

Background: The machinery for protein import into the mitochondrial intermembrane space has been studied in mammals, yeast, and plants.
Results: Protist Erv homologues are conserved sulfhydryl:cytochrome c oxidoreductases with altered mechanisms.
Conclusion: The composition and mechanism of the mitochondrial protein import machinery differs between eukaryotic lineages.
Significance: The current knowledge on mitochondrial protein import cannot be generally transferred to all eukaryotes.
Keywords: Enzyme Catalysis, Leishmania, Mitochondria, Plasmodium, Protein Evolution, Erv, Protein Import
Abstract
Mia40 and the sulfhydryl:cytochrome c oxidoreductase Erv1/ALR are essential for oxidative protein import into the mitochondrial intermembrane space in yeast and mammals. Although mitochondrial protein import is functionally conserved in the course of evolution, many organisms seem to lack Mia40. Moreover, except for in organello import studies and in silico analyses, nothing is known about the function and properties of protist Erv homologues. Here we compared Erv homologues from yeast, the kinetoplastid parasite Leishmania tarentolae, and the non-related malaria parasite Plasmodium falciparum. Both parasite proteins have altered cysteine motifs, formed intermolecular disulfide bonds in vitro and in vivo, and could not replace Erv1 from yeast despite successful mitochondrial protein import in vivo. To analyze its enzymatic activity, we established the expression and purification of recombinant full-length L. tarentolae Erv and compared the mechanism with related and non-related flavoproteins. Enzyme assays indeed confirmed an electron transferase activity with equine and yeast cytochrome c, suggesting a conservation of the enzymatic activity in different eukaryotic lineages. However, although Erv and non-related flavoproteins are intriguing examples of convergent molecular evolution resulting in similar enzyme properties, the mechanisms of Erv homologues from parasitic protists and opisthokonts differ significantly. In summary, the Erv-mediated reduction of cytochrome c might be highly conserved throughout evolution despite the apparent absence of Mia40 in many eukaryotes. Nevertheless, the knowledge on mitochondrial protein import in yeast and mammals cannot be generally transferred to all other eukaryotes, and the corresponding pathways, components, and mechanisms remain to be analyzed.
Introduction
Saccharomyces cerevisiae Erv1 (ScErv1)3 and its mammalian homologue “augmenter of liver regeneration” (ALR) assist the Mia40-dependent oxidative trapping of proteins in the mitochondrial intermembrane space. Mia40 itself oxidizes a variety of cysteine-containing substrates resulting in substrate disulfide bond formation and protein folding. Reduced Mia40 subsequently transfers an electron pair to the flavoprotein Erv1/ALR. Erv1/ALR serves as a redox switch, accepting one electron pair and transferring two single electrons separately to two molecules of cytochrome c. Accordingly, mitochondrial protein import is linked to the respiratory chain. Alternatively, Erv1/ALR was suggested to directly transfer electrons to molecular oxygen (1–6). Even though the Mia40/Erv1 system is essential in opisthokonts (a eukaryotic lineage including yeast and mammals), inactivation of Mia40 was reported to have no detectable deleterious phenotype in Arabidopsis (7). In addition, several protist lineages including apicomplexan and kinetoplastid parasites apparently have no Mia40 homologue at all, pointing to significantly altered mitochondrial import machineries (2, 8, 9). Nevertheless, recent experiments support the theory that mitochondrial protein import is functionally conserved in the course of evolution because Leishmania tarentolae marker proteins of all four mitochondrial compartments were efficiently imported into mitochondria from yeast, and opisthokont marker proteins were also successfully imported into isolated L. tarentolae mitochondria. These in organello experiments included the import of yeast and L. tarentolae homologues of Erv and small Tim proteins as substrates of the mitochondrial oxidative folding pathway (9).
The structures and mechanisms of Erv homologues from mammals (10–13), yeast (14–16), and Arabidopsis thaliana (17–19) have been studied in great detail. In contrast, very little is known about protist Erv homologues (2, 8, 9). Here, we compare Erv homologues from three different eukaryotic lineages, (i) L. tarentolae Erv (LtErv) as a member of the excavata/discicristata, (ii) Plasmodium falciparum Erv (PfErv) as a member of the chromalveolata, and (iii) ScErv1 as a member of the opisthokonta. We show that both parasite Erv homologues cannot complement the knock-out of ERV1 in yeast despite successful mitochondrial protein import. Furthermore, we characterize full-length wild type and mutant LtErv and compare its properties and enzymatic mechanism with related and non-related flavoproteins. Our findings indicate that the sulfhydryl:cytochrome c oxidoreductase activity of opisthokont Erv1/ALR is conserved in the course of evolution. On the one hand, Erv and non-related flavoproteins are intriguing examples of convergent molecular evolution resulting in similar enzyme properties. On the other hand, the exact mechanisms of Erv homologues from different eukaryotic lineages vary significantly. Considering the results from this and our previous study, the Mia/Erv pathway in opisthokonts does not seem to be strictly conserved throughout evolution. As a consequence, the knowledge on mitochondrial protein import into the intermembrane space of opisthokonts cannot be generally transferred to all other eukaryotes, and the corresponding pathways remain to be analyzed.
EXPERIMENTAL PROCEDURES
Bioinformatics Analyses and Molecular Modeling
BLAST searches and multiple sequence alignments were performed as described previously (9). Partial molecular models of PfErv and LtErv were generated based on the crystal structures of homodimeric rat ALR (PDB entry 1OQC) (10) and Erv from A. thaliana (PDB entry 2HJ3) (19). Alignments between the templates and the parasite Erv homologues were optimized manually in the Swiss-PDB Viewer (spdbv). Computations of the models were carried out at the Swiss-Model server using the “project” mode (20, 21). Very similar models were obtained for both templates. A variety of automated analyses at the server indicated a high quality of the models, and force field energies (calculated with the GROMOS96 implementation of spdbv) ranged without further energy minimization from −3.2 MJ/mol per subunit of PfErv to −4.8 MJ/mol per subunit of LtErv.
Cloning and Site-directed Mutagenesis
Untagged full-length PFERV was PCR-amplified from a P. falciparum cDNA library (strain 3D7) with perfect match primers (Metabion) that were designed based on the Plasmo DB annotation PFA0500w. The PCR product was cloned into the vector pDrive using the PCR cloning kit from Qiagen. LTERV was cloned previously (9). Constructs encoding N-terminally MRGS(H)6GS-tagged PfErv and LtErv were subcloned by PCR using the BamHI and HindIII restriction sites of the Escherichia coli expression vector pQE30 (Qiagen). Cysteine point mutations of LTERVC17S, LTERVC63S, LTERVC66S, LTERVC300S, and LTERVC304S in pQE30 were introduced by PCR using mutated primers and the QuikChange site-directed mutagenesis kit from Stratagene. A construct encoding C-terminally LE(H)6-tagged PfErvI2V was PCR-cloned via the restriction sites NcoI (causing the point mutation) and XhoI of pET28 (Novagen). The same restriction sites were used to clone untagged PFERVI2V into the yeast expression vectors pYX143 and pYX232. Untagged LTERV was subcloned from pDrive into plasmids pYX143 and pYX232 via the BamHI and XhoI restriction sites. As positive controls for complementation assays, intron-free SCERV1 was subcloned from pYX232 into pYX143 using the restriction sites EcoRI and XhoI. Correct insertions and sequences of all wild type and mutant constructs were confirmed by sequencing both strands.
Cell Culture and Western Blot Analyses
Unless otherwise indicated, S. cerevisiae cultures were grown at 30 °C in YP (1% yeast extract, 2% peptone) medium or in synthetic (S) minimal medium supplemented with or without the bases adenine and uracil and the amino acids lysine, leucine, histidine, and tryptophan (22). Media contained either 2% glucose (YPD, SD), galactose (YPGal or SGal), or lactate (SLac). The GAL-MIA40 strain expresses Mia40 under control of the galactose-induced and glucose-repressed GAL10 promoter (23). For expression of LTERV in this genetic background, the strain was transformed with plasmid pYX232-LTERV and selected on SD agar plates without tryptophan. Positive clones were grown in liquid SLac medium containing 0.5% galactose. For depletion of Mia40, the culture was shifted to tryptophan-free SLac medium containing 0.5% glucose for 30 h. For the interaction studies the plasmid pYX232-LTERV was transformed in a strain expressing Mia40 with a C-terminal His8 tag (24).
L. tarentolae promastigotes were cultured at 27 °C according to standard protocols (25) in brain heart infusion medium supplemented with hemin as described previously (9). P. falciparum strain 3D7 was cultured at 37 °C according to standard protocols (26) in RPMI medium containing 0.45% (w/v) Albumax II and human type A erythrocytes at a hematocrit of 3% under a low-oxygen atmosphere (5% CO2, 5% O2, 90% N2, and 80% humidity). For Western blot analyses P. falciparum cultures were centrifuged (3 min, 1500 × g), and erythrocytes were lysed in 0.02% saponin as described previously (27). Purified parasites were directly boiled in SDS-PAGE Laemmli buffer. The content and redox state of ScErv1, LtErv, and marker proteins were evaluated by reducing and non-reducing SDS-PAGE and Western blot analyses as described previously (9, 28, 29). For the generation of PfErv antibodies, rabbits were immunized with a synthetic peptide (NH2-CQDIQTIIDKYKTVD-COOH) corresponding to the C terminus of PfErv (Pineda Antibody Service). Antibodies were subsequently purified from sera by affinity chromatography according to established protocols (9). Approximately 2 × 108 purified malaria parasites were loaded per lane.
Complementation Assays
A YPH501-derived yeast Δerv1 knock-out strain (genotype ura3-52, lys2-801amber, ade2-101ocre, trp1-Δ63, his3-Δ200, leu2-Δ1, erv1::His3) with a copy of SCERV1 on a URA3-containing pRS426 plasmid under control of the MET25 promoter (30) was transformed with the TRP1-containing plasmids pYX232, pYX232-SCERV1, pYX232-LTERV, and pYX232-PFERV or a plasmid-free control according to a standard protocol by Ref. 31. Transformants were selected on SD agar plates without uracil, histidine, and tryptophan, and six clones of each strain were incubated in 1 ml of SD liquid medium without histidine and tryptophan overnight. Liquid cultures were subsequently diluted to an absorbance of 0.1 in sterile water, and 10-μl drops of each dilution were spotted onto SD agar plates without histidine and tryptophan containing 50 mg/liter uracil and 1 g/liter 5-fluoroorotic acid for negative selection and plasmid shuffling (32). As a control, 10-μl drops of each diluted culture were also spotted on SD agar plates without uracil, histidine, and tryptophan. In analogy, the same Δerv1 knock-out strain with an episomal copy of SCERV1 was transformed with the LEU2-containing plasmids pYX143, pYX143-SCERV1, pYX143-LTERV, and pYX143-PFERV or a plasmid-free control. After selection on SD agar plates without uracil, histidine, and leucine, six clones of each strain were incubated in 1 ml of SGal liquid medium without histidine and leucine for several days. Liquid cultures were subsequently diluted to an absorbance of 0.1 in sterile water, and 10-μl drops of each dilution were spotted onto SD and SGal agar plates without histidine and leucine containing 50 mg/liter uracil and 1 g/liter 5-fluoroorotic acid for negative selection and plasmid shuffling. All plates were incubated for up to 3 weeks at 24, 30, and 37 °C. Strains were checked for ScErv1 and LtErv by Western blot analyses.
Submitochondrial Localization Studies in Yeast
Yeast strains were grown on SD liquid medium without negative selection by 5-fluoroorotic acid. Mitochondria were purified, and proteolytic susceptibility assays with proteinase K in the presence or absence of a hypotonic buffer and/or detergent were performed as described previously (28). The membrane association of LtErv and marker proteins was analyzed by carbonate extraction and Western blotting as described (9) with yeast mitochondria replacing L. tarentolae mitochondria.
Interaction (Pulldown) Studies
Purified yeast mitochondria (1 mg) containing wild type or His-tagged Mia40 were solubilized in 250 μl of buffer (20 mm Tris/HCl, 80 mm KCl, 30 mm imidazole, pH 7.4, 1 mm phenylmethylsulfonyl fluoride) containing 0.5% (w/v) Triton X-100 at 4 °C. After a clarifying spin, the solubilized material was incubated with 30 μl of nickel-nitrilotriacetic acid agarose (Ni-NTA) beads for 1 h at 4 °C under gentle mixing. After 3 washing steps with the solubilization buffer containing 0.05% (w/v) Triton X-100, bound proteins were eluted with Laemmli buffer containing 300 mm imidazole for 5 min at 95 °C. Samples were analyzed by SDS-PAGE and immunodecoration with antibodies.
Heterologous Expression and Protein Purification
His-tagged wild type and mutant LtErv was expressed in E. coli strain SHuffle T7 Express (New England Biolabs). Overnight cultures were diluted into 6 liters of Luria-Bertani medium supplemented with 100 μg/ml ampicillin to an initial absorbance at 600 nm of ∼0.02. The cultures were grown at 25 °C to an absorbance of 0.3–0.4 and were subsequently shifted to 16 °C. Expression was induced at an absorbance of 0.5–0.7 by adding 0.1 mm isopropyl-β-d-1-thiogalactopyranoside. After 17–18 h, cultures were transferred to an ice-water bath and harvested by a single centrifugation step at 4 °C using a Beckman JS-4.2 rotor (15 min, 4000 × g). Bacteria were rapidly resuspended on ice in 60 ml of disruption buffer containing 0.25% (v/v) Tween 20, 20 mm imidazole, 300 mm NaCl, 50 mm sodium phosphate, pH 8.5 (at 4 °C). The suspension was supplemented with DNase and lysozyme (1 mg/ml) and stirred for 1 h on ice. Afterward, the suspension was sonicated and centrifuged at 4 °C (30 min, 10,000 × g). The obtained pellet was resuspended again in disruption buffer followed by another sonication and centrifugation cycle (the second extraction was found to significantly improve the overall yield). The supernatants were loaded on a column containing 0.5–0.7 ml of Ni-NTA that had been equilibrated with disruption buffer. The column was washed with 10–14 ml of 75 mm imidazole, 300 mm NaCl, 50 mm sodium phosphate, pH 8.0. Afterward, LtErv was eluted twice with 0.5–0.7 ml of elution buffer containing 400 mm imidazole, 300 mm NaCl, 50 mm sodium phosphate, pH 8.0.
LtErv was further purified by gel filtration chromatography on a HiLoad 16/60 Superdex 75 prep grade column that was connected to an ÄKTA-FPLC system (GE Healthcare). The protein was eluted in buffer containing 300 mm NaCl, 50 mm sodium phosphate, pH 7.8, with or without 2 mm dithiothreitol (DTT). FPLC fractions were detected photometrically, and protein-containing fractions were analyzed by standard reducing and non-reducing SDS-PAGE and Western blotting against the His tag.
Non-enzymatic Characterization of LtErv
Concentrations of purified wild type and mutant LtErv were determined according to Bradford with bovine serum albumin as a standard (33). In addition, protein concentrations and extinction coefficients were evaluated based on FAD absorbance: Spectra of 800-μl samples of ∼20 μm LtErv in elution buffer were recorded at 25 °C on a thermostatted Jasco V-650 UV-visible spectrophotometer. Afterward 20 μl of a 10% SDS-solution were added, and spectra were recorded over time until no further changes were observed (34). The spectra were corrected for the absorbance of elution buffer with or without 0.25% SDS. The exact concentration of FAD in the cuvette was determined with reference spectra of 12–57 μm FAD (Sigma) in elution buffer containing 0.25% SDS.
To confirm the integrity of purified LtErv, proteolytic digests of the final single SDS-PAGE band were analyzed by HPLC-MS/MS on a LTQ-Orbitrap mass spectrometer at a core facility of the Heidelberg University. The sequence coverage with trypsin and elastase was 61 and 80%, respectively. C-terminal peptides with complete y series were detected in both digests indicating that not only the N terminus (detected by Western blotting) but also the C terminus of purified LtErv was unaltered.
DTT:Cytochrome c Electron Transferase Assay
The enzymatic activity of purified wild type and mutant LtErv was analyzed according to a previously published assay with DTT as electron donor and cytochrome c as electron acceptor (15). DTT was from Fluka, and equine heart cytochrome c and bovine erythrocyte superoxide dismutase 1 (SOD) were purchased from Sigma. Stock solutions of 2 mm DTT and 1 mm cytochrome c were prepared freshly before each set of measurements in assay buffer containing 50 mm sodium phosphate, pH 8.0, supplemented with 300 mm NaCl and 2.5 mm EDTA. Initial reaction velocities were measured at 25 °C in assay buffer with 2 μm LtErv, 40 μm equine heart cytochrome c, and variable concentrations of DTT ranging from 5 μm to 1 mm. In one set of experiments S. cerevisiae cytochrome c from Sigma was used for comparison. Alternatively, variable concentrations of heart cytochrome c ranging from 2 to 40 μm were analyzed with 0.4 μm LtErv and 0.1 mm DTT. All assays were performed with or without 75 units/ml SOD to discriminate between Erv-mediated specific electron transfer and nonspecific electron transfer due to flavoprotein-dependent superoxide anion formation (13). Furthermore, activities were corrected for the spontaneous reduction of cytochrome c by DTT. All kinetics were monitored at 550 nm and evaluated by non-linear (hyperbolic) regression analyses in Sigmaplot 10.0 (Systat) using an extinction coefficient of 29 mm−1cm−1 for reduced equine cytochrome c.
RESULTS AND DISCUSSION
Kinetoplastid Erv Homologues Have Altered Domain Architectures
Based on our previous in silico analyses (9), we generated a multiple sequence alignment for Erv homologues from kinetoplastid and apicomplexan parasites (Fig. 1A). Although LtErv shares a higher sequence similarity with yeast mitochondrial Erv1 than endoplasmic reticulum Erv2 (E values of 1e−24 and 1e−16, respectively), the difference is far less obvious for PfErv (E values of 4e−18 and 6e−18, respectively). Using different bioinformatics programs listed at ExPASy, no N-terminal targeting signal was predicted for the Erv homologues from parasitic protists in contrast to Erv2 from yeast. Thus, as previously confirmed for LtErv (9), Erv homologues from kinetoplastid and apicomplexan parasites are probably located in the mitochondrial intermembrane space and not in the endoplasmic reticulum. Moreover, the acquisition of an Erv homologue in the endoplasmic reticulum seems to be a more recent, presumably fungi-specific event in the course of evolution (35).
FIGURE 1.

Structural features of Erv homologues. A, multiple sequence alignment of the conserved FAD-binding Erv/ALR domain and a C-terminal KISS domain is shown. The conserved active site cysteine residues (A), the distal cysteine residues (D), the so-called structural cysteine residues (S), and the clamp-forming cysteine residues (C) are shaded in red, blue, green, and purple, respectively. Erv homologues from opisthokonts, alveolates, kinetoplastid parasites, and plants are boxed from top to bottom. The sequence of yeast Erv2 is shown for comparison. Sc, S. cerevisiae; Nc, Neurospora crassa; Dm, Drosophila melanogaster; Ce, Caenorhabditis elegans; Hs, Homo sapiens; Py, Plasmodium yoelii; Pb, Plasmodium berghei; Pf, P. falciparum; Ta, Theileria annulata; Bb, Babesia bovis; Pt, Paramecium tetraurelia; Tt, Tetrahymena thermophila; Lm, L. major; Lb, L. braziliensis; Lt, L. tarentolae; Tc, T. cruzi; Tb, T. brucei; At, A. thaliana; Zm, Zea mays. B and C, molecular models and architectures of homodimeric PfErv and LtErv, respectively. FAD and cysteine residues are highlighted. The N and C termini are missing in the molecular models. The distance between the thiol groups of residues Cys101 and Cys129 in PfErv is almost 18 Å. Putative interactions between the active site and distal cysteine residues are indicated by dotted arrows. D, the structure of rat ALR (10) served as a template for the models and is shown for comparison (PDB entry 1OQC). Please note the additional intermolecular head-to-tail disulfide clamp between relatively conserved cysteine residues before and after the Erv/ALR domain.
The Erv domain (36) is conserved in all homologues in Fig. 1A, except for helix α5, which might be replaced by a different structural element e.g. in Erv homologues from apicomplexan parasites. Accordingly, stable and reliable molecular models for dimeric PfErv and LtErv including the FAD binding site were generated based on the structure of rat ALR (Fig. 1, B–D). The most striking feature of Erv homologues from kinetoplastid parasites is an additional C-terminal domain of almost 200 amino acids (Fig. 1, A and C). Using this sequence as an input for BLAST searches against the genomes of kinetoplastid parasites yielded Erv homologues only. Analogous searches against non-Kinetoplastida revealed no significant hits (the lowest E values were 0.002 for hypothetical proteins). Thus, the domain is specific for these Erv homologues, and we therefore termed it Kinetoplastida-specific second (KISS) domain of Erv. Bioinformatics predictions for the KISS domain suggest an αββαα secondary structure with elongated loops between each of the secondary structure elements. Of note, the KISS domain is rather variable and differs not only between Leishmania and Trypanosoma homologues but also between Trypanosoma spp. For example, a highly charged segment after the first predicted α helix in Leishmania spp. and Trypanosoma cruzi presumably forms another α helix that is missing in the Trypanosoma brucei homologue (see deletion between residues VKS and VLEL in Fig. 1A). In contrast, the N-terminal linker region between the domains as well as the C-terminal arm of the KISS domain are highly conserved. In summary, Erv homologues from apicomplexan and kinetoplastid parasites most likely adopt the typical Erv/ALR fold resulting in FAD binding and homodimer formation. The C-terminal KISS domain is a special feature of Erv homologues from Kinetoplastida.
Protist Erv Homologues Have Altered Cysteine Motifs
The proteins in Fig. 1 can be grouped according to their cysteine patterns and deviations at the N and C termini. Four different disulfide bond-forming cysteine pairs can be distinguished for Erv homologues (Fig. 1) (2): (i) a proximal or active site cysteine pair, (ii) a distal or shuttle cysteine pair, (iii) a structural disulfide bond, and (iv) a clamp disulfide bond. (i) The cysteine residues in the Y(P/A)CXXC motif at the active site are the only strictly conserved residues in addition to the FAD binding site (Fig. 1, A–D). The disulfide bond between the former residues was shown to be essential for the oxidase activity of ScErv1 (37). (ii) The distal cysteine pair is highly variable. Erv homologues from alveolates including the apicomplexan parasites have a CX(D/E)X2C motif instead of an opisthokont-specific CRXC motif at the N-terminal arm of the protein (Fig. 1, A and B). In contrast, the distal cysteine pair of Erv homologues from kinetoplastid parasites is located close to the C terminus in a CXVYC motif (Fig. 1, A and C). This architecture is analogous to Erv homologues from plants (18) and Erv2 from yeast (14) (Fig. 1A). (iii) The structural disulfide bond connecting helix α4 and the following loop cannot be formed in PfErv (Fig. 1, A and B) because the first cysteine residue is missing, and the second residue is poorly conserved in Plasmodium. Because both structural cysteine residues are found in ciliate Erv homologues (Fig. 1A), the disulfide bond was presumably lost during the evolution of apicomplexan parasites. Thus, the structural disulfide bond is not a fundamental prerequisite for proper Erv folding. (iv) The N-terminal cysteine residue of the disulfide clamp connecting both subunits in ALR is conserved among Kinetoplastida, ciliates, and most opisthokonts (Fig. 1, A, C, and D), but is absent in apicomplexan parasites, plants, and yeast (Fig. 1, A and B). The second residue is even less conserved, suggesting that the clamp disulfide bond was either lost or acquired several times throughout evolution. Please note that the cysteine residue in the CP motif before the Erv/ALR domain might form alternative intermolecular disulfide bonds, e.g. in LtErv (Fig. 1, A and C) and similar proteins, provided that the N-terminal arm has a high flexibility. Noteworthy, alternative inter- or intramolecular disulfide patterns that are poorly conserved throughout evolution are relatively common among redox proteins and can be also found, for example, in peroxiredoxins (38) and glutaredoxins (39).
In summary, structural prerequisites for electron transfer, including the proximal active site cysteine pair and a distal shuttle cysteine pair for the interaction with protein substrates, are conserved among Erv homologues. However, the distal cysteine residues are located either close to the N or the C terminus and are found in highly variable motifs regarding the type and number of spacing residues. Other cysteine residues forming a structural or a clamp disulfide bond are not strictly conserved among Erv homologues.
Protist Erv Homologues Cannot Complement a Δerv1 Yeast Strain
Even though protein import into the mitochondrial intermembrane space is functionally conserved throughout evolution, the import mechanisms are presumably altered (9). To address the question of whether protist Erv homologues can replace ScErv1 in the yeast model system, we cloned and subcloned the genes encoding PfErv and LtErv (Table 1) and performed complementation assays using alternative vector/promotor systems and culture conditions as summarized under “Experimental Procedures”. In contrast to a positive control, none of the conditions resulted in a successful complementation (Fig. 2A). Thus, the genes encoding PfErv and LtErv cannot rescue yeast in the absence of endogenous ScErv1. Whether the unsuccessful complementation is due to the essential roles of ScErv1 for protein import (1–5) or for maturation of cytosolic iron-sulfur clusters (40) remains to be studied. Moreover, a negative result from complementation assays can obviously have alternative causes such as protein mistargeting. We, therefore, performed the following controls.
TABLE 1.
Properties of SCERV1, LTERV, and PFERV
| Property | SCERV1 | LTERV | PFERV |
|---|---|---|---|
| Accession number | gi 398365231 | gi 359829081 | gi 124505837 |
| Introns | 1 | None | None |
| Amino acids | 189 | 312 | 143 |
| Molecular mass (Da)a | 21,639.4 | 34,687.7 | 17,160.7 |
| Isoelectric pointa | 8.46 | 6.50 | 8.80 |
a Calculated using the ExPASy ProtParam tool.
FIGURE 2.

Functional analyses of protist Erv homologues in yeast. A, shown are yeast complementation assays with LTERV and PFERV via plasmid shuffling in the presence of 5-fluoroorotic acid (FOA). Empty vector and episomal SCERV1 served as negative and positive controls, respectively. B, submitochondrial localization of LtErv in an LTERV-expressing yeast strain was analyzed by proteolytic susceptibility, SDS-PAGE, and immunodecoration with antibodies against the indicated proteins. PK, proteinase K; Sw, swollen mitochondria in hypotonic buffer; TX-100, Triton X-100. C, shown is a reproduction of comparative LtErv in organello import experiments using radioactively labeled protein and isolated mitochondria (mito) from L. tarentolae (Lt) and yeast (Sc) as described in Ref 9. D, shown is a carbonate fractionation of yeast mitochondria purified from an LTERV-expressing strain. Total, total mitochondrial protein; Sup, supernatant; Pel, pellet. E, shown is a steady-state level analysis of LtErv in wild type (WT) and Mia40-depleted (Mia40↓) yeast mitochondria. Different amounts of isolated mitochondria (12.5 and 25 μg) were loaded per lane for comparison. F, shown are the interaction (pulldown) studies with LtErv-containing yeast mitochondria from WT cells and cells expressing Mia40-His8. Proteins were isolated by Ni-NTA chromatography and analyzed by SDS-PAGE and immunodecoration with antibodies against the indicated proteins. Total, 10% of total proteins loaded; Sup, 100% of the unbound proteins; Bound, 100% of bound proteins loaded.
Because of the availability of a specific antibody (9), we checked whether LtErv localizes to the intermembrane space of yeast mitochondria (Fig. 2B). LtErv was clearly enriched in mitochondrial subcellular fractions (data not shown). In contrast to Tom70 (a marker protein of the outer membrane), LtErv was protected from proteolytic degradation as long as the outer mitochondrial membrane was intact. Under hypotonic conditions, the outer membrane ruptured, and LtErv was degraded like ScErv1 and the intermembrane space marker protein Tim50. Matrix proteins including the marker Hep1 were still protected unless the inner mitochondrial membrane was permeabilized by a detergent (Fig. 2B). We, therefore, conclude that LtErv was correctly targeted to the intermembrane space of yeast mitochondria. Of note, the presented in vivo localization data confirm our previous conclusions on the functional conservation of mitochondrial protein import from in organello experiments (9) (Fig. 2C) (even though analogous in organello import experiments with PfErv could not be performed because of technical difficulties).
Despite the absence of striking hydrophobic patches, LtErv was shown to be tightly membrane-associated in L. tarentolae mitochondria (9). To analyze a potential membrane association in yeast mitochondria, we performed analogous carbonate extractions (Fig. 2D). In contrast to soluble ScErv1 and Hep1, LtErv was predominantly found in the pellet fraction indicating a membrane association as determined in Leishmania. Next, we checked whether S. cerevisiae Mia40 plays a role for the import of LtErv into yeast mitochondria. Depletion of Mia40 did not affect the mitochondrial levels of LtErv but resulted in reduced levels of known import substrates of Mia40 including ScErv1, Ccs1, and Tim13 (Fig. 2E) (41–45). Other mitochondrial proteins, such as the matrix proteins Tim44 and Hep1, were not affected by the depletion of Mia40 (Fig. 2E). These results suggest that LtErv is imported into the intermembrane space of yeast mitochondria independent of Mia40. Endogenous ScErv1 interacts with Mia40 (29). When we performed pulldown experiments using a His-tagged form of Mia40 and mitochondrial lysates containing LtErv, LtErv was not co-purified with Mia40-His8, whereas a fraction of ScErv1, used as control protein, was co-isolated. Thus, an interaction between LtErv and Mia40 was not observed, in contrast to the interaction with ScErv1 (Fig. 2F). Two possible explanations are that LtErv either does not bind at all to Mia40 or that the interaction cannot be observed in the presence of competing ScErv1 due to a significantly lower affinity. At present, preliminary experiments revealed no indication that LtErv is able to oxidize Mia40 in an Erv-catalyzed Mia40 oxidation assay in vitro. In conclusion, the inability of parasite Erv homologues to complement a Δerv1 yeast strain is not due to protein mislocalization but is most likely due to different mechanistic properties. Moreover, the in vivo import of LtErv into yeast mitochondria appears to be Mia40-independent, pointing to an alternative import mechanism.
LtErv Is a Functional Sulfhydryl:Cytochrome c Oxidoreductase
Initial experiments revealed that recombinant LtErv and PfErv cannot be purified from E. coli using standard expression/purification protocols. Even though we were unable to purify soluble N- or C-terminally tagged PfErv (despite a variety of tested E. coli strains, tags, and expression protocols), we established and optimized an expression/purification protocol for membrane-associated N-terminally His-tagged full-length LtErv (Fig. 3, A and B) (see also “Experimental Procedures” for details). The purified protein was subsequently analyzed in a DTT:cytochrome c electron transferase assay (Fig. 3, C and D). The activity in the assay was comparable to ScErv1, which served as a positive control (data not shown). Depending on the purification, apparent kcat values in the presence of SOD were ∼1 s−1, and the estimated apparent Km value for DTT was ∼1–2 mm. A similar Kmapp value for DTT was determined for ALR in an oxidase assay, and a kcatapp value of 4.5 s−1 was reported for a short splice variant of ALR in stopped-flow measurements using 2 mm DTT and variable cytochrome c concentrations (13). Furthermore, the reported Kmapp value for cytochrome c of ALR around 10 μm (13) is similar to the value of LtErv around 3 μm (Fig. 3D). In the absence of SOD the apparent activity of LtErv was significantly higher, in particular, at increasing DTT concentrations (Fig. 3C). Thus, in agreement with previous reports on flavoproteins (2, 13), a background reaction due to an unspecific oxidase activity of LtErv cannot be neglected under aerobic conditions in vitro, and the addition of SOD (to remove superoxide anion as an electron donor) is a prerequisite for the correct analysis of the electron transferase activity. Nevertheless, a comparison of the activities in Fig. 3C and the low Kmapp value in Fig. 3D support the theory that cytochrome c, not O2, is the physiological electron acceptor of Erv in Kinetoplastida (2) in accordance with previous observations on ALR (13). Thus, LtErv is primarily an electron transferase (EC 1.8.2) and not an oxidase (EC 1.8.3), and the enzymatic activity of opisthokont Erv homologues is presumably conserved in other eukaryotic lineages. Of note, LtErv transferred electrons to equine and yeast cytochrome c, suggesting that the interaction with the second substrate might be either strictly conserved throughout evolution or that an electron might tunnel from reduced Erv to cytochrome c as soon as both proteins get close enough. In summary, we established the purification of full-length LtErv and confirmed that it is a functional sulfhydryl:cytochrome c oxidoreductase.
FIGURE 3.

Two step purification and enzymatic activity of recombinant His-tagged LtErv. A, LtErv was first purified by Ni-NTA chromatography. Protein-containing fractions were analyzed by reducing SDS-PAGE (15% gel, left side) and Western blotting against the His tag (right side). Recombinant LtErv has a theoretical molecular mass of 35.9 kDa. To, total cells; Pe, cell pellet after first sonication; F1/2, flow through 1 and 2; W1/2, wash 1 and 2; E1/2, eluate 1 and 2. B, eluates from panel A were further purified by gel filtration chromatography resulting in three different peaks. Peak 1 contained pure full-length LtErv. Depending on the protein preparation, an additional band of ∼30 kDa was visible in peak 2. Western blotting against the N-terminal His tag revealed that this protein species lacks the presumably flexible C-terminal arm of the KISS domain (see architecture model in Fig. 1C). Peaks 2 and 3 contained three C-terminally truncated LtErv species consisting of the conserved Erv/ALR domain (17.1 kDa) with shorter and longer fragments of the KISS domain. C, the sulfhydryl:cytochrome c oxidoreductase activity of purified LtErv was analyzed with DTT and 40 μm cytochrome c in the presence or absence of SOD. In the presence of SOD the specific electron transferase activity was measured, whereas an additional unspecific oxidase activity resulted in a higher activity in the absence of SOD. Data points were averaged from two independent protein purifications. D, shown is a variation of the cytochrome c concentration revealing Michaelis-Menten kinetics with an apparent Km value around 3 μm. Measurements were performed with 0.1 mm DTT in the presence of SOD.
Protist Erv Homologues Form Intermolecular Disulfide Bonds in Vitro and in Vivo
A second band at ∼70 kDa was detected by non-reducing SDS-PAGE upon purification of recombinant LtErv. Under reducing conditions, the 70-kDa band was converted to the single 36-kDa band of full-length monomeric LtErv (Fig. 4A). We, therefore, conclude that the 70-kDa band corresponds to disulfide-bridged dimeric LtErv. Next, we asked whether the band is also formed in E. coli and in L. tarentolae. A second band at the corresponding size was indeed observed by Western blot analyses of E. coli extracts and purified L. tarentolae mitochondria (Fig. 4B). To analyze the covalent dimerization of PfErv, a peptide antibody was generated and purified. Although a dimer could not be unambiguously assigned, Western blot analyses of P. falciparum cell extracts revealed several potential mixed disulfide bonds between PfErv and other proteins (Fig. 4C). In E. coli extracts, a band at ∼35 kDa was detected in addition to the band of monomeric recombinant PfErv around 18 kDa. Because the former band was lost upon reduction, PfErv also most likely forms disulfide-bridged homodimers. In summary, we identified (potential) intermolecular disulfide bonds for PfErv and LtErv in vitro and in vivo.
FIGURE 4.

Intermolecular disulfide bond formation of LtErv and PfErv. A, detection of disulfide-bridged homodimers of purified full-length LtErv (Fig. 3A) by non-reducing SDS-PAGE is shown. B, Western blot analyses are shown for recombinant and endogenous disulfide-bridged LtErv dimers in E. coli extracts (E. coli) and purified mitochondria from L. tarentolae (L.t.), respectively. E. coli cells were centrifuged and directly boiled in the corresponding Laemmli buffer. C, shown are Western blot analyses of endogenous and recombinant disulfide-bridged PfErv in extracts from P. falciparum and E. coli, respectively. Recombinant PfErv has a theoretical molecular mass of 18.4 kDa. Truncation of the presumably flexible N terminus of native PfErv (Table 1) results in a protein with a theoretical molecular mass of ∼13 kDa. The band labeled with an asterisk could, therefore, be either a nonspecific cross reaction or proteolytically processed PfErv. Ab, affinity purified antibody. D, dimer; D?, potential dimer; M, monomeric full-length LtErv; ME, 2-mercaptoethanol; PS, preimmune serum; S, serum.
The Enzymatic Mechanism of LtErv
To decipher which cysteine residues shuttle the electron pair from the substrate at the protein surface to the active site, we generated several LtErv mutants and determined their ability to form disulfide bridges in E. coli by Western blot analyses (Fig. 5). Long film exposures revealed two alternative LtErv dimer bands. The lower dimer band was specifically lost when either the more N-terminal residue of the active site cysteine pair (Cys63) or the more C-terminal residue of the distal cysteine pair (Cys304) was mutated. Thus, the C-terminal arm of one subunit forms a disulfide bond with the active site of a second subunit via Cys304′ and the so-called interchange residue Cys63 (Figs. 1C and 5). Another intermolecular disulfide bond resulting in the upper dimer band was formed between Cys300 of two subunits, suggesting that this residue usually interacts with the substrate thiol group resulting in substrate oxidation.
FIGURE 5.

Identification of disulfide-bridged cysteine residues in LtErv. Wild type and mutant recombinant LtErv were produced in E. coli and detected by Western blotting against LtErv. Cells were centrifuged 2 h post induction and directly boiled in the corresponding Laemmli buffer with or without 2-mercaptoethanol (ME). Free cysteine residues were blocked with iodoacetamide (IAc) to prevent boiling-induced disulfide bond formation. The point mutations in full-length LtErv are indicated at the top. A longer exposure of the right Western blot and schematic interpretations of the disulfide bond pattern are shown for clarity. D1, upper dimer band; D2, lower dimer band; M, monomer; T?, potential trimer.
Next, we purified selected cysteine mutants and wild type LtErv and compared their spectra and activities. The flavin absorbance maxima of LtErv (Table 2) shifted to 450, 376, and 271 nm upon denaturation (Fig. 6A). Thus, FAD (and not FMN) was liberated from LtErv. Calibration curves from two independent expression and purification experiments revealed flavin-to-protein ratios (μm FAD to μm LtErv) of 1.0 and 0.98. Thus, the recombinant protein was saturated with FAD. The spectral properties of LtErvC304S, LtErvC300S/C304S, and wild type enzyme were identical (Fig. 6B), whereas LtErvC300S had an additional maximum at longer wavelengths (Fig. 6C, Table 2). The positions of the maxima in the LtErvC66S spectrum resembled free FAD (Fig. 6, A and D, Table 2). In contrast, the spectrum of LtErvC63S revealed a pronounced shoulder at longer wavelengths (Fig. 6E).
TABLE 2.
Properties of recombinant wild type and mutant LtErv from Fig. 6
| Protein | λmax1 | λmax2 | λmax3 | λmax4 | ϵmax3 | kcatapp | Km,DTTapp |
|---|---|---|---|---|---|---|---|
| nm | nm | nm | nm | mm−1cm−1 | s−1 | mm | |
| LtErv (WT) a | 274 | 378 | 462 | 10.2 | 0.92 | 2.0 | |
| LtErvC304S | 274 | 378 | 462 | 10.3 | 0.85 | 2.0 | |
| LtErvC300S/C304S | 274 | 378 | 462 | 10.3 | 0.61 | 2.0 | |
| LtErvC300S b | 272 | 368 | 458 | 563 | 8.15 | 0.33 | 2.9 |
| LtErvC63S b | 274 | 364 | 458 | 9.67 | Inactive | ND | |
| LtErvC66S | 274 | 376 | 454 | 11.0 | ND | ND |
a Common ratios between the absorbances at 274 and 462 nm were 6.3–6.8.
b Spectra changed over time. ND, not determined.
FIGURE 6.

Analysis of the enzymatic mechanism of LtErv. A, UV-visible spectrum and determination of the extinction coefficient of wild type LtErv are shown. The eluate is shown in the inset. Spectra were recorded before (orange line) and after the addition of SDS (black line). The exact concentration of liberated FAD in the cuvette was determined with reference spectra of pure FAD yielding calibration curves for the absorbances at 450 and 376 nm (closed and open symbols, respectively). B, shown are normalized spectra of LtErvC304S and LtErvC300S/C304S (black lines) compared with wild type enzyme (orange line). C–E, shown are normalized spectra of freshly purified LtErv300S (blue line), LtErvC66S (green line), and LtErvC63S (red line) compared with wild type enzyme (orange line). F, shown are schematic interpretations of the spectra in panels B–E. G, shown is the sulfhydryl:cytochrome c oxidoreductase activity of wild type and mutant LtErv in the presence of SOD. Protein properties are summarized in Table 2.
The spectra can be mechanistically interpreted as follows (Fig. 6F); in LtErvC304S, LtErvC300S/C304S, and wild type enzyme the active site cysteine pair formed a disulfide bond in the proximity of FAD. In accordance with the molecular model in Fig. 1C, removal of the interchange residue Cys63 (Fig. 5) trapped an interaction between reduced residue Cys66 and FAD (Fig. 6, E and F). Because a fraction of Cys66 was deprotonated at the slightly alkaline pH, a charge-transfer complex (CTC) between the thiolate and FAD was partially formed as predicted previously (2). Furthermore, the rather minor spectral changes in LtErvC66S (Fig. 6D) indicate that the electron density of FAD is also slightly influenced by Cys66 in the fully oxidized protein. The pronounced CTC of LtErvC300S and the additional maximum at longer wavelengths are interpreted as a delocalization of conjugated electron pairs between the C-terminal cysteine residues of one subunit and the active site of a second subunit (Fig. 6F). In the absence of Cys300 and of a physiological electron acceptor, this species was trapped (Fig. 6C). Thus, the mutant probably mimics a reaction intermediate of the reductive half-reaction (2). Because of an oxidase activity of LtErv (Fig. 3C), the CTC in LtErvC300S and LtErvC66S was slowly lost over time (data not shown). A semiquinone radical, therefore, had to be formed as predicted for the catalytic mechanism (2) in accordance with observations on a short splice variant of human ALR (13) and A. thaliana Erv1 (17). A similar spectrum to the one of LtErvC300S in Fig. 6C was also recorded for ScErv1C30S (16) in agreement with an essential intermolecular disulfide bond between Cys33 and Cys130′ in vivo (15, 16).
Regarding the electron transferase activity of LtErv, replacement of the interchange residue in LtErvC63S completely abolished the enzymatic activity, whereas mutations of the distal cysteine residues in the C-terminal arm had either no or intermediate effects (Fig. 6G). Thus, DTT cannot directly reduce Cys66 or FAD, and the proximal active site cysteine pair is a prerequisite for the electron transferase activity of LtErv. However, DTT is small enough to bypass the distal cysteine residues and to directly interact with the interchange residue Cys63 during catalysis. The lower activity and the higher Kmapp value of the mutant LtErvC300S (Fig. 6G, Table 2) might be explained by a trapped conformation of the C-terminal arm partially blocking the active site of the second subunit. The results are not only in perfect agreement with the molecular model in Fig. 1C and the spectral analyses in Fig. 6 but also with DTT oxidase activities measured for the short splice variant of human ALR lacking the distal cysteine residues (as well as mutants without the clamp-forming cysteine residues) (13). Moreover, similar results were obtained for oxidase activities of ScErv1 double mutants without the N-terminal distal cysteine pair Cys30/Cys33 or the active site cysteine pair Cys130/Cys133 (37). In summary, we unraveled the disulfide bond patterns for recombinant LtErv and gained first insights into its catalytic mechanism.
Model of LtErv Catalysis
Based on the results in Figs. 4–6, we propose the following model for the sulfhydryl:cytochrome c oxidoreductase activity of LtErv (Fig. 7). During the reductive half-reaction electrons are transferred from the sulfhydryl substrate to the distal shuttle cysteine disulfide bond at the C-terminal arm of LtErv. After a conformational change, Cys304 from one subunit interacts with the interchange cysteine residue Cys63′ of another subunit. The electron pair is subsequently delocalized between the shuttle cysteine residues and the active site of two subunits (Figs. 6C and 7) before Cys300 restores the disulfide bond with Cys304 at the C-terminal arm. As a result, a protein species with a thiolate-FAD CTC is formed (Figs. 6E and 7), and the disulfide bond formation at the C-terminal arm might trigger the reversal of the conformational change. During the predicted oxidative half-reaction, two molecules of cytochrome c consecutively accept a single electron from the flavin of LtErv (Fig. 7). In summary, in analogy to A. thaliana Erv1 (18, 19), LtErv requires a C-terminal shuttle cysteine pair in contrast to ScErv1 and ALR. Because of the position of the shuttle arm, the additional KISS domain, and the altered cysteine motifs, the mechanisms of kinetoplastid and opisthokont Erv homologues differ significantly, providing a plausible explanation for the results in Fig. 2.
FIGURE 7.
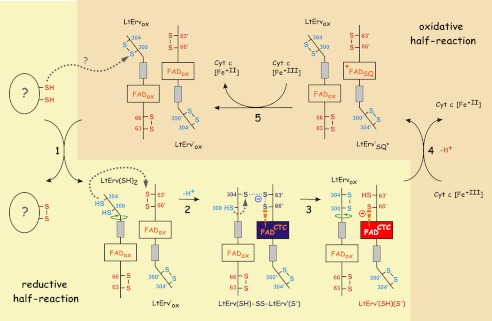
Model of LtErv catalysis. The reductive (steps 1–3) and oxidative (steps 4 and 5) half-reaction are separated. Significant structural changes due to the movement of the C-terminal arm carrying the distal shuttle cysteine residues are indicated by green arrows. Single electron transfers during the predicted oxidative half-reaction require formation of a FAD semiquinone radical (FADSQ). Color coding of the flavin corresponds to Fig. 6F. See Ref. 2 on ScErv1 catalysis for comparison.
Flavoenzymes Are Intriguing Examples of Convergent Molecular Evolution
The spectra of LtErvC304S, LtErvC300S/C304S, and wild type enzyme are similar to other oxidized flavoproteins such as glutathione reductase (46) or thioredoxin reductase (47, 48) despite completely different primary structures and protein folds (2, 35, 36). Analogous to the spectrum of borohydride-reduced glutathione reductase (46), the delocalization of the negative charge in LtErvC63S results in a thiolate-FAD CTC shoulder at longer wavelengths (Fig. 6E). Similar spectra were recorded for mutants of the interchange residue Cys130 in ScErv1 (16) and Cys145 in human ALR (49, 50). Thus, a CTC similar to many other flavoproteins also seems to be a conserved feature of the Erv family.
Erv and thioredoxin reductase both require a (flexible) arm with a distal cysteine pair to shuttle electrons between cysteine residues of a bulky protein substrate and the active site with a flavin cofactor serving as an electron switch (2). The reductive half-reaction of Erv (Fig. 7) (2) and the oxidative half-reaction of thioredoxin reductase (47, 48), therefore, share significant similarities despite completely different protein folds. As a result, the spectrum of LtErvC300S with a delocalized electron pair between the C-terminal arm of one subunit and the active site of another subunit (Fig. 6C) resembles the spectra of reduced thioredoxin reductase isoforms (47, 48). Accordingly, analogous reaction intermediates are formed (Fig. 7) (47, 48) irrespective of the position of the distal shuttle cysteine pair at the N or C terminus and the inverse flow of electrons in Erv and thioredoxin reductase. In summary, thioredoxin reductase and Erv homologues are bona fide examples for convergent molecular evolution that is presumably driven by the selection pressure of fundamental principles in redox catalysis and flavin biochemistry.
Divergent Molecular Evolution of Erv
We recently described three different scenarios for the evolution of the Mia40/Erv1 system and oxidative protein import into the mitochondrial intermembrane space; primitive eukaryotes either contained the ancestors of (i) Erv and Mia40, (ii) Erv alone, or (iii) Erv and an unknown component Z (9). Because Mia40 cannot be identified in silico in many eukaryotic lineages (8, 9), the first scenario is rather unlikely unless Mia40 was lost or its primary structure was significantly altered in the course of evolution. Considering (a) the conserved electron transferase activity with cytochrome c as an electron acceptor (Fig. 6G), (b) the conserved import of LtErv in organello and in L. tarentolae (9), and (c) the successful Mia40-independent import of LtErv in S. cerevisiae (Fig. 2), scenarios (ii) and (iii) might be favored. The tight membrane association of full-length LtErv in L. tarentolae (9) and S. cerevisiae (Fig. 2D) might be responsible for trapping LtErv in the intermembrane space of both organisms. Thus, following an established nomenclature (51), LtErv could be a special class III intermembrane space protein. Because LtErv was also membrane-associated in E. coli, the protein might have a high affinity for lipids such as cardiolipin.
Four different types of non-related shuttle arms are found in Erv homologues from opisthokonts, plants, alveolates, and Kinetoplastida. Two of them are located at the N terminus, and two of them are found at the C terminus (Fig. 8) (2, 12, 15, 16, 18, 19). Thus, potential alternative machineries for the import of proteins into the mitochondrial intermembrane space might have evolved independently in different eukaryotic lineages. The position of the shuttle arm does not seem to be a prerequisite for the interaction with Mia40 because opisthokonts and plants both have a Mia40/Erv1 couple despite opposing arm topologies (Fig. 8). However, like the protist Erv homologues (Fig. 2A), the Arabidopsis Erv homologue could not complement Erv1 in yeast (7), suggesting a complementarity of the lineage-specific shuttle arm and cysteine motifs of the Mia40/Erv couple. The exact compositions and mechanisms of the mitochondrial machineries for oxidative protein import in parasitic protists are so far completely unknown (2, 8, 9). Notably, because of the conservation of the cysteine residues in kinetoplastid homologues (Fig. 1A), the enzymatic mechanism of LtErv depicted in Fig. 7 could be also valid for other kinetoplastid parasites including highly important pathogens. Because mitochondrial protein import is expected to be essential for parasite survival, the alternative mechanisms of kinetoplastid and mammalian Erv catalysis are not only interesting from an evolutionary viewpoint but might also be attractive for inhibition studies.
FIGURE 8.
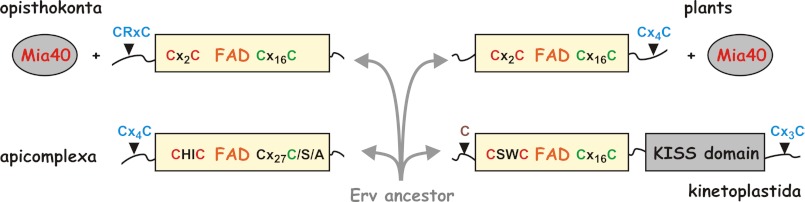
Evolution and mechanistic diversity of Erv. Representative homologues without similarities at the N and C termini (Fig. 1A) from four eukaryotic lineages are shown. The protein architectures suggest an early divergent molecular evolution that might point to significantly different import machineries for mitochondrial intermembrane space proteins.
Conclusion
We showed that neither apicomplexan nor kinetoplastid Erv can replace essential Erv1 in yeast. Likely reasons are altered cysteine patterns and protein architectures that might result in incompatible protein-protein interactions with Mia40. Furthermore, a purification protocol for recombinant full-length wild type and mutant LtErv was established, and first insights into the mechanism of kinetoplastid Erv catalysis were gained. On the one hand, the characterization of LtErv revealed that the sulfhydryl:cytochrome c oxidoreductase activity of opisthokont Erv homologues is conserved in the course of evolution, whereas the exact enzymatic mechanism is significantly altered in different eukaryotic lineages. The results are in agreement with previous in silico analyses suggesting that the Mia40/Erv1 pathway in opisthokonts is not a general feature in different eukaryotic lineages (2, 8, 9). On the other hand, the comparison of Erv properties with non-related flavoproteins revealed an intriguing example of convergent molecular evolution resulting in similar enzyme mechanisms despite completely different protein architectures.
This work was supported by Deutsche Forschungsgemeinschaft Grants DE 1431/2 (to M. D.) and SFB 594,B13 (to K. H.).
- ScErv1
- S. cerevisiae Erv1
- ALR
- augmenter of liver regeneration
- SOD
- superoxide dismutase 1
- LtErv
- L. tarentolae Erv
- PfErv
- P. falciparum Erv
- Ni-NTA
- nickel-nitrilotriacetic acid agarose
- KISS
- Kinetoplastida-specific second
- CTC
- charge-transfer complex.
REFERENCES
- 1. Chacinska A., Koehler C. M., Milenkovic D., Lithgow T., Pfanner N. (2009) Importing mitochondrial proteins. Machineries and mechanisms. Cell 138, 628–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Deponte M., Hell K. (2009) Disulphide bond formation in the intermembrane space of mitochondria. J. Biochem. 146, 599–608 [DOI] [PubMed] [Google Scholar]
- 3. Sideris D. P., Tokatlidis K. (2010) Oxidative protein folding in the mitochondrial intermembrane space. Antioxid. Redox Signal. 13, 1189–1204 [DOI] [PubMed] [Google Scholar]
- 4. Endo T., Yamano K., Kawano S. (2011) Structural insight into the mitochondrial protein import system. Biochim. Biophys. Acta 1808, 955–970 [DOI] [PubMed] [Google Scholar]
- 5. Herrmann J. M., Riemer J. (2012) Mitochondrial disulfide relay. Redox-regulated protein import into the intermembrane space. J. Biol. Chem. 287, 4426–4433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kojer K., Bien M., Gangel H., Morgan B., Dick T. P., Riemer J. (2012) Glutathione redox potential in the mitochondrial intermembrane space is linked to the cytosol and impacts the Mia40 redox state. EMBO J. 31, 3169–3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carrie C., Giraud E., Duncan O., Xu L., Wang Y., Huang S., Clifton R., Murcha M., Filipovska A., Rackham O., Vrielink A., Whelan J. (2010) Conserved and novel functions for Arabidopsis thaliana MIA40 in assembly of proteins in mitochondria and peroxisomes. J. Biol. Chem. 285, 36138–36148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Allen J. W., Ferguson S. J., Ginger M. L. (2008) Distinctive biochemistry in the trypanosome mitochondrial intermembrane space suggests a model for stepwise evolution of the MIA pathway for import of cysteine-rich proteins. FEBS Lett. 582, 2817–2825 [DOI] [PubMed] [Google Scholar]
- 9. Eckers E., Cyrklaff M., Simpson L., Deponte M. (2012) Mitochondrial protein import pathways are functionally conserved among eukaryotes despite compositional diversity of the import machineries. Biol. Chem. 393, 513–524 [DOI] [PubMed] [Google Scholar]
- 10. Wu C. K., Dailey T. A., Dailey H. A., Wang B. C., Rose J. P. (2003) The crystal structure of augmenter of liver regeneration. A mammalian FAD-dependent sulfhydryl oxidase. Protein Sci. 12, 1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Daithankar V. N., Schaefer S. A., Dong M., Bahnson B. J., Thorpe C. (2010) Structure of the human sulfhydryl oxidase augmenter of liver regeneration and characterization of a human mutation causing an autosomal recessive myopathy. Biochemistry 49, 6737–6745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Banci L., Bertini I., Calderone V., Cefaro C., Ciofi-Baffoni S., Gallo A., Kallergi E., Lionaki E., Pozidis C., Tokatlidis K. (2011) Molecular recognition and substrate mimicry drive the electron-transfer process between MIA40 and ALR. Proc. Natl. Acad. Sci. U.S.A. 108, 4811–4816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Farrell S. R., Thorpe C. (2005) Augmenter of liver regeneration. A flavin-dependent sulfhydryl oxidase with cytochrome c reductase activity. Biochemistry 44, 1532–1541 [DOI] [PubMed] [Google Scholar]
- 14. Gross E., Sevier C. S., Vala A., Kaiser C. A., Fass D. (2002) A new FAD binding fold and intersubunit disulfide shuttle in the thiol oxidase Erv2p. Nat. Struct. Biol. 9, 61–67 [DOI] [PubMed] [Google Scholar]
- 15. Bien M., Longen S., Wagener N., Chwalla I., Herrmann J. M., Riemer J. (2010) Mitochondrial disulfide bond formation is driven by intersubunit electron transfer in Erv1 and proofread by glutathione. Mol. Cell 37, 516–528 [DOI] [PubMed] [Google Scholar]
- 16. Hofhaus G., Lee J. E., Tews I., Rosenberg B., Lisowsky T. (2003) The N-terminal cysteine pair of yeast sulfhydryl oxidase Erv1p is essential for in vivo activity and interacts with the primary redox centre. Eur. J. Biochem. 270, 1528–1535 [DOI] [PubMed] [Google Scholar]
- 17. Farver O., Vitu E., Wherland S., Fass D., Pecht I. (2009) Electron transfer reactivity of the Arabidopsis thaliana sulfhydryl oxidase AtErv1. J. Biol. Chem. 284, 2098–2105 [DOI] [PubMed] [Google Scholar]
- 18. Levitan A., Danon A., Lisowsky T. (2004) Unique features of plant mitochondrial sulfhydryl oxidase. J. Biol. Chem. 279, 20002–20008 [DOI] [PubMed] [Google Scholar]
- 19. Vitu E., Bentzur M., Lisowsky T., Kaiser C. A., Fass D. (2006) Gain of function in an ERV/ALR sulfhydryl oxidase by molecular engineering of the shuttle disulfide. J. Mol. Biol. 362, 89–101 [DOI] [PubMed] [Google Scholar]
- 20. Schwede T., Kopp J., Guex N., Peitsch M. C. (2003) SWISS-MODEL. An automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guex N., Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer. An environment for comparative protein modeling. Electrophoresis 18, 2714–2723 [DOI] [PubMed] [Google Scholar]
- 22. Sherman F. (1991) Getting started with yeast. Methods Enzymol. 194, 3–21 [DOI] [PubMed] [Google Scholar]
- 23. Terziyska N., Lutz T., Kozany C., Mokranjac D., Mesecke N., Neupert W., Herrmann J. M., Hell K. (2005) Mia40, a novel factor for protein import into the intermembrane space of mitochondria is able to bind metal ions. FEBS Lett. 579, 179–184 [DOI] [PubMed] [Google Scholar]
- 24. Terziyska N., Grumbt B., Kozany C., Hell K. (2009) Structural and functional roles of the conserved cysteine residues of the redox-regulated import receptor Mia40 in the intermembrane space of mitochondria. J. Biol. Chem. 284, 1353–1363 [DOI] [PubMed] [Google Scholar]
- 25. Simpson L., Frech G. C., Maslov D. A. (1996) RNA editing in trypanosomatid mitochondria. Methods Enzymol. 264, 99–121 [DOI] [PubMed] [Google Scholar]
- 26. Trager W., Jensen J. B. (1976) Human malaria parasites in continuous culture. Science 193, 673–675 [DOI] [PubMed] [Google Scholar]
- 27. Hiller N., Fritz-Wolf K., Deponte M., Wende W., Zimmermann H., Becker K. (2006) Plasmodium falciparum glutathione S-transferase. Structural and mechanistic studies on ligand binding and enzyme inhibition. Protein Sci. 15, 281–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harner M., Neupert W., Deponte M. (2011) Lateral release of proteins from the TOM complex into the outer membrane of mitochondria. EMBO J. 30, 3232–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mesecke N., Terziyska N., Kozany C., Baumann F., Neupert W., Hell K., Herrmann J. M. (2005) A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell 121, 1059–1069 [DOI] [PubMed] [Google Scholar]
- 30. Mumberg D., Müller R., Funk M. (1994) Regulatable promoters of Saccharomyces cerevisiae. Comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 22, 5767–5768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gietz R. D., Schiestl R. H. (2007) Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 35–37 [DOI] [PubMed] [Google Scholar]
- 32. Sikorski R. S., Boeke J. D. (1991) In vitro mutagenesis and plasmid shuffling. From cloned gene to mutant yeast. Methods Enzymol. 194, 302–318 [DOI] [PubMed] [Google Scholar]
- 33. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 34. Aliverti A., Curti B., Vanoni M. A. (1999) Identifying and quantitating FAD and FMN in simple and in iron-sulfur-containing flavoproteins. Methods Mol. Biol. 131, 9–23 [DOI] [PubMed] [Google Scholar]
- 35. Thorpe C., Coppock D. L. (2007) Generating disulfides in multicellular organisms. Emerging roles for a new flavoprotein family. J. Biol. Chem. 282, 13929–13933 [DOI] [PubMed] [Google Scholar]
- 36. Fass D. (2008) The Erv family of sulfhydryl oxidases. Biochim. Biophys. Acta 1783, 557–566 [DOI] [PubMed] [Google Scholar]
- 37. Ang S. K., Lu H. (2009) Deciphering structural and functional roles of individual disulfide bonds of the mitochondrial sulfhydryl oxidase Erv1p. J. Biol. Chem. 284, 28754–28761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deponte M., Becker K. (2005) Biochemical characterization of Toxoplasma gondii 1-Cys peroxiredoxin 2 with mechanistic similarities to typical 2-Cys Prx. Mol. Biochem. Parasitol. 140, 87–96 [DOI] [PubMed] [Google Scholar]
- 39. Tamarit J., Belli G., Cabiscol E., Herrero E., Ros J. (2003) Biochemical characterization of yeast mitochondrial Grx5 monothiol glutaredoxin. J. Biol. Chem. 278, 25745–25751 [DOI] [PubMed] [Google Scholar]
- 40. Lange H., Lisowsky T., Gerber J., Mühlenhoff U., Kispal G., Lill R. (2001) An essential function of the mitochondrial sulfhydryl oxidase Erv1p/ALR in the maturation of cytosolic Fe/S proteins. EMBO Rep. 2, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gross D. P., Burgard C. A., Reddehase S., Leitch J. M., Culotta V. C., Hell K. (2011) Mitochondrial Ccs1 contains a structural disulfide bond crucial for the import of this unconventional substrate by the disulfide relay system. Mol. Biol. Cell 22, 3758–3767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Klöppel C., Suzuki Y., Kojer K., Petrungaro C., Longen S., Fiedler S., Keller S., Riemer J. (2011) Mia40-dependent oxidation of cysteines in domain I of Ccs1 controls its distribution between mitochondria and the cytosol. Mol. Biol. Cell 22, 3749–3757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gabriel K., Milenkovic D., Chacinska A., Müller J., Guiard B., Pfanner N., Meisinger C. (2007) Novel mitochondrial intermembrane space proteins as substrates of the MIA import pathway. J. Mol. Biol. 365, 612–620 [DOI] [PubMed] [Google Scholar]
- 44. Kallergi E., Andreadaki M., Kritsiligkou P., Katrakili N., Pozidis C., Tokatlidis K., Banci L., Bertini I., Cefaro C., Ciofi-Baffoni S., Gajda K., Peruzzini R. (2012) Targeting and maturation of Erv1/ALR in the mitochondrial intermembrane space. ACS Chem. Biol. 7, 707–714 [DOI] [PubMed] [Google Scholar]
- 45. Terziyska N., Grumbt B., Bien M., Neupert W., Herrmann J. M., Hell K. (2007) The sulfhydryl oxidase Erv1 is a substrate of the Mia40-dependent protein translocation pathway. FEBS Lett. 581, 1098–1102 [DOI] [PubMed] [Google Scholar]
- 46. Deponte M., Urig S., Arscott L. D., Fritz-Wolf K., Réau R., Herold-Mende C., Koncarevic S., Meyer M., Davioud-Charvet E., Ballou D. P., Williams C. H., Jr., Becker K. (2005) Mechanistic studies on a novel, highly potent gold-phosphole inhibitor of human glutathione reductase. J. Biol. Chem. 280, 20628–20637 [DOI] [PubMed] [Google Scholar]
- 47. Bauer H., Massey V., Arscott L. D., Schirmer R. H., Ballou D. P., Williams C. H., Jr. (2003) The mechanism of high Mr thioredoxin reductase from Drosophila melanogaster. J. Biol. Chem. 278, 33020–33028 [DOI] [PubMed] [Google Scholar]
- 48. McMillan P. J., Arscott L. D., Ballou D. P., Becker K., Williams C. H., Jr., Müller S. (2006) Identification of acid-base catalytic residues of high Mr thioredoxin reductase from Plasmodium falciparum. J. Biol. Chem. 281, 32967–32977 [DOI] [PubMed] [Google Scholar]
- 49. Banci L., Bertini I., Calderone V., Cefaro C., Ciofi-Baffoni S., Gallo A., Tokatlidis K. (2012) An electron-transfer path through an extended disulfide relay system. The case of the redox protein ALR. J. Am. Chem. Soc. 134, 1442–1445 [DOI] [PubMed] [Google Scholar]
- 50. Chen X., Li Y., Wei K., Li L., Liu W., Zhu Y., Qiu Z., He F. (2003) The potentiation role of hepatopoietin on activator protein-1 is dependent on its sulfhydryl oxidase activity. J. Biol. Chem. 278, 49022–49030 [DOI] [PubMed] [Google Scholar]
- 51. Herrmann J. M., Hell K. (2005) Chopped, trapped, or tacked. Protein translocation into the IMS of mitochondria. Trends Biochem. Sci. 30, 205–211 [DOI] [PubMed] [Google Scholar]