

Background: Yeast RNA polymerase II domains involved in maintenance of transcription register are unknown.
Results: We isolated and biochemically characterized RPB1 mutations leading to register loss (transcription slippage).
Conclusion: All RPB1 mutations affect slippage localize close to the active center and near the secondary pore of RNA polymerase II.
Significance: Our results shed light on the mechanism of slippage by eukaryotic RNA polymerases.
Keywords: RNA Polymerase II, RNA Synthesis, RNA-Protein Interaction, Transcription Elongation Factors, Transcription Regulation, Transcription Fidelity, Transcription Slippage
Abstract
The fidelity of RNA synthesis depends on both accurate template-mediated nucleotide selection and proper maintenance of register between template and RNA. Loss of register, or transcriptional slippage, is particularly likely on homopolymeric runs in the template. Transcriptional slippage can alter the coding capacity of mRNAs and is used as a regulatory mechanism. Here we describe mutations in the largest subunit of Saccharomyces cerevisiae RNA polymerase II that substantially increase the level of transcriptional slippage. Alleles of RPB1 (RPO21) with elevated slippage rates were identified among 6-azauracil-sensitive mutants and were also isolated using a slippage-dependent reporter gene. Biochemical characterization of polymerase II isolated from these mutants confirms elevated levels of transcriptional slippage.
Introduction
Faithful transcription of DNA into RNA is essential to the flow of biological information. In contrast to the wealth of information on the mechanisms that influence the fidelity of DNA replication or translation, relatively little is known about what controls the fidelity of transcription in vivo. The lack of understanding of transcription fidelity is largely attributable to the difficulty in isolating RNA polymerase mutants defective in the process because the error rate for translation in general is much higher than that of transcription (1). Genetic screens for RNA polymerase variants with reduced fidelity that are based on misincorporation at a single codon of a reporter gene are, by their very nature, dependent on a very sensitive assay and/or a very high substitution rate (2–4). Cells with high levels of ribonucleotide misincorporation would be expected to be sick or unviable. Mutations of rpb1 that result in reduced fidelity have been identified from indirect screens based on sensitivity to drugs that alter the nucleotide pools such as 6-azauracil (5, 6) or mycophenolic acid (7). The rpb1-E1103G allele increases the level of misincorporation and the ability to extend mispaired bases and confers a dependence on the transcription proofreading factor TFIIS (5, 6). This synthetic lethality supports the view that TFIIS has a role in editing misincorporation errors made by Pol II (8) and confirms the expectation that elevated error rates of transcription can be catastrophic.
We describe here genetic screens using a different approach, one based on homopolymeric tracts of nucleotides, in which RNA polymerases exhibit higher inherent error rates in maintaining DNA and RNA register (transcription slippage) during elongation (9). Additional observations of transcriptional slippage on homopolymeric tracts have been reported in bacteriophages (10), prokaryotes (11–14), viruses (15), and eukaryotes (16, 17). A clear demonstration of in vivo transcriptional slippage during elongation was provided by Wagner et al. (18), who showed that Escherichia coli RNA polymerase made slippage RNA products from templates with a homopolymeric tract greater than 9 nucleotides. They showed that a run of 11 adenine or thymine residues near the beginning of an out of frame lacZ gene allowed a level of β-galactosidase expression ∼30% of the in-frame gene and subsequently demonstrated that the β-galactosidase activity was a result of transcriptional slippage.
Wagner et al. (18) also showed that the level of transcriptional slippage on a homopolymeric tract was greatly reduced in yeast (Saccharomyces cerevisiae) compared with E. coli. They could not detect slippage length heterogeneity in cDNA made from yeast lacZ reporter mRNA containing 11A tract and observed only 0.8% of the level of β-galactosidase for the 11A(+1) out of frame reporter compared with the 11A in-frame reporter. The (+1) indicates a 1-nucleotide insertion relative to the reading frame of lacZ.
To explore the mechanism of yeast Pol II slippage, we created sensitive reporter genes with homopolymeric runs that disrupted their reading frame and used them as tools to screen for Pol II2 variants with reduced accuracy. We report here the isolation and characterization of mutations in the largest Pol II subunit Rpb1 that increase transcriptional slippage and thus reduce the fidelity of transcription. Using an approach similar to that described here, E. coli RNA polymerase mutants, which altered slippage during transcription elongation, were isolated and characterized in the accompanying paper (19).
EXPERIMENTAL PROCEDURES
Yeast Strains and Techniques
The yeast strains we used were described in Ref. 5 and are listed in Table 1. Media, including synthetic complete (SC), were made according to Amberg et al. (20). The isolation of the rpb1-N488D and rpb1-S751F alleles was described in Ref. 5. Briefly, a mutagenized pool of a CEN vector carrying RPB1 (pJS670) was transformed into yeast cells with a conditional RPB1 under the control of a doxycycline-regulated promoter. We identified several plasmids carrying elongation defective rpb1 alleles from strains that were sensitive to 6-azauracil in the presence of doxycycline so that the chromosomal allele was repressed. Isolation of the rpb1-M487I mutation from mutagenized pools of a variant of pJS670 was performed as described below. Oligonucleotide-based site-directed mutagenesis using a Lambda Red recombineering technique was used as described (21). To create strains for purification of Pol II variants carrying mutation, rpb1 alleles had been integrated into the chromosome at its normal position by transformation to replace a doxycycline-regulated RPB1 gene (GRY3030). The doxycycline regulated RPB1 allele was marked by the kanMX gene conferring resistance to G418 (22). The resulting transformants were doxycycline-resistant and G418-sensitive. The integrated alleles were sequenced to confirm the presence of the mutations.
TABLE 1.
Plasmids and yeast strains
| Plasmid/strain | Genetic markers |
|---|---|
| pJS571 | URA3 CEN malE 10A In-frame-lacZ |
| pJS593 | URA3 CEN malE 10A In-frame-HIS3 |
| pJS725 | URA3 2-micron RPB1 |
| pJS670 | LEU2 CEN RPB1 |
| pJS757 | LEU2 CEN RPB1 |
| GRY2566 | MATa his3-Δ1 leu2::PADH1:MBP-11A- (+1)-HIS3 lys2-Δ0 met15-Δ0 ura3-Δ0 rpb1::kanMX [pJS725] |
| GRY3030 | MATa his3 leu2-Δ1 lys2-Δ0 met15-Δ0 trp1-hisG URA3::CMV-tTA RPB3::6XHis ptet07-RPB1::kanMX |
| GRY3038 | MATa his3 leu2-Δ1 lys2-Δ0 met15-Δ0 trp1-hisG pep4::HIS3 prb1-Δ1.6R URA3::CMV-tTA RPB3::6XHis rpb1-N488D |
| GRY3154 | MATa his3 leu2-Δ1 lys2- Δ0 met15-Δ0 trp1-hisG pep4::HIS3 prb1-Δ1.6R URA3::CMV-tTA RPB3::6XHis rpb1-S751F |
| GRY3177 | MATa his3 leu2 lys2- Δ0 met15-Δ0 trp1 pep4::HIS3 prb1-Δ1.6R rpb1::kanMX RPB3::6XHis [pJS725] |
Plasmids and Slippage Reporter Fusions
Several plasmids were used in this study. pJS725 is a URA3-based 2-μm vector carrying the RPB1 ORF, as well as 594 bases upstream and 233 bases downstream of the ORF. pJS670 is a LEU2-based CEN vector carrying the RPB1 ORF, as well as 594 bases upstream and 501 bases downstream of the ORF. pJS757 is a related plasmid in which a PvuII site in the backbone was mutated. The plasmids carrying rpb1 mutants are derivatives of pJS670 and pJS757. pJS571 is a URA3 CEN vector carrying a promoter region 926 bases upstream of the ADH1 ORF, designated PADH1. The PADH1 promoter drives the expression of a fusion protein of the maltose-binding protein and β-galactosidase (see Fig. 1A). The fusion is made of codons 28–392 of the malE gene of E. coli fused to codon 10 (Val) through the end of the lacZ gene of E. coli. Between the malE and lacZ sequences, there is a linker sequence that encodes a tract of adenosines in mRNA (see Fig. 1B). The linkers were inserted as double-stranded oligonucleotides into the NheI and BamHI sites and confirmed by sequencing. pJS593 is a URA3-based CEN vector with a fusion between the malE and HIS3 genes. It has 193 bases upstream of the HIS3 ORF that functions as a promoter, PHIS3, driving a fusion of malE and HIS3 ORFs. The ORFs are separated by the in-frame oligonucleotide linker sequence shown in Fig. 1B. pJS594 is an out of frame derivative of pJS593 and contains an 11A(+1) oligonucleotide linker sequence.
FIGURE 1.
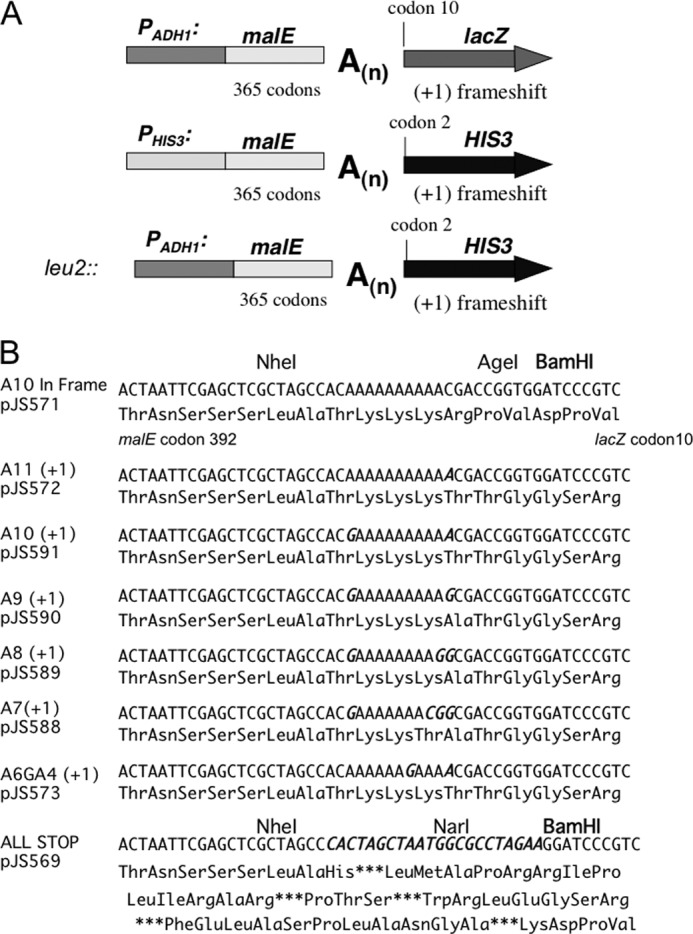
Constructs of slippage strains. A, transcription slippage reporters. The open reading frame (codons 28–392) for the maltose-binding protein (malE) was fused out of frame via a homopolymeric tract of adenosines in the coding strand to the lacZ gene (starting at codon 10) to provide a quantifiable reporter of transcription slippage. Similar fusions with the HIS3 gene (starting at codon 2) provided plasmid-based and chromosomal insertion (at the leu2 locus) reporters for transcription slippage that can be used as a screen for mutations that cause increased slippage. B, transcription slippage substrates. Tracts of adenosines were inserted between the malE and lacZ open reading frames as diagrammed in Fig. 1A. The first codon shown (Thr) is codon 392 of malE. The last codon shown for the in-frame linker (Val) is codon 10 of lacZ. The +1 frame substrates all have the same number of total bases between the two open reading frames. Base substitutions made to create shorter A tracts are italicized.
A chromosomal version of the malE–HIS3 fusion slippage reporter was made by inserting it as a substitution for the LEU2 locus. A region from 153 bases upstream of the LEU2 ORF to 23 bases downstream of the LEU2 ORF was deleted and replaced with a KanMX cassette (22), followed by the 926-base PADH1 promoter, and then codons 28–392 of the malE gene of E. coli fused to codon 2 (Thr) of HIS3 through 27 bases downstream of the HIS3 ORF. In the yeast strain GRY2566, the malE and HIS3 ORFs are separated by the linker oligonucleotide 11A(+1) shown in Fig. 1B and hence are out of frame.
β-Galactosidase Assay
Measurement of β-galactosidase activity was done using the yeast Galacto-Star assay system from Applied Biosystems (Grand Island, NY) according to the manufacturer's manual. This assay uses a chemiluminescent substrate. The cells were grown in deep 96-well plates to saturation in 0.6 ml of synthetic complete medium lacking uracil to select for the lacZ reporter plasmid. The cells were then diluted 25-fold into YEPD medium at 30 °C for 7 h. The cells were pelleted by centrifugation, and the liquid was poured off. The pellets were frozen by floating the plates on liquid nitrogen. The β-galactosidase units were normalized to protein concentration as determined by a Bradford assay. The assays were done on derivatives of GRY2568 in which the URA3-based plasmid pJS725 was replaced by the wild type RPB1 gene on pJS757 (or mutant derivatives), and then the lacZ reporter plasmids related to pJS571 were introduced.
Pol II Purification and RNA and DNA Oligonucleotides
Pol II carrying hexahistidine-tagged Rpb3 and E. coli RNA polymerase (RNAP) were purified as described (23, 24). The DNA and RNA oligonucleotides for ternary elongation complex (TEC) assembly were from IDT (Coralville, IA): RNA9, 5′-AUC GAG AGG-3′; NDS79 (nontemplate DNA strand), 5′-CCT ATA GGA TAC TTA CAG CCA TCG AGA GGG ACA CGG CGA ATA GCC ATC CCA ATC CAC ACG TCC AAC GGG GCA AAC CGT A-3′; and TDS76 (template DNA strand), 5′-GGT TTG CCC CGT TGG ACG TGT GGA TTG GGA TGG CTA TTC GCC GTG TCC CTC TCG ATG GCT GTA AGT ATC CTA TAG G-3′. The oligonucleotides for the slippage assays were: NDS50A11 (nontemplate DNA strand), 5′-GGT ATA GGA TAC TTA CAG CCA TCG AGA GGG AAA AAA AAA AAC GAG GCG AA-3′; and TDS50A11 (template DNA strand), 5′-TTC GCC TCG TTT TTT TTT TTC CCT CTC GAT GGC TGT AAG TAT CCT ATA CC-3′.
Assembly of TECs and Transcription in Vitro
Assembly of the TEC9 (number indicates the 9-nt RNA length) from RNA and DNA oligonucleotides, immobilization on Ni2+-nitrilotriacetic acid-agarose beads, TEC purification, and elution with imidazole were performed as described (23). For the bulk elongation assay, the 5′-labeled RNA9 and nonlabeled TDS76 and NDS79 DNA oligonucleotides were used, and transcription assay was performed as described (25) after TEC9 purification and its elution from the beads with imidazole. To assay transcription elongation at 10 μm NTP, TEC20 was obtained by the addition of 10 μm each ATP, CTP, and GTP for 1 min at room temperature followed by the addition of 10 μm UTP. To analyze transcription elongation at 200 μm NTP, TEC20 was obtained by the addition of 200 μm each ATP, CTP, and GTP to the TEC9 for 3 min followed by the addition of 200 μm UTP. After the indicated time intervals, the reaction was stopped with gel loading buffer (20 mm Tris-HCl, pH 7.9, 6 m urea, 25 mm EDTA). For the processive slippage assay, TEC9 was obtained with the nonlabeled RNA9 primer and TDS50A11 and NDS50A11 oligonucleotides.
The immobilized TECs by slippage mutant RNA polymerases from yeast were purified by washing with transcription buffer (20 mm Tris-HCl, pH 7.9, 5 mm MgCl2, 3 mm β-mercaptoethanol) containing 40 mm KCl (TB40), and the RNA was labeled by incubation of TEC9 with 0.3 μm [α-32P]GTP (3000 Ci/mmol; PerkinElmer Life Sciences), resulting in TEC10. ATP (10 μm) was added to TEC10 for indicated time, and the reaction was stopped by the addition of the gel loading buffer. The transient slippage assay was similar to the processive slippage assay except that three times the amount of Pol II, Ni2+-nitrilotriacetic acid-agarose, and RNA and DNA oligonucleotides were used for the TEC assembly, and the RNA was labeled with [α-32P]CTP downstream from the A11 tract. The RNA in TEC9 was elongated with the nonlabeled GTP (5 μm, 4 min) to form TEC10, and the beads were washed five times with 1 ml of TB40 followed by the addition of 10 or 100 μm ATP and CTP (100 μm) and 1.3 μm [α-32P]CTP (3000 Ci/mmol; PerkinElmer Life Sciences) in TB containing 150 mm KCl for 8 min to obtain TEC22. The reaction was stopped by washing the NTPs off the beads with TB40 and by the addition of the gel loading buffer. In all the assays described above, the RNA products were resolved by denaturing polyacrylamide gel electrophoresis and analyzed by phosphorimaging (GE Healthcare). For comparison of slippage pattern at 11A tract by E. coli RNAP and Pol II (WT and S751F), the reaction was performed at 100 μm ATP to better approximate the in vivo conditions.
RESULTS AND DISCUSSION
Rationale
We created sensitized reporters for detection of Pol II slippage during transcription elongation based on the inclusion of homopolymeric runs. The reporters were based on the lacZ and HIS3 genes and encoded runs of adenines upstream of the reporter open reading frame. It has been reported that the fidelity of transcription during the initiation phase is lower than during elongation (26). Stuttering or reiterative synthesis at promoters can reflect an initial barrier to transition to the elongation phase and is used as a regulatory mechanism (11, 27). The slippage tracts in the reporters used by Wagner et al. (18) were located near the transcription start. To focus on transcriptional slippage in the elongation phase, we placed the slippery site well downstream of the start of the transcript between the open reading frame for the maltose-binding protein (malE) and lacZ (Fig. 1). The maltose-binding protein was chosen because it includes a large open reading frame (372 codons) and is a relatively well tolerated N-terminal addition to many proteins (28). We adopted the convention used by Wagner et al. (18) that reporters that are out of frame because of an additional base are designated “(+1),” and the reporter's names refer to the homopolymeric run in the nontemplate strand. The reporters with slippery A tracts in the (+1) frame are shown in Fig. 1.
Homopolymeric Tract Length-dependent Transcriptional Slippage
We measured the level of lacZ activity from plasmid-based reporters with different adenosine tract lengths (Fig. 1B). Note that the total number of bases in all of the (+1) reporters is the same, and only the length of the homopolymeric tract changes. Similar to the results reported by Wagner et al. (18), we observed less than 1% of the lacZ activity from a 11A(+1) reporter as compared with the in-frame 10A construct. Note that the opportunities for ribosome slippage are the same in the 11A, 10A, and 9A reporters, but the level of lacZ expression is reduced as the A tract gets shorter (Fig. 2). These data are consistent with transcriptional slippage, rather than tRNA slippage, as the source of the lacZ activity. Shorter A(+1) tracts showed decreasing lacZ activity, consistent with the observation that the RNA-DNA hybrid in the Pol II elongation complex is 8–9 base pairs. To maintain the same length RNA-DNA hybrid, slippage on substrates less than 9 bases of a homopolymeric run would require a mismatch in the RNA-DNA hybrid or the looping out of bases in the template or RNA.
FIGURE 2.
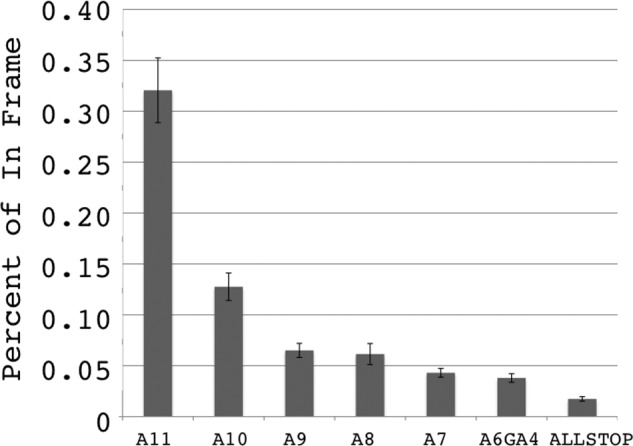
Length dependence for transcription slippage. Shown are the percentages of lacZ activity from out of frame (+1) reporter plasmids relative to expression from the in-frame plasmid pJS571 in strains related to GRY2954. The values are the averages of eight samples for each plasmid with standard deviation.
Testing rpb1 Mutants in Transcriptional Slippage Assays
We previously described the isolation of novel rpb1 alleles from a screen for sensitivity to 6-azauracil (5). 6-Azauracil unbalances the nucleotide pool and has been used to isolate mutations that confer transcription defects. Genetic and biochemical characterization of one of those alleles (rpb1-E1103G) have demonstrated that it causes an increase in the misincorporation of bases during transcription (5, 6). We tested the same collection of rpb1 alleles for elevated transcriptional slippage using the plasmid based lacZ reporter constructs described above. The rpb1-N488D allele gave the most dramatic results with a 7-fold higher level of lacZ activity from the malE-11A(+1)-lacZ reporter (Fig. 3). The rpb1-S751F allele also gave an elevated level of lacZ activity suggestive of elevated transcriptional slippage. We also note that loss of the transcription fidelity factor, TFIIS encoded by DST1, or loss of the ability of TFIIS to bind to Pol II caused by the rpb1-E1230K mutation (29) does not increase transcriptional slippage as monitored by lacZ expression.
FIGURE 3.
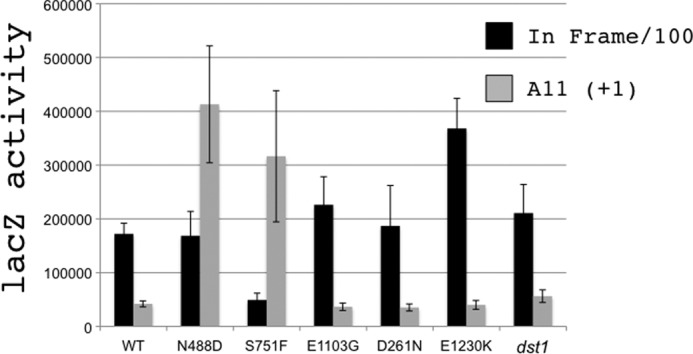
Transcription slippage in rpb1 mutants. Activity of lacZ from in-frame A10 plasmid (black bars diluted by 100-fold) and out of frame A11(+1) reporter plasmid pJS572 (gray bars) is shown, as well as the assay on 6-azauracil-sensitive mutants of rpb1 (5, 6). A strain carrying the dst1Δ::natMX4 deletion of the gene encoding TFIIS is also shown. The values are the averages of eight samples with standard deviation.
We also created slippage reporter plasmids based on the HIS3 gene (Fig. 1A). These reporter genes had promoter of the HIS3 gene and the same malE upstream region and homopolymeric slippage linkers as the lacZ reporter but were fused to the HIS3 open reading frame. Cells carrying the in-frame construct (pJS593) are His+, whereas those with the (+1) out of frame reporters are His− auxotrophs. We introduced the PHIS3:malE-11A(+1)-HIS3 reporter plasmid (pJS594) into strains carrying the various rpb1 mutations and then tested for their ability to grow in the absence of histidine. Cells with the rpb1-N488D allele carrying the malE-11A(+1)-HIS3 reporter grew significantly better on synthetic complete plates lacking histidine (SC-His) than did the RPB1 cells (Fig. 4). This phenotype provides a direct screening method for the isolation of mutants with lowered fidelity of transcription. It is noteworthy that this screen has a bias against mutations that significantly reduce cell growth rates. For example, rpb1-S751F, which makes cells less healthy, does not pass this screen. We also note that the loss of TFIIS in the dst1 mutant or the loss of the ability of TFIIS to bind to Rpb1 caused by the rpb1-E1230K mutation (29) does not lead to growth on SC-His in this assay.
FIGURE 4.
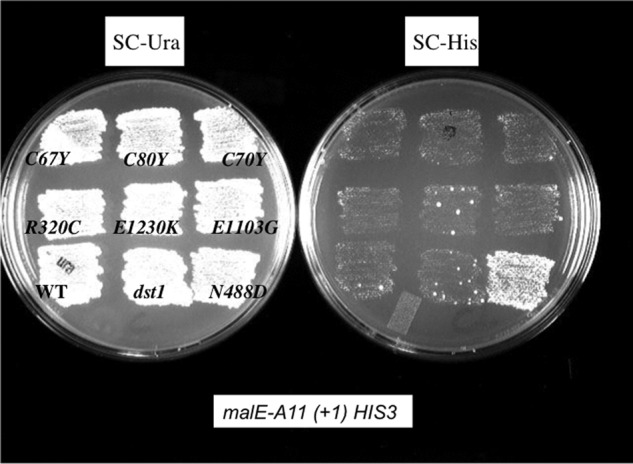
Genetic screen for slippage-prone rpb1 mutations. The Petri plates show a comparison of various rpb1 mutants for their ability to suppress an out of frame A11(+1) version of the malE HIS3 fusion slippage reporter (pJS594). The SC-Ura plate (synthetic complete medium lacking uracil) selects for the plasmid pJS594. The SC-His plate (SC lacking histidine) selects for expression of HIS3. Wild type RPB1 and a strain carrying the dst1Δ::natMX4 deletion of the gene encoding TFIIS are also shown.
Isolation of rpb1 Mutations That Cause Increased Transcriptional Slippage
Strain GRY2566 carries a chromosomal version of the malE-11A(+1) HIS3 reporter inserted at the leu2 gene and the rpb1-Δ0 deletion allele of rpb1, as well as a wild type copy of RPB1 on a URA3-based plasmid (pJS725). We transformed that strain with a mutagenized pool of a variant (pJS670) of the CEN LEU2 vector carrying RPB1, pJS757, and removed the URA3-based plasmid (pJS725) by counter selection on FOA plates (30). Control experiments with the rpb1-N488D allele demonstrated that the direct screen of colonies was too subtle, so we made patches of the transformants carrying the mutagenized plasmid pool before testing their ability to make histidine. The two-step screening procedure allowed manifestation of subtle differences in growth phenotype. From 1400 such patches, we identified one transformant with a weak His+ phenotype. The sequence of the plasmid recovered from that strain identified it as rpb1-M487I. The recovery of a novel allele in the codon next to the founding member of this class of error-prone rpb1 mutants validated this approach. When the rpb1-M487I allele was tested in the malE-11A(+1)-lacZ slippage reporter assay, it showed 7.8-fold more slippage than RPB1 (Table 2).
TABLE 2.
Quantitation of slippage in rpb1 variants
The values are expressed as ratios of β-galactosidase activity of an indicated variant to wild type from the malE-11A(+1)-lacZ slippage reporter plasmid (pJS572). The data are median values (seven or more independent samples).
| Met-487 | Asn-488 | Gly-750 | Ser-751 | Lys-752 | |
|---|---|---|---|---|---|
| Gly | 2.0 | 2.2 | WT (1.0) | 2.0 | |
| Ala | 2.4 | 1.1 | 0.8 | 1.0 | 1.5 |
| Val | 4.4 | 3.4 | 0.7 | 6.5 | |
| Leu | 7.9 | 0.9 | Dead | Dead | |
| Ile | 7.8 | Dead | 0.8 | Dead | sick |
| Ser | 0.6 | 0.9 | 0.6 | WT (1.0) | Dead |
| Thr | 3.0 | 2.3 | 1.1 | ||
| Cys | 2.4 | 2.4 | |||
| Met | WT (1.0) | 7.0 | Dead | ||
| Asp | Dead | 7.0 | Dead | ||
| Glu | 2.0 | 0.6 | 0.9 | ||
| Asn | Dead | WT (1.0) | 0.7 | ||
| Gln | 3.4 | 0.6 | |||
| Lys | Dead | WT (1.0) | |||
| Arg | 0.7 | 1.2 | 2.9 | ||
| His | Dead | 2.3 | 0.7 | sick | |
| Phe | 5.1 | Dead | 0.9 | 4.6 | |
| Tyr | Dead | Dead | 1.6 | Dead | |
| Trp | Dead | Dead | |||
| Pro | Dead | 0.6 | 2.5 | Dead |
Site-directed Mutagenesis of the RPB1-M487, RPB1-N488, and RPB1-S751 Codons
To make libraries of codon substitutions at RPB1-M487 and RPB1-N488, we used recombineering (31) on a LEU2 CEN RPB1 plasmid (pJS757) to create an rbp1-N488Q allele. This substitution created a unique PvuII site in the plasmid (pJS761) spanning codons 488 and 489. A second round of recombineering based site-directed mutagenesis was used to insert substitutions at codons 487 or 488. A library of oligonucleotides with all 64 substitutions at codon 487 was co-electroporated along with pJS761 into the bacteria and selected for ampicillin resistance. The transformants were pooled, cut with PvuII, and retransformed into a DH5α bacterial strain. Plasmids were sequenced to identify the substitutions and were transformed into yeast carrying rpb1-Δ0 and the URA3-based RPB1 plasmid (pJS725). This process gave 15 different amino acid substitutions at codon 487 (Table 2).
We then determined whether the mutant allele of rpb1 was viable by asking whether the wild type RPB1 URA3-based plasmid was dispensable as evidenced by growth on FOA plates (30). Those that could not grow on FOA plates were assumed to have mutations that could not support growth of yeast under these conditions and are indicated as dead in Table 2. Those mutants that survived without the wild type copy were transformed with the malE-11A(+1)-lacZ slippage reporter plasmid (pJS572) and monitored for lacZ expression levels (Table 2). Note that the majority of the viable substitutions at codon Met-487 result in elevated slippage. A similar substitutions library was made for codon Asn-488, and again, most viable substitutions at Asn-488 exhibit elevated slippage. Substitution libraries for codons Gly-750, Ser-751, and Lys-752 revealed that codon Gly-750 did not have a major role in controlling slippage, whereas some substitutions in codons Ser-751 did promote slippage. As expected for the highly conserved codon Lys-752, most substitutions are very sick or not viable.
Elongation Properties of the Mutant Polymerases in Vitro
We performed biochemical characterization of the purified 12-subunit Pol II core enzymes for five mutations exhibiting strong slippage phenotype in vivo (rpb1-M487V/I, M488D/Q, and S751F). The strains with mutant rpb1 alleles also carried a functional chromosomal hexahistidine-tagged Rpb3 subunit, which allowed immobilization of Pol II on Ni2+-nitrilotriacetic acid-agarose beads for transcription assays in the solid phase or in solution after elution with imidazole as was described previously (25, 32). The purified enzymes were assembled into TECs with a synthetic RNA-DNA scaffold coding for the 56-nt runoff RNA. A bulk elongation rate was tested in a single-round runoff formation assay. All of the mutants showed reduced rates of the runoff formation. Pol II from rpb1-M487S, which did not show increased slippage in vivo, in a more stringent assay proved to be only slightly slower than the WT (Fig. 5A). A single nucleotide incorporation assay (data not shown) confirmed that all mutants exhibited reduced rates for a single phosphodiester bond formation with the strongest inhibition observed with the S751F mutant. The slowest S751F mutant carried a bulky substitution explaining its strong effect on elongation. Note that the elongation defects in these mutants was partially due to the increased pausing at the U21, A26, and U27 positions of the template with the smaller effect on transcription between the pauses.
FIGURE 5.

Elongation and slippage properties of the rpb1 mutants in vitro. A, a time course of runoff RNA formation in a single round in vitro elongation assay. TEC20 by the WT Pol II and six rpb1 mutants was chased with 200 μm of 4 NTPs for 10, 20, 40, 90, 180, 360, 720, and 1440 s. The effect of rpb1-M487S mutation on elongation rate was determined at a lower 10 μm NTPs for 10, 20, 40, 90, and 180 s to slow down the elongation rate for more reliable comparison with WT Pol II (bottom left panel). The graph on the right shows quantification of the rates at 200 μm NTPs. Note that the M487S mutant showed the smallest elongation defect compared with the other Rpb1 mutants. B, the processive slippage assay. Slippage at the end of 11A was performed by incubation of the RNA-labeled TEC10 with 10 μm ATP for 10, 20, 30, 40, and 60 s as described in the text. Arrowheads mark time-dependent progression of the center of the slippage products distribution. Note that a fraction of TEC was arrested at +20/+22 positions because of Pol II backtracking near the end of 11A tract. This fraction was resistant to slippage by multiple AMP additions and to elongation with four NTPs.
Processive Transcription Slippage at the End of an 11A Tract in Vitro
We evaluated the in vitro slippage for the rpb1 mutants by using the RNA-DNA scaffold containing a tract of 11 adenines in the transcript to match the sequence of the 11A cassette of the malE-11A(+1)-lacZ reporter. For each mutant, we assembled TEC carrying the 9-nt RNA primer (TEC9, the number stands for the length of the RNA in TEC) hybridized to the template strand immediately upstream from the homopolymeric tract. The nascent RNA was elongated by 1 nucleotide by incorporation of [α-32P]GMP to form the 3′-labeled TEC10 stalled at the first adenine of the tract because of the lack of ATP. Upon ATP addition, transcription continued to the end of the 11A tract followed by a stop because of the lack of the next cognate CTP (Fig. 5B). One would expect TEC21 to be formed in this reaction because of the lack of CTP (the next template-encoded NTP). Instead, slippage during passage through the tract and continuous slippage at the end of the tract generated a spectrum of slippage products all longer than 21 nucleotides, which were progressively elongated to the longer species upon prolonged incubation with ATP. This assay allowed estimation of slippage for each mutant Pol II as a rate of progression of the center of the slippage products distribution at different time of incubation with ATP (Fig. 5B, indicated by the triangles). This assay did not measure slippage under physiological conditions where all four NTPs are available. Instead, it was designed to monitor slippage events leading to insertions, which provided important information on the catalytic rate of AMP addition by slippage.
Relative to the wild type Pol II, the M487I, M487S, M487V, and N488D mutants showed a substantial 1.5–2.5-fold increase, and the N488Q mutant showed a mild increase of the slippage rate. The S751F mutant showed significantly decreased slippage. Note that our assay for the processive slippage involved low 10 μm ATP concentration. It is noteworthy, that elongation rate is not directly correlated with slippage as measured in this processive slippage assay. For example, N488D and S751F are both slower in elongation (Fig. 5A), N488D exhibits elevated slippage and S751F reduced slippage in this assay. The decreased slippage of the S751F mutant in vitro was in an apparent disagreement with the increased expression of malE-11A(+1)-lacZ reporter in the mutant cells. As shown below, this is because the in vivo reporter was designed to look for one base deletion (or two base additions), whereas the processive slippage assay detects multiple additions. M487S mutant showed decreased AMP base deletion by slippage in vivo (Table 2) and increased multiple AMP insertions in vitro (Fig. 5B). Note that M487S mutant was also most similar to the WT in the bulk elongation assay compared with the other slippage mutants (Fig. 5A).
Slippage on malE-11A(+1)-lacZ reporter was significantly reduced on the A tract shorter than 9 nucleotides and on the interrupted A tract (Fig. 2). Accordingly, we did not observe any processive slippage at the ends of A5U1A5 (11 nt), A8 (8 nt), and (UA)6 (12 nt) tracts (data not shown). These results strongly argued that the processive AMP incorporation at the end of 11A tract in Fig. 5B was due to Pol II slippage as opposed to misincorporation of AMP for CMP. Note that the sensitivity of our in vitro assay was not sufficient to detect slippage below 3–5% of the total RNA in TEC21 stalled at the end of the tract.
Capturing Slippage during Unimpeded Transcription through an 11A Tract
The processive slippage assay of Fig. 5B did not mimic in vivo conditions in two key ways. First, transcription through the 11A tract in vivo occurs without stopping of Pol II at the end of the tract as imposed by the CTP deprivation in vitro. Second, the 1A deletion for which malE-11A(+1)-lacZ reporter was designed escapes detection in the processive slippage assay. To detect both insertions and deletions and to prevent the idling of Pol II at the end of the A tract, we modified the assay of Fig. 5B to enable detection of deletions and insertions during unimpeded elongation. This assay involved simultaneous addition of the unlabeled ATP and [α-32P]CTP to TEC10 to generate a 3′-labeled transcription complex with 22-nt RNA (TEC22) stopped one base pair downstream from the 11A tract following the CMP incorporation (Fig. 6A). In this assay, RNA in TEC9 was elongated with unlabeled GTP to generate TEC10 as shown in Fig. 5B followed by the removal of the nonincorporated GTP. After adding ATP and labeled CTP, the TEC10 transcribes to the end of the 11A tract, incorporates the labeled CMP, and comes to a stall because of the lack of the next cognate GTP (Fig. 6A, TEC22). Only the TECs that transcribed past the homopolymeric tract incorporate radioactive CMP into the nascent RNA, which instantly stops slippage and also ensures that the shorter RNA species result from slippage, but not from transcription pausing and arrest within the 11A tract. The 3′-labeled RNA slippage products were resolved from the regular 22-nt labeled RNA by urea-PAGE, and their amount (insertions and deletions separately) was normalized to the amount of the correct 22-nt products (Fig. 6A, summarized in the table panel).
FIGURE 6.

Transient slippage during unimpeded transcription through the 11A tract. A, scheme on the top depicts the RNA and DNA sequences of the 3′ labeled TEC22 halted 1 nt downstream from the 11A tract by the labeled CMP incorporation and GTP deprivation. The autoradiogram shows the pattern of deletions and insertions in the 11A tract generated by WT Pol II and its mutant variants during transient slippage. The phosphorimaging scans of the corresponding lanes of the autoradiogram are shown to the right. Black dots indicate radioactivity peaks corresponding to the intact 11A tract in the nascent RNAs. The table at the bottom summarizes the effect of rpb1 mutations on slippage efficiency, directionality, and the bulk elongation rate. The numbers in the second and third columns represent the fraction of deletions and insertions including 9A, 10A, 12A, and 13A products, normalized to the amount of the correct 22-nt products. The fourth column shows the ratio between insertions and deletions for each mutant. The fifth column presents the effect of the mutations on the elongation rate of RNAP on heteropolymeric DNA reproduced from the results of Fig. 5A. The downward arrows indicate the decreased elongation rate detected for all six mutants. The M487S mutant exhibited the smallest decrease in the elongation rate compared with the wild type (indicated by an asterisk). The last column shows the normalized slippage by the mutants at the 11A(+1) sequence of the lacZ reporter in vivo (reproduced from Table 2). B, in vitro slippage pattern at the 11A tract by WT E. coli RNAP, WT yeast Pol II, and rpb1-S751F mutant of Pol II. All of the symbols and conditions are the same as in A, except the reaction was performed at 100 μm ATP to match the in vivo conditions. The Rpb1-S751F mutant generated insertions and deletion with the similar efficiency at 100 μm ATP as opposed to the low 10 μm ATP (A), where it was making more deletions than insertions. The decreased slippage at 100 μm ATP was due to the reduced Pol II pausing in the 11A tract at high NTP, which shortened the “time window” for slippage.
The result of Fig. 6A demonstrated that all rpb1 mutations that we analyzed profoundly increased deletions of 1–2 adenines from the 11A tract. The S751F and N488D mutants had the largest (∼3-fold) increase of the single AMP deletion compared with the WT. The increased fraction of RNAs containing 10A tract is consistent with the enhanced slippage phenotype of the mutant in the cells carrying the malE-11A(+1)-lacZ reporter also designed to detect the single AMP deletions. Notably, all other rpb1 slippage mutations identified with the in vivo reporter also showed a bias for making more deletions in vitro (Fig. 6A, compare lane 1 with lanes 3, 6, and 7; also Ins/Del ratio in the table panel). In contrast, the M487S mutant, which exhibited decreased slippage in vivo (Table 2) and slightly decreased Pol II elongation rate in the bulk transcription assay (Fig. 5A), showed a relatively minor and proportional increase in both the deletions and insertions of single AMP compared with the other mutants.
Bacterial and Yeast RNA Polymerases Have Different Efficiency and Directionality of Slippage
The previous reports on slippage in E. coli and yeast that employed similar out of frame lacZ reporters showed significantly less slippage in yeast compared with bacteria (18). To test whether the same difference is observed in vitro, we compared a pattern and efficiency of the transient slippage for E. coli RNAP and yeast Pol II on the 11A template. The E. coli RNAP slippage at the 11A tract was significantly higher than slippage by the yeast WT Pol II. Even the most slippage-prone yeast mutant, rpb1-S751F, exhibits much less slippage than the wild type bacterial RNA polymerase (Fig. 6B, lanes 1–3 and the gel scans on the left). Importantly, E. coli RNAP generated more insertions than deletions, whereas Pol II generated substantially smaller bias toward insertions and showed a much lower yield of slippage. With a shorter 9A tract, WT Pol II made exclusively 1A deletion and no insertions (data not shown), whereas E. coli RNAP kept making 1A insertions and deletions (19).
Summary Remarks
Accurate transcription requires both the ability to avoid the incorporation of the noncognate NMP and the ability to maintain proper register between the transcribed strand of the DNA and the nascent RNA. We previously described a mutation in the largest subunit of Pol II that elevates the level of misincorporation of nucleotides (6). Here we describe the isolation and characterization of mutations that increase the level of transcriptional slippage. The effects on elongation were consistent with the close proximity of the Rpb1-M487, -N488, and -S751 residues to the active center of Pol II (Fig. 7A).
FIGURE 7.

Location of Rpb1-M487, -N488, and -S751 residues in the structure of TEC by Pol II. A, post-translocated TEC structure of S. cerevisiae Pol II and location of the amino acid residues in Rpb1 subunit carrying slippage mutations characterized in vitro (in red) (Protein Data Bank code 2E2H). Template DNA/front end DNA duplex (gray) and RNA (black) are shown as a space fill model, and NTP (sticks model) in the active center is shown at the 3′ end of the RNA. The corresponding residues in the β′ subunit of E. coli RNAP are shown (in brackets). The other amino acid residues carrying mutations described in this work are highlighted in yellow. The inset shows the zoomed structure of the active center of yeast Pol II made by the protein loops where all mutations affecting slippage are localized. The catalytic Mg2+ ion (Mg-B) and rpb1-K752 adjacent to rpb1-S751 residue are shown in green and yellow, respectively. B, protein alignment of the catalytic and pyrophosphate-binding loops in E. coli (Ec) and yeast (Sc) RNA polymerases. The mutated residues are highlighted in red. Rpb1-K752 constitutes a part of the NTP-binding site where it protrudes toward the γ phosphate of the incoming substrate.
On most transcription templates, the register of the RNA and DNA is preserved at least in part by the specificity of base pairing. Moving the RNA relative to the transcribed strand would result in mispaired bases in the 8–9-base pair RNA-DNA hybrid formed in elongating RNA polymerase. However, on homopolymeric tracts longer than the RNA-DNA hybrid, movement of the RNA relative to the template does not create mispairs. Therefore, the register of the RNA and template must be maintained by interactions between the RNA-DNA hybrid and the RNA polymerase. At the same time, the normal process of elongation requires the RNA polymerase to translocate downstream on the DNA, and proper register must be maintained during that process. Similarly, RNA polymerase must maintain register when it backtracks upstream on the template at intrinsic sequence-dependent pause sites or at pauses caused by the RNA polymerase encountering DNA-binding proteins. Backtracking is also a step in transcript editing stimulated by TFIIS that allows the removal of misincorporated bases. Fig. 8 shows a cartoon of normal elongation maintaining register through the translocation process. It consists of the repeating cycles of forward translocation of the enzyme along DNA to vacate the active site (indicated by an asterisk) for the next incoming substrate (ATP) followed by the substrate binding, bond formation, and another round of translocation. This cycle includes several steps at which failures to maintain RNA-DNA register could result in frameshifts. One model for slippage that results in a transcript that is one base longer than the template is diagrammed on the left side of Fig. 8, in which the transcript at the pretranslocation step moves backward relative to the template and RNA polymerase. This process brings the new 3′ end to the active site and allows the incorporation of a second base at the same template position, resulting in a transcript with an extra base. This slippage error could be viewed as an aberrant translocation step in which the transcript moved, but the template did not. Iterative repetitions of this process would create the extended ladder of incorporation seen in Fig. 5B. The right side of Fig. 8 shows a model that generates a transcript that is one base shorter than the template. In this model, the slippage occurs when the TEC is in the post-translocation step and involves precocious movement of the RNA relative to the template so that it comes to occupy the pretranslocation position. Subsequent translocation places the 3′OH at the active site and in position to generate a transcript missing a base.
FIGURE 8.
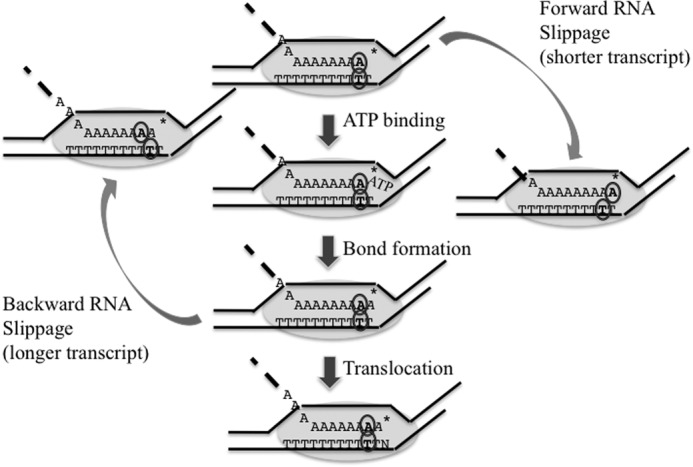
A model for transcription slippage. Down the center is the normal elongation cycle of NTP selection, bond formation, and translocation with the template and RNA kept in register. A base in the template and RNA are circled to show maintenance of register. The active site is designated by an asterisk. The next template base after translocation is shown as N. One mechanism to generate a shorter transcript is for the RNA to move forward relative to the template so that it adopts the pretranslocation stage (right side of figure). One mechanism to generate a longer transcript is for the RNA to move backward relative to the template after the bond formation step so that it adopts the post-translocation state (left side of figure).
The close proximity of the mutations affecting slippage to the RNA-DNA hybrid in yeast and bacterial RNA polymerase (Ref. 33; also see the accompanying article in Ref. 19) strongly indicates that physical integrity of the hybrid is crucial for maintaining transcription register. However, the details of how the RNA slips relative to the DNA are yet to be determined. Anikin et al. (9) provide excellent descriptions of possible slippage mechanisms. The first mechanism involves transient separation of the RNA and DNA strands of the 9-bp RNA-DNA hybrid in the catalytic cleft of the enzyme allowing their shift of one or more bases followed by restoration of the hybrid at the new position within a homopolymeric tract. This process should not generate any mismatches in the hybrid leading to the insertions or deletions depending on the shift directionality. It is unlikely, within the confines of the channel surrounding the RNA-DNA hybrid during elongation, that the strands completely dissociate and then reanneal. The near absence of slippage on dinucleotide repeats again argues for a slippage being via a mechanism that involves single base steps. The second model proposes that the shift may occur without the strand separation by flipping out of one or more bases in the RNA or DNA strand at the active center of RNA polymerase followed by propagation of the flipped base through the catalytic cleft of the enzyme to the upstream end of the RNA-DNA hybrid. This model is based on the reported ability of RNA polymerase to accommodate bulky base analogs, DNA lesions, and unpaired/mispaired bases in the RNA-DNA hybrid without dissociation of the enzyme from DNA. As an alternative to the bulge propagation model, strand slippage was proposed to occur by transient formation of shared cross-strand hydrogen bonds between the adjacent bases in the RNA-DNA hybrid. The bond sharing at dA-dT homoduplexes requires the minimal energy cost because these sequences exhibit high propeller twist supporting the hydrogen bond sharing (9). The latter mechanism was supported by structural analysis of complexes of HIV RT at the polypurine tract, where a noncanonical form of the hybrid carrying slipped and mismatched bases was observed (34).
Mutations that alter the level of transcriptional slippage are expected to have consequences to the cell. Genes with homopolymeric runs within the open reading frames would give rise to transcripts that shift into alternative reading frames. Changes in the slippage rates might also influence gene expression levels by altering the efficiency of promoter escape or termination (35). The growth rates of the slippage-prone mutants are generally slower than the WT strain with the rpb1-S751F allele, causing the most severe growth defect. The Rpb1-M487 residue is conserved among the yeast and E. coli RNA polymerase, whereas the E. coli enzyme contains alanines at the positions corresponding to Asn-488 and Ser-751 (Fig. 7B).
The Rpb1-M487 and the Rpb1-N488 residues are located below the active site in the catalytic loop and near the position of the magnesium ions and the phosphates of the incoming NTP. These residues are also close to the template DNA base involved in base pairing with the NTP (Fig. 7A). Their role in maintaining proper register of the RNA and template strand may reflect stabilization of the 3′ end of the transcript. Rpb1-S751 is located in the secondary pore in Pol II connecting the active center with the enzyme surface. This residue is also in close proximity to the active center, making a protein loop together with the conserved Lys-752 residue interacting with the β and γ phosphates of the incoming NTP and the catalytic magnesium ion (Fig. 7A; pyrophosphate-binding loop). This loop may suppress slippage by restricting flexibility of the active center and not allowing the 3′ end of the RNA to freely disengage from the base pairing with template. Interestingly, the S751F mutation mostly promotes deletions during slippage, which require shifting the RNA forward relative to the DNA. Normally, the secondary pore in Pol II accommodates the 3′ end of the RNA extruded from the active center during backtracking. The mutation might facilitate the 3′ RNA shifting during slippage. Indeed, recent report on bacteriophage T7 RNA polymerase indicates that the 3′ RNA extrusion during backtracking may constitute a part of the slippage mechanism (36). In addition, a short distance 1–2-bp backtracking from the end of 11A tract induces pausing of RNA polymerase without moving the enzyme away from the homopolymeric sequence. The pausing may provide an extra time for shift of the RNA-DNA hybrid to occur in the backtracked complex.
The approach described here utilizing homopolymeric runs as at-risk sequences for transcription errors to identify error-prone RNA polymerases should be of general use in identifying features of the elongation complex that control fidelity. The in vitro data support the in vivo phenotype of the yeast Pol II mutants. Because 11A(+1) out of frame reporters were used in the isolation scheme, a slip resulting in a one base deletion would be the most effective way to restore the in-frame of the reporters. The isolation scheme would in fact bias for the mutant Pol II with enhanced deletion slippage products. This could also explain why E. coli RNAP slippage mutants were biased toward making insertion slippage products when a 9A(−1) reporter was used for isolation (19). Alternatively, the difference in the largest subunit of yeast Pol II as described in this manuscript and the second largest subunit of E. coli RNAP (rpoB) as described in Ref. 19 could also contribute the difference in the directionality of slippage observed in those mutant RNAPs. Further studies are warranted to address these issues.
Acknowledgments
We thank our colleagues for discussions and comments.
This work was supported, in whole or in part, by the National Institutes of Health Intramural Research Program. This work was also supported by the National Cancer Institute and the Center for Cancer Research.
- Pol II
- polymerase II
- SC
- synthetic complete
- RNAP
- RNA polymerase
- TEC
- ternary elongation complex(es)
- nt
- nucleotide(s)
- CEN
- centromere.
REFERENCES
- 1. Kramer E. B., Farabaugh P. J. (2007) The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA 13, 87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koyama H., Ito T., Nakanishi T., Kawamura N., Sekimizu K. (2003) Transcription elongation factor S-II maintains transcriptional fidelity and confers oxidative stress resistance. Genes Cells 8, 779–788 [DOI] [PubMed] [Google Scholar]
- 3. Nesser N. K., Peterson D. O., Hawley D. K. (2006) RNA polymerase II subunit Rpb9 is important for transcriptional fidelity in vivo. Proc. Natl. Acad. Sci. U.S.A. 103, 3268–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shaw R. J., Bonawitz N. D., Reines D. (2002) Use of an in vivo reporter assay to test for transcriptional and translational fidelity in yeast. J. Biol. Chem. 277, 24420–24426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Malagon F., Kireeva M. L., Shafer B. K., Lubkowska L., Kashlev M., Strathern J. N. (2006) Mutations in the Saccharomyces cerevisiae RPB1 gene conferring hypersensitivity to 6-azauracil. Genetics 172, 2201–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kireeva M. L., Nedialkov Y. A., Cremona G. H., Purtov Y. A., Lubkowska L., Malagon F., Burton Z. F., Strathern J. N., Kashlev M. (2008) Transient reversal of RNA polymerase II active site closing controls fidelity of transcription elongation. Mol. Cell 30, 557–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kaplan C. D., Larsson K. M., Kornberg R. D. (2008) The RNA polymerase II trigger loop functions in substrate selection and is directly targeted by α-amanitin. Mol. Cell 30, 547–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thomas M. J., Platas A. A., Hawley D. K. (1998) Transcriptional fidelity and proofreading by RNA polymerase II. Cell 93, 627–637 [DOI] [PubMed] [Google Scholar]
- 9. Anikin M., Molodtsov D., Temiakov D., McAllister (2010) Transcript slippage and recoding, in Recoding: Expansion of Decoding Rules Enriches Gene Expression (Atkins J. F., Gesteland R. F., eds) pp. 409–434, Springer, New York [Google Scholar]
- 10. Macdonald L. E., Zhou Y., McAllister W. T. (1993) Termination and slippage by bacteriophage T7 RNA polymerase. J. Mol. Biol. 232, 1030–1047 [DOI] [PubMed] [Google Scholar]
- 11. Baranov P. V., Hammer A. W., Zhou J., Gesteland R. F., Atkins J. F. (2005) Transcriptional slippage in bacteria: distribution in sequenced genomes and utilization in IS element gene expression. Genome Biol. 6, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Larsen B., Wills N. M., Nelson C., Atkins J. F., Gesteland R. F. (2000) Nonlinearity in genetic decoding. Homologous DNA replicase genes use alternatives of transcriptional slippage or translational frameshifting. Proc. Natl. Acad. Sci. U.S.A. 97, 1683–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Penno C., Hachani A., Biskri L., Sansonetti P., Allaoui A., Parsot C. (2006) Transcriptional slippage controls production of type III secretion apparatus components in Shigella flexneri. Mol. Microbiol. 62, 1460–1468 [DOI] [PubMed] [Google Scholar]
- 14. Xiong X. F., Reznikoff W. S. (1993) Transcriptional slippage during the transcription initiation process at a mutant lac promoter in vivo. J. Mol. Biol. 231, 569–580 [DOI] [PubMed] [Google Scholar]
- 15. Ratinier M., Boulant S., Combet C., Targett-Adams P., McLauchlan J., Lavergne J. P. (2008) Transcriptional slippage prompts recoding in alternate reading frames in the hepatitis C virus (HCV) core sequence from strain HCV-1. J. Gen. Virol. 89, 1569–1578 [DOI] [PubMed] [Google Scholar]
- 16. Linton M. F., Pierotti V., Young S. G. (1992) Reading-frame restoration with an apolipoprotein B gene frameshift mutation. Proc. Natl. Acad. Sci. U.S.A. 89, 11431–11435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Linton M. F., Raabe M., Pierotti V., Young S. G. (1997) Reading-frame restoration by transcriptional slippage at long stretches of adenine residues in mammalian cells. J. Biol. Chem. 272, 14127–14132 [DOI] [PubMed] [Google Scholar]
- 18. Wagner L. A., Weiss R. B., Driscoll R., Dunn D. S., Gesteland R. F. (1990) Transcriptional slippage occurs during elongation at runs of adenine or thymine in Escherichia coli. Nucleic Acids Res. 18, 3529–3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou Y. N., Lubkowska L., Hui M., Court C., Chen S., Court D. L., Strathern J. N., Jin D. J., Kashlev M. (2013) J. Biol. Chem. 288, 2700–2710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Amberg D. C., Burke D., Strathern J. N. (eds). (2005) Methods in Yeast Genetics, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 21. Sawitzke J. A., Thomason L. C., Costantino N., Bubunenko M., Datta S., Court D. L. (2007) Recombineering. In vivo genetic engineering in E. coli, S. enterica, and beyond. Methods Enzymol. 421, 171–199 [DOI] [PubMed] [Google Scholar]
- 22. Mnaimneh S., Davierwala A. P., Haynes J., Moffat J., Peng W. T., Zhang W., Yang X., Pootoolal J., Chua G., Lopez A., Trochesset M., Morse D., Krogan N. J., Hiley S. L., Li Z., Morris Q., Grigull J., Mitsakakis N., Roberts C. J., Greenblatt J. F., Boone C., Kaiser C. A., Andrews B. J., Hughes T. R. (2004) Exploration of essential gene functions via titratable promoter alleles. Cell 118, 31–44 [DOI] [PubMed] [Google Scholar]
- 23. Komissarova N., Kireeva M. L., Becker J., Sidorenkov I., Kashlev M. (2003) Engineering of elongation complexes of bacterial and yeast RNA polymerases. Methods Enzymol. 371, 233–251 [DOI] [PubMed] [Google Scholar]
- 24. Kireeva M. L., Hancock B., Cremona G. H., Walter W., Studitsky V. M., Kashlev M. (2005) Nature of the nucleosomal barrier to RNA polymerase II. Mol. Cell 18, 97–108 [DOI] [PubMed] [Google Scholar]
- 25. Kireeva M. L., Kashlev M. (2009) Mechanism of sequence-specific pausing of bacterial RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 106, 8900–8905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pal M., Luse D. S. (2003) The initiation-elongation transition. Lateral mobility of RNA in RNA polymerase II complexes is greatly reduced at +8/+9 and absent by +23. Proc. Natl. Acad. Sci. U.S.A. 100, 5700–5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Turnbough C. L., Jr. (2008) Regulation of bacterial gene expression by the NTP substrates of transcription initiation. Mol. Microbiol. 69, 10–14 [DOI] [PubMed] [Google Scholar]
- 28. Fox J. D., Waugh D. S. (2003) Maltose-binding protein as a solubility enhancer. Methods Mol. Biol. 205, 99–117 [DOI] [PubMed] [Google Scholar]
- 29. Wu J., Awrey D. E., Edwards A. M., Archambault J., Friesen J. D. (1996) In vitro characterization of mutant yeast RNA polymerase II with reduced binding for elongation factor TFIIS. Proc. Natl. Acad. Sci. U.S.A. 93, 11552–11557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boeke J. D., LaCroute F., Fink G. R. (1984) A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast. 5-Fluoro-orotic acid resistance. Mol. Gen. Genet. 197, 345–346 [DOI] [PubMed] [Google Scholar]
- 31. Yu D., Ellis H. M., Lee E. C., Jenkins N. A., Copeland N. G., Court D. L. (2000) An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 97, 5978–5983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kireeva M. L., Lubkowska L., Komissarova N., Kashlev M. (2003) Assays and affinity purification of biotinylated and nonbiotinylated forms of double-tagged core RNA polymerase II from Saccharomyces cerevisiae. Methods in enzymology 370, 138–155 [DOI] [PubMed] [Google Scholar]
- 33. Strathern J. N., Jin D. J., Court D. L., Kashlev M. (2012) Isolation and characterization of transcription fidelity mutants. Biochim. Biophys. Acta 1819, 694–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sarafianos S. G., Das K., Tantillo C., Clark A. D., Jr., Ding J., Whitcomb J. M., Boyer P. L., Hughes S. H., Arnold E. (2001) Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 20, 1449–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Turnbough C. L., Jr. (2011) Regulation of gene expression by reiterative transcription. Curr. Opin. Microbiol. 14, 142–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu X., Martin C. T. (2009) Transcription elongation complex stability. The topological lock. J. Biol. Chem. 284, 36262–36270 [DOI] [PMC free article] [PubMed] [Google Scholar]