

Background: Neurons express two Na,K-ATPase isoforms, the ubiquitous α1 and neuron-specific α3.
Results: α3 is important for control of membrane potential and is fully responsible for restoration of large [Na+]i increases.
Conclusion: α1 and α3 are required for basal neuronal function, but α3 controls restoration of [Na+]i following sustained discharge.
Significance: Conditions associated with defect α3 function are likely aggravated by suprathreshold neuronal activity.
Keywords: Electrophysiology; Fluorescence; Na,K-ATPase; Neurons; Protein Kinase A (PKA); Intracellular Sodium Imaging; Ouabain; Pump Current
Abstract
Most neurons co-express two catalytic isoforms of Na,K-ATPase, the ubiquitous α1, and the more selectively expressed α3. Although neurological syndromes are associated with α3 mutations, the specific role of this isoform is not completely understood. Here, we used electrophysiological and Na+ imaging techniques to study the role of α3 in central nervous system neurons expressing both isoforms. Under basal conditions, selective inhibition of α3 using a low concentration of the cardiac glycoside, ouabain, resulted in a modest increase in intracellular Na+ concentration ([Na+]i) accompanied by membrane potential depolarization. When neurons were challenged with a large rapid increase in [Na+]i, similar to what could be expected following suprathreshold neuronal activity, selective inhibition of α3 almost completely abolished the capacity to restore [Na+]i in soma and dendrite. Recordings of Na,K-ATPase specific current supported the notion that when [Na+]i is elevated in the neuron, α3 is the predominant isoform responsible for rapid extrusion of Na+. Low concentrations of ouabain were also found to disrupt cortical network oscillations, providing further support for the importance of α3 function in the central nervous system. The α isoforms express a well conserved protein kinase A consensus site, which is structurally associated with an Na+ binding site. Following activation of protein kinase A, both the α3-dependent current and restoration of dendritic [Na+]i were significantly attenuated, indicating that α3 is a target for phosphorylation and may participate in short term regulation of neuronal function.
Introduction
The maintenance of a steep Na+ gradient across the plasma membrane is essential for the function and survival of all eukaryotic cells. The ubiquitous integral plasma membrane protein, Na,K-ATPase, which actively exports three Na+ ions and imports two K+ ions for each ATP hydrolyzed, is mainly responsible for maintenance of the transmembrane Na+ gradient and is also a major determinant of neuronal resting membrane potential (1, 2). The Na,K-ATPase exists as a heterotrimeric α-β-γ protein complex, where α is the catalytic ion-transporting subunit (3). In neurons, the Na+ gradient across the plasma membrane varies as a result of activity-dependent transient influx via voltage- and ligand-gated ion channels. Neurons express two α isoforms: α1, which is ubiquitous, and α3, which has a very restricted expression (4, 5). Although disease mutations of α3 give rise to serious neurological symptoms such as rapid onset dystonia parkinsonism (6), cognitive deficits (7, 8), mood disturbances (9, 10), and alternating hemiplegia of childhood (11, 12), the relative role of the two α isoforms at the cellular level in neurons is not completely understood.
Previous studies in cell expression systems have shown that α3 has a lower Na+ affinity than α1, and it was hypothesized that the α3 isoform is specifically required for rapid restoration of large transient increases in [Na+]i (13, 14). To test this concept and to examine the relative importance of α1 and α3 in maintaining the membrane potential and Na,K-ATPase dependent current, we have used a combination of Na+ imaging and electrophysiological approaches. The majority of this study was performed on rat hippocampal neurons in primary culture, expressing both α1 and α3 isoforms, but to test the generality of our findings, some studies were performed on cultured striatal neurons and on cortical slices.
It has been reported that repetitive activation of excitatory synapses in the hippocampus that result in a long lasting increase in synaptic strength are associated with 20–40 mm increases in [Na+]i (15). To mimic this situation of suprathreshold neuronal activity, Na+ recordings were performed in dendrites following a transient large increase in [Na+]i. The energy cost for turn-over of Na,K-ATPase is estimated to be ∼50% of total brain energy consumption (16) and because several lines of evidence suggest that the majority of this Na+ current dependent energy is expended postsynaptically rather than presynaptically (17), the Na+ imaging studies were performed on dendrites.
Short term changes in Na,K-ATPase activity can substantially impact neuronal activity. It is known from studies in cell free systems that Na,K-ATPase activity can be regulated by protein kinase A (PKA)3 phosphorylation (18, 19). To assess the modulatory capacity of neuronal Na,K-ATPase, we also recorded Na,K-ATPase specific currents and the capacity of the neuron to restore [Na+]i following PKA activation.
The digoxin analog ouabain, a steroid hormone consisting of a steroid core with a lactone ring and a sugar moiety, which has been used for centuries to treat heart disease, is a highly specific ligand to Na,K-ATPase (20–22) with dose-dependent and isoform-specific functions. Because α3 is much more sensitive to the inhibitory effect of ouabain than α1 (IC50 values are 0.1 and 32 μm, respectively, (23, 24)), the relative contribution of the two isoforms in maintaining [Na+]i during neuronal activity can be estimated by performing studies in the presence or absence of 0.1–1.0 μm ouabain.
EXPERIMENTAL PROCEDURES
All animal procedures had received prior approval by the local ethical committee, Stockholms Norra Djurförsöksetiska Nämnd and were carried out in accordance with the European Communities Council Directive (86/609/EEC).
Cell Culture
Cultures of hippocampal and striatal neurons were prepared from embryonic day 18.5 Sprague-Dawley rat (NOVA SCb) embryos of both sexes. Hippocampus and striatum were dissected, incubated for 10 min at 37 °C in Hank's balanced salt solution containing 20 mm HEPES and 0.25% trypsin, and then dissociated in minimum essential media (Invitrogen) by mechanical triturating using a fire-polished Pasteur pipette. The cells were plated on glass coverslips previously coated with poly-ornithine (80 μg/ml, overnight) and incubated for three hours in minimum essential media supplemented with 10% horse serum, 2 mm l-glutamine, and 1 mm sodium pyruvate. The cells were then cultured in neurobasal medium containing B27 and 0.5 mm l-glutamine. Cells were maintained in culture for 2–3 weeks before experiments, and culture medium was changed twice a week.
Immunocytochemistry
Primary hippocampal neurons were fixed in 10% TCA (Merck: 10% (w/v) in H2O) for 10 min at 4 °C. The cells were permeabilized in 0.25% Triton X-100 for 5 min at room temperature, followed by blocking in 10% bovine serum albumin (BSA). Primary antibody incubation in 5% BSA was performed for 4 h at room temperature or overnight at 4 °C. The Na,K-ATPase α1 isoform was stained using the α6F monoclonal antibody (1:10, ∼2.7 μg/ml) developed by Douglas M. Fambrough and obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biology (Iowa City, IA). The MA3-915 monoclonal antibody from Thermo Scientific was used to stain for Na,K-ATPase α3 (1:2000, ∼0.5 μg/ml). Alexa Fluor 546-conjugated donkey anti-mouse secondary antiserum was used for detection. Glial fibrillary acidic protein (GFAP) was used to label astrocytes (Santa Cruz Biotechnology, sc-6170, 1:250) with Alexa Fluor 633-conjugated donkey anti-goat for detection. Microtubule-associated protein 2 (MAP-2) was used to stain for neuronal morphology (Millipore, AB5622, 1:1000) with Alexa Fluor 488-conjugated donkey anti-rabbit for detection. In some experiments, cells were first transfected with mCherry-PSD-95 using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations. After 48 h, these cells were then fixed using 4% paraformaldehyde, blocked in 10% BSA for 30 min, permeabilized in 0.25% Triton X-100 for 5 min, and subsequently stained for MAP-2 for 3 h at room temperature followed by staining with Alexa Fluor 488-conjugated goat anti-rabbit secondary antiserum. All secondary antibodies were purchased from Invitrogen and used at a 1:500 dilution. Confocal images were acquired using a Zeiss 5 Live Duo confocal laser microscope (Zeiss) using a Plan-Apochromat 63×/1.40 numerical aperture objective.
Immunoprecipitation
Hippocampal tissue from 20-day-old Sprague-Dawley rats was homogenized and centrifuged at 12,000 × g for 30 min at 4 °C. Supernatant protein concentration was measured by Bio-Rad DC protein assay (Bio-Rad). Tissue lysate (800 μg) was subject to immunoprecipitation as described previously (25). The antibodies, 2-μg each, used for precipitation were anti Na,K-ATPase α1 (Developmental Studies Hybridoma Bank), anti Na,K-ATPase α3 (Upstate), and anti-postsynaptic density 95 (PSD-95) (Abcam). Proteins were eluted with Laemmli sample buffer and resolved by Western blot. The antibodies used for immunoblotting were as follows: rabbit anti Na,K-ATPase α1 (Upstate), rabbit anti Na,K-ATPase α3 (Upstate), and rabbit anti-PSD-95 (Abcam).
Imaging Equipment
Low-light level fluorescence imaging was performed on an upright epifluorescence microscope (Axioskop 2 FS Plus, Carl Zeiss) using a 40×/1.3 numerical aperture oil immersion objective lens. Fluorescence excitation wavelengths were selected using a monochromator (Polychrome IV, TILL Photonics), and fluorescence was detected using a 12-bit cooled CCD camera (ORCA ER, Hamamatsu). Image acquisition and time series were computer-controlled using MetaFluor (Universal Imaging).
Dye Loading and Imaging
Recordings were performed on primary hippocampal cultures on day in vitro 21–24. Cells were loaded at 35 °C with the acetoxymethyl ester derivative forms of fluorescent probes in the loading solution (160 mm NaCl, 5.4 mm KCl, 1.3 mm CaCl2, 0.81 mm MgSO4, 0.78 mm NaH2PO4, 20 mm HEPES, 20 mm glucose, pH 7.4, and 1% BSA) and then placed in a heated chamber for rapid exchange of solutions continuously warmed at 35 °C. Recording solution contained 125 mm NaCl, 26 mm NaHCO3, 4 mm KCl, 1 mm MgSO4, 1.25 mm NaH2PO4, 2 mm CaCl2, 10 mm glucose and was continuously bubbled with carbogen (5% CO2, 95% O2) resulting in a pH of 7.4. The K+-free solution (0 [K+]o) had the same composition, except that the NaCl and KCl concentrations were 129 mm and 0 mm, respectively. The Na+-sensitive cytosolic ANG1 (Asante NaTRIUM Green 1) was loaded at 1 μm for 25 min, and its fluorescence was excited at 490 nm and collected above 500 nm. Between 0 and 60 mm, the ANG1 fluorescence was linearly correlated with Na+ (R2 = 0.99).
Cytosolic Na+ Calibration
At the end of each experiment, neurons were superfused with Na+ calibration solutions containing stepwise increasing concentrations of Na+ in the presence of 3 μm gramicidin, 10 μm monensin, and 1 mm ouabain. Na+ calibration solutions contained [Na+ + K+] = 165 mm, 136 mm gluconate, 0.81 mm MgSO4, 0.78 mm KH2PO4, 20 mm HEPES, 1.3 mm CaCl2, pH adjusted to 7.2 with KOH.
Data Analysis
The calibrated Na+ data were analyzed using custom software in MATLAB (MathWorks). Data were smoothed using a 7-point moving average. The Na+ extrusion rate was then quantified by fitting the recovery slope to a bi-exponential equation. The apparent Vmax is taken as the absolute value of the maximum derivative of the fitted function.
Electrophysiology
Whole-cell patch clamp recordings were performed on hippocampal and striatal cultures on day in vitro 13–17. Cultures were maintained at 31 ± 1 °C and continuously perfused with extracellular recording solution (110 mm NaCl, 20 mm HEPES, 25 mm NaHCO3, 4 mm KCl, 1.2 mm MgCl2, 1 mm NaH2PO4, 1.5 mm CaCl2, 10 mm glucose, and pH adjusted to 7.4). The internal pipette solution contained 120 mm K+-gluconate, 24 mm KCl, 4 mm NaCl, 4 mm MgCl2, 0.16 mm EGTA, 10 mm HEPES, 4 mm K2-ATP, pH 7.2 adjusted with KOH. Neurons were visualized using differential interference contrast on an Axiovert 135 microscope (Carl Zeiss) and selected for whole-cell patch clamp if the cell soma was phase-bright and exhibited a minimum of three major primary dendrites. Recordings were performed using an Axopatch 200B amplifier (Molecular Devices) and pClamp software (version 8.2). Compensations for slow and fast capacitive currents, but not liquid junction potential, were performed. The signal was filtered at 2 kHz and sampled at 10 kHz. The extracellular solution for Na,K-ATPase current recordings contained 140 mm NaCl, 3 mm MgCl2, 0.33 mm NaH2PO4, 5 mm HEPES, 2 mm BaCl2, 2 mm CsOH, 2 mm NiCl2, 0.2 mm CdCl2, 15 mm mannitol, and 5 mm glucose with pH adjusted to 7.4 using NaOH. The K+ concentration in the high-K+ solution was either 8 or 20 mm using KCl. The internal pipette solution for Na,K-ATPase current recordings contained 95 mm NaMeSO4, 20 mm tetramethyl ammonium aspartate, 10 mm HEPES, 4 mm MgATP, 5 mm di sodium-phosphocreatine, 20 mm tetraethylammonium chloride, 5 mm EGTA, 5 mm glucose with pH adjusted to 7.2 using NaOH.
PKA Pathway Activation
The activation of the PKA pathway was induced by superfusing cells for 5–10 min with a solution containing dibutyryl cAMP (10 μm, membrane permeable cAMP analog), isobutylmethylxanthine (100 μm, phosphodiesterase inhibitor), FK506 (0.1 μm inhibits protein phosphatase 2B) and cantharidin (0.1 μm inhibits protein phosphatase 1 and 2A).
Extracellular Multiunit Recordings
Three- to 13-week-old male Sprague-Dawley rats (Charles River) were deeply anesthetized with an intraperitoneal injection of pentobarbital and then decapitated. The brain was dissected and immediately submerged in ice-cold, oxygenated slicing buffer containing 234 mm sucrose, 26 mm NaHCO3, 1.25 mm NaHPO4, 2 mm CaCl2, 2 mm MgSO4, 2.5 mm KCl, and 10 mm glucose. 300-μm thick sagittal slices of the somatosensory cortex were cut on a vibratome (Leica) and transferred to an interface style recording chamber (Fine Science Tools). The interface chamber was oxygenated, heated to 35 °C, and continuously perfused with oxygenated buffers to enable the occurrence of spontaneous rhythmic network activity in the form of the cortical slow oscillations (26, 27). For the first 30 min, slices were perfused with a 50:50 mixture of slicing medium and artificial cerebrospinal fluid (124 mm NaCl, 26 mm NaHCO3, 1.25 mm NaHPO4, 2.5 mm KCl, 2 mm CaCl2, 2 mm MgSO4, and 10 mm glucose) and then with only artificial cerebrospinal fluid for 60 min. Perfusion solution was switched to modified artificial cerebrospinal fluid containing 124 mm NaCl, 26 mm NaHCO3, 1.25 mm NaHPO4, 3.5 mm KCl, 1.0 mm CaCl2, 1.0 mm MgSO4, and 10 mm glucose for the recording. Extracellular multiunit recordings were performed using low resistance (<1 megohms of tungsten microelectrodes) (Fredrick Haer Corporation), sampled at 20 kHz, band pass filtered between ∼0.3 and 10 kHz using an AC/DC differential amplifier (AM Systems), and collected through an analog-digital convertor (Micro 1401, Cambridge Electronic Design) before acquisition with Spike 2 software (Cambridge Electronic Design). The cortical slow oscillation is defined as having a frequency of 0.1–1 Hz (26, 27). Base-line recordings were made for at least 15 min, with ouabain subsequently added via the bath.
Materials
Hank's balanced salt solution, trypsin, polyornithine, Neurobasal B27, and Lipofectamine were purchased from Invitrogen. Asante NaTRIUM Green 1 was purchased from TEFLabs. All other compounds were purchased from Sigma.
RESULTS
Expression of Na,K-ATPase Isoforms α1 and α3 in Hippocampal Neurons
The majority of studies were performed on rat hippocampal neurons and cultured for 14–21 days. The expression of both α1 and α3 in these neurons was documented with immunocytochemistry using isoform-specific antibodies and markers for astrocytes (GFAP) and neuronal dendrites (MAP-2) (Fig. 1, A–J). Na,K-ATPase α1 was expressed in virtually every cell profile within the culture, including both those with neuronal and non-neuronal morphologies (Fig. 1, A–D). The Na,K-ATPase α3 immunoreactivity was exclusively restricted to neuronal like cells, characterized by an enlarged soma and dendritic arborizations (Fig. 1, E–H). Confocal analysis indicated that both α1 and α3 are strongly expressed in the plasma membrane of soma and dendrites (Fig. 1, I and J). The maturity of the cultures was verified by using PSD-95-mCherry as a marker for spines and MAP-2 as a marker for dendrites. The cells displayed well developed dendritic arborizations and a high density of dendritic spines (Fig. 1K). Both Na,K-ATPase α1 and α3 were found to co-immunoprecipitate with the PSD-95 protein, a marker for dendritic spines and excitatory synapses (Fig. 1, L and M).
FIGURE 1.

Expression of Na,K-ATPase α1 and α3 isoforms at the plasma membrane of hippocampal neurons. A–D, primary culture of hippocampus stained with antibodies against α1, GFAP, and MAP-2. α1 immunoreactivity (A) is found in the majority of cells, including neuronal (MAP-2, B and D) and non-neuronal cells (GFAP, C and D). Filled and open arrowheads point out astrocytes and neurons, respectively. E–H, hippocampal culture stained with antibodies against α3 (E), GFAP (F), and MAP-2 (G). Only cells of neuronal morphology (MAP-2, F and H) display α3-specific signal (open arrowhead in H). I and J, confocal sections showing clear α immunoreactive signal in plasma membrane of soma and dendrites for α1 (I) and α3 (J). K, primary neurons transfected with PSD-95-mCherry to visualize dendritic spines (red) and stained with antibodies against MAP-2 (green) to visualize dendritic processes. L and M, co-immunoprecipitation of PSD-95 and Na,K-ATPase α1 and α3 from the rat hippocampus. Representative co-immunoprecipitation (IP) of PSD-95 and NKA α1, α3 protein complex using mouse α1, α3 mAb (L), and mouse monoclonal PSD-95 mAb (M). Mouse IgG (IgG) was used as a negative control. Input is whole lysate.
Relative Contribution of Na,K-ATPase α1 and α3 Isoforms in Maintaining Basal [Na+]i and Membrane Potential
The capacity of the two isoforms to set basal [Na+]i was assessed using live cell imaging. The cells were perfused successively with a low concentration of ouabain (1 μm), which should selectively inhibit the Na,K-ATPase α3, followed by a high concentration of ouabain (1 mm), which should completely inhibit both isoforms. Consistent with previous reports (28, 29), the basal [Na+]i in the soma of neurons was found to be 10.9 ± 1.6 mm (mean ± S.E., n = 27 cells from seven independent experiments). Selective blockade of the Na,K-ATPase α3 with 1 μm ouabain resulted in a modest increase in [Na+]i, which generally reached a plateau within 5 min. When 1 mm was added to the perfusion medium, there was a rapid and much more pronounced additional increase of [Na+]i, indicating that α1 plays a major role in maintaining basal [Na+]i (Fig. 2, A and B).
FIGURE 2.

Inhibition of the Na,K-ATPase α3 increases [Na+]i in dendrites and depolarizes hippocampal neurons. A, example traces of [Na+]i recording in a neuron before and during successive perfusion of 1 μm and 1 mm ouabain. Calibration using ionophores are performed at the end of each experiment. B, box plot of increases in [Na+]i due to ouabain (data from 21 cells from five experiments). C, whole cell current clamp recording of a hippocampal neuron showing the depolarization resulting from treatment with 1 μm ouabain. D, membrane potential of hippocampal neurons before and after superfusion of 1 μm ouabain (n = 6 neurons from six experiments). *, p < 0.05; ***, p < 0.001, using paired t test.
We next tested what impact inhibition of Na,K-ATPase α3 activity would have on the resting membrane potential of neurons measured using whole-cell current clamp recordings. Under basal conditions, the neurons exhibited spontaneous action potential firing (Fig. 2C). The administration of an α3-selective ouabain concentration to the bath consistently resulted in rapid and robust depolarization (Fig. 2, C and D). Notably, the depolarization resulted initially in an increased firing rate of the cells, which was followed by a complete loss of firing when the membrane potential became too depolarized.
Relative Contribution of α1 and α3 Isoforms to Na,K-ATPase Activity in Sodium Loaded Neurons
To directly measure the Na,K-ATPase activity within a single neuron, we used a protocol (30, 31) specifically customized to isolate the Na,K-ATPase-dependent current (Ip). Neurons were patch clamped in the whole-cell configuration with command potential clamped at 0 mV during experiments. The pipette solution contained tetramethyl ammonium as a replacement for K+, tetraethylammonium to block voltage-gated K+ channels, phosphocreatine, and ATP in excess to ensure continued pump function. The Na+ concentration in the pipette solution was set to 95 mm using NaMeSO4 to maximize turnover of both Na,K-ATPase α1 and α3. In the extracellular buffer, we added Ba2+ to block inwardly rectifying K+ channels, Cs+ to replace K+ and to block voltage-gated K+ channels, and Cd2+ and Ni2+ to block voltage-gated Ca2+ channels. The Na+/Ca2+ exchanger was blocked by excluding Ca2+ from the buffers and by adding Ni2+ to the extracellular solution. We measured the current change when cells were superfused with a solution containing a high concentration of K+ compared with the base-line current when there is no extracellular K+ (Fig. 3A). This procedure was repeated twice so that each neuron could serve as its own control. The average amplitude of the first K+-evoked Ip was 0.10 ± 0.01 nA. Under control conditions, the current ratio, the second K+-evoked Ip to the first K+-evoked Ip, reached on average 66 ± 4%. In neurons exposed to 1 μm ouabain before and during the second superfusion with high K+, the Ip was almost abolished (11 ± 3% of first K+-evoked Ip). Treating the cells with 0.1 μm ouabain, which should inhibit ∼60% of Na,K-ATPase α3 activity, caused a significant and strong decrease of the current ratio (44 ± 9%) (Fig. 3B).
FIGURE 3.
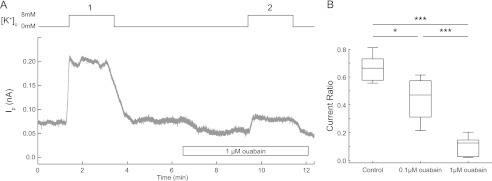
Major contribution of the Na,K-ATPase α3 to total Na,K-ATPase Ip in hippocampal neurons. Hippocampal neurons were patch clamped and Na,K-ATPase-specific current was recorded under voltage-clamped configuration in hippocampal neurons. Na,K-ATPase was activated by superfusing a K+-containing buffer (8 mm). A, example trace of an Ip recording in the absence or the presence of ouabain in hippocampal neurons. B, box plot of Ip ratio (proportion of second K+-evoked Ip to first K+-evoked Ip) in the presence and absence of ouabain. The Ip was measured as the difference in current recorded at 0 and 8 mm extracellular K+. Control, n = 7 neurons; 0.1 μm ouabain, n = 4 neurons; 1.0 μm ouabain, n = 6 neurons. *, p < 0.05; ***, p < 0.001, using one-way ANOVA with Bonferroni correction.
Importance of Postsynaptic Na,K-ATPase α3 in Neurons Challenged with a Transient Increase in [Na+]i
Suprathreshold neuronal activity will result in large [Na+]i increases in postsynaptic structures (15, 32). Here, we examined the relative capacity of the Na,K-ATPase α1 and α3 isoforms to restore a transient dendritic increase of [Na+]i. The sodium-sensitive dye, ANG, used in these experiments was particularly suitable for the recording of [Na+]i in small structures, such as dendrites, because of its strong fluorescence response (high quantum efficiency and high dynamic range, Fmax/Fmin). An example of ANG-loaded cells can be seen in Fig. 4A. To mimic the transient increase in [Na+]i reached during situations of suprathreshold neuronal activity, cells were perfused with a K+-free buffer to inhibit pump mediated extrusion of sodium. When the extracellular buffer was changed back to normal K+-containing buffer, pump activity was resumed, and the subsequent Na+ extrusion was determined. Typical recording are shown in Fig. 4B. Recordings were performed in a secondary dendritic branch, located typically 20–30 μm away from the soma. In control conditions, the Na+ extrusion rate was relatively high during the initial phase of the recovery but decreased as [Na+]i approached base-line levels (Fig. 4B). In the presence of the α3-inhibiting ouabain concentration (1 μm), there was a profound decrease of the initial Na+ clearance rate (0.12 ± 0.02 mm/s versus 0.57 ± 0.11 mm/s in controls, Fig. 4, C and D). The recordings lasted for at least 10 min. During this period, none of the dendrites that had been exposed to the α3-inhibiting ouabain concentration recovered to the initial [Na+]i, suggesting that α1 alone was not able to handle the high [Na+]i increase. The [Na+]i recovery was also found to be significantly decreased in the presence of the partially inhibitory ouabain concentration 0.1 μm (0.14 ± 0.04 mm/s, n = 13 dendrites from six experiments).
FIGURE 4.
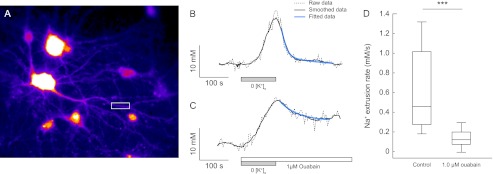
The Na,K-ATPase α3 is responsible for Na+ clearance in hippocampal neurons. A, cells loaded with Asante Natrium Green-1, white boxes show typical dendritic recording region. B and C, typical traces of calibrated [Na+]i recordings measured in single dendrites of hippocampal neurons challenged by a transient superfusion with K+-free buffer. Na+ clearance was measured in the absence (B) or presence (C) of 1 μm ouabain. Biexponential fitting of smoothed data (black traces) are represented as blue traces. D, box plot of Na+ clearance rates in the absence (0.57 ± 0.11 mm/s) or presence of 1 μm ouabain (0.12 ± 0.02 mm/s). The horizontal line within the box is the median value. Control, n = 12 dendrites from six experiments; 1 μm ouabain, n = 19 dendrites from five experiments. ***, p < 0.001 using one-way ANOVA with Bonferroni correction.
Na,K-ATPase α3-dependent Functional Effects Can Be Modified by the Protein Kinase A Signaling Pathway
The Na,K-ATPase α1 and α3 isoforms have a consensus site for PKA phosphorylation (33), but functional effects of PKA activation on Na,K-ATPase activity in intact neurons have not yet been demonstrated. Here, we tested whether the α3 mediated effects on Na,K-ATPase generated current and on the capacity to restore a transient increase of dendritic [Na+]i, could be modulated by activation of the PKA signaling pathway. To do so, neurons were incubated with dibutyryl cAMP, a membrane permeable cAMP analog; isobutylmethylxanthine, a phosphodiesterase inhibitor; FK506, a protein phosphatase 2B inhibitor; and cantharidin, a protein phosphatase 1 and 2A inhibitor. In combination, these compounds should act to increase the intracellular availability and slow the degradation of cAMP, a second messenger that activates the PKA pathway. After 5–10 min of preincubation, the Ip ratio and the Na+ clearance rate were measured as described in Fig. 3 (Ip recordings) and Fig. 4 (Na+ clearance data). When the PKA signaling pathway was activated, the Ip ratio was significantly decreased compared with control (Fig. 5A). Consistent with the Ip recordings, we found that activation of the PKA signaling pathway significantly decreased the Na+ clearance rate (Fig. 5B).
FIGURE 5.

Modulation of Na,K-ATPase α3 activity by activation of the PKA pathway. Hippocampal neurons were superfused with a solution containing cAMP analogs, inhibitors of cAMP degrading enzymes, and phosphatase inhibitors. A, box plot of the Ip ratio (proportion of second K+-evoked Ip to first K+-evoked Ip) during control and PKA-stimulating conditions. The Ip was measured as the average net current recorded during 8 mm extracellular K+. Control, n = 6 neurons; PKA activation, n = 9 neurons. B, box plot of Na+ clearance rate in the presence of vehicle (dimethyl sulfoxide, 0.11%) or drugs activating the PKA pathway. Dimethyl sulfoxide, n = 19 dendrites from four experiments; PKA, n = 15 dendrites from four experiments. *, p < 0.05, using one-way ANOVA with Bonferroni correction.
Importance of Na,K-ATPase α3 in Striatal Cultures and Cortical Slices
To evaluate the generality of our findings, we also performed whole-cell membrane current recordings on primary cultures of striatum. The dose-response relationship of the Ip ratio in response to ouabain treatment in striatal neurons showed a striking resemblance to the results obtained from hippocampal cultures (Fig. 6 cf. Fig. 3, A and B). To further assess the global relevance and potential functional consequences of α3 inhibition, we also studied whether a low ouabain concentration could alter network dynamics in the cerebral cortex. Cortical slice preparations can spontaneously express rhythmic ensemble activity in the form of slow (<1 Hz) oscillations (Fig. 7A) (26). In the presence of 1 μm ouabain, slow oscillation frequency significantly decreased by ∼50% over prolonged application, whereas a higher dose of ouabain (10 μm) completely abolished the slow oscillation (Fig. 7B).
FIGURE 6.
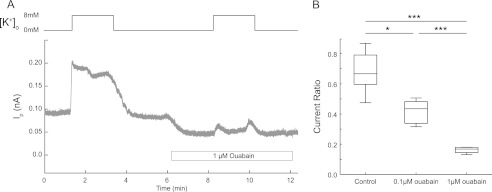
Functional role of Na,K-ATPase α3 in striatum. Na,K-ATPase-dependent current measured in striatal neurons. A, example trace of Ip recording in the absence or presence of ouabain. B, box plot of the Ip ratio (proportion of second K+-evoked Ip to first K+-evoked Ip). The Ip was measured as the difference in current recorded at 0 and 8 mm extracellular K+. Control, n = 11 neurons; 0.1 μm ouabain, n = 5 neurons; 1 μm ouabain, n = 5 neurons. *, p < 0.05; ***, p < 0.001, using one-way ANOVA with Bonferroni correction.
FIGURE 7.

Inhibition of Na,K-ATPase α3 decreases spontaneous slow oscillations in cortex. A, recording of spontaneous neuronal network oscillation in somatosensory cortex. The bottom graph illustrates the analysis result of data using the Spike software (version 2). B, graph showing group data of extracellular multiunit recordings of spontaneous slow oscillations in rat cortical slices measured as a proportion of initial frequency. The frequency of network activity decreases significantly when 1 μm ouabain is applied to the bath. In the presence of 10 μm ouabain, oscillations are completely abolished. Control, n = 4; 1 μm ouabain, n = 6; 10 μm ouabain, n = 4 experiments. *, p < 0.05; ***, p < 0.001, using one-way ANOVA with Bonferroni correction.
DISCUSSION
Na,K-ATPase α3 is specifically expressed in neurons (1, 4, 5), but its physiological importance in cells co-expressing α1 and α3 is still incompletely understood. In this study, where the majority of protocols were performed on neurons from hippocampus, we show that α3 plays an important role for control of resting membrane potential and that the function of α3 is essential when the intracellular sodium concentration is increased to levels that can be expected to occur in postsynaptic structures during high neuronal activity (15). Hippocampus is the center for memory consolidation (34). Mice heterozygous for the Na,K-ATPase α3 isoform display learning and memory deficits (8), indicating the functional importance of Na,K-ATPase α3 in this region of the brain.
The neurons were studied after 14–21 days in vitro. At that time, virtually all neurons expressed both α1 and α3 and exhibited a high degree of arborization and spine density. Overviews of hippocampal cultures stained for α1 or α3 indicated that virtually all neurons express both isoforms. Our results are in agreement with previous studies using cryosections of adult rat (5) and monkey brain (35). We could confirm that α3 is expressed in soma, axons, and dendrites of hippocampal neurons (36). In a recent comprehensive study of the adult rat brain (37), most brain regions were found to express α3. Both hippocampus and striatum appeared to express high levels of α3, whereas more moderate levels were observed in cortex. The existence of different catalytic isoforms were initially observed in rat brain preparations that displayed a bimodal inhibition of the pump to the ouabain analog strophanthidin (38, 39), and in the current study, recordings of [Na+]i in hippocampal neurons performed in the presence of 1 μm and 1 mm ouabain provided functional evidence for the co-expression of α1 and α3.
The results from this study point to an important postsynaptic role of α3. Both α1 and α3 isoforms were found to interact with PSD-95, a postsynaptic marker of excitatory synapses. Excitatory synaptic activity is associated with a postsynaptic Na+ influx via NMDA and AMPA receptors. In a study of postsynaptic Na+ signals, Rose and Konnerth (15) noted that although subthreshold synaptic activity did not result in a measureable increase in the Na+ concentration in spines and dendrites, suprathreshold stimulation caused a transient 20–40 mm rise in [Na+]i. Na,K-ATPase α3 has in cell expression systems been shown to have a much lower Na+ affinity than α1 (33 and 12 mm, respectively (14)) and has therefore been suggested to be better suited to deal with activity-related transient increases in [Na+]i than α1 (1, 13). Here, we provide experimental evidence for this hypothesis. We show that inhibition of α3 almost abolishes the capacity of dendrites to restore [Na+]i following increases of 20–40 mm. In fact, none of the dendrites studied were found to recover to the initial basal [Na+]i after full inhibition of α3.
Because dystonia is the major manifestation of Na,K-ATPase α3 mutations in rapid onset dystonia parkinsonism, Ip recordings were also performed on neurons derived from striatum, the center for control of motor activity, and the results support the role of α3 in this region. The finding that α3 in hippocampal and striatal neurons contribute to >90% of the pump current under high [Na+]i further emphasize the important role of α3.
Although most studies in transfected cell lines support the difference in Na+ affinity between the α1 and α3 isoforms, no such differences were found for rat isoforms expressed in frog oocytes or yeast, or for α3 purified from pineal gland (40). The discrepancies in results obtained from intact cells and in cell-free systems might imply that interacting proteins play an important role for setting isoform Na+ affinity. The recovery of intracellular Na+ levels reached after suprathreshold neuronal activity requires increased pump activity and consequently increased energy consumption. It is interesting to note that α3 has a higher ATP affinity relative to α1 (41) and should therefore be functionally more resistant to hypoxia and ischemia.
The important role of α3 for postsynaptic function raises the question whether α3 may be the target for phosphorylation and short term regulation. Short term regulation of postsynaptic α3 would have an impact on neuronal plasticity because the level of Na,K-ATPase activity plays a role for the after hyperpolarization that follows repetitive firing, both directly (42) and, likely also by affecting the activity of Na+-activated K+ channels (43), such as Slick (44) and Slack (45), which are highly sensitive to [Na+]i. Previous studies on cell-free or cell expression systems (18, 19) have indicated that α isoforms can be functionally regulated by PKA phosphorylation on a well conserved serine residue in the cytoplasmic loop between transmembrane segments 8 and 9, a site that may be of particular importance for Na+ transport (33, 46, 47). Our observations that the Ip and the restoration of high, transient increases of dendritic [Na+]i, were almost entirely dependent on α3, made it possible to evaluate the functional response of this isoform to PKA activation. We demonstrate that PKA activation decreases the Ip and attenuates the capacity of the neuron to restore transient large [Na+]i increases in hippocampal neurons, suggesting a PKA-dependent regulation of α3. These findings imply that Na,K-ATPase α3 should be considered as a modulator of synaptic strength and as a target for neurotransmitters, such as dopamine, acting on their postsynaptic receptors.
Under basal conditions, both isoforms were found to contribute to the maintenance of [Na+]i and an appropriate transmembrane Na+ gradient, which enables action potential discharge in the first place. The effects of α3 inhibition on the [Na+]i recorded under basal conditions were relatively modest in comparison with the dramatic effects of α3 inhibition recorded in Na+-loaded cells. However, even small changes in [Na+]i and transmembrane Na+ gradient have a large effect on membrane potential. In line with this, we found that α3 inhibition consistently depolarized hippocampal neurons, an action that may also explain the disruption of organized network activity that we observed in cortical slices.
Taken together, the results from this study suggest that α3 plays a unique and essential role following suprathreshold synaptic activity. These findings may help explain the severe neurological consequences of human α3 mutations. Two rare conditions, rapid onset dystonia parkinsonism and alternating hemiplegia of childhood, have been associated with a variety of missense mutations in the ATP1A3 gene (6, 12). The majority of the rapid onset dystonia parkinsonism mutations have been shown to result in a large reduction in Na+ affinity and pump efficiency. In rapid onset dystonia parkinsonism, stressful events generally precede the onset of dystonic symptoms and in alternating hemiplegia of childhood, which is also associated with dystonia, the hemiplegic symptoms are episodic. It is an intriguing possibility that the sudden onset of symptoms is related to loss of capacity of the mutated α3 to restore intracellular Na+ concentration after a period of suprathreshold activity in groups of neurons.
Acknowledgments
We thank Thomas Liebmann and Nina Illarionova for preparing and maintaining the primary neuronal cultures.
This work was supported by grants from the Swedish Research Council (to A. A. and C. B.), Familjen Erling Perssons Stiftelse (to A. A.), The Torsten Söderberg Foundation (to A. A.), and Rut and Arvid Wolffs stiftelse (to C. B.).
- PKA
- protein kinase A
- GFAP
- glial fibrillary acidic protein
- MAP-2
- Microtubule-associated protein 2
- PSD-95
- postsynaptic density 95
- ANOVA
- analysis of variance.
REFERENCES
- 1. Dobretsov M., Stimers J. R. (2005) Neuronal function and α3 isoform of the Na/K-ATPase. Front. Biosci. 10, 2373–2396 [DOI] [PubMed] [Google Scholar]
- 2. Blanco G., Mercer R. W. (1998) Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am. J. Physiol. 275, F633–650 [DOI] [PubMed] [Google Scholar]
- 3. Kaplan J. H. (2002) Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 71, 511–535 [DOI] [PubMed] [Google Scholar]
- 4. Hieber V., Siegel G. J., Fink D. J., Beaty M. W., Mata M. (1991) Differential distribution of (Na,K)-ATPase α isoforms in the central nervous system. Cell. Mol. Neurobiol. 11, 253–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McGrail K. M., Phillips J. M., Sweadner K. J. (1991) Immunofluorescent localization of three Na,K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na,K-ATPase. J. Neurosci. 11, 381–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Carvalho Aguiar P., Sweadner K. J., Penniston J. T., Zaremba J., Liu L., Caton M., Linazasoro G., Borg M., Tijssen M. A., Bressman S. B., Dobyns W. B., Brashear A., Ozelius L. J. (2004) Mutations in the Na+/K+-ATPase α3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron 43, 169–175 [DOI] [PubMed] [Google Scholar]
- 7. Lingrel J. B., Williams M. T., Vorhees C. V., Moseley A. E. (2007) Na,K-ATPase and the role of α isoforms in behavior. J. Bioenerg. Biomembr. 39, 385–389 [DOI] [PubMed] [Google Scholar]
- 8. Moseley A. E., Williams M. T., Schaefer T. L., Bohanan C. S., Neumann J. C., Behbehani M. M., Vorhees C. V., Lingrel J. B. (2007) Deficiency in Na,K-ATPase α isoform genes alters spatial learning, motor activity, and anxiety in mice. J. Neurosci. 27, 616–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kirshenbaum G. S., Saltzman K., Rose B., Petersen J., Vilsen B., Roder J. C. (2011) Decreased neuronal Na+,K+-ATPase activity in Atp1a3 heterozygous mice increases susceptibility to depression-like endophenotypes by chronic variable stress. Genes Brain Behav. 10, 542–550 [DOI] [PubMed] [Google Scholar]
- 10. Kirshenbaum G. S., Clapcote S. J., Duffy S., Burgess C. R., Petersen J., Jarowek K. J., Yücel Y. H., Cortez M. A., Snead O. C., 3rd, Vilsen B., Peever J. H., Ralph M. R., Roder J. C. (2011) Mania-like behavior induced by genetic dysfunction of the neuron-specific Na+,K+-ATPase α3 sodium pump. Proc. Natl. Acad. Sci. U.S.A. 108, 18144–18149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heinzen E. L., Swoboda K. J., Hitomi Y., Gurrieri F., Nicole S., de Vries B., Tiziano F. D., Fontaine B., Walley N. M., Heavin S., Panagiotakaki E., European Alternating Hemiplegia of Childhood (AHC) Genetics Consortium, Biobanca e Registro Clinico per l'Emiplegia Alternante (I.B.AHC) Consortium, European Network for Research on Alternating Hemiplegia (ENRAH) for Small and Medium-sized Enterpriese (SMEs) Consortium, Fiori S., Abiusi E., Di Pietro L., Sweney M. T., Newcomb T. M., Viollet L., Huff C., Jorde L. B., Reyna S. P., Murphy K. J., Shianna K. V., Gumbs C. E., Little L., Silver K., Ptáček L. J., Ferrari M. D., Bye A. M., Herkes G. K., Whitelaw C. M., Webb D., Lynch B. J., Uldall P., King M. D., Scheffer I. E., van den Maagdenberg A. M., Sisodiya S. M., Mikati M. A., Goldstein D. B. (2012) De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat. Genet. 44, 1030–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosewich H., Thiele H., Ohlenbusch A., Maschke U., Altmüller J., Frommolt P., Zirn B., Ebinger F., Siemes H., Nürnberg P., Brockmann K., Gärtner J. (2012) Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: a whole-exome sequencing gene-identification study. Lancet Neurol. 11, 764–773 [DOI] [PubMed] [Google Scholar]
- 13. Munzer J. S., Daly S. E., Jewell-Motz E. A., Lingrel J. B., Blostein R. (1994) Tissue- and isoform-specific kinetic behavior of the Na,K-ATPase. J. Biol. Chem. 269, 16668–16676 [PubMed] [Google Scholar]
- 14. Zahler R., Zhang Z. T., Manor M., Boron W. F. (1997) Sodium kinetics of Na,K-ATPase α isoforms in intact transfected HeLa cells. J. Gen. Physiol. 110, 201–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rose C. R., Konnerth A. (2001) NMDA receptor-mediated Na+ signals in spines and dendrites. J. Neurosci. 21, 4207–4214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Attwell D., Laughlin S. B. (2001) An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145 [DOI] [PubMed] [Google Scholar]
- 17. Hallermann S., de Kock C. P., Stuart G. J., Kole M. H. (2012) State and location dependence of action potential metabolic cost in cortical pyramidal neurons. Nat. Neurosci. 15, 1007–1014 [DOI] [PubMed] [Google Scholar]
- 18. Cheng X. J., Fisone G., Aizman O., Aizman R., Levenson R., Greengard P., Aperia A. (1997) PKA-mediated phosphorylation and inhibition of Na+-K+-ATPase in response to β-adrenergic hormone. Am. J. Physiol. 273, C893–901 [DOI] [PubMed] [Google Scholar]
- 19. Fisone G., Cheng S. X., Nairn A. C., Czernik A. J., Hemmings H. C., Jr., Höög J. O., Bertorello A. M., Kaiser R., Bergman T., Jörnvall H. (1994) Identification of the phosphorylation site for cAMP-dependent protein kinase on Na+,K+-ATPase and effects of site-directed mutagenesis. J. Biol. Chem. 269, 9368–9373 [PubMed] [Google Scholar]
- 20. Schatzmann H. J. (1953) Cardiac glycosides as inhibitors of active potassium and sodium transport by erythrocyte membrane. Helv. Physiol. Pharmacol. Acta 11, 346–354 [PubMed] [Google Scholar]
- 21. Cornelius F., Mahmmoud Y. A. (2009) Interaction between cardiotonic steroids and Na,K-ATPase. Effects of pH and ouabain-induced changes in enzyme conformation. Biochemistry 48, 10056–10065 [DOI] [PubMed] [Google Scholar]
- 22. Lingrel J. B. (2010) The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Ann. Rev. Physiol. 72, 395–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sweadner K. J. (1985) Enzymatic properties of separated isozymes of the Na,K-ATPase. Substrate affinities, kinetic cooperativity, and ion transport stoichiometry. J. Biol. Chem. 260, 11508–11513 [PubMed] [Google Scholar]
- 24. Urayama O., Sweadner K. J. (1988) Ouabain sensitivity of the α3 isozyme of rat Na,K-ATPase. Biochem. Biophys. Res. Commun. 156, 796–800 [DOI] [PubMed] [Google Scholar]
- 25. Khan F., Spicarová Z., Zelenin S., Holtbäck U., Scott L., Aperia A. (2008) Negative reciprocity between angiotensin II type 1 and dopamine D1 receptors in rat renal proximal tubule cells. Am. J. Physiol. Renal Physiol. 295, F1110–1116 [DOI] [PubMed] [Google Scholar]
- 26. Sanchez-Vives M. V., McCormick D. A. (2000) Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat. Neurosci. 3, 1027–1034 [DOI] [PubMed] [Google Scholar]
- 27. Steriade M., McCormick D. A., Sejnowski T. J. (1993) Thalamocortical oscillations in the sleeping and aroused brain. Science 262, 679–685 [DOI] [PubMed] [Google Scholar]
- 28. Rose C. R., Ransom B. R. (1997) Regulation of intracellular sodium in cultured rat hippocampal neurones. J. Physiol. 499, 573–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Diarra A., Sheldon C., Church J. (2001) In situ calibration and [H+] sensitivity of the fluorescent Na+ indicator SBFI. Am. J. Physiol. Cell Physiol. 280, C1623–C1633 [DOI] [PubMed] [Google Scholar]
- 30. Munakata M., Fujimoto M., Jin Y. H., Akaike N. (1998) Characterization of electrogenic Na/K pump in rat neostriatal neurons. Brain Res. 800, 282–293 [DOI] [PubMed] [Google Scholar]
- 31. Hamada K., Matsuura H., Sanada M., Toyoda F., Omatsu-Kanbe M., Kashiwagi A., Yasuda H. (2003) Properties of the Na+/K+ pump current in small neurons from adult rat dorsal root ganglia. Br. J. Pharmacol. 138, 1517–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rose C. R., Kovalchuk Y., Eilers J., Konnerth A. (1999) Two-photon Na+ imaging in spines and fine dendrites of central neurons. Pflugers Arch. 439, 201–207 [DOI] [PubMed] [Google Scholar]
- 33. Poulsen H., Morth P., Egebjerg J., Nissen P. (2010) Phosphorylation of the Na+,K+-ATPase and the H+,K+-ATPase. FEBS Lett. 584, 2589–2595 [DOI] [PubMed] [Google Scholar]
- 34. McKenzie S., Eichenbaum H. (2011) Consolidation and reconsolidation: two lives of memories? Neuron 71, 224–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cameron R., Klein L., Shyjan A. W., Rakic P., Levenson R. (1994) Neurons and astroglia express distinct subsets of Na,K-ATPase α and β subunits. Brain Res. Mol. Brain Res. 21, 333–343 [DOI] [PubMed] [Google Scholar]
- 36. Pietrini G., Matteoli M., Banker G., Caplan M. J. (1992) Isoforms of the Na,K-ATPase are present in both axons and dendrites of hippocampal neurons in culture. Proc. Natl. Acad. Sci. U.S.A. 89, 8414–8418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bøttger P., Tracz Z., Heuck A., Nissen P., Romero-Ramos M., Lykke-Hartmann K. (2011) Distribution of Na/K-ATPase α3 isoform, a sodium-potassium P-type pump associated with rapid-onset of dystonia parkinsonism (RDP) in the adult mouse brain. J. Comp. Neurol. 519, 376–404 [DOI] [PubMed] [Google Scholar]
- 38. Marks M. J., Seeds N. W. (1978) A heterogeneous ouabain-ATPase interaction in mouse brain. Life Sci. 23, 2735–2744 [DOI] [PubMed] [Google Scholar]
- 39. Sweadner K. J. (1979) Two molecular forms of (Na+ + K+)-stimulated ATPase in brain, separation, and difference in affinity for strophanthidin. J. Biol. Chem. 254, 6060–6067 [PubMed] [Google Scholar]
- 40. Shyjan A. W., Ceña V., Klein D. C., Levenson R. (1990) Differential expression and enzymatic properties of the Na+,K+-ATPase α3 isoenzyme in rat pineal glands. Proc. Natl. Acad. Sci. U.S.A. 87, 1178–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Blanco G., Sánchez G., Mercer R. W. (1995) Comparison of the enzymatic properties of the Na,K-ATPase α3β1 and α3β2 isozymes. Biochemistry 34, 9897–9903 [DOI] [PubMed] [Google Scholar]
- 42. Kim J. H., Sizov I., Dobretsov M., von Gersdorff H. (2007) Presynaptic Ca2+ buffers control the strength of a fast post-tetanic hyperpolarization mediated by the α3 Na+/K+-ATPase. Nat. Neurosci. 10, 196–205 [DOI] [PubMed] [Google Scholar]
- 43. Dryer S. E. (1994) Na+-activated K+ channels: a new family of large-conductance ion channels. Trends Neurosci. 17, 155–160 [DOI] [PubMed] [Google Scholar]
- 44. Bhattacharjee A., Joiner W. J., Wu M., Yang Y., Sigworth F. J., Kaczmarek L. K. (2003) Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J. Neurosci. 23, 11681–11691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yuan A., Santi C. M., Wei A., Wang Z. W., Pollak K., Nonet M., Kaczmarek L., Crowder C. M., Salkoff L. (2003) The sodium-activated potassium channel is encoded by a member of the Slo gene family. Neuron 37, 765–773 [DOI] [PubMed] [Google Scholar]
- 46. Toustrup-Jensen M. S., Holm R., Einholm A. P., Schack V. R., Morth J. P., Nissen P., Andersen J. P., Vilsen B. (2009) The C terminus of Na+,K+-ATPase controls Na+ affinity on both sides of the membrane through Arg935. J. Biol. Chem. 284, 18715–18725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Poulsen H., Nissen P., Mouritsen O. G., Khandelia H. (2012) Protein kinase A (PKA) phosphorylation of Na+/K+-ATPase opens intracellular C-terminal water pathway leading to third Na+-binding site in molecular dynamics simulations. J. Biol. Chem. 287, 15959–15965 [DOI] [PMC free article] [PubMed] [Google Scholar]