residues W1113.35, D742.50, and N2957.46 was very stable (Fig. 3C). Analysis of the trajectory revealed that an H-bond between the side chains of W1113.35 and D742.50 was present 83.6% of the time in this inactivable receptor. The higher apparent stability of this interaction within the N111W-AT1 receptor is likely due to the fact that the H-bond donor of the tryptophan is in the ϵ position and therefore closer to D742.50 than the H-bond donor of the asparagine (which is in δ position), hence leading to a stronger H-bond. To test this assumption, we evaluated the distribution of the length of the H-bonds formed between the side chains of residues W1113.35 and D742.50 throughout the simulation. Compared with the H-bond formed between N1113.35 and D742.50, the average distance of the H-bond between W1113.35 and D742.50 is smaller by 0.04 Å (2.93 versus 2.97 Å). More strikingly, this H-bond is shorter than 2.8 Å 27.7% of the time for the N111W-AT1 receptor compared with 16.1% for the AT1 receptor (data not shown). In addition, we observed that an H-bond between the side chains of D742.50 and N2957.46 was persistent (95.9% of the time), whereas the side chains of W1113.35 and N2957.46 made an H-bond only 0.3% of the time. However, the side chains of D742.50 and N461.50 were engaged in an H-bond 18.9% of the time. It is interesting to note that the χ1 angle of D742.50 remained in a gauche (−) conformation (Fig. 4A). This interaction can be explained by the fact that in the N111W-AT1 receptor, the χ2 angle of residue N461.50 adopted a gauche (+) orientation 51.7% of the time during the MD simulation (Fig. 4B), which brings the Nδ2 atom close to the carboxylate of D741.50. Thus, it appears that the mutation of residue N1113.35 to a tryptophan stabilizes its interaction with the side chain of residue D742.50 while at the same time allowing the side chain of D742.50 to interact more frequently with the side chain of N461.50 while keeping its χ1 angle in a gauche (−) conformation.
The N111W Mutation Stabilizes the Hydrophobic Core between TMD3 and TMD7
MD simulations revealed that both the AT1 receptor and the N111W-AT1 receptor display a similar binding pocket with the hydrophobic core stabilized in a “closed,” packed state (Fig. 7C). Indeed, the number of water molecules in the vicinity of the hydrophobic core (Fig. 8) and the distance between TMD3 and TMD7 (Fig. 10) in the N111W-AT1 receptor are comparable with those observed in the AT1 receptor. In addition, the highly conserved W2536.48, known as the tryptophan toggle switch, adopts a very similar conformation in both the AT1 receptor and the N111W-AT1 receptor. However, in contrast to the AT1 receptor, the introduced aromatic tryptophan residue at position 1113.35 of the N111W-AT1 receptor provides additional hydrophobic interaction to the hydrophobic core. Notably, the MD trajectories suggest that residue W1113.35 forms π-stacking interactions with residue F772.53, which is also involved in aromatic interactions with Y2927.43 (Fig. 13). The indole moiety of W111 apparently stabilizes the hydrophobic cluster involving L1123.36, A2917.42, and W2536.48 at the bottom of the hydrophobic core (Fig. 11C). Consequently, the substitution of the asparagine at position 111 for a tryptophan stabilizes the interaction with residue D742.50 and adds favorable hydrophobic interaction to the hydrophobic core. Both effects are consistent with a stabilization of an inactive state.
FIGURE 13.

Snapshot of the 84-ns MD simulation of the N111W-AT1 receptor showing the aromatic residues within the hydrophobic core. Residues N111W3.35 (green), F772.53 (blue),Y2927.43 (red), and W2536.48 (orange) are shown.
The Hydrophobic Core Stabilizes the Inactive Conformation of the Receptor
To evaluate the contribution of F772.53 to the stability of the hydrophobic core, it was substituted for an alanine, and the functional properties of mutant receptors were evaluated (Table 3). Binding experiments revealed that the mutant receptors were efficiently expressed and exhibited high binding affinities (Table 2). Fig. 12C shows that the N111W-AT1 receptor was very poorly activated, producing a maximal level of IP1 (36.5 ± 2.4 nm) severalfold lower than that of the AT1 receptor. The double mutant F77A/N111W-AT1 receptor was much more efficient with a maximal IP1 production of 118.3 ± 5.9 nm, corresponding to 50% of that of the AT1 receptor. Moreover, the basal (45.9 ± 6.6 nm) and AngII-induced (281.6 ± 8.4 nm) activities of the single mutant F77A-AT1 receptor were both slightly higher than that of the AT1 receptor. These results suggest that weakening the hydrophobic core partially rescues the functional properties of a receptor harboring the N111W mutation. More generally, the results highlight the importance of the hydrophobic core for the stabilization of the inactive state of the AT1 receptor.
DISCUSSION
Using an approach combining MD simulations and structure-function relationships, we have unveiled a molecular mechanism that could explain the constitutive activity of the N111G-AT1 receptor and the inactivity of the N111W-AT1 receptor. MD simulations suggested that the N111G mutation leads to the disruption of an H-bond network observed in the homology model of the AT1 receptor, presumably in its inactive form. This H-bond network involves the stabilization of the D742.50 side chain by the side chains of residues N1113.35 and N2957.46 (the N111-D74-N295 network). On the other hand, the N111G mutation leads to the formation of a new H-bond network where the side chain of D742.50 interacts with the side chains of N2957.46 and N461.50 (the N46-D74-N295 network). These results suggest that the reorientation of D742.50 from N1113.35 to N461.50 is a molecular switch involved in the constitutive activation of the receptor. The substitution of N46 for a glycine abolished the constitutive activity and strongly reduced the agonist-induced activation, supporting the importance of N461.50 in the constitutive and agonist-induced activity of the AT1 receptor. A MD simulation approach using bacteriorhodopsin as template previously proposed that in the inactive conformation of AT1 receptor the residue N1113.35 forms an H-bond with residue Y2927.43 (65). In our model using CXCR4 receptor as template, this interaction was never observed. Indeed, our model suggests that Y2927.43 and N1113.35 are too distant to form an H-bond. In our MD simulations, residue Y2927.43 points upward toward the extracellular side as χ1 is maintained in a gauche (−) conformation in all three simulations. With χ1 in a trans or gauche (+) conformation, the side chain of Y2927.43 would clash with the backbone of TMD1 or TMD7, respectively, except in the N111G-AT1 receptor where TMD7 is locally distorted. In that case, Y2927.43 could potentially interact with D742.50 if it were to adopt a trans conformation.
MD simulations of the N111W-AT1 receptor suggested that tryptophan111 stabilizes the W111-D74-N295 network and the hydrophobic core involving residues F77, V108, L112, W253, I288, A291, and F292. We propose that these are the basis for stabilizing the inactive state of the N111W-AT1 receptor. In support of this assertion, the substitution of residue F772.53 for an alanine provided some agonist-induced activity to the double mutant F77A-N111W-AT1 receptor, thus partially rescuing the inactivity conferred by the N111W mutation. Interestingly, this hydrophobic cluster is unstable and opened during the MD simulation of the constitutively active N111G-AT1 receptor. These results suggest that the N1113.35-D742.50-N2957.46 H-bond network and the hydrophobic core act in concert to stabilize the inactive state of the AT1 receptor. The results reported here were obtained from a limited simulation time (84 ns) and in that regard furnish information exclusively on the local effects of the mutations. Much longer simulations are needed to evaluate the consequences of the changes in the H-bond network and hydrophobic core on the transmission of the information across the whole receptor and to the cytosolic domains engaging the interaction with the G protein and other effectors.
The results obtained with the MD simulations are consistent with the results obtained in previous experiments using the substituted cysteine accessibility method to probe for differences in the solvent accessibility of residues delimiting the binding pocket of the AT1 that have shown that residue A2917.42 becomes more accessible to the solvent (18). Furthermore, photolabeling experiments have shown that position 77 was more accessible to a photoreactive AngII analog in the N111G-AT1 receptor than in the wild type AT1 receptor (12, 13). These experiments also showed that TMD7 in both the AT1 receptor and the N111G-AT1 receptor could be photolabeled by the antagonist [Sar1,Bpa8]AngII from residues 2937.43 through 2977.48. This supports our results from the MD simulation of the N111G-AT1 receptor suggesting that TMD7 can be locally distorted. Moreover, the N111G-AT1 receptor is more permissive than the AT1 receptor to accommodate AngII analogs with bulky modifications (66). These results suggest that the region of the receptor encompassing the hydrophobic core is opened and becomes part of the ligand binding pocket when the hydrophobic core is disrupted. Oliveira et al. (67) proposed a similar mechanism involving the expansion of the seven-transmembrane bundle upon activation of bovine rhodopsin and the AT1 receptor. Our results suggest that this expansion through the disruption of the hydrophobic core has an impact on the orientation of residue W2536.48. The reorientation of the conserved W2536.48, known as the “tryptophan toggle switch” has been suggested to be a crucial part of the activation mechanism of several GPCRs (7, 8, 64, 68, 69). However, recent crystal structures of the β1-adrenergic receptor and β2-adrenergic receptor with a bound agonist as well as of a photoactivated constitutively active mutant of rhodopsin feature this tryptophan side chain in the same rotamer as their “inactive” counterpart (70–73). This indicates that the conformation of this side chain is not the sole determinant for activation. In accordance, we observed in our simulations with the AT1 receptor that the indole moiety of W6.48 packed between small hydrophobic residues at position 7.42 (A2917.42) and 3.36 (L1123.36). Hence, the reorientation appeared to be the result of an increased hydration of the ligand binding pocket caused by the N111G mutation, resulting in an unfavorable environment for the hydrophobic portion of the indole moiety. A similar event could occur in other GPCRs and promote a movement of W6.48 while not causing it to be stabilized in a different rotamer. We therefore suggest that the disruption of the hydrophobic core within the N111G-AT1 receptor influences the conformation and the mobility of the tryptophan toggle switch. Notably, a recent study suggests that W6.48 in the human CXCR1 receptor may adopt many different configurations (74).
The mechanisms of stabilization of the inactive state unveiled here might shed light on previous studies performed on the AT1 receptor. Notably, it was suggested that only the size of residue N1113.35 was responsible for its role as a conformational switch, not its polarity or potential to form H-bonds (26). Our results actually suggest that both properties can have an impact on the stabilization of the inactive state as position 1113.35 can stabilize the inactive state either via its polar interaction with D742.50 or by contacting the hydrophobic core.
Interestingly, an interaction between residues N3.35 and D2.50 is present in the crystal structures of the CXCR4 (14) and the opioid receptors (2–5). Moreover, the crystal structures of the opioid receptors show an interaction between D2.50 and S7.46. As such, the three residues corresponding to the N1113.35-D742.50-N2957.46 network within the AT1 receptor form a similar H-bond network in the crystal structures of the opioid receptors. Whether this H-bond network is involved in the activation of opioid receptors is still an open question. However, considering that the μ-, κ-, and δ-opioid receptors possess some constitutive activity (75) and knowing that the substitution of residue N2957.46 for a serine produces a pseudo-constitutively active AT1 receptor (16, 76), it is tempting to suggest that it is. Moreover, the recently released structure of the CXCR1 receptor suggests that in its active state an H-bond is formed between the side chains of residues N571.50 and D852.50 (74). This H-bond is analogous to the N461.50-D742.50 H-bond observed in the N111G-AT1 receptor.
Previous work by Balmforth et al. (76) proposed that the side chains of N1113.35 and N2957.46 would form an H-bond in the inactive conformation of the AT1 receptor and that it would be broken during activation, which are coherent with the results from our simulations. Furthermore, Perlman et al. (77) suggested that the formation of an H-bond between the side chains of D712.50 and N431.50 was necessary for the activation of the thyrotropin-releasing hormone receptor. MD simulations of this receptor featured a local perturbation of the helicity of TMD7 as observed in MD simulation of the N111G-AT1 receptor. Interestingly, mutation N43A considerably hampered the activation of the thyrotropin-releasing hormone receptor, implying a similar role for residue N1.50 in both the AT1 receptor and the thyrotropin-releasing hormone receptor. However, the role played by this highly conserved N1.50 in the activation mechanism of the AT1 receptor appears to be different from its role in the activation mechanism of the β2-adrenergic receptor. Indeed, the loss of the polar side chain decreased the constitutive and the agonist-induced activity of the AT1 receptor, whereas it did the opposite to the β2-adrenergic receptor (24). Furthermore, the MD simulations suggested direct side chain-side chain interactions between residues D742.50-N2957.46 and D742.50-N461.50 in the AT1 receptor, whereas the corresponding interactions are water-mediated in the β2-adrenergic receptor (24). Thus, depending on the receptor, the conserved polar residues forming the H-bond networks can act in different ways and through different means.
In conclusion, MD simulations using homology models and structure-function relationships revealed key interactions associated with the activation of the AT1 receptor and with the stabilization of its inactive state. The H-bond network involving N1113.35, D742.50, and N2957.46 is associated with the inactive state, whereas the H-bond network involving D742.50, N461.50, and N2957.46 is associated with the activation of the AT1 receptor. Furthermore, the stabilization of a hydrophobic cluster above the N111-D74-N295 H-bond network is associated with the stabilization of the inactive state of the receptor. These results highlight the importance of the formation of an H-bond between residues D74 and N46 in the activation process of the AT1 receptor.
This work was supported in part by the Canadian Institutes of Health Research.
- GPCR
- G protein-coupled receptor
- TMD
- transmembrane domain
- AT1
- angiotensin II type 1
- AngII
- angiotensin II
- CXCR4
- CXC chemokine receptor type 4
- MD
- molecular dynamics
- IP1
- inositol phosphate
- Bpa
- p-benzoyl-l-phenylalanine
- Sar
- sarcosine.
REFERENCES
- 1. Katritch V., Cherezov V., Stevens R. C. (2012) Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci. 33, 17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Manglik A., Kruse A. C., Kobilka T. S., Thian F. S., Mathiesen J. M., Sunahara R. K., Pardo L., Weis W. I., Kobilka B. K., Granier S. (2012) Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu H., Wacker D., Mileni M., Katritch V., Han G. W., Vardy E., Liu W., Thompson A. A., Huang X. P., Carroll F. I., Mascarella S. W., Westkaemper R. B., Mosier P. D., Roth B. L., Cherezov V., Stevens R.C. (2012) Structure of the human κ-opioid receptor in complex with JDTic. Nature 485, 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thompson A. A., Liu W., Chun E., Katritch V., Wu H., Vardy E., Huang X. P., Trapella C., Guerrini R., Calo G., Roth B. L., Cherezov V., Stevens R. C. (2012) Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 485, 395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Granier S., Manglik A., Kruse A. C., Kobilka T. S., Thian F. S., Weis W. I., Kobilka B. K. (2012) Structure of the δ-opioid receptor bound to naltrindole. Nature 485, 400–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Worth C. L., Kleinau G., Krause G. (2009) Comparative sequence and structural analyses of G-protein-coupled receptor crystal structures and implications for molecular models. PLoS One 4, e7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schwartz T. W., Frimurer T. M., Holst B., Rosenkilde M. M., Elling C. E. (2006) Molecular mechanism of 7TM receptor activation—a global toggle switch model. Annu. Rev. Pharmacol. Toxicol. 46, 481–519 [DOI] [PubMed] [Google Scholar]
- 8. Holst B., Nygaard R., Valentin-Hansen L., Bach A., Engelstoft M. S., Petersen P. S., Frimurer T. M., Schwartz T. W. (2010) A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J. Biol. Chem. 285, 3973–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lebon G., Warne T., Edwards P. C., Bennett K., Langmead C. J., Leslie A. G., Tate C. G. (2011) Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474, 521–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Gasparo M., Catt K. J., Inagami T., Wright J. W., Unger T. (2000) International Union of Pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 52, 415–472 [PubMed] [Google Scholar]
- 11. Auger-Messier M., Clement M., Lanctot P. M., Leclerc P. C., Leduc R., Escher E., Guillemette G. (2003) The constitutively active N111G-AT1 receptor for angiotensin II maintains a high affinity conformation despite being uncoupled from its cognate G protein Gq/11α. Endocrinology 144, 5277–5284 [DOI] [PubMed] [Google Scholar]
- 12. Arsenault J., Cabana J., Fillion D., Leduc R., Guillemette G., Lavigne P., Escher E. (2010) Temperature dependent photolabeling of the human angiotensin II type 1 receptor reveals insights into its conformational landscape and its activation mechanism. Biochem. Pharmacol. 80, 990–999 [DOI] [PubMed] [Google Scholar]
- 13. Clément M., Cabana J., Holleran B. J., Leduc R., Guillemette G., Lavigne P., Escher E. (2009) Activation induces structural changes in the liganded angiotensin II type 1 receptor. J. Biol. Chem. 284, 26603–26612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu B., Chien E. Y., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., Stevens R.C. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bihoreau C., Monnot C., Davies E., Teutsch B., Bernstein K. E., Corvol P., Clauser E. (1993) Mutation of Asp74 of the rat angiotensin II receptor confers changes in antagonist affinities and abolishes G-protein coupling. Proc. Natl. Acad. Sci. U.S.A. 90, 5133–5137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feng Y. H., Zhou L., Qiu R., Zeng R. (2005) Single mutations at Asn295 and Leu305 in the cytoplasmic half of transmembrane α-helix domain 7 of the AT1 receptor induce promiscuous agonist specificity for angiotensin II fragments: a pseudo-constitutive activity. Mol. Pharmacol. 68, 347–355 [DOI] [PubMed] [Google Scholar]
- 17. Pérodin J., Deraët M., Auger-Messier M., Boucard A. A., Rihakova L., Beaulieu M. E., Lavigne P., Parent J. L., Guillemette G., Leduc R., Escher E. (2002) Residues 293 and 294 are ligand contact points of the human angiotensin type 1 receptor. Biochemistry 41, 14348–14356 [DOI] [PubMed] [Google Scholar]
- 18. Boucard A. A., Roy M., Beaulieu M. E., Lavigne P., Escher E., Guillemette G., Leduc R. (2003) Constitutive activation of the angiotensin II type 1 receptor alters the spatial proximity of transmembrane 7 to the ligand-binding pocket. J. Biol. Chem. 278, 36628–36636 [DOI] [PubMed] [Google Scholar]
- 19. Martin S. S., Boucard A. A., Clément M., Escher E., Leduc R., Guillemette G. (2004) Analysis of the third transmembrane domain of the human type 1 angiotensin II receptor by cysteine scanning mutagenesis. J. Biol. Chem. 279, 51415–51423 [DOI] [PubMed] [Google Scholar]
- 20. Clément M., Martin S. S., Beaulieu M. E., Chamberland C., Lavigne P., Leduc R., Guillemette G., Escher E. (2005) Determining the environment of the ligand binding pocket of the human angiotensin II type I (hAT1) receptor using the methionine proximity assay. J. Biol. Chem. 280, 27121–27129 [DOI] [PubMed] [Google Scholar]
- 21. Martin S. S., Holleran B. J., Escher E., Guillemette G., Leduc R. (2007) Activation of the angiotensin II type 1 receptor leads to movement of the sixth transmembrane domain: analysis by the substituted cysteine accessibility method. Mol. Pharmacol. 72, 182–190 [DOI] [PubMed] [Google Scholar]
- 22. Domazet I., Holleran B. J., Martin S. S., Lavigne P., Leduc R., Escher E., Guillemette G. (2009) The second transmembrane domain of the human type 1 angiotensin II receptor participates in the formation of the ligand binding pocket and undergoes integral pivoting movement during the process of receptor activation. J. Biol. Chem. 284, 11922–11929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Domazet I., Martin S. S., Holleran B. J., Morin M. E., Lacasse P., Lavigne P., Escher E., Leduc R., Guillemette G. (2009) The fifth transmembrane domain of angiotensin II Type 1 receptor participates in the formation of the ligand-binding pocket and undergoes a counterclockwise rotation upon receptor activation. J. Biol. Chem. 284, 31953–31961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nygaard R., Valentin-Hansen L., Mokrosinski J., Frimurer T. M., Schwartz T. W. (2010) Conserved water-mediated hydrogen bond network between TM-I, -II, -VI, and -VII in 7TM receptor activation. J. Biol. Chem. 285, 19625–19636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Noda K., Feng Y. H., Liu X. P., Saad Y., Husain A., Karnik S. S. (1996) The active state of the AT1 angiotensin receptor is generated by angiotensin II induction. Biochemistry 35, 16435–16442 [DOI] [PubMed] [Google Scholar]
- 26. Feng Y. H., Miura S., Husain A., Karnik S. S. (1998) Mechanism of constitutive activation of the AT1 receptor: influence of the size of the agonist switch binding residue Asn111. Biochemistry 37, 15791–15798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thomas W. G., Qian H., Chang C. S., Karnik S. (2000) Agonist-induced phosphorylation of the angiotensin II (AT1A) receptor requires generation of a conformation that is distinct from the inositol phosphate-signaling state. J. Biol. Chem. 275, 2893–2900 [DOI] [PubMed] [Google Scholar]
- 28. Le M. T., Vanderheyden P. M., Szaszák M., Hunyady L., Vauquelin G. (2002) Angiotensin IV is a potent agonist for constitutive active human AT1 receptors. Distinct roles of the N- and C-terminal residues of angiotensin II during AT1 receptor activation. J. Biol. Chem. 277, 23107–23110 [DOI] [PubMed] [Google Scholar]
- 29. Miura S., Zhang J., Boros J., Karnik S. S. (2003) TM2-TM7 interaction in coupling movement of transmembrane helices to activation of the angiotensin II type-1 receptor. J. Biol. Chem. 278, 3720–3725 [DOI] [PubMed] [Google Scholar]
- 30. Nikiforovich G. V., Mihalik B., Catt K. J., Marshall G. R. (2005) Molecular mechanisms of constitutive activity: mutations at position 111 of the angiotensin AT1 receptor. J Pept. Res. 66, 236–248 [DOI] [PubMed] [Google Scholar]
- 31. Lee C., Hwang S. A., Jang S. H., Chung H. S., Bhat M. B., Karnik S. S. (2007) Manifold active-state conformations in GPCRs: agonist-activated constitutively active mutant AT1 receptor preferentially couples to Gq compared to the wild-type AT1 receptor. FEBS Lett. 581, 2517–2522 [DOI] [PubMed] [Google Scholar]
- 32. Miura S., Kiya Y., Kanazawa T., Imaizumi S., Fujino M., Matsuo Y., Karnik S. S., Saku K. (2008) Differential bonding interactions of inverse agonists of angiotensin II type 1 receptor in stabilizing the inactive state. Mol. Endocrinol. 22, 139–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhuiyan M. A., Hossain M., Miura S., Nakamura T., Ozaki M., Nagatomo T. (2009) Constitutively active mutant N111G of angiotensin II type 1 (AT1) receptor induces homologous internalization through mediation of AT1-receptor antagonist. J. Pharmacol. Sci. 111, 227–234 [DOI] [PubMed] [Google Scholar]
- 34. Bhuiyan M. A., Hossain M., Nakamura T., Ozaki M., Nagatomo T. (2010) Internalization of constitutively active N111G MUTANT of AT1 receptor induced by angiotensin II-receptor antagonists candesartan, losartan, and telmisartan: comparison with valsartan. J. Pharmacol. Sci. 112, 459–462 [DOI] [PubMed] [Google Scholar]
- 35. Yan L., Holleran B. J., Lavigne P., Escher E., Guillemette G., Leduc R. (2010) Analysis of transmembrane domains 1 and 4 of the human angiotensin II AT1 receptor by cysteine-scanning mutagenesis. J. Biol. Chem. 285, 2284–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guillemette G., Escher E. (1983) Analysis of the adrenal angiotensin II receptor with the photoaffinity labeling method. Biochemistry 22, 5591–5596 [DOI] [PubMed] [Google Scholar]
- 37. Ballesteros J. A., Weinstein H. (1995) in Methods in Neurosciences (Sealfon S. C., ed) Vol. 25, pp. 366–428, Academic Press, Waltham, MA [Google Scholar]
- 38. Zhang Y. (2007) Template-based modeling and free modeling by I-TASSER in CASP7. Proteins 69, Suppl. 8, 108–117 [DOI] [PubMed] [Google Scholar]
- 39. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 41. Berendsen H. J. C., van der Spoel D., van Drunen R. (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 [Google Scholar]
- 42. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
- 43. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 44. van der Spoel D., Hess B. (2011) GROMACS—the road ahead. WIREs Comput. Mol. Sci. 1, 710–715 [Google Scholar]
- 45. Kandt C., Ash W. L., Tieleman D. P. (2007) Setting up and running molecular dynamics simulations of membrane proteins. Methods 41, 475–488 [DOI] [PubMed] [Google Scholar]
- 46. Lemkul J. A., Bevan D. R. (2009) Perturbation of membranes by the amyloid β-peptide: a molecular dynamics study. FEBS J. 276, 3060–3075 [DOI] [PubMed] [Google Scholar]
- 47. de Vries A. H., Mark A. E., Marrink S. J. (2004) The binary mixing behavior of phospholipids in a bilayer: a molecular dynamics study. J. Phys. Chem. B 108, 2454–2463 [Google Scholar]
- 48. Lemkul J. A., Bevan D. R. (2008) A comparative molecular dynamics analysis of the amyloid β-peptide in a lipid bilayer. Arch. Biochem. Biophys. 470, 54–63 [DOI] [PubMed] [Google Scholar]
- 49. Berweger C. D., van Gunsteren W. F., Müller-Plathe F. (1995) Force field parametrization by weak coupling. Re-engineering SPC water. Chem. Phys. Lett. 232, 429–436 [Google Scholar]
- 50. Berger O., Edholm O., Jähnig F. (1997) Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 72, 2002–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tieleman D. P. (2004) The molecular basis of electroporation. BMC Biochem. 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tieleman D. P., Leontiadou H., Mark A. E., Marrink S. (2003) Simulation of pore formation in lipid bilayers by mechanical stress and electric fields. J. Am. Chem. Soc. 125, 6382–6383 [DOI] [PubMed] [Google Scholar]
- 53. MacCallum J. L., Tieleman D. P. (2006) Computer simulation of the distribution of hexane in a lipid bilayer: spatially resolved free energy, entropy, and enthalpy profiles. J. Am. Chem. Soc. 128, 125–130 [DOI] [PubMed] [Google Scholar]
- 54. Anézo C., de Vries A. H., Höltje H., Tieleman D. P., Marrink S. (2003) Methodological issues in lipid bilayer simulations. J. Phys. Chem. B 107, 9424–9433 [Google Scholar]
- 55. Werner T., Morris M. B., Dastmalchi S., Church W. B. (2012) Structural modelling and dynamics of proteins for insights into drug interactions. Adv. Drug Deliv. Rev. 64, 323–343 [DOI] [PubMed] [Google Scholar]
- 56. Nose S. (1984) A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 [Google Scholar]
- 57. Hoover W. G. (1985) Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 [DOI] [PubMed] [Google Scholar]
- 58. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 59. Corpet F. (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10881–10890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ehrhardt C., Schmolke M., Matzke A., Knoblauch A., Will C., Wixler V., Ludwig S. (2006) Polyethylenimine, a cost-effective transfection reagent. Signal Transduct. 6, 179–184 [Google Scholar]
- 61. Cheng Y., Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 62. Kulińska K., Kuliński T., Lyubartsev A., Laaksonen A., Adamiak R. W. (2000) Spatial distribution functions as a tool in the analysis of ribonucleic acids hydration—molecular dynamics studies. Comput. Chem. 24, 451–457 [DOI] [PubMed] [Google Scholar]
- 63. Bhattacharya S., Hall S. E., Li H., Vaidehi N. (2008) Ligand-stabilized conformational states of human β2 adrenergic receptor: insight into G-protein-coupled receptor activation. Biophys. J. 94, 2027–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nygaard R., Frimurer T. M., Holst B., Rosenkilde M. M., Schwartz T. W. (2009) Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol. Sci. 30, 249–259 [DOI] [PubMed] [Google Scholar]
- 65. Joseph M.-P., Maigret B., Bonnafous J.-C., Marie J., Scheraga H. A. (1995) A computer modeling postulated mechanism for angiotensin II receptor activation. J. Protein Chem. 14, 381–398 [DOI] [PubMed] [Google Scholar]
- 66. Fillion D., Lemieux G., Basambombo L. L., Lavigne P., Guillemette G., Leduc R., Escher E. (2010) The amino-terminus of angiotensin II contacts several ectodomains of the angiotensin II receptor AT1. J. Med. Chem. 53, 2063–2075 [DOI] [PubMed] [Google Scholar]
- 67. Oliveira L., Costa-Neto C. M., Nakaie C. R., Schreier S., Shimuta S. I., Paiva A. C. (2007) The angiotensin II AT1 receptor structure-activity correlations in the light of rhodopsin structure. Physiol. Rev. 87, 565–592 [DOI] [PubMed] [Google Scholar]
- 68. Ahuja S., Smith S. O. (2009) Multiple switches in G protein-coupled receptor activation. Trends Pharmacol. Sci. 30, 494–502 [DOI] [PubMed] [Google Scholar]
- 69. Colson A. O., Perlman J. H., Jinsi-Parimoo A., Nussenzveig D. R., Osman R., Gershengorn M. C. (1998) A hydrophobic cluster between transmembrane helices 5 and 6 constrains the thyrotropin-releasing hormone receptor in an inactive conformation. Mol. Pharmacol. 54, 968–978 [DOI] [PubMed] [Google Scholar]
- 70. Warne T., Edwards P. C., Leslie A. G., Tate C. G. (2012) Crystal structures of a stabilized β1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure 20, 841–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Deupi X., Edwards P., Singhal A., Nickle B., Oprian D., Schertler G., Standfuss J. (2012) Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. Proc. Natl. Acad. Sci. U.S.A. 109, 119–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Standfuss J., Edwards P. C., D'Antona A., Fransen M., Xie G., Oprian D. D., Schertler G. F. (2011) The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature 471, 656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the β2-adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Park S. H., Das B. B., Casagrande F., Tian Y., Nothnagel H. J., Chu M., Kiefer H., Maier K., De Angelis A. A., Marassi F. M., Opella S. J. (2012) Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature 491, 779–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sadée W., Wang D., Bilsky E. J. (2005) Basal opioid receptor activity, neutral antagonists, and therapeutic opportunities. Life Sci. 76, 1427–1437 [DOI] [PubMed] [Google Scholar]
- 76. Balmforth A. J., Lee A. J., Warburton P., Donnelly D., Ball S. G. (1997) The conformational change responsible for AT1 receptor activation is dependent upon two juxtaposed asparagine residues on transmembrane helices III and VII. J. Biol. Chem. 272, 4245–4251 [DOI] [PubMed] [Google Scholar]
- 77. Perlman J. H., Colson A. O., Wang W., Bence K., Osman R., Gershengorn M. C. (1997) Interactions between conserved residues in transmembrane helices 1, 2, and 7 of the thyrotropin-releasing hormone receptor. J. Biol. Chem. 272, 11937–11942 [DOI] [PubMed] [Google Scholar]