

Abstract
Introduction:
Liquisolid technique is used in delivery of lipophilic and poorly water soluble drugs through oral route. It involves dissolving water insoluble drugs in nonvolatile solvents and converting into acceptably flowing and compressible powders. The objective of the present work was to enhance the dissolution rate of ketoprofen using microcrystalline cellulose as carrier, aerosil 200 as coating material, and polyethylene glycol as nonvolatile water miscible liquid vehicle.
Materials and Methods:
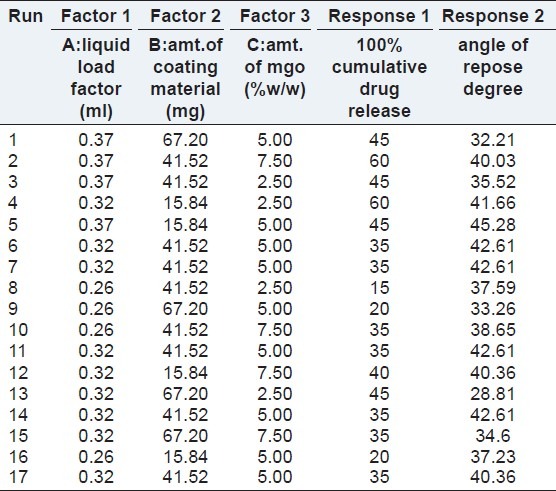
The drug concentration was kept constant in all formulations at 40% w/w. Optimization was carried out using Box–Behnken design by selecting liquid load factor, amount of coating material, and amount of magnesium oxide as independent variables; cumulative percentage drug release and angle of repose were considered as dependent variables.
Results:
The Fourier transform infrared (FTIR) and differential scanning calorimetry (DSC) studies revealed that there was no possible interaction between drug and tablet excipients. Prepared ketoprofen liquisolid tablets were evaluated for hardness, weight variation, friability, in-vitro disintegration time, drug content uniformity, and in-vitro dissolution studies. The optimized formulation yielded the response values, which were very close to the predicted values. The accelerated stability studies conducted showed that liquisolid tablets were not affected by ageing and there were no appreciable changes in the drug content.
Keywords: Box-Behnken design, dissolution, ketoprofen, liquisolid compacts
INTRODUCTION
Synthesis of poorly water soluble drugs is gaining lot of importance nowadays. The poor dissolution rate of such water-insoluble drugs confronts a major obstacle in development of pharmaceutical dosage forms.[1] The drugs which are poorly water soluble will be released at a slow rate owing to their limited solubility within gastrointestinal tract. The rate determining step in drug absorption is rate of drug dissolution. Enhancing the rate of dissolution or solubility of poorly water soluble drugs is a major challenge.[2] Formulation methods are targeted at dissolution enhancement of poorly soluble drug substances. Different techniques used to enhance the dissolution of water insoluble drugs are particle size reduction, use of surfactant as solubilizing agent, drug complex with hydrophilic carrier, pro-drug approach, and formulation of drug as solid solution to improve the dissolution rate by decreasing the crystallinity.[3–7] Among these the most promising method for promoting dissolution is the use of liquisolid compacts.[8–16] The term ‘liquisolid systems’ (LS) is a powdered form of liquid drug formulated by converting liquid lipophilic drug or drug suspension or solution of water-insoluble solid drug in suitable nonvolatile solvent systems, into dry looking, nonadherent, free-flowing, and readily compressible powdered mixtures by blending with selected carrier and coating materials.[17]
Cellulose, starch, and lactose are used as the carrier materials, whereas silica powder is used as the coating material. The good flow and compression properties of LS may be attributed due to large surface area and fine particle size of these carrier and coating materials. Hence LS compacts containing water-insoluble drugs expected to display enhanced dissolution characteristics and consequently improved oral bioavailability.[18] In the present investigation, ketoprofen a water insoluble drug was formulated into LS compacts using a liquid vehicle and studied for its pre- and postcompression parameters. Optimization of formulation was carried out using Box–Behnken design by selecting liquid load factor, amount of coating material and amount of magnesium oxide as independent variables and cumulative percentage drug release, angle of repose as dependent variables and the effect of formulation variables was studied.
MATERIALS AND METHODS
Ketoprofen and micro crystalline cellulose were purchased from Yarrow Chem Products, Mumbai. Methanol and propylene glycol were purchased from Ranbaxy Fine Chemicals Ltd., New Delhi. Glycerin and aerosil 200 were purchased from Himedia Laboratories Pvt, Ltd., Mumbai. Crosscarmellose sodium was gifted from Anglo French Drugs and Industries. Ltd., Bangalore. Magnesium oxide was purchased from NR Chem, Mumbai. Polyethylene glycol 400 (PEG 400) was purchased from E. Merck (India) Ltd., Mumbai. All other reagents and chemicals were of analytical grade.
Solubility studies
Solubility studies of ketoprofen were carried out in different nonvolatile solvents namely glycerine, propylene glycol, Tween 80, PEG 400, and distilled water. Saturated solutions were prepared by adding excess amount of drug in a 10 ml volumetric flask with liquid vehicle. The containers were sealed and kept in water shaker bath for 48 h at ambient temperature under constant vibration. After 48 h, the solutions were filtered through 0.45 μm Millipore filter, diluted suitably and analyzed spectrophotometrically at 260 nm. Three determinations were carried out for each sample to calculate the solubility of ketoprofen in each vehicle and percent weight of ketoprofen in its saturated solution with the solvent under investigation was calculated.
Experimental design of ketoprofen liquisolid compacts
The optimization of ketoprofen liquisolid compacts was carried out by taking into consideration the liquid load factor, amount of coating material and amount of magnesium oxide as formulation variables and the cumulative percentage drug release(t100%) in minutes and angle of repose. The experimental runs or formulation design were based on Box–Behnken designs using response surface methodology and utilized to evaluate the response variables. The responses were subjected to multiple regression analysis to find out the relationship between the factors used and the responses obtained. The independent variables selected for the analysis were liquid load factor, amount of coating material, and amount of magnesium oxide. The responses subjected for the analysis were cumulative percentage drug release and angle of repose. The effect of formulation variables on the response variables were statistically evaluated by applying one way analysis of variance (ANOVA) at 0.05 level using Design Expert 8.04 trial version (Stat Ease, USA). The design was evaluated by quadratic model, which bears the form of following equation:
Y= b0 + b1X1 + b2X2 + b3X1X2 + b4X12 + b5X22
Where y is the response variable, b0 the constant and b1, b2, b3…b5 is the regression coefficient. X1 and X2 stand for the mail effect; X1X2 are the interaction terms that shows how response changes when two factors are simultaneously changed. X12, X22 are quadratic terms of the independent variables. The experimental runs are shown in Tables 1 and 2.
Table 1.
Variables operating range for ketoprofen liquisolid compacts

Table 2.
Formulation trials as per Box–Behnken design
Preparation of liquisolid tablets
Liquisolid systems of ketoprofen (LS-1 to LS-15) were prepared and compressed into cylindrical tablets each containing 50 mg drug, using the rotary tablet press machine (Rimek Press, Ahmedabad). All liquisolid formulations contained microcrystalline cellulose as the carrier and aerosil 200 as coating material at different powder excipient ratio (R). PEG 400 was used as the liquid vehicle to prepare the liquid medications with a fixed 50% (w/w) drug concentration. Different liquid load factor, Lf, 0.263, 0.325, and 0.372 w/w were employed and varying percentage of magnesium oxide 2.5%, 5%, and 7.5% (w/w) was used as a flow activator. Finally, 5% cross carmellose sodium was used as a disintegrant and 1% magnesium oxide as lubricant in all systems. Ketoprofen was dispersed in PEG 400 and the mixture of microcrystalline cellulose and aerosil 200 were added to mixture with continuous stirring in a mortar. Crosscarmellose sodium and magnesium oxide were added to above mixture and the final powder blend was subjected to compression with flat circular punches using rotary tablet press machine.
Drug excipient compatibility studies
Fourier transform infrared studies
Compatibility studies of pure drug and excipients were carried out using Fourier Transformed Infrared Spectrophotometer (Shimadzu, Japan FT-IR 8400-S) in the range of 400–4000/cm by KBr disc method. A base-line correction was made using dried potassium bromide and then the spectrum of the pure ketoprofen and excipients was obtained.
Differential scanning calorimetry studies
DSC studies was performed using differential scanning calorimeter (Mettler 7, Germany) in order to assess the thermal behavior of the ketoprofen and the liquisolid compacts prepared. The thermal behavior of the samples was investigated at a scanning rate of 10°C/min, covering a temperature range of 0–300°C.
Pre-and postcompression studies of ketoprofen liquisolid compacts
Flow properties are the important concern in the formulation and industrial production of tablet dosage form. All the prepared liquisolid powders undergo the precompression studies such as bulk density, tapped density, compressibility index, Hausner's ratio, and angle of repose. The compressed liquisolid tablets of ketoprofen were characterized for their weight variation, hardness, friability, and disintegration time.[19]
In-vitro dissolution studies
Dissolution studies were carried out by using USP type II apparatus (USP XXIII Dissolution Test Apparatus) using 900 ml of phosphate buffer pH 7.4 as dissolution medium. Temperature of the dissolution medium was maintained at 37 ± 2°C. Aliquots of sample (2 ml) were withdrawn from the dissolution apparatus at regular predetermined time intervals and the samples were replaced with fresh dissolution medium. Absorbance of the collected samples after suitable dilution with phosphate buffer pH 7.4 was measured at 260 nm by using UV/Visible Spectrophotometer (Shimadzu-1700, Japan). Percentage of drug released was calculated from the standard calibration curve.
Stability studies
The tablet formulations were packed in aluminum foil and were placed in the stability test chamber and subjected to stability studies at accelerated testing (40 ± 2°C/75 ± 5% RH) for 6 months. The tablets were checked for physical appearance, hardness, and friability and disintegration time, drug content and in-vitro dissolution studies at the interval of 2 months. The shelf life was predicted by using similarity factor (f2) analysis according to International Conference on Harmonisation (ICH) guidelines.[20]
RESULTS AND DISCUSSION
Solubility studies in various non-volatile solvents
The drug solubility in the non-volatile solvents is an important parameter in formulation of liquisolid compacts. As greater the solubility, the more would be the drug particles dissolved in the liquid vehicle prior to the adsorption onto the carrier materials. The solubility of ketoprofen in the nonvolatile liquid vehicles glycerin, propylene glycol, Tween 80, PEG 400 and water was determined. The amounts of ketoprofen dissolved were 3.6 mg/ ml in water, 3000 mg/ml in PEG 400, 1000 mg/ml in propylene glycol, 1250 mg/ml in Tween 80, and 20 mg/ml in glycerin. The results showed that greater solubility of the drug in all the vehicles was achieved in comparison with water. The solubility results showed that ketoprofen had significantly more solubility in PEG 400 as compared with other nonvolatile solvents used in this study. As the aim of this study was to enhance the dissolution rate of drug, PEG 400 was selected as the suitable non-volatile solvent in the preparation of ketoprofen liquisolid compacts.
Drug-polymer compatibility studies
FTIR studies
Infrared (IR) spectrum of ketoprofen showed characteristic absorption peaks at 3051.18, 1697.24, and 1654.81/cm denoting stretching vibrations of aromatic C–H stretching, C = O stretching of acid and C = O stretching of ketone, respectively. The absorption peaks at 1589.23 and 1446.51/cm were due to C = C stretching of aromatic ring [Figure 1a]. Similar absorption peaks were observed in the IR spectra of physical blend of drug and excipients [Figures 1b–d], showed that there was no shift or disappearance of the characteristic parameter prelated to and was very much in conformity with the standard reference spectra, but with lower intensity substantiating the compatibility of the drug and the polymers used.
Figure 1.
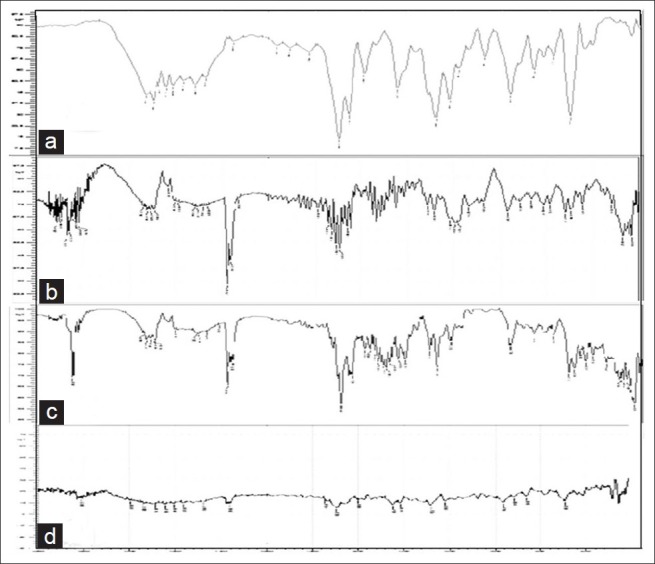
Infra red spectrum of ketoprofen and excipients. (a) Infra red spectrum of ketoprofen. (b) Infra red spectrum of avicel PH-101. (c) Infra red spectrum of aerosil 200. (d) Infra red spectrum of magnesium oxide
DSC studies
Figure 2 revealed the thermal behaviors of the pure components together with the thermal behavior of the final liquisolid system prepared. Ketoprofen [Figure 2a] demonstrated a sharp characteristic endothermic peak at 96.60°C corresponding to its melting temperature, which signified that ketoprofen was in pure crystalline state. The thermal behavior of micro crystalline cellulose in Figure 2b did not show any sharp peaks; proving that the carrier material was almost in an amorphous state. The thermo gram of liquisolid compact as shown in Figure 2c displayed a sharp endothermic peak at 96.58°C that might correspond to the melting and decomposition of the whole liquisolid system. The DSC thermo gram of physical mixture in Figure 2d showed an endothermic peak at 68.64°C. These studies confirmed that there was no change in crystallinity and there was no interaction between the drug and excipients in the formulation.
Figure 2.
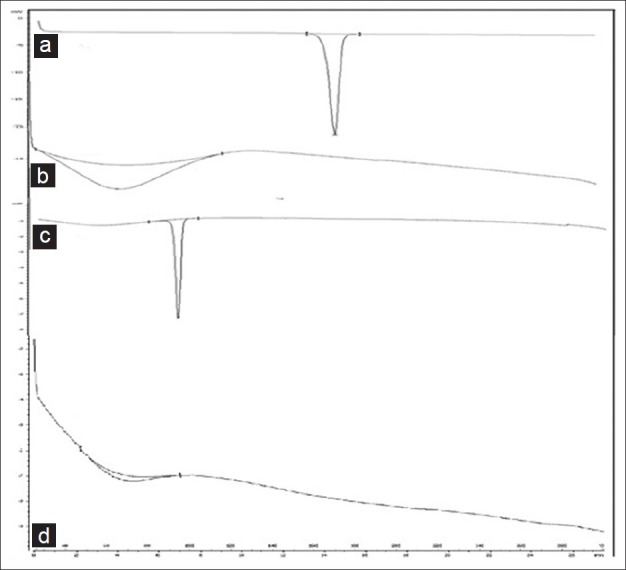
DSC thermograms of ketoprofen and liquisolid compact. (a) DSC thermogram of ketoprofen. (b) DSC thermogram of avicel PH-101. (c) DSC thermogram of optimized liquisoild compact. (D)DSC thermogram of physical mixture of ketoprofen and excipients
Evaluation of liquisolid compacts of ketoprofen
The values of bulk density were found to be in the range from 0.291 to 0.336 g/cc; the tapped density was in the range of 0.362–0.457 g/cc, which indicated that the powder blends were possessing good flow properties. Angle of repose is a characteristic related to interparticulate friction or resistance to movement between particles. The angle of repose for the formulation blends was carried out and LS-1, LS-3, LS-9, LS-13, and LS-15 showed good flow properties whereas formulations LS-4, LS-6, LS-7, LS-11, and LS 14 showed moderate flow properties. Carr's index is a useful parameter in reflecting the interparticulate friction. The powder flowability of ketoprofen liquisolid formulations was determined using Carr's index and the values ranged between 11% and 30%. Hausner's ratio was calculated for all the ketoprofen liquisolid formulations batches and it was found to be between 1.1 and 1.4 indicating that all the formulations possess good flow property and were in accordance with the limit of <1.25 for good flow. All the batches prepared posses hardness in the range of 3–5 kg/cm2 to ensure good handling characteristics. Friability was found to be less than 1% ensuring that all the batches were mechanically stable without any change in surface hardness. The disintegration time for the prepared liquisolid compacts was found between 3 and 5 min. It was observed that there was a direct relationship between disintegration time and amount of carrier material used. As the amount of carrier increased the disintegration time decreased and this was observed in the formulation LS-8, LS-9, LS-10, and LS-11.
Drug content
The drug content was performed for all the 17 formulations and only few tablets had met IP content uniformity criteria, in which individual content was between 76% and 100% of the average content and that indicated uniformity of mixing or homogeneous distribution throughout the batch.
In-vitro dissolution study
In-vitro drug release studies were performed in phosphate buffer pH 7.4 for all the prepared formulations by using USP dissolution test apparatus-Type II, Rotating Paddle method. The graphs showing drug release profile for formulations are shown in the Figures 3 and 4. In-vitro dissolution studies showed that the formulation containing increased amount of carrier material showed 100% drug release within 20 min. Formulations LS-1, LS-2, LS-3, LS-4, LS-12, and LS-13 showed 100% drug release within 60 min as this formulation contained very little amount of carrier. From the obtained results, it was also observed that there is a relationship between the powder excipient ratio and the in-vitro release. Liquisolid compacts with lower R-values contained relatively smaller amounts of powder admixture (carrier + coating materials) and the amounts of nonvolatile solvent available per powder substrate are relatively higher. In contrast, liquisolid compacts with higher R-values contain higher amounts of powder admixture and the amounts of nonvolatile solvent available per powder substrate are relatively smaller. Therefore, the liquisolid tablets with low R-values showed relatively poor dissolution.
Figure 3.
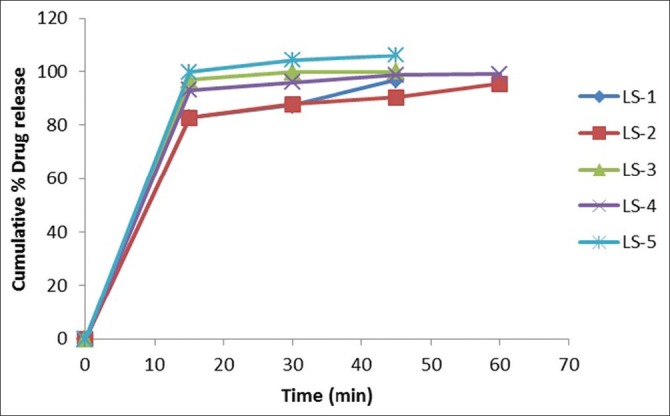
In-vitro drug release studies of LS 1-5
Figure 4.
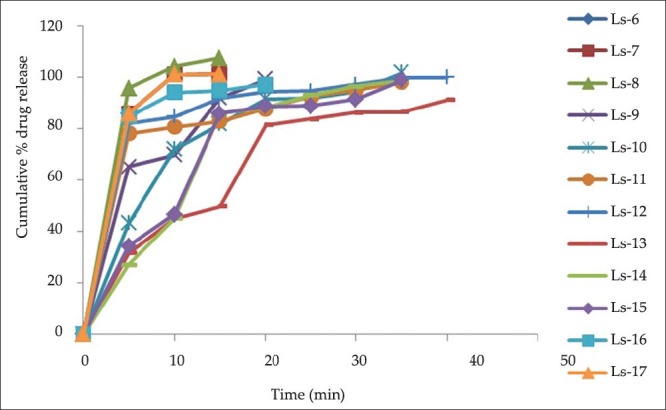
In-vitro drug release studies of LS 6-17
Optimization of ketoprofen liquisolid formulation
Optimization was carried out by Box–Behnken design employing response surface methodology by taking into consideration liquid load factor, amount of coating material, and amount of magnesium oxide as independent variables and the influence of these variables on the responses, cumulative percentage drug release and angle of repose respectively. The Box–Behnken design was adopted to analyze the relationships between multiple variables with a reduced number of experimental runs. According to this design, 17 formulations were prepared as mentioned in Table 2.
Effect of formulation variables on cumulative percentage drug release
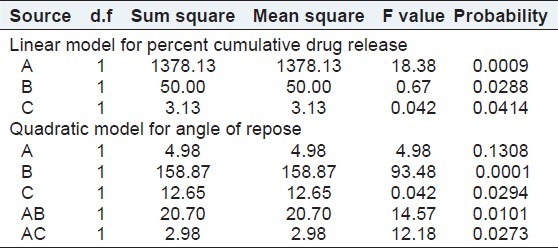
The results of formulations as per the design when fitted into various models, a linear model was found to be significant for cumulative percent drug release with F value of 6.36 and P value 0.0069. In this model factors A and B (liquid load factor and amount of aerosil 200)significantly affected the cumulative percentage drug release, whereas factor C (amount of magnesium oxide) do not have significant effect on the response. The model equation is as follows: cumulative drug release = -36.02304 + 240.82569× A-0.097352× B + 0.25000. The effect of both the factors A and B can be explained with the help of the 3D response surface plot as shown in Figure 5. As the liquid load factor and concentration of aerosil increased, the cumulative percent drug release increased.
Figure 5.
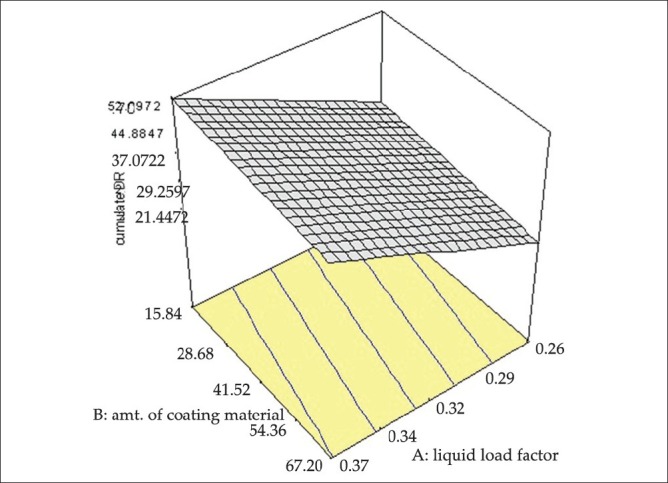
3D Response surface graph of cumulative percentage drug release
Effect of formulation variables on angle of repose
The results of formulations as per the design when fitted into various models, quadratic model was found to be significant for angle of repose with F value of 17.83 and P value 0.0010. In this model factor B (amount of aerosil 200) significantly affected the angle of repose, whereas, the factors A and C (liquid load factor and amount of magnesium oxide) do not have significant effect on angle of repose. The model equation is as follows: Angle of repose = -42.58705 + 432.45607× A + 0.62982× +1.226815.12160E-003× +6.33028 + 0.027609. The effect of factor B can be explained with the help of the 3D response surface plot as shown in Figure 6. As the concentration of aerosil increased, the angle of repose of the liquisoild compacts increased. The results of ANOVA are shown in Table 3.
Figure 6.
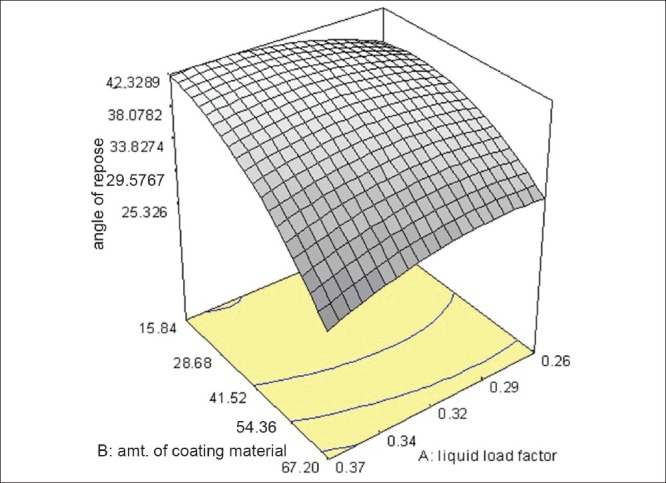
3D Response surface graph of angle of repose
Table 3.
Summary of ANOVA for formualtion variables from Box–Behnken design
The results when analyzed and optimized had generated numerical optimized solutions based on these experimental design. From the numerical optimization results, a solution was selected randomly, coded as OPLS and considered as optimized liquisoild compact formulation. The results of predicted observation and actual experimentation are shown in Table 4 which confirmed the closeness of the predicted results with that of the observed results and it was observed that the response was almost similar to the response predicted by the software.
Table 4.
Optimized formulation (Predicted vs Actual)

Precompression evaluation for optimized formulation OPLS
The values of bulk density was found to be 0.326 gm/cc; the tapped density was found to be 0.479 gm/cc, which indicated that the powder blends were having good flow properties. The compressibility index and Hausner's ratio are measures of the propensity of a powder to be compressed, which reflects the interparticle friction. In a free-flowing powder, such interactions are generally less significant, these differences are reflected in the compressibility index. The powder flowability of optimized liquisolid formulations determined using Carr's index was found to be 31.81%. Hausner's ratio was calculated and found to be 1.466 indicating that all the optimized formulation OPLS possesses good flow property. Angle of repose is a characteristic related to interparticulate friction or resistance to movement between particles. The angle of repose for the optimized formulation OPLS was carried out, the results are shown in Table 5. It revealed that the optimized formulation OPLS was having good flow properties and was found to be in accordance with the limit.
Table 5.
Precompression and postcompression results of optimized formulation OPLS
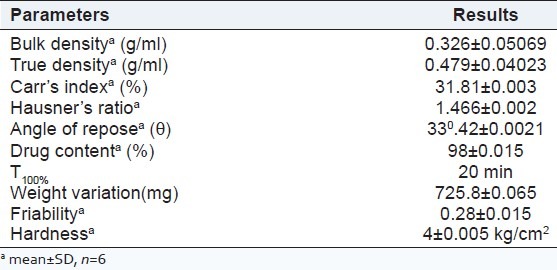
Post-compression studies
The mean hardness of each liquisolid tablet was determined and was found to be 4 kg/cm2 and the required hardness was produced to ensure good handling characteristics.
Weight variation was carried out for the optimized formulation OPLS and was found to be within the average weight variation limits. This ensures that all the formulations were within the IP specifications. Friability was found to be below 1% ensuring that all the batches were mechanically stable without any change in surface hardness. The drug content was performed for the optimized formulation OPLS and the results showed maximum drug entrapment, which ensures uniform mixing during formulation [Table 5].
In-vitro dissolution studies
The In-vitro drug release data for optimized formulation OPLS was performed in pH phosphate buffer 7.4 and the predicted drug release mechanism is depicted in Figure 7. The optimized formulation showed 100% drug release within 20 min, which correlated with the predicted data. The optimized formulation showed peppas as a best fit model with r2 value of 0.9127. The release rate followed the nonfickian diffusion mechanism as its n value was found to be 0.0712 with ‘k’ value of 77.372.
Figure 7.
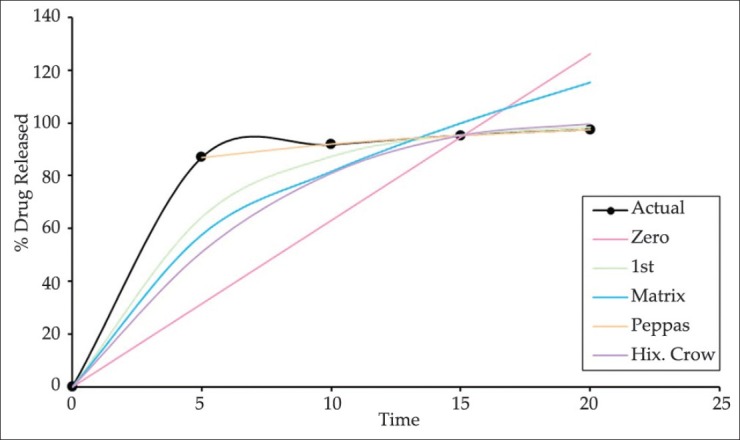
In-vitro drug release profile of optimized formulation by using various kinetic models
Stability studies
The accelerated stability studies of liquisolid tablets were performed as per the ICH guidelines to investigate whether the liquisolid tablets is affected during storage conditions. In order to study the effect of aging on physico–chemical parameters and dissolution profile of ketoprofen liquisolid compacts, Optimized formulation OPLS were kept at 40°C with 75% relative humidity for a period of 6 months. The physical appearance, hardness, friability, disintegration time, drug content, and dissolution rate were measured for these tablets at the end of 2, 4, and 6 months. The results showed that there was no significant difference between the initial and aged liquisolid tablets. This indicated that the physico–chemical parameters and dissolution of liquisolid compacts was not affected by aging.
CONCLUSION
In the present study, the potential of liquisolid systems to improve the dissolution properties of water-insoluble drug was investigated using ketoprofen as the model drug. Optimization of ketoprofen liquisolid compacts was carried out using Box–Behnken design by selecting liquid load factor, amount of coating material and amount of magnesium oxide as independent variables and cumulative percentage drug release and angle of repose as dependent variables. The results showed that solubility of water insoluble drug ketoprofen was increased to greater extent thereby improving its dissolution rate. Thus liquisolid technology shall be used to improve the release rate of poorly water soluble drugs that will make the dosage form will be cost effective.
ACKNOWLEDGEMENTS
The authors are thankful to Gokula Education Foundation, Bangalore for providing necessary facilities to carry out the research work. The authors thank Indian Institute of Science (IISc) for providing facilities to carry out DSC studies.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Hitendra SM, Manoj RD, Surendra GG, Ashwini DR, Hannan TS. Enhanced dissolution rate of glipizide by a liquisolid technique. Int J Pharm Sci Nan. 2011;3:1205–13. [Google Scholar]
- 2.Vijaykumar Nagabandi, Ramarao T, Jayaveera KN. Formulation development and evaluation of liquisolid systems to improve the dissolution rate of ketoproefen. Int J Bio Res. 2011;2:530–41. [Google Scholar]
- 3.Javadzadeh Y, Siahi-Shadbad MR, Barzegar JM, Nokhodchi A. Enhancement of dissolution rate of piroxicam using liquisolid compacts. Farmaco. 2005;60:361–5. doi: 10.1016/j.farmac.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Yadav VB, Yadav AV. Improvement of solubility and dissolution of indomethacin by liquisolid and compaction granulation technique. J Pharm Sci. 2009;1:44–51. [Google Scholar]
- 5.Tiong N, Elkordy AA. Effects of liquisolid formulations on dissolution of naproxen. Eur J Pharm Biopharm. 2009;73:373–84. doi: 10.1016/j.ejpb.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Spireas S, Bolton SM. Liquisolid systems and methods of preparing same. US Patent 6,423,339. 2002 [Google Scholar]
- 7.Spiro S, Srinivas S. Enhancement of prednisolone dissolution properties using liquisolid compacts. Int J Pharm. 1998;166:177–88. [Google Scholar]
- 8.Babatunde A, Amal AN, Ebtessam AE, Sahar E. Liquisolid systems to improve the dissolution of furosemide. Sci Pharm. 2010;1:1–20. doi: 10.3797/scipharm.0912-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Javadzadeh Y, Jafari-Navimipour B, Nokhodchi A. Liquisolid technique for dissolution rate enhancement of a high dose water-insoluble drug (Carbamazepine) Int J Pharm. 2007;341:26–34. doi: 10.1016/j.ijpharm.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 10.Spiro S, Srinivas S, Rakesh G. In-vitro release evaluation of hydrocortisone liquisolid tablets. J Pharm Sci. 1998;87:867–72. doi: 10.1021/js970346g. [DOI] [PubMed] [Google Scholar]
- 11.Fahmy RH, Kassem MA. Enhancement of famotidine dissolution rate through liquisolid tablets formulation: In-vitro and in-vivo evaluation. Eur J Pharm Biopharm. 2008;69:993–1003. doi: 10.1016/j.ejpb.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 12.Amrit BK, Indrajeet DG, Avinash HH, Pandurang ND, Satish BB. Dissolution rate enhancement of fenofibrate using liquisolid tablet technique. Lat Am J Pharm. 2009;28:219–25. [Google Scholar]
- 13.Yadav AV, Shete AS, Dabke AP. Formulation and evaluation of orodispersible liquisolid compacts of aceclofenac. Ind J Pharm Edu Res. 2010;44:227–35. [Google Scholar]
- 14.Sanjeev RG, Ravindra J. Formulation and characterization of atorvastatin calcium liquisolid compacts. Asian J Pharm Sci. 2010;5:50–60. [Google Scholar]
- 15.Elsay Khalid M, Sajmy Ahmed M, Fetouh Mohamed I. Formulation and evaluation of rofecoxib liquisolid tablets. Int J Pharm Sci Rev Res. 2010;3:135–42. [Google Scholar]
- 16.Boghra Rikisha, Patel Anuradha, Desai Hetal, Jadhav Anil. Formulation and evalution of irbesartan liquisolid tablets. Int J Phar Sci Rev. 2011;9:32–7. [Google Scholar]
- 17.Ajit KS, Nagesh H, Aloorkar MS, Jayashree B, Gaja Liquisolid Systems: A review. Int J PhamaSci Nano. 2010;3:795–802. [Google Scholar]
- 18.SambasivaRao A, Naga AT. Liquisolid technology: An overview. Int J Res Pharm Biomed Sci. 2011;2:401–9. [Google Scholar]
- 19.Subraamanyam CV. Text book of Physical Pharmaceutics. 1st ed. New Delhi: Vallabh Prakashan; 1998. pp. 225–8. [Google Scholar]
- 20.Grimm W. Extension of the international conference on harmonization tripartite guideline for stability testing of new drug substances and products to countries of climatic zones III and IV. Drug Dev Ind Pharm. 1998;24:313–25. doi: 10.3109/03639049809085626. [DOI] [PubMed] [Google Scholar]