

Background: Hsp100 chaperones cooperate with Hsp70 chaperones to disaggregate and reactivate heat-denatured proteins.
Results: Mutations in the interface region between NBD1 and M domains of Hsp100 result in a hyperactive protein toxic to the cell.
Conclusion: The interaction between M and NBD1 domains is crucial for regulation of Hsp100 activity.
Significance: A novel important aspect of the Hsp100 mechanism of action is described.
Keywords: Chaperone Chaperonin, Heat Shock Protein, Protein Aggregation, Protein Denaturation, Stress Response, Hsp100 Chaperone, Hsp70 Chaperone, Protein Disaggregation, Thermotolerance
Abstract
Hsp100 chaperones cooperate with the Hsp70 chaperone system to disaggregate and reactivate heat-denatured aggregated proteins to promote cell survival after heat stress. The homology models of Hsp100 disaggregases suggest the presence of a conserved network of ionic interactions between the first nucleotide binding domain (NBD1) and the coiled-coil middle subdomain, the signature domain of disaggregating chaperones. Mutations intended to disrupt the putative ionic interactions in yeast Hsp104 and bacterial ClpB disaggregases resulted in remarkable changes of their biochemical properties. These included an increase in ATPase activity, a significant increase in the rate of in vitro substrate renaturation, and partial independence from the Hsp70 chaperone in disaggregation. Paradoxically, the increased activities resulted in serious growth impediments in yeast and bacterial cells instead of improvement of their thermotolerance. Our results suggest that this toxic activity is due to the ability of the mutated disaggregases to unfold independently from Hsp70, native folded proteins. Complementary changes that restore particular salt bridges within the suggested network suppressed the toxic effects. We propose a novel structural aspect of Hsp100 chaperones crucial for specificity and efficiency of the disaggregation reaction.
Introduction
The yeast Saccharomyces cerevisiae Hsp104 chaperone, a member of the AAA+ superfamily and the Hsp100 family, is crucial for development of thermotolerance and propagation of yeast prions (1, 2). At the physiological temperature the expression of HSP104 gene is not essential, and no clear phenotype is associated with it (1). The importance of Hsp104 in thermotolerance is due to its ability to solubilize and refold, in cooperation with the Hsp70 chaperone system, proteins trapped in aggregates formed during heat stress (3). Both the disaggregation and prion propagation processes are dependent on the common mechanism of substrate threading through the central channel of Hsp104 (4, 5). The importance of channel integrity for the activity of Hsp100 chaperones was shown by mutagenesis studies (6, 7).
Hsp104 and its orthologs (ClpB in Escherichia coli) involved in disaggregation consist of an N-terminal domain and two nucleotide binding domains (NBD1 and NBD2)5 (8, 9). Despite their diverse functions, AAA+ superfamily members share important features: oligomeric structure, a homologous Walker-type nucleotide binding domain(s), and the capability of utilizing energy from ATP hydrolysis to remodel their substrates (8). The structure and orientation of the domains were determined through analysis of the crystal structure of monomeric ClpB from Thermus thermophilus, the ortholog of Hsp104, and cryoelectron microscopy reconstitution of its oligomer. ClpB forms a two-tiered hexameric ring with the NBD1 and NBD2 domains forming the top and bottom rings, respectively, and surrounding the narrow axial pore (10, 11). The NBD1 domain contains an additional coiled-coil region of four α helices protruding from the hexamer, called the M domain (10). This coiled-coil region resembles a two-wing propeller in which each wing is formed by two α helices twisted around each other and stabilized by leucine zipper-like hydrophobic interactions (12). The presence of the M domain differentiates chaperones involved in disaggregation from the other family members (ClpA, ClpX, HslU) that function as regulatory subunits of proteases (13, 14). The only exception is ClpC, a member of the Bacillus subtilis Hsp100 family. Although it is involved in proteolysis, it has an M domain too. However, this domain is much shorter than the analogous domains in disaggregating chaperones and consists of two α helices only (15).
Because the M domain is characteristic of disaggregating chaperones, substantial efforts have been made to understand its role in the disaggregation process. Yet its function and even its localization within the Hsp100 hexamer remain elusive. Cryoelectron microscopy studies of ClpB and Hsp104 suggest that these proteins, despite considerable sequence similarity, may adopt diverse hexameric structures with the M domains located on either the exterior (10, 16) or interior of the complex (17, 18). The former structure is compact with a narrow pore, whereas the latter is a far more open, basket-like structure (17).
The M domain has been subjected to a number of genetic and biochemical analyses that support the hypothesis of its exterior localization. The mobility of this domain was shown to be essential for disaggregation (10, 19, 20). In agreement with the aforesaid, molecular dynamics studies point out the exceptional mobility of this domain within the modeled hexamers (21). Additionally, the cross-linking studies between NBD1 and the M domain have suggested that its position regulates the ATPase cycle (19). In a recent mutagenesis-based study (12) it was reported that the stability of both wings of the coiled-coil regions is critical for Hsp100 function. Other studies have shown the M domain of ClpB to couple ClpB threading activity with Hsp70 chaperone activity, suggesting that Hsp70 shuffles polypeptides to the entrance of the ClpB channel through interaction with this domain (20). More to the point, genetic screens have identified amino acids lying in the wing-1 region as essential for the function of Hsp104 in yeast and Hsp101 in Arabidopsis (22, 23).
In the present study we have focused on the regions of Hsp104 and ClpB where interactions between residues of helix 2 in wing-1 and specific regions of the NBD1 domain are predicted by our homology modeling to take place. One of the amino acids plausibly involved in such interactions is aspartic acid in position 184. Asp-184 was identified in an earlier genetic screen to be critical for prion propagation and thermotolerance in the yeast S. cerevisiae (24). Further work established the importance of this residue in Hsp100 disaggregase activity (25). However, the actual mechanism in which this residue participates has not been clear. Based on the results of our previous in silico structural studies (21) and the data presented here, we put forward the hypothesis that this residue is part of a novel regulatory element of Hsp104 that consists of ionic interactions between the face-to-face surfaces of the M and NBD1 domains.
EXPERIMENTAL PROCEDURES
Homology Modeling
The homology model of S. cerevisiae Hsp104 was built using the T. thermophilus ClpB crystal structure (PDB entry 1QVR_A) (10). In silico mutations, insertions, and/or deletions necessary to obtain the Hsp104 model were performed using standard AMBER tools (26) and the PyMOL program (27). The C terminus was truncated, as it is irrelevant for nucleotide binding. The three-dimensional models of ATP were built using the coordinates of NTP (PDB entry 1QVR_A). The force parameters of the PB-O3B and O3B-PG bonds and all the angles in which the O3B atom participates were obtained from the AMBER database. Non-standard fragments were parameterized according to Ref. 28 and standard AMBER tools (29). Finally, the entire full-atom model of Hsp104 was energy-minimized.
Protein Purification
Previously published protocols were used for the purification of DnaK, DnaJ, GrpE (30), ClpB (31), GFP (32), RepA1–70-GFP (33), and GroELtrap (D87K) (34). Recombinant firefly luciferase was purchased from Promega. Protein concentrations were determined with the Bio-Rad Protein Assay using BSA as a standard.
Hsp104 protein and its variants were purified from E. coli cells carrying the appropriate gene on a pET5a plasmid. The purification procedure involved Q-Sepharose, Hi-Trap Heparin-Sepharose and Superdex 200 chromatography columns.
The Ssa1 protein was purified from S. cerevisiae cells transformed with the p416TEF-HisSSA1 plasmid. Briefly, the constitutively expressing cells were lysed, and the supernatant was applied onto nickel-nitrilotriacetic acid resin. The His-Ssa1-containing fractions eluted by imidazole gradient were applied onto ATP-agarose, eluted with 5 mm ATP, and subsequently concentrated on nickel-nitrilotriacetic acid resin.
The Ydj1 protein was purified from an E. coli strain transformed with the pBAD-YDJ1 plasmid. Overexpression was induced by 0.2% arabinose. The cell lysate was applied onto DEAE-Sepharose resin. Ydj1-containing fractions were subsequently applied onto hydroxyapatite and Q-Sepharose resin.
Biochemical Analysis
Hsp104 ATPase activity was assayed using the pyruvate kinase/lactate dehydrogenase coupled assay described by Nørby (35). In vitro renaturation of thermally denatured GFP was evaluated as described (32). GFP fluorescence was measured using a Beckman Coulter DTX880 microplate reader. In vitro renaturation of urea-denatured luciferase was performed as described (36). The luminescence of luciferase was analyzed in a Berthold Sirius luminometer.
Site-specific Mutagenesis and in Vivo Experiments
The desired point mutations were introduced into pRS313-HSP104 (for in vivo experiments) or pET5a-HSP104 by PCR site-specific mutagenesis and subsequently confirmed by direct sequencing of the HSP104 gene. Purified plasmids were introduced into S. cerevisiae strain W303hsp104Δ by lithium acetate transformation. The replica plating experiment was performed as described (37). Briefly, 200 colonies were grown on SC plates. Replicas were made on SC and SC (−His) plates. Plating efficiency was calculated as the percentage of colonies growing on the selective plate relative to the number on the non-selective (mother) plate. The experiments were repeated at least three times.
For drop-tests, equal numbers of yeast cells W303hsp104Δ transformed with the pRS313 plasmid-carrying variants of the HSP104 gene were serially diluted and spotted onto the SC (−His) plate. The desired point mutations were introduced into pGB2-clpB (for in vivo experiments) or pET20b-clpB by PCR site-specific mutagenesis and subsequently confirmed by direct sequencing of the clpB gene.
E. coli MC4100ΔclpB cells transformed with variants of pGB2-clpB were grown at 30 °C in LB to an A600 of 0.6. Cells were serially diluted and spotted onto LA (Luria agar, Novagene) plates. Plates were incubated overnight at 37 or 42 °C.
Microscopy Evaluation
S. cerevisiae cells W303hsp104Δ carrying the HSP104 gene or its mutants on a pRS313 plasmid were harvested in log phase and washed 5 times with PBS. Phalloidin staining was done according to Tessarz et al. (38). Cells were evaluated under an Olympus BX51 fluorescence microscope.
E. coli MC4100ΔclpB cells transformed with variants of the pGB2-clpB plasmid were grown at 30 °C in LB to an A600 of 0.4. Then the cultures were transferred to 37 or 42 °C for 3 h. Cells were harvested and suspended in PBS. Plasma membrane vital stain FM4–64 (Invitrogen) was used according to the manufacturer's protocol. Cells were evaluated under an Olympus BX51 fluorescence microscope.
In Vivo Luciferase Renaturation
E. coli MC4100ΔclpB transformed with the pLux plasmid and variants of the pGB2-clpB plasmid were grown at 30 °C in LB medium to an A600 of 0.4. Expression of luxAB genes was induced by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside for 1 h. The cultures were transferred to 42 °C for 30 min to induce the heat shock response. Subsequently tetracycline (25 μg/ml) was added to stop de novo protein synthesis, and luciferase was thermally inactivated at 45 °C for 35 min. The cells were transferred to 30 °C to allow luciferase reactivation. At the indicated time points 500-μl aliquots of the cultures were mixed with 17 μl of 10% n-decanal, and the luminescence of luciferase was analyzed in a Berthold Sirius luminometer.
Template Unwinding Assay
Helicase unwinding assays were performed as previously described (39). The supercoiled plasmid DNA template pKD19L1 (300 ng) containing the RK2 replication origin was used. Proteins were added in the following amounts: DnaA (10 ng), DnaB (600 ng), DnaC (120 ng), TrfA G254D/S267L (250 ng), TrfA (500 ng), HU (5 ng), gyrase (120 ng), and SSB (230 ng). The reactions were incubated at 32 °C for 20 min, then stopped by the addition of EDTA and SDS. DNA samples were analyzed on a 1% agarose gel.
RepA-GFP Unfolding Experiments
The spectrofluorimetric assay was carried out using 0.25 μm RepA-GFP, 6 μm Hsp104 variant, 35 μm GroELtrap, 10 mm ATP, and the creatine kinase/phosphocreatine ATP regeneration system as previously described (33, 40). The level of GFP fluorescence was monitored in a PerkinElmer Life Sciences LS50B spectrofluorimeter at 395-nm excitation and 510-nm emission wavelength.
For GroELtrap complex analysis, the reaction mixtures prepared in 100 μl containing 1.14 μm RepA-GFP, 14 μm GroELtrap, and 2 μm Hsp104 variant were applied on top of 3 ml or 20–45% glycerol gradients and centrifuged for 16 h as described (40). Gradients were fractionated (10 fractions) and analyzed by SDS-PAGE followed by Western blot for GFP and Coomassie Brilliant Blue for GroELtrap.
RESULTS
Analysis of the homology model of the S. cerevisiae Hsp104 monomer indicates that the Asp-184 residue, known to be critical for prion propagation and thermotolerance, may constitute a part of an ionic interaction network. We suggest that the network comprises three charged amino acids that connect the NBD1 domain and the coiled-coil M region (Fig. 1A). The Asp-184 residue comes into contact with lysine in position 358, which is located on the opposite lobe of the ATP-binding cleft in NBD1. The Lys-358 residue in turn is engaged in interaction with aspartic acid 484, which is located in the M domain. The sequence alignment for the NBD1-M interface (Fig. 1B) shows that all three residues, Asp-184, Lys-358, and Asp-484, are highly conserved in 498 analyzed non-redundant sequences of disaggregating Hsp100 chaperones. In particular, (i) Asp-184 is conserved in 100% of the analyzed sequences, (ii) basic amino acid residues are found in all sequences in the position corresponding to Lys-358, and (iii) the position corresponding to Asp-484, despite the very low overall conservation of the M domain, is occupied by an acidic amino acid in 99% of the sequences analyzed, indicating high conservation of charges at these loci (Fig. 1C). In view of the aforementioned data, we suggest that the triad of conserved residues, namely Asp-184, Lys-358, and Asp-484, is responsible for NBD1-M interdomain communication and possibly the transmission of conformational changes between the highly mobile coiled-coil M domain and the catalytic NBD1 domain. To verify our hypothesis, we have evaluated the effects of point mutations at each position. Site-directed mutagenesis of the HSP104 gene under control of its native promoter was performed on the centromeric pRS313 plasmid. Three mutants were created in which the charge of the analyzed amino acid was changed to the opposite one (D184K, K358E, and D484K). Initially, we intended to transform the S. cerevisiae hsp104Δ strain with the respective pRS313 plasmids to analyze the thermotolerance of the mutants. Yet, remarkably, yeast expressing the plasmid-borne Hsp104 K358E or Hsp104 D484K variants showed growth arrest on selective medium (−His) in drop-test experiments (Fig. 2A). Replica-plating experiments revealed that only a small percentage of cells carrying the pRS313 plasmid coding for either of these variants was able to form colonies on selective medium after growth on non-selective medium compared with cells expressing wild type Hsp104 or the D184K mutant (Fig. 2B). This experiment shows that during growth on non-selective medium the plasmid coding for both the selection marker gene and the HSP104 gene variant is lost when either the K358E or D484K mutation in HSP104 gene is present on the plasmid. No such effect was observed for wild type or the D184K version of HSP104. The rapid loss in the absence of selective pressure of the plasmid coding for the K358E and D484K Hsp104 variants suggests that these Hsp104 variants are toxic to the cell. This result was additionally confirmed by analysis of Hsp104 protein levels in selective and non-selective conditions. In yeasts cultured on a non-selective medium, the level of Hsp104 K358E or Hsp104 D484K was below the limit of detection by Western blot analysis. When yeast cells were grown under selective pressure, the conditions in which the presence of the plasmid is obligatory, the level of Hsp104 was similar for all the analyzed mutants (data not shown). However, under these conditions, much slower growth was observed in liquid cultures for the toxic variants (data not shown). Taken together, the results of drop-tests, replica plating, and protein level analysis experiments indicate that the K358E and D484K Hsp104 variants are toxic to yeast cells.
FIGURE 1.

Putative ionic interactions between M and NBD1 domains. A, the homology model of S. cerevisiae Hsp104 is shown. Domains are color coded as follows: cyan, N-terminal domain; green, NBD1; magenta, coiled-coil M domain; yellow, NBD2. ATP molecules are shown. The circled magnified fragment shows the arrangement of the indicated charged amino acid residues in an energy-minimized structure. B, shown is sequence alignment of the regions containing the Asp-184, Lys-358, and Asp-484 residues (colored red for negative, blue for positive charge). The sequences of S. cerevisiae (Sc) and Candida albicans (Ca) Hsp104 and T. thermophilus (Tht) and E. coli (Eco) ClpB proteins are shown. C, shown is schematic representation of the putative NBD1-M domain interface. The percentage of residue conservation shown in parentheses was calculated using the ConSurf server for 498 non-redundant sequences from UNIREF90 database.
FIGURE 2.

Analysis of the toxicity of the HSP104 mutants. A, shown are drop-tests of yeast W303hsp104Δ strain transformed with pRS313 plasmid-carrying variants of the HSP104 gene. Equal amounts of yeast cells (106 cells/ml) were serially diluted and spotted onto the SC (−His) plate. B, shown is replica plating efficiency of S. cerevisiae hsp104Δ cells transformed with the pRS313 plasmid carrying each particular variant of the HSP104 gene. Data are the means (±S.D.) of at least three independent experiments.
To identify the underlying cellular defects leading to the growth arrest of cells expressing Hsp104 mutant variants, we have tested for possible morphological alterations. Cells expressing Hsp104 K358E or D484K displayed a significant change in cell size distribution compared with cells expressing wt Hsp104 or its D184K variant (Fig. 3A) or cells carrying the empty pRS313 plasmid (data not shown). A substantial percentage of yeast cells expressing Hsp104 K358E or D484K had diameters exceeding 13 μm. These enlarged cells were frequently found lysed. Conversely, no cells larger than 13 μm in diameter were observed for yeast carrying the wt Hsp104 or empty plasmid.
FIGURE 3.

Morphology of S. cerevisiae W303 hsp104Δ cells carrying variants of the HSP104 gene. A, the upper panel shows representative microscopic images of cells. For toxic K358E and D484K mutants, besides an apparent increase of size, numerous lysed cells were also observed. Lower panel, histograms of cell diameters for respective HSP104 mutants (∼100 cells were measured) are shown. B, fluorescence microphotographs of S. cerevisiae cells stained with TRITC-phalloidin to visualize actin structures are shown.
Such a phenotype is typical for mutations affecting the actin cytoskeleton (41, 42). To further test our observation, we examined the actin cytoskeleton using TRITC-labeled phalloidin. In contrast to the cells expressing wt Hsp104, those expressing the K358E or D484K variants were found to have severely impaired integrity of their actin cytoskeleton (Fig. 3B).
Subsequently, we tested whether removal of the charged groups in Hsp104 instead of such a drastic change as charge reversal results in cell toxicity. In yet another series of mutagenesis studies, we exchanged each charged amino acid with a polar residue. Thus, three novel HSP104 mutants, namely D184S, K358S, and D484S, were created. Hsp104 K358S, but not the D484S and D184S variants, conferred toxicity to the host cell (Fig. 2B). A similar toxic phenotype was observed for the Hsp104 K358A mutant (Fig. 2B).
Together, the results suggest that the amino acids 358 and 484 are crucial for the in vivo functioning of Hsp104. The lysine at position 358, which in our model forms ionic interactions with aspartic acid at positions 184 and 484, seems to be the key element of the network as not only a change of the charge, but also substitutions with a polar (serine) or small hydrophobic amino acid (alanine) result in the toxic effect.
To better understand the bases of the observed toxic effect, we purified the mutants for biochemical analyses. CD spectra were recorded, and the ability to form hexamers was analyzed for all mutants. No major differences were found when compared with wild type Hsp104 (data not shown), except for the impaired hexamerization of the D184K mutant. However, biochemical assays revealed drastic effects on protein function. The ATPase activity of the Hsp104 D184K variant was severely diminished, whereas the two “toxic” variants (K358E and D484K) were characterized by activity considerably higher (10–15-fold) than wild type (Fig. 4A). We have also examined the ability of our Hsp104 variants to disaggregate heat-aggregated proteins in collaboration with the Hsp70 chaperone system (Ssa1, Ydj1 proteins). To our surprise, thermally denatured GFP was reactivated much more efficiently by the K358E and D484K toxic variants compared with wild type Hsp104, whereas the D184K variant remained, as expected from in vivo studies (43, 44), inactive in disaggregation (Fig. 4B). Similar results were obtained from disaggregation studies of firefly luciferase (data not shown), indicating that these toxic mutants are “hyperactive.”
FIGURE 4.

Biochemical properties of the Hsp104 variants. A, shown is ATPase activity of Hsp104 and its variants. Data are the means (±S.D.) of three independent experiments. B, renaturation of thermally denatured GFP by purified variants of Hsp104 and wild type protein at 2 μm concentration is shown. Reactivation was performed in the presence of the Hsp70 chaperone system (Ssa1 (3 μm) and Ydj1 (1 μm)). C, shown is Hsp70-independent reactivation of thermally denatured GFP by variants of Hsp104 at 2 μm concentration. The amount of native GFP used in experiments in panels B and C corresponds to 8 × 106 fluorescence (absorbance units (a.u.)).
As Hsp100 disaggregases require the presence of the Hsp70 chaperone system for their activity, we tested whether the activity of our hyperactive mutants is still dependent on Hsp70. Both hyperactive variants, K358E and D484K, were able to reactivate aggregated GFP in the absence of Hsp70 quite efficiently (Fig. 4C). As expected, wt Hsp104 and the D184K variant were inactive under such conditions (Fig. 4C). Although the disaggregating activity of the hyperactive K358E and D484K mutants is stimulated by the Hsp70 system, in its absence the mutants retain a level of activity similar to wild type Hsp104 operating in the presence of Hsp70 (Fig. 4, B and C).
Because the triad of charged amino acids is extremely conserved in all disaggregating chaperones and strong effects of the mutations were observed in our in vivo and in vitro studies, we decided to perform a similar series of experiments for ClpB, the E. coli orthologous disaggregase. Site-directed mutagenesis of the clpB gene under control of its native promoter was performed on the low copy pGB2 plasmid. Three clpB mutants were created in which the charge of the analyzed amino acid was changed to the opposite one (D179K, R353E, and E481R, respectively) (Fig. 5A). The influence of these clpB mutants on the growth of E. coli clpBΔ was analyzed in drop-test experiments. No major differences in the growth of the mutants and the wt bacteria at 37 °C were observed. However, we noticed that the clpB R353E mutant forms smaller colonies (Fig. 5B). When the plates were incubated at 42 °C, the temperature at which ClpB expression is triggered by its heat shock inducible promoter, a strong thermosensitive-like phenotype was observed for the clpB R353E mutant (Fig. 5B). Only a small fraction of the cells was able to grow under heat shock conditions, suggesting that increased expression of the mutated clpB gene inhibits growth. Microscopic observations revealed that bacterial cells expressing R353E clpB form short filaments at 37 °C that became substantially longer when the cells are cultured at 42 °C (Fig. 5C).
FIGURE 5.

Analysis of the toxicity of clpB mutants. A, shown is a schematic representation of the clpB mutants. B, shown are drop-tests of the E. coli MC4100 ΔclpB strain transformed with the pGB2 plasmid-carrying variants of the clpB gene. Equal amounts of cells were serially diluted (Dil.) and spotted onto the plate and grown at 37 or 42 °C. C, shown is morphology of the E. coli MC4100 Δclp cells carrying the indicated variants of the clpB gene on a pGB2 plasmid. The cells were stained with plasma membrane FM4–64 vital stain.
For a comprehensive comparison with Hsp104, we purified the ClpB mutants for biochemical analyses. Their CD spectra were recorded, and no major differences were found compared with wild type ClpB (data not shown). The ATPase activity of the ClpB D179K variant was severely diminished, whereas the “thermosensitive” variant (R353E) possessed considerably higher activity (∼20-fold) than wild type ClpB (Fig. 6A). The ATPase activity of the E481R variant was similar to that of the wild type protein (Fig. 6A). Subsequently, we examined the ability of ClpB variants to disaggregate aggregated proteins in collaboration with the Hsp70 chaperone system (DnaK, DnaJ, and GrpE). Denatured luciferase was disaggregated and reactivated 3-fold faster by the thermosensitive variant (R353E) compared with wild type ClpB (Fig. 6B). The E481R variant was less active than wild type ClpB, whereas the D179K variant was inactive with respect to disaggregation (Fig. 6B). Our results show clearly that the thermosensitive R353E mutant is hyperactive in a way similar to the homologous yeast chaperone. Accordingly, we have analyzed whether the activity of our hyperactive ClpB R353E protein is still dependent on the DnaK, DnaJ, and GrpE chaperones. The R353E variant was able to reactivate aggregated luciferase in the absence of DnaK and its co-chaperones (Fig. 6C), albeit the efficiency of reactivation was not high. As expected, wt ClpB and its D179K and E481R variants were inactive under such conditions (Fig. 6C).
FIGURE 6.

Biochemical properties of the purified ClpB variants. A, shown is ATPase activity of ClpB variants. B, shown is renaturation of thermally denatured luciferase by purified variants of ClpB and wild type protein at 2 μm concentration. Reactivation assays were performed in the presence of the Hsp70 chaperone system (DnaK (1 μm), DnaJ (0.2 μm), and GrpE 0.1 μm)). C, shown is an Hsp70-independent reactivation of thermally denatured luciferase by variants of ClpB at 6 μm concentration. Data are the means (±S.D.) of three independent experiments.
Taken together, the results obtained for homologous mutations in Hsp104- and ClpB-disaggregating chaperones clearly indicate that there is a correlation between an increase in disaggregation activity, in vitro independence from the Hsp70 system in disaggregation, and the toxic effect observed in vivo. One may have expected that the elevated disaggregation activity would be beneficial to the cells challenged by heat shock conditions. However, the observed toxic effect was severe for yeast. It was less pronounced in bacteria as the latter was observed during prolonged growth at high temperature. Therefore, we decided to use the bacterial system to analyze how the toxic ClpB variant (R353E) influences the disaggregation process after a relatively short exposure of the cells to heat shock conditions. Firefly luciferase was expressed in E. coli cells carrying plasmid-borne clpB mutants. After a relatively short heat shock exposure at 45 °C, the activity of luciferase was analyzed. The toxic, hyperactive R353E clpB mutant was as effective in disaggregation and recovery of heat-inactivated luciferase as the wild type clpB or E481R mutant (Fig. 7A). The clpB D179K mutant was inactive in this experiment similarly to the control cells transformed with an empty plasmid (Fig. 7A).
FIGURE 7.

In vivo activity of clpB mutants. A, shown is luciferase reactivation in E. coli cells carrying the pGB2 plasmid with indicated clpB variants and a luciferase-coding plasmid. The increase in culture luminescence was analyzed. Time 0 corresponds to the end of the heat shock (35 min at 45 °C). Subsequent increase in luminescence was expressed as a percentage of the maximum level of luminescence before heat shock (∼10 million counts). Data are the mean (±S.D.) of three independent experiments. B, shown is thermotolerance of E. coli MC4100 ΔclpB cells transformed with either empty pGB2 or plasmid carrying wild type clpB or its R353E variant. Exponentially growing E. coli cells were transferred to 42 °C to induce the heat shock response. After this, the cells were transferred to 50 °C. At indicated time points cells were serially diluted and spotted onto the LA plates.
We have also analyzed the ability of E. coli cells expressing clpB mutant genes to develop thermotolerance and survive short extreme heat shock. The toxic hyperactive clpB R353E mutant plasmid was able to partially restore thermotolerance compared with the empty plasmid (Fig. 7B). Thus, the clpB R353E mutation is beneficial and not toxic to cells after a relatively short exposure to heat shock conditions. On the other hand, the growth of the cells carrying the clpB R353E mutant is inhibited during continuous incubation at high temperature. Our results suggest that the beneficial function of this clpB mutant is related to disaggregation of aggregates and the harmful effect observed for prolonged growth at high temperature has a different nature.
More insight into the mechanism of the toxicity of disaggregases was provided by the K358E and D484K Hsp104 yeast chaperone variants that efficiently slow down the growth of E. coli when overexpressed in the BL21 strain (Fig. 8). This effect was not observed in DH5α in which transcription of pET5a plasmid-encoded genes is not possible (data not shown). Taking into account the fact that the E. coli Hsp70 system (DnaK, DnaJ, GrpE) cannot cooperate in the disaggregation reaction with Hsp104 (3, 36, 45), our results point out that the harmful activity of the disaggregase variants is Hsp70-independent.
FIGURE 8.

The growth of bacterial colonies after the transformation of pET5a plasmid carrying analyzed HSP104 mutants into BL21(DE3) E. coli strain. The photographs were taken 24 and 40 h after the transformation.
Wild type ClpB and Hsp104 have never been reported to unfold native proteins alone, in the absence of the Hsp70 chaperone system under normal conditions. Such independent activity of mutated disaggregases might be harmful to the cell if it results in unfolding of native proteins.
To evaluate the effect of the Hsp100 mutants on native proteins, we decided to implement the experimental approach designed by Wickner and co-workers (33) in which a GFP fusion protein containing a fragment of RepA (amino acids residues 1–70) is unfolded and captured by the mutant chaperonin GroELtrap. GroELtrap captures unfolded proteins in the molten-globule state (34, 46) and prevents refolding but does not bind native GFP (47). When RepA-GFP was incubated in the presence of hyperactive Hsp104 K358E and GroELtrap, a significant decrease in GFP fluorescence was observed (Fig. 9A), suggesting that the hyperactive Hsp104 mutant is able to unfold the RepA-GFP fusion protein. Conversely, no decrease was observed for wild type Hsp104 (Fig. 9A). When wild type GFP was used in a similar experiment, the unfolding effect observed for hyperactive Hsp104 mutant was marginal (data not shown). Thus, some feature of the RepA1–70 fragment provides a recognition signal for substrate engagement and allows the hyperactive Hsp104 variant to unfold a stable domain of GFP. The fact that the observed decrease of fluorescence results from RepA-GFP unfolding and capturing of the unfolded polypeptides by GroELtrap was confirmed by sedimentation analysis. After sedimentation, a substantial fraction of RepA-GFP co-sedimented with heptameric GroELtrap when the hyperactive Hsp104 K358E mutant was present, whereas no such effect was observed for wild type Hsp104 (Fig. 9B). Thus, we conclude that hyperactive, but not wild type Hsp104 is able to unfold highly stable GFP protein possessing the N-terminal fragment of RepA, required for substrate engagement.
FIGURE 9.

Hyperactive Hsp100 variants unfold native proteins. A, unfolding of RepA1–70-GFP by the Hsp104 K358E variant and wt Hsp104 is shown. At time 0 the indicated variant of Hsp104 was added to the fluorimetric cuvette containing RepA1–70-GFP, and GroELtrap. GFP fluorescence was recorded. A.U., absorbance units. B, RepA1–70-GFP polypeptide was recognized by GroELtrap as a result of hyperactive Hsp104 variant unfolding of native protein. Sedimentation (16-h) of protein complexes formed after the addition of Hsp104 variants to the reaction containing RepA1–70-GFP and GroELtrap. Fractions were analyzed by SDS-PAGE followed by a Western blot for RepA1–70-GFP and Coomassie Brilliant Blue for GroELtrap. C, hyperactive ClpB R353E, but not wild type ClpB, activates dimeric TrfA in the absence of the Hsp70 chaperone system. TrfA replication protein activity was determined using a reconstituted purified protein system and a helicase-dependent DNA unwinding assay with a supercoiled plasmid DNA template. Formation of FI*, an extensively unwound covalently closed circular (CCC) DNA form, depends upon TrfA-dependent origin opening and helicase loading. Chaperone proteins (ClpB and variants (2 μm), ClpX (1.6 μm), DnaK (2.2 μm), DnaJ (0.4 μm), and GrpE (1.2 μm)) were present in the reactions as indicated.
A similar experimental approach was applied to the TrfA protein, a native substrate of the ClpB chaperone. Synergistic action of ClpB and DnaK, DnaJ, and GrpE activates the TrfA protein, the replication initiation factor of RK2 plasmid, by converting inactive dimers to an active monomer form (48). The replication activity of TrfA can be determined in vitro by analyzing FI* formation, an extensively unwound covalently closed circular DNA replication intermediate (37). Using this assay we analyzed the ability of the hyperactive R353E ClpB variant to catalyze TrfA monomerization in the absence of the DnaK chaperone system. The R353E ClpB variant, as opposed to wild type ClpB, was fully active in the TrfA activation process performed in the absence of the DnaK system (Fig. 9C). This result suggests that the hyperactive mutant is not only capable of disaggregating aggregates on its own but is able to act unaided on properly folded dimeric TrfA protein. Additional in vivo results (Fig. 7) suggest that the toxic R353E effect is not related to the disaggregation reaction.
The above experimental results directly showing the unfolding of RepA-GFP fusion protein by the hyperactive Hsp104 mutant and the Hsp70-independent monomerization of the dimeric TrfA replication initiation protein by the hyperactive ClpB mutant suggest that the capacity to unfold even properly folded native proteins might be the reason for the observed in vivo toxicity of these mutants.
Our model implies that ionic interactions formed by the three charged amino acids connect the NBD1 domain and the coiled-coil M region. Because all the yeast and bacterial Hsp100 chaperone mutations, except E481R clpB, which change the charge within the analyzed region, resulted in either an inactive or hyperactive toxic protein, we have decided to recreate the ionic interactions that had been disrupted in single mutants. Therefore, we have performed a second round of site-directed mutagenesis to determine whether the intragenic suppression occurs. Thus, in addition to the initial K358E mutation in HSP104, we have also reversed the charge of the putative interacting amino acid partner. Accordingly, two additional HSP104 mutants, namely D184K/K358E and K358E/D484K, were created (Fig. 10A). Neither of the two novel variants resulted in the toxic effect observed in K358E and D484K mutants in drop-test experiments (Fig. 10B). More to the point, the size and morphology of the yeast cells were normal, and no major differences were found in replica plating experiments (data not shown). Next, because both compensatory mutants D184K/K358E and K358E/D484K were able to suppress toxic phenotype, we analyzed the ability of these double mutants to provide thermotolerance. Both the D184K/K358E and K358E/D484K mutants showed partial thermotolerance compared with wild type HSP104 (data not shown), suggesting that they have regained some activity in vivo.
FIGURE 10.

Compensatory mutations, which potentially recreate ionic interactions, suppress the toxic effect observed for both HSP104 and clpB mutants. A, schematic representation of the analyzed amino acids in Hsp104 and (ClpB) variants is shown. B, drop-tests of yeast W303hsp104Δ strain transformed with the pRS313 plasmid carrying variants of the HSP104 gene are shown. Equal amounts of yeast cells (106 cells/ml) were serially diluted (Dil.) and spotted onto the SC (−His) plate. C, shown are drop-tests of E. coli MC4100 ΔclpB strain transformed with the pGB2 plasmid carrying variants of the clpB gene. Equal amounts of cells were serially diluted and spotted onto the plate and grown at 4 °C.
Similar compensatory mutations were introduced into bacterial clpB. Starting from the hyperactive, thermosensitive R353E clpB mutant, the double mutants D179K/R353E and R353E/E481R were constructed (Fig. 10A). Neither double mutant displayed the thermosensitive phenotype of the R353E clpB mutant (Fig. 10C). Next, we analyzed the thermotolerance properties of these mutants. The D179K/R353E mutant exhibits the same level of thermotolerance as the wild type strain despite the fact that when acting individually, the D179K mutant does not tolerate extreme heat shock (50 °C), and the R353E variant is only partially tolerant (Fig. 7B and data not shown). The other double mutant, R353E/E481R, did not show thermotolerance, in contrast to the single mutants R353E and E481R that tolerate the elevated temperature (Fig. 7B and data not shown). The fact that toxic effects due to the single mutations can be suppressed by specific intragenic mutations in both yeast and bacterial Hsp100 disaggregases substantiates the existence of the postulated ionic interactions.
Next we purified the Hsp104 and ClpB double mutants for biochemical analyses. Both D184K/K358E Hsp104 (Fig. 11A) and D179K/R353E ClpB (Fig. 11B) variants were active in the disaggregation reaction performed in the presence of the respective Hsp70 chaperone system. Independence from the Hsp70 system was assessed using the luciferase disaggregation assay, which resulted in only trace amounts being renatured (∼0.1% for the Hsp104 variant and below detection level for the ClpB variant). Additionally, the D179K/R353E ClpB variant does not have the ability to catalyze TrfA monomerization in the absence of the DnaK chaperone system (Fig. 11C). Thus, our compensatory mutations result in the loss of the hyperactivity observed in the K358E Hsp104 and R353E ClpB toxic variants as well as restore the dependence on the Hsp70 chaperone system. We also purified the compensatory mutants K358E/D484K Hsp104 and R353E/E481R ClpB and showed that both are inactive in the disaggregation reaction despite the fact that at least one of them, K358E/D484K Hsp104, is partially active in vivo (data not shown).
FIGURE 11.

Biochemical analysis of the purified compensatory Hsp104 and ClpB variants. A, shown is renaturation of thermally denatured GFP by purified compensatory D184K/K358E Hsp104 variant and wild type protein. Reactivation was performed in the presence of the Hsp70 chaperone system (Ssa1 and Ydj1). The amount of native GFP used in the experiment corresponded to 8 x 106 fluorescence. A.U., absorbance units. B, shown is renaturation of thermally denatured luciferase by purified compensatory D179K/R353E ClpB variant (1.8 μm) and wild type ClpB (1.8 μm). Reactivation was performed in the presence of the Hsp70 chaperone system (DnaK (1 μm), DnaJ (0.2 μm), and GrpE 0.1 μm)). C, the compensatory D179K/R353E ClpB, as opposed to the R353E ClpB variant, is not able to activate dimeric TrfA in the absence of the Hsp70 chaperone system. TrfA replication protein activity was determined using a reconstituted purified protein system and a helicase-dependent DNA unwinding assay with a supercoiled plasmid DNA template. The concentration of chaperone proteins was as in Fig. 8. CCC, covalently closed circular.
The observed restitution of the strict Hsp70 dependence and the substantial suppression of the in vitro hyperactivity by restoring postulated ionic interactions in our double mutants are in complete concordance with the in vivo observations presented earlier, affirming the importance of the circuit constituted by the analyzed residues. However our observations do not provide evidence that residues 184, 358, and 484 in Hsp104 in fact directly interact. To address this issue, we created a series of Hsp104 mutants in which the residues in question were replaced by cysteines. Three single cysteine variants (D184C, K358C, and D484C) and two double cysteine variants (D184C/K358C and K358C/D484C) were created. The respective proteins were purified, and their mobility was analyzed under reducing and non-reducing conditions in SDS-PAGE. All analyzed proteins migrated similarly in SDS-PAGE under reducing conditions (Fig. 12). However, under non-reducing conditions, both the D184C/K358C and K358C/D484C double cysteine variants displayed bands corresponding to proteins migrating more slowly due to the formation of disulfide bridges (Fig. 12). No such effect was observed with the single cysteine variants. This shows that the analyzed residues are indeed in very close proximity to one another and are able to directly interact.
FIGURE 12.

Residues 184, 358, and 484 lie close enough to each other in the Hsp104 structure to form disulfide bridges in the respective double cysteine variants. Mobility of the Hsp104 cysteine variants was analyzed by SDS-PAGE followed by Coomassie Brilliant Blue staining. Proteins were incubated in 5 mm iodoacetamide (10 min) before electrophoresis to block the cysteines that do not form disulfide bridges. To better visualize the mobility differences, the separation time was substantially increased. Shown is SDS-PAGE under reducing conditions (left penal) and under non-reducing conditions (right panel). βME, β-mercaptoethanol.
DISCUSSION
The findings presented in this work demonstrate a novel structural aspect of the Hsp100 disaggregases. We propose that a network of ionic interactions on the interface between the NBD1 and M domains is crucial for disaggregase activity.
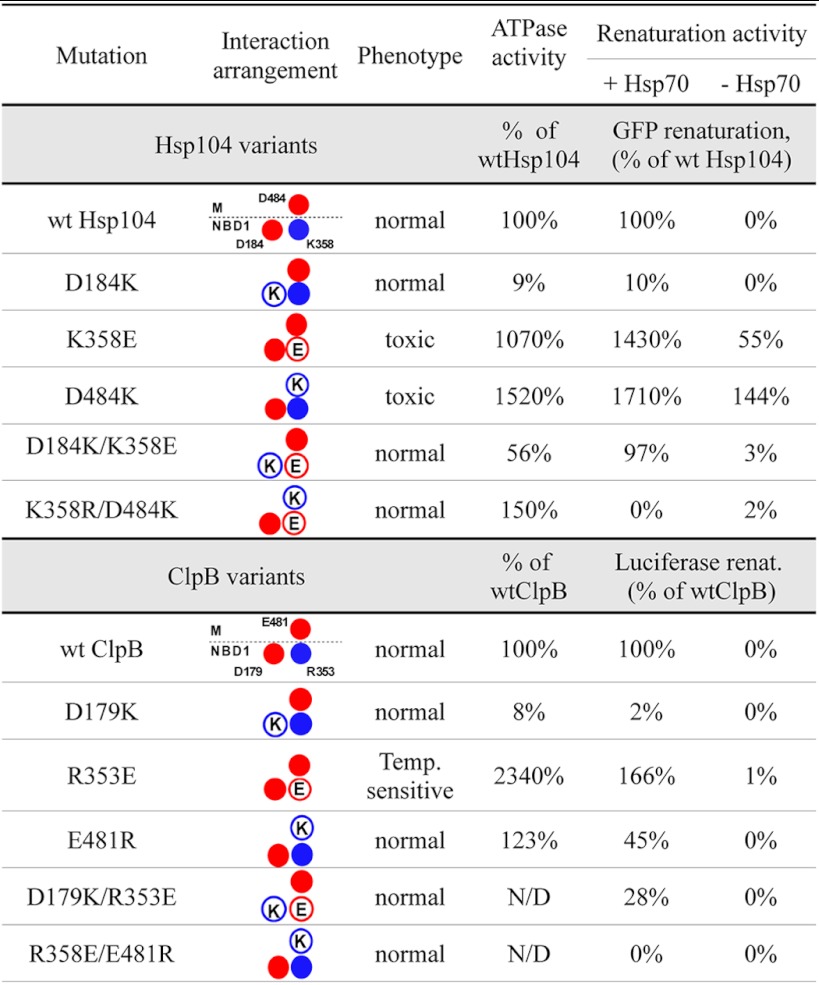
Disruption of these postulated ionic interactions in the yeast Hsp104 chaperone imposes a serious growth impediment on cells under selective pressure. Rapid loss of the corresponding mutant Hsp104-encoding plasmids in the absence of selective pressure was observed. The toxic effects of analogous mutations in bacterial clpB were also observed; however, these were less severe. Purified toxic variants of Hsp104 and ClpB displayed unexpected biochemical properties, namely a significant increase in both ATPase activity and the substrate disaggregation rate. Thus, these mutants constitute much more efficient disaggregating machines compared with their wild type counterparts, at least from the biochemical point of view. Their properties are summarized in Table 1. Because the major function of Hsp104 and ClpB has been defined as the disaggregation of aggregated proteins following heat stress, it is surprising that the increase in efficiency is not beneficial to the cell and does not confer a more favorable phenotype. This paradox can be resolved by assuming that it is not the increased disaggregation ability but rather the loss of regulation and/or specificity of this process that is toxic to cells. In agreement with the aforementioned statement, toxic mutants were partially active in disaggregation in the absence of the Hsp70 system, which works upstream of Hsp100 and has been suggested to initiate the disaggregation (4, 32, 40). More to the point, the hyperactive Hsp104 K358E variant was able to efficiently unfold, in the absence of Hsp70 chaperone system, the monomeric RepA-GFP fusion protein, suggesting that the toxic effect may result from uncontrolled unfolding of native proteins. Also the hyperactive R353E ClpB variant was able to efficiently monomerize, in the absence of DnaK, the dimeric TrfA protein. TrfA is a natural native substrate of the DnaK and ClpB bi-chaperone system for which the DnaK chaperone requirement was shown to be strict (48). It is difficult to speculate about the specificity and the level of undesirable unfolding activity in the living cell without performing proteomic studies. It seems, however, rational to assume that the toxic effect exerted by mutated disaggregases is connected to the unfolding of proteins important for the cell. Recent studies using a dominant-negative variant of Hsp104 that degrades substrate proteins instead of remodeling them suggest that the Spa2 protein, a polarisome component, is a substrate of Hsp104 (38). The possible increased unfolding of proteins responsible for cytoskeleton organization explains the morphological changes, growth arrest, and frequently observed cell lysis as well as changes in phalloidin fluorescent foci in affected cells shown in our study. However, although these are easily detectable changes, they are not necessarily the only process that is disturbed, and unfolding of other proteins may possibly further contribute to the observed toxic phenotype.
TABLE 1.
Properties of analyzed Hsp104 and ClpB variants
The ATPase activity of wt Hsp104 was 15.3 min−1. The disaggregation and reactivation rate was measured in GFP disaggregation assay (31,080 fluorescence units/min for wild type Hsp104). The renaturation rate of Hsp104 in the absence of Hsp70 system is expressed as percent of wild type Hsp104 rate measured in the presence of Hsp70. The ATPase activity of wt ClpB104 was 6.9 min−1. The disaggregation and reactivation rate was measured in luciferase disaggregation assay (increase of 1.93% of native luciferase activity/min for wild type ClpB). The renaturation rate of ClpB in the absence of Hsp70 system is expressed as percent of wild type ClpB rate measured in the presence of Hsp70.
We speculate that in vivo the disaggregation activity of Hsp104/ClpB must be controlled in a very precise manner. We believe that disaggregases have evolved not only to be maximally efficient but that they must also be effectively controlled. The unique M subdomain, which was shown to be responsible for interaction with Hsp70 chaperones (20, 49, 50) and which interacts with the NBD1 domain, may play a crucial role in this regulation.
The ionic interactions between the M and NBD1 domains constitute an important aspect of the Hsp100-mediated disaggregation mechanism. Understanding the principles of its action will improve our knowledge about the structure-function relationship of this class of chaperones. Accordingly, we have performed detailed in vitro studies of the Hsp104/ClpB variants that have alterations in the NBD1-M domain interface. Our results confirm the importance of the interaction between charged residues within this region and suggest that the conserved, positively charged Lys-358/Arg-353 residue plays a critical role in control of the disaggregating activity. The close proximity of the analyzed residues, deduced from in silico modeling, was confirmed by constructing cysteine variants in which disulfide bridges were formed between residues 358 and 184 and between residues 358 and 484. ATPase activity and disaggregation data show that in our hyperactive variants disruption of the ionic interactions within the triad of residues does not simply decouple ATP hydrolysis from the production of useful force for threading substrates. Rather, it is the fully functional working cycle that is stimulated in hyperactive variants.
Our hypothesis is in complete accordance with the current state of knowledge regarding the role of the M domain. It was shown that Hsp70 chaperones act upstream of Hsp100 in the disaggregation reaction (4, 32). The M domain of Hsp104 was shown to be the site of Hsp70 interaction (20, 49, 50) and must undergo conformational changes to allow Hsp100 to disaggregate (10, 20). This possibly leads to changes in the pattern of interactions within the 184–358-484 (179–353-481 for ClpB) triad of the native protein, which in turn releases the power generated by the ATPase active site. The interaction triad may be artificially fixed in a state where there is no contact between Lys-358 (Arg-353) and Asp-484 (Glu-481) as in the case of the K358E (R353E) or D484K mutation. Such a situation possibly emulates the state of activation by Hsp70 even in the absence of this chaperone. Recently, it was shown that the Hsp104 chimera harboring T4 lysozyme within the M domain gains the ability to solubilize heat-aggregated substrate in the absence of Hsp70 (16). We propose that alteration of the mobility of the M domain in the chimera may influence M-NBD1 interdomain communication as suggested for our hyperactive mutants. On the other hand, it was shown by Doyle and co-workers (33) that the balance between ATP binding and hydrolysis is crucial for regulation of “holding” and “unfolding” activity of the Hsp100s. Disturbing this balance by the addition of a nucleotide analog (ATPγS) caused disaggregation of some substrates to become Hsp70-independent. We postulate that the regulatory role of Hsp70 is carried out by means of regulating ATP binding site properties. This would be in accordance with the putative role of residues 184, 358, and 484; that is, their regulatory role in the ATPase cycle is dependent on the position of the M domain that may be influenced by Hsp70 binding (20, 49, 50).
In line with our structural model of the interaction site, the reversal of charges in positions 358 and 484 should result in no electrostatic attraction between the two lobes of NBD1 (residues 184, 358, both negative). Thus the NBD1-M interdomain salt bridge (residues 358 and 484, opposite charges), not having a competing counterpart, would be effectively strengthened. It was shown (10) that fixing the M domain close to NBD1 by introducing a disulfide bridge in this region for ClpB, inhibits the activity of the protein. Therefore, the observed lack of disaggregation activity in the K358E/D484K Hsp104 (R353E/E481R in ClpB) mutant can be explained by the stabilization of the M domain in contact with NBD1. This presents another argument for the validity of our proposed model.
The interpretation of Hsp100 mutant properties must, however, take into account the fact that “compensatory” mutations result in significant change of the protein sequence. The “triad” concept is obviously a simplified view, and residues 184 (179), 358 (353), and 484 (481) do interact with other amino acids in their neighborhood. Thus, other effects such as steric disturbance or triad interaction with other residues may influence protein activity in other ways.
Our findings cast a new light into the complex process of Hsp100/Hsp70 disaggregation. The Hsp70 system, shown to act during the first steps of the process and even to renature some model protein aggregates, is thought to prepare aggregates by extracting loose ends of polypeptide chains. These ends are presented to Hsp100 to be threaded through the hexameric central pore. Although we support this model of action, our results suggest that the role of Hsp70 is not only to present substrates but also to “switch on” the Hsp104 molecular motor by interaction with the M domain. This, in turn, initiates ATP energy expenditure by the disaggregase, resulting in generation of the unfolding force. This model also identifies a role for Hsp70 in distinguishing between native and non-native protein. Hsp100 possesses enough intrinsic power to unfold even stable native domains such as GFP (provided that the recognition signal is present), and thus this activity must be directed toward aggregated proteins only and not other cellular structures. Our hyperactive Hsp100 variants, being in a permanently “switched on” state without Hsp70 control, have a detrimental effect on the cell, most probably due to native protein unfolding, causing morphological defects, growth arrest, and even cell lysis. The recent results of the Bukau laboratory (51) showing that the ClpB protein co-localizes with aggregates only in the presence of the DnaK protein support our hypothesis that tight control of Hsp100 disaggregating activity and directing Hsp100 toward aggregates are novel functions of the Hsp70 system.
How the state of the interdomain triad affects disaggregation and ATPase cycles at the atomic level remains to be elucidated. This may be difficult as the M domain is not structurally well defined presumably due to its inherent mobility. In silico analysis of the molecular models is restricted by the fact that it allows one to see only a single “snapshot” of protein conformational space. Despite these limitations of the model, both the in vivo and in vitro results demonstrating the presence of a network of NBD1-M ionic interactions reveal an important novel aspect of the Hsp100 molecular machine. Moreover, the toxicity of those interface mutants that gained hyperactivity has taught us that the precise control of disaggregase activity is critical for the cell.
Acknowledgments
We thank Drs. Debbie Ang and Beata S. Lipska for discussions and critical reading of the manuscript. We thank Drs. Bernd Bukau, Elisabeth Craig, Susan Wickner, and Sabina Kedzierska for the plasmids used in the study.
This work was supported by Foundation for Polish Science under the TEAM 2009–3/5 Programme (to N. L., S. Z., A. J., W. P., E. M., and A. W.).
- NBD
- nucleotide binding domain
- M
- middle
- TRITC
- tetramethylrhodamine Isothiocyanate
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- SSB
- single-stranded DNA binding protein
- HU
- histone like protein.
REFERENCES
- 1. Sanchez Y., Lindquist S. (1990) HSP104 required for induced thermotolerance. Science 248, 1112–1115 [DOI] [PubMed] [Google Scholar]
- 2. Chernoff Y. O., Lindquist S. L., Ono B., Inge-Vechtomov S. G., Liebman S. W. (1995) Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 268, 880–884 [DOI] [PubMed] [Google Scholar]
- 3. Glover J. R., Lindquist S. (1998) Hsp104, Hsp70, Hsp40. A novel chaperone system that rescues previously aggregated proteins. Cell 94, 73–82 [DOI] [PubMed] [Google Scholar]
- 4. Weibezahn J., Tessarz P., Schlieker C., Zahn R., Maglica Z., Lee S., Zentgraf H., Weber-Ban E. U., Dougan D. A., Tsai F. T., Mogk A., Bukau B. (2004) Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell 119, 653–665 [DOI] [PubMed] [Google Scholar]
- 5. Tessarz P., Mogk A., Bukau B. (2008) Substrate threading through the central pore of the Hsp104 chaperone as a common mechanism for protein disaggregation and prion propagation. Mol. Microbiol. 68, 87–97 [DOI] [PubMed] [Google Scholar]
- 6. Lum R., Tkach J. M., Vierling E., Glover J. R. (2004) Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J. Biol. Chem. 279, 29139–29146 [DOI] [PubMed] [Google Scholar]
- 7. Schlieker C., Weibezahn J., Patzelt H., Tessarz P., Strub C., Zeth K., Erbse A., Schneider-Mergener J., Chin J. W., Schultz P. G., Bukau B., Mogk A. (2004) Substrate recognition by the AAA+ chaperone ClpB. Nat. Struct. Mol. Biol. 11, 607–615 [DOI] [PubMed] [Google Scholar]
- 8. Ammelburg M., Frickey T., Lupas A. N. (2006) Classification of AAA+ proteins. J. Struct. Biol. 156, 2–11 [DOI] [PubMed] [Google Scholar]
- 9. Barends T. R., Werbeck N. D., Reinstein J. (2010) Disaggregases in 4 dimensions. Curr. Opin. Struct. Biol. 20, 46–53 [DOI] [PubMed] [Google Scholar]
- 10. Lee S., Sowa M. E., Watanabe Y. H., Sigler P. B., Chiu W., Yoshida M., Tsai F. T. (2003) The structure of ClpB. A molecular chaperone that rescues proteins from an aggregated state. Cell 115, 229–240 [DOI] [PubMed] [Google Scholar]
- 11. Diemand A. V., Lupas A. N. (2006) Modeling AAA+ ring complexes from monomeric structures. J. Struct. Biol. 156, 230–243 [DOI] [PubMed] [Google Scholar]
- 12. Watanabe Y. H., Nakazaki Y., Suno R., Yoshida M. (2009) Stability of the two wings of the coiled-coil domain of ClpB chaperone is critical for its disaggregation activity. Biochem. J. 421, 71–77 [DOI] [PubMed] [Google Scholar]
- 13. Doyle S. M., Wickner S. (2009) Hsp104 and ClpB. Protein disaggregating machines. Trends Biochem. Sci. 34, 40–48 [DOI] [PubMed] [Google Scholar]
- 14. Zolkiewski M. (2006) A camel passes through the eye of a needle. Protein unfolding activity of Clp ATPases. Mol. Microbiol. 61, 1094–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang F., Mei Z., Qi Y., Yan C., Hu Q., Wang J., Shi Y. (2011) Structure and mechanism of the hexameric Mec-ClpC molecular machine. Nature 471, 331–335 [DOI] [PubMed] [Google Scholar]
- 16. Lee S., Sielaff B., Lee J., Tsai F. T. (2010) CryoEM structure of Hsp104 and its mechanistic implication for protein disaggregation. Proc. Natl. Acad. Sci. U.S.A. 107, 8135–8140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wendler P., Shorter J., Plisson C., Cashikar A. G., Lindquist S., Saibil H. R. (2007) Atypical AAA+ subunit packing creates an expanded cavity for disaggregation by the protein-remodeling factor Hsp104. Cell 131, 1366–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wendler P., Shorter J., Snead D., Plisson C., Clare D. K., Lindquist S., Saibil H. R. (2009) Motor mechanism for protein threading through Hsp104. Mol. Cell 34, 81–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Watanabe Y. H., Takano M., Yoshida M. (2005) ATP binding to nucleotide binding domain (NBD)1 of the ClpB chaperone induces motion of the long coiled-coil, stabilizes the hexamer, and activates NBD2. J. Biol. Chem. 280, 24562–24567 [DOI] [PubMed] [Google Scholar]
- 20. Haslberger T., Weibezahn J., Zahn R., Lee S., Tsai F. T., Bukau B., Mogk A. (2007) M domains couple the ClpB threading motor with the DnaK chaperone activity. Mol. Cell 25, 247–260 [DOI] [PubMed] [Google Scholar]
- 21. Zietkiewicz S., Slusarz M. J., Slusarz R., Liberek K., Rodziewicz-Motowidło S. (2010) Conformational stability of the full-atom hexameric model of the ClpB chaperone from Escherichia coli. Biopolymers 93, 47–60 [DOI] [PubMed] [Google Scholar]
- 22. Schirmer E. C., Hofmann O. R., Kowal A. S., Lindquist S. (2004) Dominant gain of function mutations in Hsp104p reveal crucial roles for the middle region. Mol. Biol. Cell 15, 2061–2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee U., Wie C., Escobar M., Williams B., Hong S. W., Vierling E. (2005) Genetic analysis reveals domain interactions of Arabidopsis Hsp100/ClpB and cooperation with the small heat shock protein chaperone system. Plant Cell 17, 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jung G., Jones G., Masison D. C. (2002) Amino acid residue 184 of yeast Hsp104 chaperone is critical for prion-curing by guanidine, prion propagation, and thermotolerance. Proc. Natl. Acad. Sci. U.S.A. 99, 9936–9941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nowicki L., Leźnicki P., Morawiec E., Litwińczuk N., Liberek K. (2012) Role of a conserved aspartic acid in nucleotide binding domain 1 (NBD) of Hsp100 chaperones in their activity. Cell Stress Chaperones 17, 361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Case D. A., Darden T. A., Cheatham. T,E, III., Simmerling C. L., Wang J., Duke R. E., Luo R., Merz K. M., Pearlman D. A., Crowley M., Walker R. C., Zhang W., Wang B., Hayik S., Roitberg A., Seabra G., Wong K. F., Paesani F., Wu X., Brozell S., Tsui V., Gohlke H., Yang L., Tan C., Mongan J., Hornak V., Cui G., Beroza P., Mathews D. H., Schafmeister C., Ross W. S., Kollman P. A. (2006) AMBER 9, University of California, San Francisco [Google Scholar]
- 27. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA [Google Scholar]
- 28. Meagher K. L., Redman L. T., Carlson H. A. (2003) Development of polyphosphate parameters for use with the AMBER force field. J. Comput. Chem. 24, 1016–1025 [DOI] [PubMed] [Google Scholar]
- 29. Wang J., Wang W., Kollman P. A., Case D. A. (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 25, 247–260 [DOI] [PubMed] [Google Scholar]
- 30. Zylicz M., Ang D., Liberek K., Georgopoulos C. (1989) Initiation of lambda DNA replication with purified host- and bacteriophage-encoded proteins. The role of the dnaK, dnaJ, and grpE heat shock proteins. EMBO J. 8, 1601–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Woo K. M., Kim K. I., Goldberg A. L., Ha D. B., Chung C. H. (1992) The heat-shock protein ClpB in Escherichia coli is a protein-activated ATPase. J. Biol. Chem. 267, 20429–20434 [PubMed] [Google Scholar]
- 32. Zietkiewicz S., Krzewska J., Liberek K. (2004) Successive and synergistic action of the Hsp70 and Hsp100 chaperones in protein disaggregation. J. Biol. Chem. 279, 44376–44383 [DOI] [PubMed] [Google Scholar]
- 33. Doyle S. M., Shorter J., Zolkiewski M., Hoskins J. R., Lindquist S., Wickner S. (2007) Asymmetric deceleration of ClpB or Hsp104 ATPase activity unleashes protein-remodeling activity. Nat. Struct. Mol. Biol. 14, 114–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fenton W. A., Kashi Y., Furtak K., Horwich A. L. (1994) Residues in chaperonin GroEL required for polypeptide binding and release. Nature 371, 614–619 [DOI] [PubMed] [Google Scholar]
- 35. Nørby J. G. (1988) Coupled assay of Na+,K+-ATPase activity. Methods Enzymol. 156, 116–119 [DOI] [PubMed] [Google Scholar]
- 36. Krzewska J., Langer T., Liberek K. (2001) Mitochondrial Hsp78, a member of the Clp/Hsp100 family in Saccharomyces cerevisiae, cooperates with Hsp70 in protein refolding. FEBS Lett. 489, 92–96 [DOI] [PubMed] [Google Scholar]
- 37. Shifrine M., Phaff H. J., Demain A. L. (1954) Determination of carbon assimilation patterns of yeasts by replica plating. J. Bacteriol. 68, 28–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tessarz P., Schwarz M., Mogk A., Bukau B. (2009) The yeast AAA+ chaperone Hsp104 is part of a network that links the actin cytoskeleton with the inheritance of damaged proteins. Mol. Cell. Biol. 29, 3738–3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Konieczny I., Helinski D. R. (1997) Helicase delivery and activation by DnaA and TrfA proteins during the initiation of replication of the broad host range plasmid RK2. J. Biol. Chem. 272, 33312–33318 [DOI] [PubMed] [Google Scholar]
- 40. Zietkiewicz S., Lewandowska A., Stocki P., Liberek K. (2006) Hsp70 chaperone machine remodels protein aggregates at the initial step of Hsp70-Hsp100-dependent disaggregation. J. Biol. Chem. 281, 7022–7029 [DOI] [PubMed] [Google Scholar]
- 41. Hartwell L. H. (1971) Genetic control of the cell division cycle in yeast. IV. Genes controlling bud emergence and cytokinesis. Exp. Cell Res. 69, 265–276 [DOI] [PubMed] [Google Scholar]
- 42. Norden C., Liakopoulos D., Barral Y. (2004) Dissection of septin actin interactions using actin overexpression in Saccharomyces cerevisiae. Mol. Microbiol. 53, 469–483 [DOI] [PubMed] [Google Scholar]
- 43. Ferreira P. C., Ness F., Edwards S. R., Cox B. S., Tuite M. F. (2001) The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol. Microbiol. 40, 1357–1369 [DOI] [PubMed] [Google Scholar]
- 44. Jung G., Masison D. C. (2001) Guanidine hydrochloride inhibits Hsp104 activity in vivo. A possible explanation for its effect in curing yeast prions. Curr. Microbiol. 43, 7–10 [DOI] [PubMed] [Google Scholar]
- 45. Miot M., Reidy M., Doyle S. M., Hoskins J. R., Johnston D. M., Genest O., Vitery M. C., Masison D. C., Wickner S. (2011) Species-specific collaboration of heat shock proteins (Hsp) 70 and 100 in thermotolerance and protein disaggregation. Proc. Natl. Acad. Sci. U.S.A. 108, 6915–6920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Martin J., Langer T., Boteva R., Schramel A., Horwich A. L., Hartl F. U. (1991) Chaperonin-mediated protein folding of groEL through a “molten globule” like intermediate. Nature 352, 36–42 [DOI] [PubMed] [Google Scholar]
- 47. Weber-Ban E. U., Reid B. G., Miranker A. D., Horwich A. L. (1999) Global unfolding of a substrate protein by a the Hsp100 chaperone ClpA. Nature 401, 90–93 [DOI] [PubMed] [Google Scholar]
- 48. Konieczny I., Liberek K. (2002) Cooperative action of Escherichia coli ClpB protein and DnaK chaperone in the activation of a replication initiation protein. J. Biol. Chem. 277, 18483–18488 [DOI] [PubMed] [Google Scholar]
- 49. Doyle S. M., Hoskins J. R., Wickner S. (2007) Collaboration between the ClpB AAA+ remodeling protein and the DnaK chaperone system. Proc. Natl. Acad. Sci. U.S.A. 104, 11138–11144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sielaff B., Tsai F. T. (2010) The M-domain controls Hsp104 protein remodeling activity in an Hsp70/Hsp40-dependent manner. J. Mol. Biol. 402, 30–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Winkler J., Tyedmers J., Bukau B., Mogk A. (2012) Hsp70 targets Hsp100 chaperones to substrates for disaggregation and prion fragmentation. J. Cell Biol. 198, 387–404 [DOI] [PMC free article] [PubMed] [Google Scholar]