

Abstract
The regulatory requirements of various countries of the world vary from each other. Therefore, it is challenging for the companies to develop a single drug which can be simultaneously submitted in all the countries for approval. The regulatory strategy for product development is essentially to be established before commencement of developmental work in order to avoid major surprises after submission of the application. The role of the regulatory authorities is to ensure the quality, safety, and efficacy of all medicines in circulation in their country. It not only includes the process of regulating and monitoring the drugs but also the process of manufacturing, distribution, and promotion of it. One of the primary challenges for regulatory authority is to ensure that the pharmaceutical products are developed as per the regulatory requirement of that country. This process involves the assessment of critical parameters during product development.
Keywords: Development, drug, generic, global, regulatory
INTRODUCTION
The pharmaceutical industry is one of the highly regulated industries, with many rules and regulations enforced by the government to protect the health and well-being of the public. Therefore, the aim of the pharmaceutical industry is to identify and develop a generic drug product which can be tailor made to meet the diverse market requirements. As per global market trend, it is estimated that approximately $150 billion worth of drugs will be off-patented during the period 2010 to 2017, which will serve as a platform for pharmaceutical companies to develop generic drugs.[1] The pharmaceutical industry in India has shown a remarkable growth which in turn has risen the economy of India.[2] After the introduction of the product patent regime in India, there was a need for pharmaceutical companies both in India and abroad to explore newer markets. Indian pharma majors are entering new markets with global ambitions, mergers and acquisitions are in focus with a reason to enter new market. For sustained growth over the next few decades, firms have to concentrate on generic drug products. “Diseases that cannot be cured, diseases that have to be managed, provide great opportunities for generic drugs.” Government has the responsibility to protect their citizens. It is the responsibility of national governments to establish regulatory authorities with strong guidelines for quality assurance and drug regulations in the respective territories. Somewhat parallel with the ongoing harmonization and movement toward creating a common market for medicines inside the EU, the need for wider harmonization was felt by officials from Japan, EU, and US during International Conference of Drug Regulatory Authorities (ICDRA) organized by world health organization (WHO). The informal discussions had led to a need of the harmonization of requirements relating to the new innovative drugs and also subsequently paved the wayto the establishment of International Conference on Harmonization of Technical Requirements for the Registration of Pharmaceuticals for Human Use (ICH), a collaborative initiative between the EU, Japan, and the United States with observers from WHO, EFTA, and Canada. Efforts to harmonize various elements of drug regulatory activities have been initiated by various inter-governmental organizations at regional and inter-regional level in the past decade. The driving force behind these efforts has been the increase in global trade in pharmaceutical products, and growth in the complexity of technical regulations related to drug efficacy, safety, and quality.
Status as of today: Due to the emerging regulatory needs of pharmaceutical sector, the drug evaluation for the control of drug quality and trade has become highly sophisticated. Regulatory guidelines and standard tools provide a basis for implementation of laws, whereas laws provide a legal basis for drug control. The world covers more than 100 countries, where most of them have established pharmaceutical legislations and regulatory requirements. For worldwide regulatory dossier submissions, it is a pre-requisite requirement to have a knowledge of country specific guidelines and norms. Therefore, it is very important to analyze the differences and commonness between the regulatory requirements and pharmaceutical legislations of different countries of the world. The Pharmaceutical market based on the diversity in the regulation region and marketing interest can be divided into two groups: Regulated and emerging markets. The regulated market involves those countries where there are defined regulatory requirements set by the regulatory bodies of that country and the emerging market countries are those who still lag behind in putting forward the well defined regulations for drugs. United States (US) and the EU are the biggest and the most potential markets for in the world and are categorized under the regulated markets, whereas ROW (Rest of the World) market includes all the emerging markets like Brazil (LATAM), Tanzania (Africa), Russia (CIS), Hong Kong (ASIA), etc.
GENERIC DRUG DEVELOPMENT
To make a generic product, formulator must know in detail the exact regulatory requirements of each concerned country where the drug is intended to be filed. Generic drug product development uses a different approach and strategy compared to that used to develop an innovator drug product containing a new chemical entity. Generic drug product manufacturers must formulate a drug product that will have the same therapeutic efficacy, safety, and performance characteristics as of its branded counterpart. The key factor is that the generic drug product must meet all the necessary criteria to be therapeutically equivalent to the innovator drug product. Therapeutically equivalent means that the drug product shows pharmaceutical equivalence as well as bioequivalence. Table 1 shows regulatory requirement for generic drug product development in some selected countries.
Table 1.
Comparison of regulatory requirement in selected countries during generic product development

The decision to proceed with the development of a generic drug product should therefore be based on well-researched data that primarily indicate market value together with a sound knowledge of patent expiry dates, predicted market share, and growth rate for the product, amongst others. The predicted profitability of the new generic product will require strategic planning for the subsequent launch timing, which must take into account the expected generic price and knowledge of anticipated competitors, such as who they are and when they are expected. According to Hamrell R.Michael “The Drug Price Competition and Patent Term Restoration Act” in 1984 changed the regulatory climate for generic drugs. This law allowed for the approval of generic “me-too” copies of many approved drug after the patent had expired.[3] As per Kathy Redmond the regulatory agencies have a responsibility to ensure that high-quality, safe, and effective medicines are made available to patients in a timely manner.
Despite the fact that all regulators worldwide share the same aims, they do not adopt a consistent approach to drug approval requirements, and as a result, medicines are often approved quicker in some countries than others.[4] Therefore, there is need for a harmonized drug regulation globally.
FILING A GENERIC DRUG APPLICATION
When a dossier is ready as per the regulatory requirement of the respective country, it is submitted to the regulatory agency of that country. Various regulatory agencies worldwide are tabulated in the Table 2.
Table 2.
Illustrates the regulatory authorities of various countries
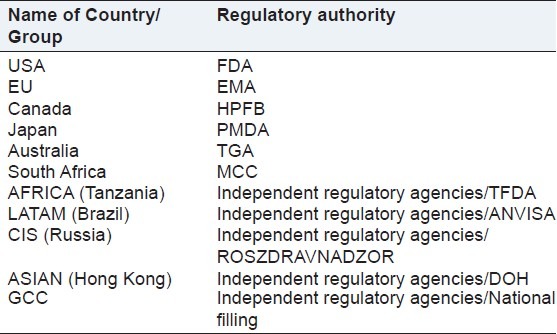
Food and Drug Administration(FDA), European Medicines Agency (EMA), Pharmaceutical and Medical Devices Agency (PMDA), Therapeutic Goods Administration (TGA), Medicines Control Council (MCC), Tanzania Food and Drugs Authority (TFDA), AgênciaNacional De VigilânciaSanitária (National Health Surveillance Agency) (ANVISA), Commonwealth Independent States (CIS), Department of Health (DOH), The Gulf Co-Operation Council (GCC).
United States of America
USA is the major market for the pharmaceutical industry. The USA has evolved from no regulations in the 18th century to one of the highly regulated and admired regulatory authority in the world. The food and drug administration (FDA) within the U.S. Department of Health and Human Services regulates the drug approval system in United States with help of six product centers including Center for Drug Evaluation and Research (CDER).[5] Drug registration in USA is majorly categorized by two types of applications: New Drug Application (NDA) and Abbreviated New Drug Application (ANDA). ANDA is filled for generic drug products; those require marketing authorization and are of exact or close copies of already approved drugs.[6] The ANDA approval process is depicted in Figure 1[7] Indeed, the way this country regulates drugs typically has been born out of adversity, out of events that have killed and injured thousands. The evolution of the current drug regulatory system in USA is recognized globally as the gold standard for drug safety and efficacy. During 1990, FDA began work to develop standards for the exchange of electronic information critical to the agency's mission. This recognized both the inefficiency of paper for transferring mass quantities of data and the need to develop a harmonized format that would be usable by FDA as well as its counterparts in the European Union and Japan. Consequently, firms are now able to submit paperless product applications and related material to world regulatory agencies more efficiently, while each review authority maintains its own high standards for product evaluation. Because all drugs have some risk, FDA task force advised the agency to make more systematic use of the principles of risk management in the way FDA oversees drug development and marketing.
Figure 1.
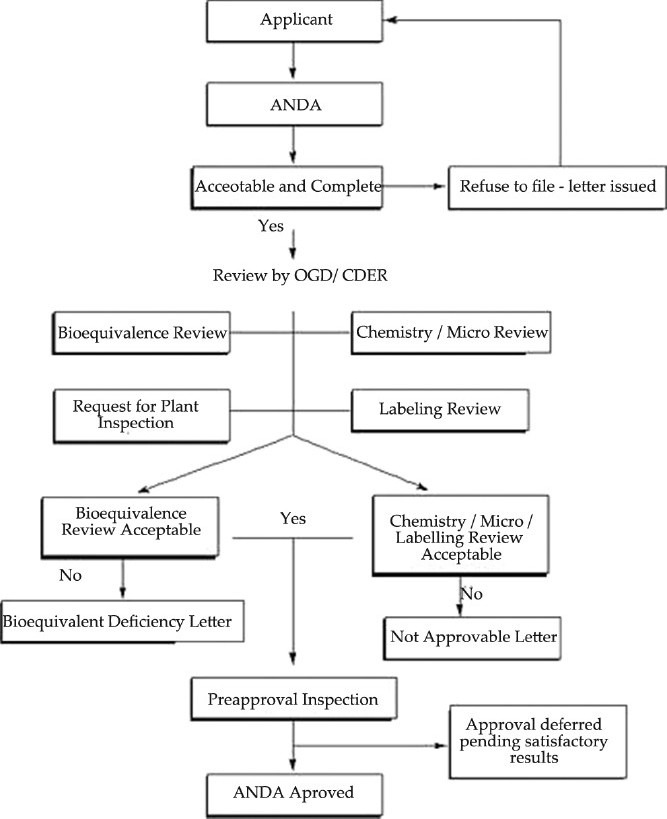
Approval process of ANDA[11]
European Union
The EU has one of the most highly regarded regulatory systems in the world. The system comprises of European parliament, the council of ministers, and the European Commission. EU consists of 27 member states: Austria, Belgium, Bulgaria, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Ireland, Italy, Latvia, Lithuania, Luxemburg, Malta, Netherlands, Poland, Portugal, Romania, Slovakia, Slovenia, Spain, Sweden, and the United Kingdom and three countries which are member of European Free Trade Agreement (EFTA) Iceland, Norway, and Liechtenstein.[8] These EFTA members are those countries which were unable to join rest of the 27 member states as common market. These three EFTA member countries along with 27 EU member states, comprises of the European Economic Area (EEA). The European Medicines Agency is a decentralizedagency of the European Union, located in London.[8] The Agency is responsible for the scientific evaluation of medicines developed by pharmaceutical companies for use in the European Union and applications for European marketing authorizations for both human and veterinary medicines (centralized procedure). Under the centralized procedure, companies submit a single marketing-authorization application to the Agency. Once granted by the European Commission, a centralized (or “Community”) marketing authorization is valid in all European Union (EU) and EEA-EFTA states (Iceland, Liechtenstein and Norway). The European parliament approves the laws together with the council of ministers. The council of ministers is the voice of Member states and is responsible for enactment of directives.
Legal basis for applications in Europe
The eligibility and the requirements are set in the commission regulation (EC) No 726/2004 and defined in articles 8 and 10 are of the Directive 2001/83/EC. The Figure 2[10] represents the types of application filed in Europe.
Figure 2.
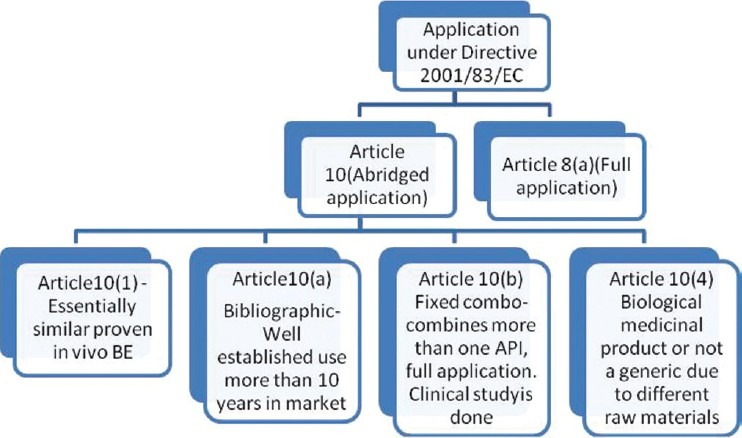
Types of Application filed in Europe[11]
Types of submission procedure
To market a generic medicinal product in European Economic Area (EEA) which consists of 27 member states and 3 EFTA countries, a marketing authorization has to be issued. European medicines Agency (EMA formerly known as EMEA) regulates the medicinal products marketing authorization through various committees. Different types of submissions for receiving Marketing authorization in Europe are given below in Table 3.
Table 3.
Different types of procedures for marketing authorization applications in europe

In case of Generic drug products, generally the decentralized procedure is followed whereas in case of the new drug products the application for marketing authorization is always submitted through a centralized procedure.
Brazil (LATAM)
Brazil's pharmaceutical market is the 11th largest in the world and second in Latin America after Mexico since the devaluation of 2001.[11] Brazil's market is clearly a key market to drive the global development of any pharmaceutical company with international ambitions and may have located regional headquarters in the country. The regulatory framework is considerably improved and makes Brazil a preferred gateway to other Latin American markets. The federal regulatory agency responsible for pharmaceutical product registration in Brazil is ANVISA (National Sanitary Vigilance agency), which was established in 1999.[12] The 1999 Law (The Generics Law) and the ANVISA regulate the implementation of generic pharmaceuticals policy in Brazil, establishes the technical standards and defines the concepts of bioavailability, bioequivalent drugs, innovators, reference drugs, and similar. According to the Brazilian legislation, all the pharmaceutical products must be registered with ANVISA before coming to market in Brazil. Product registration in Brazil is a laborious exercise, and is to be requested by the local Brazilian based office of the foreign company or its distributor in Brazil. The registration is valid for 5 years and can be renewed continuously for the same period. Law must complete the registration process within 90 days after the registration is requested, or denied. For registration purposes, ANVISA classifies the products in various categories. The medications for human use are divided into three distinct areas i.e., New Product, Similar Product, Generic Product.
Tanzania (AFRICA)
African medicines regulatory authorities (MRAs) role is to ensure that the pharmaceutical products those are needed, are registered in their country: This process is called “registration,” “marketing approval,” “marketing authorization” or “product licensing”, and involves assessment of product information submitted by the manufacturer (the product ‘dossier’) to make sure that it is safe and effective for use by local patients. Assessment of generic drugs is relatively simple. This is because the regulator only needs to establish two key points. First, generic drug product is bioequivalent to and thus therapeutically interchangeable with the comparator product. Secondly, product meets comparable sustainable quality standards to that of the innovator product. Every country of the African region has its own regulatory framework. Drug product registration was gradually introduced in Tanzania under the Tanzania Food, Drugs and Cosmetics Act 2003, to have a smooth transition, beginning with 1-year provisional registration taken as a notification from 1998. This gave ample time for the Pharmacy Board to prepare guidelines to assist applicants and evaluators to respectively submit and evaluate correctly the required information. Following the preparation of the guidelines, the first application was received in 1997 and the first product was registered in April 1999.[13] All documents shall be in Kiswahili or English. Applications that do not comply to requirements prescribed in these guidelines will be rejected and returned to the applicant at his own cost. All ingredients used in the formulation of generic medicinal products must comply with specifications prescribed either in the USP (United States pharmacopoeia), BP (British pharmacopoeia), EP (European pharmacopoeia), and International orJapanese pharmacopoeia. In-house specifications shall only be accepted if the limits are tighter than those prescribed in those pharmacopoeias and other specifications may be accepted if they are validated.
Russia (CIS)
According to some estimates, Russia is poised to be among the top five Global pharmaceutical markets in terms of value in the next five years.[13] Today, Russia stands at the threshold of becoming a major force in the global pharmaceutical market. Russia is a member country of “The Commonwealth of Independent States” (CIS) founded in 1991, which is a regional organization whose participating countries are former Soviet Republics, formed after the dissolution of the Union of Soviet Socialist Republics (USSR). The regulatory processes in CIS countries are led and supervised by Regulatory Agencies closely collaborating with or operating within the respective Ministries of Health. Figure 3 depicts the scheme of registration process. Each of the CIS countries has established individual registration guidelines. Registration in RUSSIA is a national procedure. Estimated duration of procedure is up to 24 months. Documentation is done in Russian language in format compliant with Russian requirements. Recommended submission of a bioequivalence study is carried out in certified research organizations within the Russian Federation's territory. Original and generic products pass the same stages of registration. Original products must pass through all registration procedures while the generic products are exempted from some of them. For example, original product must undergo clinical trials in Russia. For generic products, bio-equivalence studies can be conducted in any other countries and not only in Russia.
Figure 3.
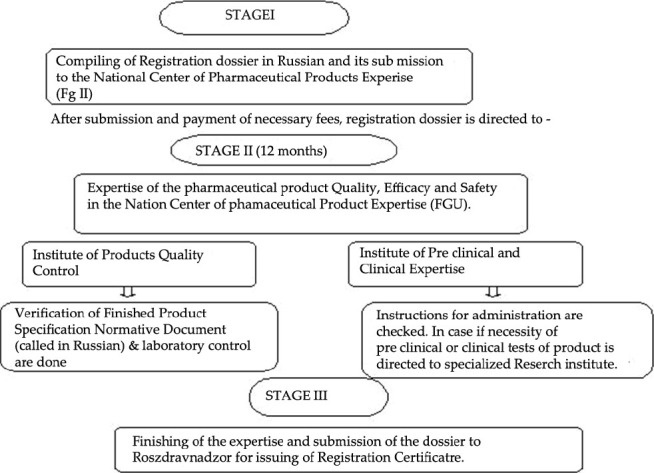
Scheme of the registration process
Hong Kong (ASIA)
Hong Kong's market for pharmaceuticals drugs is about $1.5 billion.[15] As a part of developed economy in Asia, it still lags behind other advanced economies of the OECD in medicines regulation.[16] The pharmaceutical regulatory agency in Hong Kong remains conservative in outlook but is facing similar strain of challenge from pharmaceutical sector despite the issues raised are of plain trade and business. The HA's adoption of purchasing policy favoring use of bulk contract and generic substitution has undercut the market for multinational pharmaceutical companies represented bythe Hong Kong Association of the Pharmaceutical Industry (HKAPI). This alongside the difficulty of listing new drugs in the HA Drug Formulary, the delay in new drug registration application submitted to the Hospital Authority (HA) Drug Formulary, the delay in new drug registration applications submitted to the pharmacy and poisons Board (PPB), and Intellectual property rights issues,[17] have provoked outcries about deterioration in business environment for the pharmaceutical trade sector that calls for government policy changes. Hong Kong's pharmaceutical regulatory body and its pharmaceutical business sector evidently lag behind international developments in number of ways. The PPB has not gained membership of Pharmaceutical Inspection Cooperation Scheme (PICS) that facilitates signing of Mutual Recognition Agreement with regulatory bodies in developed countries. This lack of International harmonization of GMP standard makes it difficult for local pharmaceutical manufacturers to go down the path of becoming exporters of medicines.
CONCLUSION
Although there is a continuous process of harmonization taking place all around the world, still we see a huge challenge, which is yet to be overcome by the Pharmaceutical industry in case of generic drug development and filing. This is due to the heterogeneity in the regulatory landscape of the various countries. Therefore, to meet these challenges, a lot of strategic planning is required before the development of any generic drug product.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Stephanie Sutton Global Market Boom for Generic Drugs. ON The Electronic Newsletter of Pharmaceutical Technology. 2012. [Last accessed on 2012 Jan 19]. Available from: http://www.pharmtech.com/pharmtech/News/Global-Market-Boom-for-Generic-Drugs/ArticleStandard/Article/detail/756488 .
- 2.Srinivasan R. Indian pharmaceutical industry: Evaluation of current scenario and future trends. [Last accessed on 2012 Jun 10]. Available from: http://www.tejas-iimb.org/interviews/13.php .
- 3.Hamrell MR. 2. Vol. 14. California: ON Clinical Research and Regulatory Affairs; 1997. [Last accessed on 2012 Jun 10]. An Update on the Generic Drug Approval Process; pp. 139–54. Available from: http://www.informahealthcare.com/doi/abs/10.3109/10601339709019635?journalCode=crr . [Google Scholar]
- 4.Redmond K. The US and European Regulatory Systems: A Comparison: ON JAmbul Care Manage. [Last accessed on 2011 Nov];2004 27:105–14. doi: 10.1097/00004479-200404000-00005. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15069987 . [DOI] [PubMed] [Google Scholar]
- 5. [Last accessed on 2012 Apr]. Available from: http://www.fda.gov/AboutFDA/CentersOffices/OrganizationCharts/ucm135674.htm/
- 6.Praveen K, Ramesh T, Saravanan D. ON Pharma Times. Goa: Sanofi-Synthelabo (India) Limited; 2011. Regulatory perspective for entering global pharma markets; p. 43. [Google Scholar]
- 7.Leon S, Kanfer I. Generic drug product development Solid Oral Dosage forms. New York: Marcel Dekker Inc; 2005. Introduction to Generic drug product development; p. 8. [Google Scholar]
- 8. [Last accessed on 2012 Jan]. Available from: http://www.wikipedia.org/wiki/European_Union .
- 9. [Last accessed on 2012 Jan]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/about_us/general/general_content_000235.jsp and mid .
- 10.Committee For medicinal product for Human use (CHMP) London, UK: European Medicines Agency; 2012. EMEA-Scientific Guidelines on Quality. [Google Scholar]
- 11.Arzeno N, Diaz R, Gonzalez S. Brazil's Generic Drug Manufacturing Success and the policies that permitted it. Final Project. 2004. [Last accessed on 2012 Feb]. Available from: http://www.ocw.mit.edu/courses/ electrical-engineering-and-computer-science/6-901-inventions-and-patents-fall-2005/projects/brazil_gen_drug.pdf .
- 12. [Last accessed on 2012 Feb]. Available from: http://www.anvisa. gov .
- 13. [Last accessed on 2012 May]. Available from: http://www.tfda.or.tz/function. php .
- 14.Sheftelevich Y, Satish TC. Drug Registration in Russia and the New Law. [Last accessed on 2012 Jan]. Available from: http://www.biomedconsult.com/201009focusrussia.pdf .
- 15. [Last accessed on 2012 Apr]. Available from: http://www.pacificbridgemedical.com/services/regulatory/registration/hongkong-drugs .
- 16.Benjamin Tak-Yuen Chan. Pharmaceutical Policy in Hong Kong: Defining an Evolving Area of Study. [last cited on 2009 Oct 12]. Available from http://papers.ssrn.com/sol3/papers.cfm?abstract_id=1487662 .
- 17.Hardacre S. IP issues faced by the pharmaceutical industry in Hong Kong ON Presentation in the Conference on Intellectual Property in HK and Mainland China, Best Practices and International Impact, March 2007. [Last accessed on 2012 Feb]. Available from: http://www.delhkg.cec.eu. int/en/doc/Mr%20Steve%20Hardacre.pdf .