

Abstract
Clinical studies comparing the response and side effects of various opioids have not been able to show robust differences between drugs. Hence, recommendations of the regulatory authorities have been driven by costs with a general tendency in many countries to restrict physician's use of opioids to morphine. Although this approach is recognized as cost-effective in most cases there is solid evidence that, on an individual patient basis, opioids are not all equal. Therefore it is important to have an armamentarium of strong analgesics in clinical practice to ensure a personalized approach in patients who do not respond to standard treatment. In this review we highlight differences between opioids in human studies from a pharmacological, experimental, clinical and health economics point of view. We provide evidence that individuals respond differently to opioids, and that general differences between classes of opioids exist. We recommend that this recognition is used to individualize treatment in difficult cases allowing physicians to have a wide range of treatment options. In the end this will reduce pain and side effects, leading to improved quality of life for the patient and reduce the exploding pain related costs.
Keywords: costs of illness, meta-analysis, opioids, pain, receptors
Introduction
Pain is the most common reason for individuals seeking medical care in the Western World and opioids are increasingly used to treat different types of pain. Although opioids have many side effects and their use has been associated with dependence, increasing misuse and mortality, no other strong analgesics can substitute for these drugs. Randomized studies provide little evidence that, at equi-analgesic doses, commonly used opioids differ markedly in their side effects. In head to head comparisons, most studies have failed to show relevant differences between drugs on a population level. In fact major variability between consumption of different opioids between countries is seen, even though they share borders and their populations are thought to have the same genetic and cultural background. This is illustrated in Table 1 using data from ‘The International Narcotics Control Board’. Hence, one can speculate that selection of opioids is driven by local traditions in medical practice rather than rational pharmacotherapy [1]. Accordingly, there has been pressure from regulatory authorities to restrict opioid use to the cheapest drugs. On the other hand, clinical experience and case reports suggest that on an individual patient level, some patients respond to certain opioids but are intolerant to others. Furthermore, pharmacological, experimental and clinical data support that there are major differences between opioids, and that, for example, switching from one opioid to another results in improvement of symptoms or less side-effects in more than 50% of patients [2]. Hence, both the National Cancer Institute, the American Pain Society, the British Pain Society and The European Association for Palliative Care recommend that several opioids should be available for the clinicians to ensure an optimal and individualized treatment approach [3, 4]. In this light it is of major concern that some national recommendations appear to neglect the fact that inter-individual variability in pharmacodynamics and pharmacokinetics results in some patients responding more favourably to one opioid than to another. The same trend has been seen, for example, in depression which shares many pathogenic mechanisms with pain, where the effect of treatment is markedly increased if the whole spectrum of available drugs is used [5].
Table 1.
Estimated requirements of narcotic drugs for 2011 in four European countries: Denmark, the Netherlands, United Kingdom and Germany. Numbers refer to total of estimated grams according to The International Narcotics Control Board –http://www.incb.org. The difference in populations should be taken into consideration (factors 15:5:1.5:1). Note that some medications (e.g. oripavine, etorphine) may not necessarily be used therapeutically
| Opioid | Countries | |||
|---|---|---|---|---|
| Denmark | Netherlands | United Kingdom | Germany | |
| Alfentanil | 350 | 500 | 6 000 | 2 300 |
| Codeine | 1 800 000 | 450 000 | 63 000 000 | 5 506 000 |
| Concentrate of poppy straw | – | – | 90 000 000 | – |
| Dextromoramide | – | 30 000 | 15 000 | 5 |
| Dextropropoxyphene | 100 000 | 1 000 | 2 000 000 | 2 005 000 |
| Dihydrocodeine | – | – | 14 700 000 | 170 000 |
| Dihydroetorphine | – | 1 | – | 1 |
| Diphenoxylate | 1 000 | – | – | 40 010 |
| Ethylmorphine | 500 | 3 000 | – | 250 |
| Etorphine | 12 | 1 | 50 | 1 |
| Fentanyl | 10 000 | 32 000 | 175 000 | 400 000 |
| Heroin | 55 000 | 225 000 | 100 000 | 55 000 |
| Hydrocodone | 10 000 | 10 | 1 000 | 200 |
| Hydromorphone | 7 000 | 5 000 | 30 000 | 400 000 |
| Ketobemidone | 50 000 | – | – | 250 |
| Levorphanol | 1 | – | – | – |
| Methadone | 200 000 | 350 000 | 3 370 000 | 3 600 000 |
| Methadone intermediate | – | – | 3 500 000 | – |
| Morphine | 300 000 | 250 000 | 9 500 000 | 1 850 000 |
| Morphine-N-oxide | 1 | – | – | 1 |
| Nicomorphine | 5 000 | 1 000 | – | – |
| Norcodeine | 1 | – | – | 1 |
| Opium | 60 000 | 10 000 | – | 300 000 |
| Oripavine | 1 | – | – | 1 |
| Oxycodone | 400 000 | 350 000 | 3 000 000 | 2 200 000 |
| Oxymorphone | 1 | 500 | – | 25 000 |
| Pethidine | 75 000 | 25 000 | 1 000 000 | 2 000 000 |
| Pethidine intermediate | – | – | 1 500 000 | – |
| Pholcodine | 1 | 80 000 | 1 000 000 | 1 |
| Piritramide | – | 14 000 | – | 146 000 |
| Remifentanil | 1 200 | 420 | 4 000 | 5 500 |
| Sufentanil | 12 | 100 | 500 | 700 |
| Thebaine | 1 | 200 000 | 25 000 025 | 3 104 000 |
| Tilidine | – | – | – | 49 020 000 |
Opioid prescriptions have increased dramatically over the past 20 years and some opioids have become very popular. This is likely to be due to solid marketing from the pharmaceutical industry rather than due to scientific knowledge. Hence, reported data have often been of poor quality and may have been biased for marketing purposes. It is therefore the responsibility of the scientific community to question these data and move towards more evidence based practice, e.g. reporting of data spanning from basic research to clinical trials that looks into differences between opioids and the inter-individual response to treatment with these drugs. The aim of the current review is to highlight differences between opioids from a pharmacological, experimental and clinical point of view. Although the data are supported by abundant animal experiments, we have mainly restricted the literature to human studies. Even though clinical pain studies in humans are biased by many confounders they are still considered the gold standard, and the examples we have used in the more basic sections are mainly included to support the bridge from basic science to the clinical situation. Due to the major costs of pain and its treatment to society, health economic considerations are also taken into consideration.
Pharmacology of opioids
The chemical structure of opioids is subdivided into those based on (i) the 4,5-epoxymorphinan ring, such as morphine, codeine, oxymorphone, oxycodone, buprenorphine,hydromorphone and hydrocodone, (ii) the phenylpiperidines such as alfentanil, fentanyl and sufentanil and (iii) the diphenylheptylamines such as methadone (Figure 1). Although these compounds differ in chemical structure, physicochemical properties and in pharmacokinetics they have one common feature, which is their interaction with the mu (μ) opioid receptor as the primary target. Despite this similarity, large differences in the clinical responses (efficacy, effectiveness, toxicity and safety) are seen between the classes of opioids. Furthermore, there are also major inter-individual variations in the response to the single opioid. An important cause of this inter-individual variability is thought to be of pharmacogenetic nature. To facilitate understanding of individual responses in pharmacokinetics of opioids which may be reflected in the clinic as an insufficient response and/or intolerable side effects, the general aspects of pharmacokinetics (absorption, distribution, metabolism and elimination) will be explained in the following sections.
Figure 1.
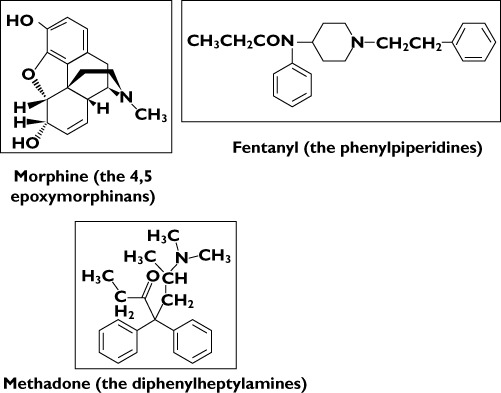
Chemical structural formulae for examples of drugs belonging to the three main classes of opioids
Absorption
The majority of opioids i.e. morphine, oxycodone, hydromorphone, methadone, ketobemidone, tramadol, tapentadol, fentanyl, sufentanil, buprenorphine and codeine all show a high gastrointestinal permeability, and thus they are readily and completely absorbed from the gastrointestinal tract following oral administration. However, the bioavailability of fentanyl, sufentanil and buprenorphine is very low and highly variable since these opioids are subjected to high hepatic first pass metabolism [6–10]. As a consequence they are not available in pharmaceutical formulations intended for oral administration. Recent research has revealed that low and variable bioavailability seen after oral administration may partly be explained by the substances being substrates for transporters present in the intestinal epithelium [11]. Drug transporters are present all over the body in the gastrointestinal tract, in the kidneys, in hepatocytes and at the blood–brain barrier. The two main families of drug transporters of relevance to opioid pharmacokinetics are (i) the ATP binding cassette (ABC) efflux transporters [e.g. P-glycoprotein (P-gp)], which restrict the passage and (ii) the solute carrier (SLC) influx transporters which facilitate the passage [12]. An example of the SLC influx transporters is OCT1 (SLC22A1) which has recently been linked to tramadol M1 metabolite pharmacokinetics [13]. The P-gp transporters are subject to naturally occurring genetic variations [14], whereas the genetic variation in SLC transporters is not yet fully elucidated. Clinical studies on SLC transporters in peripheral tissue do not yet exist [15], but it should be expected that genetic differences may explain variation in opioid absorption and explain insufficient responses on the individual level. Also as stated above the classes of opioids are expected to have different affinities for the transporter systems and this may explain more generalized variability in their clinical effects.
Distribution
After being absorbed the opioids distribute throughout the body tissue i.e. the site of main action within the central nervous system (CNS). To reach the CNS opioids have to cross the blood–brain barrier. Fentanyl, morphine and methadone have been shown to be substrates for the P-gp efflux transporters (Table 2) [16, 17]. The clinical aspects of fentanyl have been confirmed in a single human study, where an increased respiratory depression was seen in patients with a decreased P-gp expression [18]. Oxycodone is not a substrate for the P-gp efflux transporters, but it is substrate for the SCL influx transporters [19, 20], thus being actively transported into the brain. Most of the research elucidating the transport of opioids across the blood–brain barrier has been done in rodents and thus caution should be taken when extrapolating the findings to humans. However, as for the absorption, this may play a role in treatment heterogeneity.
Table 2.
Chemical and biological differences between commonly available opioids
| References | Opioids | Chemical structure class | Drug (+) or prodrug (–) | Receptor binding | Receptor internalization | Log P values (pH 7.4) | Drug P-gp efflux transporters | ||
|---|---|---|---|---|---|---|---|---|---|
| μ | κ | δ | |||||||
| [41, 43, 45, 55, 56, 61] | Codeine | 1 | – | + | – | 1.19 | – | ||
| [11, 43, 47, 50, 57] | Fentanyl | 2 | + | +++ | + | 4.12 | Substrate | ||
| [41, 46] | Hydromorphone | 1 | NA | +++ | – | – | – | ||
| [11, 41, 43, 50, 51, 57] | Methadone | 3 | + | +++ | + | 1.82 | Substrate | ||
| [11, 43, 47, 50, 57] | Morphine | 1 | + | +++ | + | – | –0.21 | Substrate | |
| [42, 47, 51] | Oxycodone | 1 | ± | + | ++ | – | 1.26 | Non-substrate | |
| [11, 41, 47, 52, 53] | Tilidine | 2 | – | + | NA | – | – | ||
| [41, 49, 54] | Tramadol | 2 | – | + | NA | 1.35 (pH 7.0) | Non-substrate | ||
| [41, 44, 48, 54] | Tapentadol | 2 | + | +++ | – | 2.87 | – | ||
Chemical structure class: 1:Class 1: 4,5-epoxymorphinan ring; 2: Class 2: phenylpiperidines; 3: Class 3: diphenylheptylamines. Receptor binding: += agonist. The number of symbols indicates potency. Receptor internalization: ± induce/do not induce receptor internalization. Log P = Partition coefficient. Drug P-gp efflux transporters are analyzed by using using cell cultures or in vivo animal techniques. NA, not available.
Metabolism
After absorption most opioids undergo first pass metabolism in the liver and here there are major differences between classes of drugs as well as individual differences in responses to the same opioid. The chemical class phenylpiperidines are metabolized by CYP3A4. This enzyme has many genetic polymorphisms but until recently none has been shown to be of major clinical relevance. However, a newly discovered single nucleotide polymorphism may change this view [21]. On the other hand, there is no evidence of the CYP3A4 metabolites of the phenylpiperidines being pharmacologically active [22, 23].
Within the class 4,5-epoxymorphinans (which additionally are alkyl esters at the 3-phenolic hydroxyl group i.e. codeine, hydrocodone and oxycodone) drugs are subject to O-dealkylation, catalyzed by CYP2D6 enzymes. In this way codeine is metabolized to morphine [22, 24], hydrocodone to hydromorphone and oxycodone to oxymorphone [22, 25, 26]. These metabolites are analgesic and often possess higher potency at the μ receptor than their parent compounds. For opioids belonging to this class, individual difference in metabolism may therefore result in unpredictable clinical responses. Up to 10% of Caucasians (and varying proportions of other populations) lack CYP2D6 activity. These patients experience little analgesia from codeine. CYP2D6 gene duplication on the other hand, found in 3% of Caucasians, is associated with ultrarapid metabolism of codeine to morphine [27]. These patients are more susceptible to both the beneficial and adverse effects of codeine. There have been a number of case reports of fatal neonatal opioid toxicity in children who were breastfed by CYP2D6 ultrarapid metabolizing mothers [28]. However, genetic variation in CYP2D6 has not been found to be associated with clinical variability in response to the strong opioid oxycodone, but it has been reported to affect experimental pain responses [29, 30]. Tramadol is metabolized to the active metabolite M1 (O-desmethyl tramadol). In this demethylation process CYP2B6 and CYP2C19 also play an active role, and hence individual differences in these enzymes may explain clinical findings of individual tolerability and effect [31]. Apart from the examples above, the possible impact of genetic polymorphism on opioid kinetics and dynamics have been thoroughly reviewed by Somogyi et al. [12]. In conclusion, with regard to influencing heterogeneity in the overall clinical opioid response, the most important genetic polymorphism remains that of CYP2D6, although more clinical studies are needed to reach a final conclusion.
Opioids of the 4,5-epoxymorphinan class are also subject to N-dealkylation. This results in nor-derivatives, which bind to the μ receptors, but show lower affinities than the parent compounds. The N-dealkylation is mainly catalyzed by CYP3A4, which as mentioned above does not show any genetic polymorphism but, however, it shows clinically relevant variation between subjects. Finally, 4,5-epoxymorphinans containing free hydroxyl groups (e.g. morphine, codeine, hydromorphone, buprenorphine) are subject to glucuronidation. The glucuronides formed at the 6-aliphatic hydroxyl group i.e. codeine-6-glucuronide [32] and morphine-6-glucuronide [33] are in general believed to be analgesically active. The glucuronidation is primarily mediated by the UDP glucuronosyltranferase UGT2B7. This enzyme shows genetic polymorphism. However the in vitro and in vivo functional significance is not obvious [34, 35] and results from studies on the clinical impact of this polymorphism are contradictory [36–38]. Furthermore, some metabolites such as morphine-3-glucuronide are thought to be toxic.
Methadone, a representative of the diphenylheptylamines, is metabolized by cytochrome P450 (CYP) isoforms to a stable metabolite 2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrilidine. The CYP isoform involved in the metabolism of methadone is thought to be CYP3A4, but probably also CYP2B6, CYP2D6 and CYP2B19 are involved [12, 39]. Genetic polymorphisms for these enzymes are described above.
Even though many aspects of metabolism are still disputed, individual differences in the complex metabolism of opioids will invariably be the case for some patients, and thus some opioids may not alleviate pain whereas others have more effects than expected (and more side effects). As a consequence there has been a growing interest in the pharmacogenetics of strong opioids, i.e. whether an individual's genetic makeup may influence response to morphine and other opioids. In the past year a number of papers have been published exploring genetic factors associated with variation in daily opioid dose, opioid side effects and pain relief [40–42]. Hence, genetic testing has the potential to facilitate the choice of the right dose of the right opioid for each individual patient. To date however, the functional and clinical applicability of these data are uncertain and any apparently positive results have yet to be tested and replicated in a prospective population. Until then selection of opioids, such as fentanyl, with low inter-subject variations within the pharmacokinetic parameters and with few or no active metabolites will be a rational approach.
Excretion
The vast majority of opioids are excreted as metabolites though the kidneys. Thus for substances transformed to pharmacologically active metabolites, decreased kidney function may influence the overall clinical effect due to accumulation of the metabolites and this may explain differences in effects and side effects between opioids. The best described examples of clinically relevant active metabolites are morphine-6-glucuronide and morphine-3-glucuronide. As stated above the 6-glucuronide is thought to be an active analgesic like the parent compound, morphine. Although the potency ratio to morphine still remains to be established it plausible that in steady-state conditions and in patients with impaired kidney function it contributes more to analgesia. Results from some studies have suggested the 3-glucuronide possess anti-analgesic and excitatory effects. However results from other studies have failed to prove that. Thus clinical relevance of accumulation of this metabolite in patients with impaired kidney function still remains controversial [43].
Receptor binding
The drugs also differ with respect to their receptor binding [12, 44–61] (Table 2). For the majority of the commonly used opioids interaction with the μ receptor is the most important. Additionally interaction with the kappa (κ) receptors might also be essential for overall effects of some opioids like buprenorphine [62] and oxycodone [55, 56]. Buprenorphine (or its metabolite norbuprenorphine) may also have effect on the delta (δ) receptors involved in hyperalgesia [63]. Although data about receptor binding are based on both animal and human studies, interaction with the N-methyl-D-aspartate receptor may in humans contribute to the analgesic effects of opioids like methadone and ketobemidone [64, 65]. For tramadol the serotonin and norepinephrine re-uptake blocking effects seem to play a major role in the analgesic effects [66]. Tapentadol has additional analgesia due to increased downstream inhibition from the brainstem on the spinal cord that is facilitated via norepinephrine re-uptake inhibition [67]. However, even for opioids acting primarily by binding to the μ receptors there are differences in the achieved analgesic profile, this being mainly due to differences in affinity to and efficacy at the receptor and thus the overall potency. Along this line sufentanil, fentanyl and buprenorphine are being regarded as high potency opioids, methadone, oxycodone, morphine, ketobemidone and hydromorphone as medium potency opioids and codeine, hydrocodone, tramadol and tapentadol as low potency opioids. Further, as outlined above some of the opioids (morphine, codeine, tramadol, buprenorphine, hydromorphone, oxycodone and hydrocodone) have metabolites, which also bind to the receptors and thus might contribute to the overall analgesia. The best known and most elucidated example is again morphine-6-glucuronide [44]. Due to differences in up-regulation of receptors and variability in the pain picture (e.g. neuropathic pain and hyperalgesia) opioids will have different effects from a theoretical point of view.
Experimental pain studies
Basic physiological studies in animals have shed light on many pharmacological mechanisms and a series of animal studies has shown that major differences between opioids exist [68]. Although of major importance, the scope of this paper is not to review animal studies and for a recent review of this literature, the reader is referred to [69]. It should be stated, however, that data from opioids in animal studies cannot be uncritically translated to man. First of all animal studies are mainly based on motor reflexes or behavioural responses and such data can only partly be interpolated to human pain, which is a net result of complex sensory, affective and cognitive processing. Second, there are major differences between the effects of drugs across species (and even strains), and this limits generalization of findings. As many of these models are also optimized for success the construct validity is often limited [70], and in fact, only one analgesic (ziconotide) has ever gone from bench to bedside on the basis of animal models alone [71].
Therefore, knowledge about how opioids interfere with the human pain system is highly warranted, and clinical studies on different patient groups have attempted to explore mechanisms and effects. A major shortcoming is that clinical assessment of pain and effect of analgesics in patient studies is confounded by many factors such as general malaise, fear and anxiety, psychomotor impairment, cognitive disturbances, social consequences of disease and co-medications. Moreover, sedative properties of opioids make pain evaluation difficult [72]. For example, if a new analgesic under investigation has only tranquillizer properties, this can also decrease pain indirectly, as the effect on fear and worrying in general may improve the situation, leading to increased mobility and a better social situation. This will again lead to increased production of endogenous opioids (endorphins) and, indirectly, improvement of the pain although the drug (at least hypothetically) has no direct effects on the pain system per se. On the other hand, complex psychosocial settings may also overshadow the effect of a true analgesia rendering the patient to rate pain as constant. This may explain the discrepancy between doctors' impressions of differentiated effects of analgesics in individual patients and the limited proof for this in clinical trials [73].
Experimental methods to evoke and assess pain under controlled circumstances are advantageous as they encompass many of the above problems and offer the opportunity to demonstrate analgesic effects objectively [72]. Using such models, the investigator can control the experimentally induced pain (including the nature, localization, intensity, frequency and duration of the stimulus), and provide quantitative measures of the psychophysical, behavioural, neurophysiologic or imaging responses [74] (Figure 2). The methods are also very specific on different levels on the pain system. Hence, they are able to activate various nerve pathways and reflect pain responses at certain levels of the neuraxis. In combination with neurophysiological assessment and neuroimaging the methods have improved sensitivity and added value by explaining drug mechanisms [70, 72, 75]. Newer methods in general have high reliability and are robust across individuals. In evaluation of analgesics most studies have relied on models in the skin, but from a clinical perspective deep pain from, for example, muscles and viscera is more interesting, and reliable and valid pain models from these tissues have also been developed [74, 76]. Experimental studies have also shown that models with tonic, deep pain are often more sensitive to opioids, especially when intensities above the pain threshold are used [72, 77, 78]. The sensitivity of the models has been used in pharmacogenetic research where variants of genes such as OPRM1 (μ-opioid receptor gene), COMT (catechol-O-methyltransferase), GCHR (guanosine triphosphate cyclohydrolase 1) and MC1R (melanocortin 1 receptor) have been associated with response to morphine and its metabolites. This association, however, has not been convincingly replicated in the more complex clinical settings [41, 79]. The models are also suitable for combining pharmacokinetics and pharmacodynamics that can be used to study concentration–effect relationships of opioids and their metabolites in detail. Hence, human experimental pain models may bridge the gap between animal and clinical studies. It should be stressed, however, that the more complex experimental models may be difficult to use and are only available in the most advanced laboratories which limits their general use.
Figure 2.
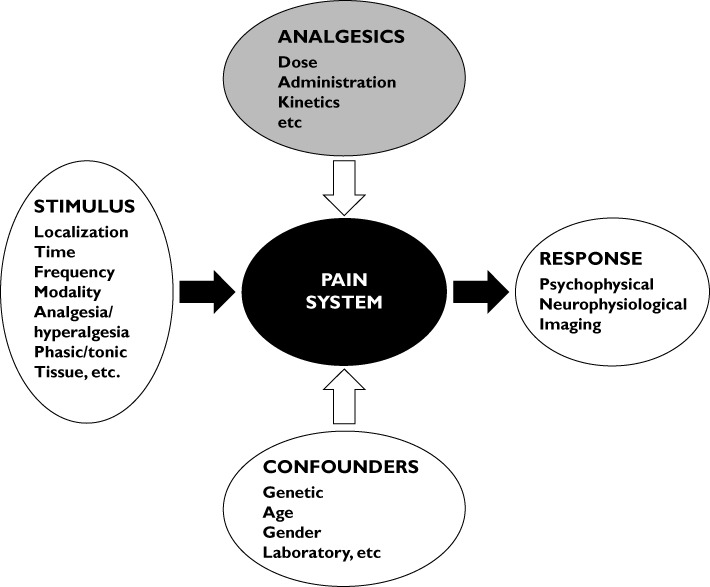
The concept for experimental pain induction using a variety of controlled stimulations of the pain system (black box) with different methods for assessment of the evoked response. The robustness of the system can be challenged by different confounders and modulated by analgesics and experimental procedures
Experimental models have been used for decades to evaluate opioid mechanisms and to monitor their effect in clinical settings (for review see Staahl et al. [72]). To exemplify the use of experimental models it has been claimed that opioids have different binding profiles to receptor types and that κ-agonists can be effective in the treatment of visceral pain [80, 81]. Animal data have suggested that the antinociceptive effects of oxycodone are mediated by μ- and possibly κ-opioid receptors, the latter being present mainly on visceral peripheral afferents [56]. In an experimental study in healthy volunteers equi-analgesic doses of oxycodone, morphine (considered a selective μ-agonist) and placebo were compared in a multi-modal, multi-tissue model. The two opioids alleviated skin and muscle pain to a similar extend confirming equi-analgesia, but oxycodone was more effective in visceral pain [78]. The peripheral effect of oxycodone on the oesophagus was later confirmed in a pharmacodynamic–pharmacokinetic study [82]. The same model was then repeated in patients with chronic pancreatitis. Contrasting the situation in healthy volunteers the chronic inflammation and pain should hypothetically result in a generalized up-regulation of κ-receptors in the CNS as seen in animal studies, and therefore the peripheral effects on viscera should have minor contribution [83]. As hypothesized the efficacy of oxycodone on all three tissues was in this study significantly enhanced and better than morphine and placebo [84]. A subsequent phase III study in patients with pain due to pancreatic cancer showed no differences between the two opioids, but this was not surprising as the study was underpowered considering the many limitations of clinical studies listed above [85].
Hyperalgesia and allodynia are features dominating pain in the clinic and these conditions can also be mimicked in experimental models serving as a translational bridge to the clinical situation. In these models the effects of opioids have been consistently reported [86, 87]. Koppert et al. have developed a model where intradermal electrical stimulation results in ongoing pain and secondary mechanical hyperalgesia [88]. Using this model they were able to demonstrate that buprenorphine had antihyperalgesic effects as compared with the traditional μ-agonists fentanyl and alfentanil [86]. We developed a model eliciting generalized hyperalgesia by perfusion of acid and capsaicin in the oesophagus. After hyperalgesia was developed, oxycodone (with theoretical effect on upregulated κ receptors in the CNS) decreased pain to a higher degree than morphine and placebo in skin, muscle and viscera [87]. Buprenorphine is an opioid with an agonistic effect at the μ receptors and variable effect at the κ and δ receptors. However, there is preclinical evidence the its active metabolite norbuprenorphine may act as a δ receptor agonist with antihyperalgesic effects [89, 90]. We tested the effect of equi-analgesic patches of buprenorphine, fentanyl and placebo over a week in a model where superficial, deep and hyperalgesic pain was evoked. Buprenorphine attenuated bone associated pain and hyperalgesia to mechanical stimulation (in a first degree sunburn erythema area evoked with ultra violet B light) when compared with placebo, whereas findings for fentanyl were not different from placebo [91]. Hence, these differentiated properties support clinical observations that different opioids often show variable effect in individual patients.
Meta-analysis, mixture models and utility functions to unravel differences between opioids
While drugs may significantly differ in their pharmacokinetics and dynamics, the outcome of clinical trials comparing one drug with another in actual patients may not reach significance of differences in either efficacy or toxicity/safety. Apart from technical issues (including quality of the study, sample size, statistical approach), this is certainly related to the large variability in the effectiveness and safety to treatment across the patient population (i.e. heterogeneity of treatment effect [92]) and the overall results are difficult to extrapolate to individual patients [93].
High variability in study populations is also true for pain patients enrolled in clinical trials on opioids. Hence, comparative studies on efficacy but also adverse events seldom show differences, despite variability in opioid potency, onset/offset times, dosing and routes of administration. Along this line Kalso et al. analyzed 15 randomized, double-blind, placebo controlled studies on long term use of strong opioids for efficacy and safety in chronic non-cancer patients [73]. Conditions (neuropathic and nociceptive pain), opioids (morphine, oxycodone, methadone), doses and routes of administration (intravenous, oral) varied considerably among the 15 included studies. The results indicated that while the opioids did alleviate both nociceptive and neuropathic pain, there was a large individual variation in responses, with a mean pain relief from any opioid of only 30%. Hence, such data support that in pain studies, the heterogeneity of treatment effect plays a major role and may explain why no differences were found between opioids [92, 93].
Systematic reviews
One of the main problems of conventional meta-analyses is the large heterogeneity among studies. A Cochrane review on morphine [94] concludes that morphine remains the gold standard for moderate to severe cancer pain. However, this is based on evidence showing that morphine is as effective as but not superior to other opioids. Looking into adverse effects, a recent meta-analysis on transdermal opiates (fentanyl and buprenorphine) vs. long acting oral morphine in the treatment of moderate to severe cancer pain also concluded that there were no differences in the overall adverse effect profile [95]. Although meta-analyses are important within many aspects of medicine the above considerations call for cautious interpretations. Not only are the populations of distinct studies difficult to compare, treatment paradigms (see below) and end points chosen are often dissimilar causing difficulty in the interpretation. Regarding treatment paradigms, many meta-analyses include studies using both a parallel randomized design and crossover designs. Although many crossover trials have been performed to evaluate opioids, parallel studies are to be preferred. This is especially the case when strong analgesics are compared with placebo, since the differences in efficacy and side-effects are so obvious that blinding is difficult or non-existing after the first period in the crossover trials.
We recently performed a systematic review to address the issue of whether one opioid (morphine) is superior to other strong opioids with respect to efficacy and tolerability [96]. Only studies with parallel design were included. In contrast to previous meta-analyses, we performed a network meta-analysis which pools the effect estimates of different treatments, even when there are no direct comparisons. For example, studies comparing opioid A to opioid B, opioid A to placebo and opioid C to placebo are included to compare the relative efficacy of opioid A to opioid B and to placebo. Fifty-six studies were included with an important limitation of the analysis the large heterogeneity in study population and intervention, but with the main strength the inclusion of only parallel randomized controlled trials. No significant differences in pain relief were observed between morphine and oxycodone, methadone and oxymorphone, but this was the expected outcome as doses normally are selected to be equi-analgesic. However, compared with morphine, patients on buprenorphine were more likely to discontinue treatment due to lack of effect. A decreased risk for discontinuation due to adverse effects relative to morphine was observed for patients on fentanyl, methadone and buprenorphine. Despite the improved methodology, large variations in study populations and interventions precluded significance of differences in either efficacy or toxicity/safety among the opioids tested. The study, however, indicated that there are differences between clinically available opioids even when pooling heterogenic studies.
New models
A different approach is required to obtain and quantify objectively meaningful differences between opioid analgesics. One such approach is mixture modelling coupled to a time series analysis of pain relief data [97]. The mixture model objectively determines the probability of an analgesic effect by subdividing the patient population into various response groups with different response probabilities. This type of analysis is attractive as it rapidly allows subdivision of analgesic responses into subgroups that may be defined a priori. Furthermore, a placebo response may be quantified among subgroups. Since the efficacy of the drug varies between subgroups, linking these subgroups to clinically relevant factors such as underlying disease characteristics (e.g. duration and severity of disease), patient characteristics (e.g. weight, gender, age, genetic make-up) as well as a multitude of other covariates will increase our insight into the efficacy of opioid analgesics within each of the different subgroups. Meaningful differences in efficacy among opioid analgesics will or will not (when they do not exist) be revealed using this approach.
A different but even more sophisticated approach in the assessment of differential opioid effects is to compare the efficacy vs. safety and toxicity of opioids by construction of a utility function. Utility (or safety) functions assess the net clinical effect of drug exposure. The utility of drug effect may be defined as the probability of obtaining the desired effect minus the probability of obtaining a side effect and is based on acquired pharmacokinetic and pharmacodynamic data [98, 99]. Hence, despite the fact that no clear differences are observed in analgesic efficacy between opioids, the utility functions can provide evidence for a clear separation in terms of utility or efficacy/safety balance. This technique is applicable in humans in experimental and clinical settings (see Figure 3 on the utility function of morphine) [100]. It allows dose and drug selection (which opioid suits this patient best and what dose provides an optimal probability for desired compared with undesired effects). As more mechanistic biomarkers become available to assess drug effect (related to analgesia and toxicity/safety), utility functions may evolve from descriptive to predictive tools and could enable an informed choice of opioid treatment in specific patient subpopulations.
Figure 3.
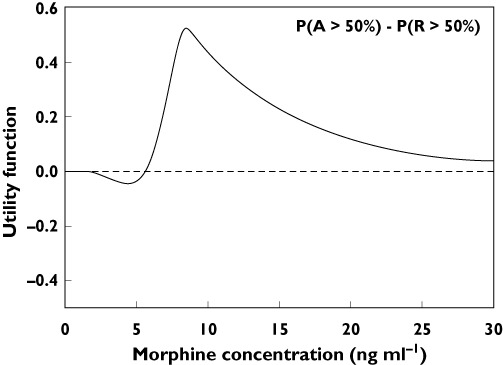
Utility function of morphine showing the probability of an analgesic effect [P(A)] greater than 50% [P(A > 50%)] minus the probability of toxicity (in this case respiratory depression) greater than 50% [P(R > 50%)]. A negative value indicates that the probability for toxicity is larger than the probability for analgesic efficacy. The reverse is true for a positive value of the utility function. For morphine at low dose the probability for respiratory depression exceeds that of analgesia, at doses >5 ng ml−1 the probability for analgesia is greater. At high morphine concentrations no differences in probability are apparent (value of the utility function approaches zero). Analgesia and respiratory data are obtained in healthy volunteers
Clinical perspectives
Morphine has traditionally been the recommended choice of strong opioid for cancer pain [101]. This initial recommendation was based on expert opinion, availability, familiarity and price. In recent years there has been growing availability of other alternative strong opioids, including but not limited to, oxycodone, buprenorphine, hydromorphone, methadone, alfentanil and fentanyl. The choice of strong opioid in the management of severe pain varies widely [102]. There have been relatively few clinical studies comparing morphine and alternative opioids in terms of efficacy, toxicity and tolerability. The studies which have been carried out have been small, with subsequent limited scope for analyses or conclusion. Although traditional μ agonists may be relative comparable, opioids with effects on other receptor systems have become available. For example tapentadol also has effects on the endogenous pain modulation system (see pharmacology section) [67]. Tramadol has some of the same effects as tapentadol with additional action on serotonin re-uptake. Tramadol, however, is considered a weak analgesic.
The World Health Organization analgesic ladder was established over 20 years ago in order to direct the management of cancer pain. This ladder represents a sequential approach, with morphine and other strong opioids being the predominant pharmaceutical agents used in the management of moderate to severe pain. Given the paucity of high quality evidence upon which to base recommendations, clinical guidelines as to the use of opioids in the management of cancer pain (or other types of pain) are derived largely from expert consensus and based on systematic reviews of the existing literature [4]. For any particular patient with cancer-related pain, there is no fixed or predetermined dose of strong opioids. Instead the central dogma of cancer pain management is individualized drug therapy.
Side effects
Common side effects of strong opioids include confusion, drowsiness, hallucinations, bad dreams, dry mouth, nausea, vomiting and constipation. Pruritus, sweating, opioid-induced hyperalgesia, myoclonus and delirium are less common. In clinical trials of patients with chronic back pain and osteoarthritis, when compared with oxycodone, naloxone/oxycodone combinations as well as tapentadol appears to have similar efficacy in terms of analgesic action but possibly fewer gastrointestinal side-effects [103]. These studies were sponsored by pharmaceutical companies and have yet to be confirmed independently. Respiratory depression is rare if strong opioids are titrated carefully according to individual patient response. Side effects may become apparent as the opioid dose is increased. These toxicities may often be managed with other medications, e.g. antiemetics/antipsychotics [104, 105] but sometimes they persist or become intolerable and thus dose-limiting. In head to head studies most strong opioids are equally tolerated [4], but there are inter-individual differences between these drugs with respect to side effects. Although it has been claimed that patch formulations can reduce side effects this is still an open question since the studies were not sufficiently powered [106]. As stated above tapentadol may have less opioid induced bowel dysfunction than other opiods. Opioids also affect the endocrine and immune systems as well as cognitive function [107]. In vitro data have suggested that some opioids such as tramadol and buprenorphine have less immunosuppressive effects although the clinical relevance is still controversial [108].
Individual difference in response
Despite there being no significant evidence on a population level of major differences between morphine and other strong opioids in terms of efficacy or tolerability, there is growing recognition of marked variation between individual patients in response to individual opioids for cancer pain. Not all patients respond well to each strong opioid. For example, up to 30% of cancer patients on morphine fail to achieve adequate analgesia, despite escalating dose and/or experience intolerable or dose-limiting side effects. These patients have been referred to as ‘morphine non-responders’[109]. A similar pattern of response variability is seen with other strong opioids [110–112]. Clinical experience and a number of studies have demonstrated variability in terms of the level of analgesia achieved, the side effects experienced and the daily dose of opioid required. The two case studies listed below are not unusual for clinical practice.
Case study 1
A 61-year-old woman with an inoperable leiomyosarcoma was seen by the palliative care team because of a constant dragging pelvic pain. She was unable to take oral medication due to nausea and vomiting and was on a continuous 24 h subcutaneous syringe driver of 60 mg of morphine sulphate. She had incomplete pain control and was also experiencing visual hallucinations, moderate confusion and drowsiness. Her renal function and serum calcium were within the normal range. She had no evidence of infection. She was opioid switched to oxycodone in the syringe driver, initially at a dose of 20 mg over 24 h. The dose was titrated according to effect over the following days. Within 48 h, her pain was well controlled and she was no longer experiencing intolerable opioid side effects. She was eventually discharged home on modified release oxycodone 40 mg twice a day orally.
Case study 2
A 48-year-old woman with advanced breast cancer and extensive bone metastases was admitted with uncontrolled pain in her lumbar spine. She had received radiotherapy to this area 4 months previously. Her doctor had started her on modified release oxycodone 30 mg twice a day. She was also on gabapentin, paracetamol and diclofenac. Her doctor had also prescribed immediate release oral oxycodone to be taken as required. She was reluctant to take the immediate release oxycodone as she did not feel that it was effective and made her feel drowsy. Radiological imaging demonstrated bony metastases with no compromise of the spinal cord. She was opioid switched to immediate release morphine sulphate 20 mg every 4 h. The next day the pain was much improved and she had minimal drowsiness. She required no further dose titration and was converted to modified release morphine sulphate 60 mg twice a day.
There may be a number of factors, both opioid-related and non-opioid related, which may contribute to this inter-individual variability in response to opioids (see also section about pharmacology). Some cancer pain (especially predominately neuropathic pain) may not be entirely opioid-responsive. Non-cancer pain studies have demonstrated associations between differences in pain sensitivity and analgesic response to opioids and age [113, 114]. Gender, psychological distress, sleep quality and renal function may also influence outcome [115–117]. Apparent side effects of morphine and other opioids may therefore be due to pharmacokinetic, pharmacodynamic or genetic differences, individual patient comorbidities (including biochemical imbalances) and concomitant medications. However, as shown in the cases the inconsistent effectiveness of opioids between patients (switch from morphine to oxycodone and vice versa) often makes the clinical situation difficult and unpredictable.
In clinical settings where inter-individual differences between opioids exist, it makes therapeutic sense that clinicians have access to several strong opioids to optimize analgesic control. In some circumstances switching from one opioid to another may be a useful approach to improve analgesia and reduce opioid side effects [4] (Figure 4). Evidence from randomized controlled trials is lacking in this area [2, 118]. The data from the first randomized controlled trial comparing response to morphine and oxycodone in opioid naïve patients and positive response with opioid switching are due to be presented and published this year. The study details can be seen at http://www.controlled-trials.com/ISRCTN65155201.
Figure 4.
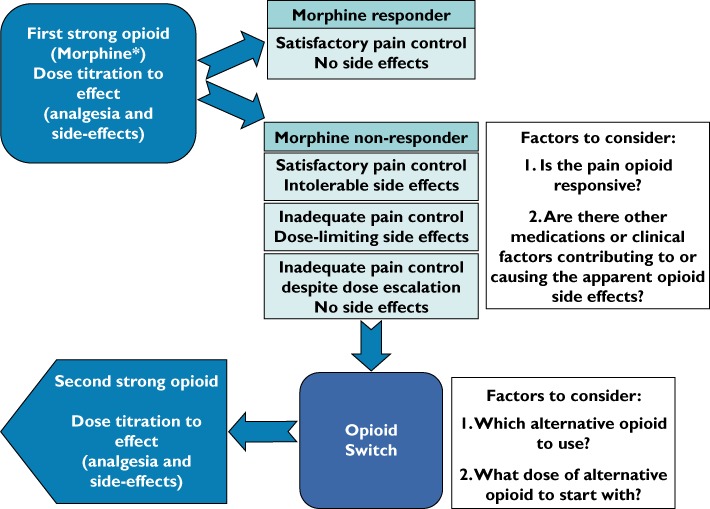
Opioid switching in cancer pain: Clinical considerations. *Morphine is the WHO first line strong opioid of choice but an opioid switch may be made between any two strong opioids
Opioid switching
Existing data support the efficacy of this practice, with some studies suggesting that up to 80% of patients achieve a successful outcome after opioid switching [110, 119, 120]. There appear to be two broad groups of patients for whom opioid switching may be beneficial in terms of improving analgesia and minimizing side effects. In the first group, opioid switching is required shortly after initiation of the original opioid, when the dose is relatively low. Some of the inter-individual variability in opioid response in these patients may be associated with differences in pharmacokinetic and pharmacodynamic pathways. In the second group, response to the initial opioid may be satisfactory until a later stage when the patient has been on opioids for some time, or as the opioid dose is titrated higher [121]. It is likely that the mechanisms underlying the effectiveness of opioid switching in each of these two groups is different.
The most likely reasons why patients who have been stable on an opioid for some time require escalating opioid doses are disease progression and the development of drug tolerance. Tolerance describes the apparent reduction in response to a drug after repeated administration. In terms of opioids, tolerance can develop to both the analgesic action as well as the central side effects of the drug (tolerance to the peripheral opioid side effects including constipation is less common). Tolerance to the analgesic effects of the opioid and disease progression both necessitate the requirement for further increased opioid doses in order to achieve adequate analgesia. If the patient does not develop tolerance to the side effects of the opioid as the dose increases, these toxicities may become dose-limiting, thus prompting an opioid switch. The rationale for switching these patients to an alternative strong opioid is based on the phenomenon of incomplete cross tolerance. Incomplete cross tolerance is thought to be due to the existence of a number of different μ-opioid receptor subtypes, differential location of these subtypes and varying action of strong opioids at these subtypes [122–124]. Thus the degree to which the patient is tolerant to the analgesic action and side effects of one μ-opioid agonist may be different when another μ-opioid agonist is used.
Clinically, incomplete cross tolerance may facilitate the use of lower doses of the alternative opioid, which may allow adequate analgesia without intolerable side effects. Incomplete cross tolerance presents a particular challenge in the use of opioids and opioid switching for cancer pain, namely dose conversions between the different strong opioids. Equi-analgesic dose conversion tables exist but these were mostly derived from single dose studies and therefore the applicability is unclear [125]. Differences in the required dose of the alternative opioid may be influenced by the reason for opioid switching. Patients with pain may need higher doses than patients who are being switched because of intolerable side effects. There is no available method or model to predict the dose of the second opioid. The general recommendation is to use the available equi-analgesic dose tables as reference tables only, to reduce the equivalent dose of the second opioid by at least 33–50% and to titrate according to effect. Hence, experience with opioid switching emphasizes the importance of individual differences in the response to the various opioid drugs and suggests that the most favourable opioid in an individual cannot be predicted [126].
Opioid switching to and from methadone is more complicated and should only be carried out under close specialist supervision [127]. On the other hand methadone is very different from the more traditional opioids as it has a fairly long half-life and lacks active metabolites. This is advantageous in the setting of individual variation in genetic phenotype and also in renal and liver failure [44]. When administered after treatment with another opioid, its potency increases and observational studies suggest that most patients benefit when an unsatisfactory regimen is switched to methadone [128].
Cost drivers
Pain has been identified as the most common reason for individuals seeking medical care in the US and costs more than $ 100 billion (75 billion €) each year in health care, compensation and litigation. The disability associated with pain presents a significant and costly liability to workers, employers and society and about 14% of workers who are in pain take time off from their jobs [129]. Currently, only limited knowledge exists on the costs associated with treatment of pain as a symptom of an underlying medical condition rather than a treatment of the disease itself. In a recent study by Gustavsson et al. [130] the costs associated with treatment of nine diagnosis groups related to chronic pain were estimated. It was found that the total annual direct cost to the Swedish health care system ranged from approximately 6000 to 2800 € for cancer and headache respectively, using current exchange rates. On the other hand the annual cost of prescribing pain medication alone for the same diseases only varied from 73 to 45 €. In a US health care setting the drug costs related to low back pain have been estimated to be $ 8752 (6590 €) on an annual basis per patient [131]. Further, in another study by Berger et al. [132] the total annual drug cost per patient with painful neuropathic disorder was estimated to be 880 €. Due to the high prevalence of chronic pain it imposes a significant demand on the scarce resources available in health care systems worldwide. As the following review of the literature indicates caution should be taken when restricting access to pain relieving medication as an avenue for obtaining cost savings as the costs of pain medication are relatively low and the costs of not being compliant, discontinuing as well as managing side effects are relatively high ceteris paribus.
Costs of opioids
The costs of opioids and other analgesics are estimated to be only 1.2–2.8% of total direct costs for cancer and intervertebral disc disorders [130]. Figure 5 shows the cost per defined daily dosage (DDD) based on public list prices of different opioids in Denmark (Danish Medical Agency 2011), Germany (Rote Liste 2011) and the United Kingdom (National Health Service 2011). The dosage of oxycodone and morphine is 10 mg and the release of fentanyl and buprenorphine patches is 50 µg h−1 and 52.5 µg h−1 respectively.
Figure 5.
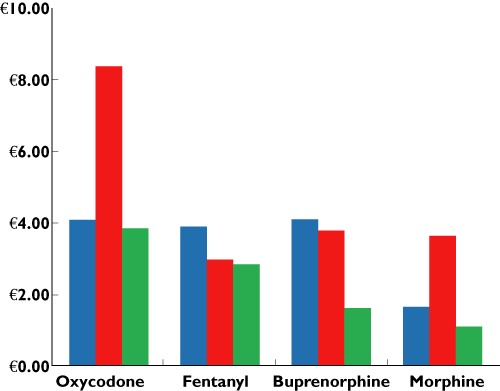
Prices of commonly available opioids based on the defined daily dose. For comparability the cheapest opioid was chosen from the public prices available on the following internet sites: http://www.medicinpriser.dk; http://www.nhsbsa.nhs.uk/prescriptions; http://www.rote-liste.de.  , Denmark;
, Denmark;  , Germany;
, Germany;  , UK
, UK
As can be seen the low costs of daily treatment with pain reliving medicines support the findings by Gustavsson and coworkers [130] and also shows that this holds across different health care systems. However, there are substantial differences in costs for the individual drugs with morphine being the cheapest drug in most countries.
Costs of side effects
Side effects of opioid treatment in the management of chronic pain are well known and these can lead to treatment discontinuation [133] and non-compliance [134] adding a costly secondary effect. The direct annual costs to the Danish health care system related to side effects in patients receiving long term treatment with opioids due to non-cancer related pain on the CNS and on the gastrointestinal tract can be seen in Figure 6.
Figure 6.
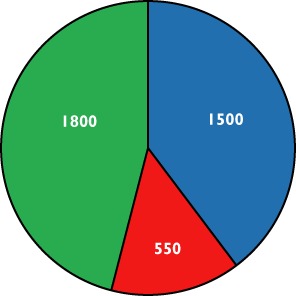
The yearly direct costs to the Danish health care system relating to managing of opioid induced side effects. Data are extrapolated and adopted from Annemans, converted and rounded off to €[143].  , central nervous system;
, central nervous system;  , constipation;
, constipation;  , nausea and vomiting
, nausea and vomiting
In another recent study by Kwong et al. [135] the costs of gastrointestinal event related claims were estimated following outpatient treatment in US non-cancer patients using oral opioids. The analysis included opioid-naive patients who received a new prescription of oxycodone or hydrocodone containing immediate release oral products. The results showed that compared with patients without any gastrointestinal symptoms, the additional adjusted mean total health care costs for patients with a gastrointestinal related event ranged from approximately 3700 to 27 200 € over a 90 day follow-up period. Compared with the cost estimates in Figure 7, the costs in the study by Kwong et al. [135] are quite high. The costs in Figure 7 are mean estimates of over all patients with or without opioid related adverse events, whereas the estimates in the study of Kwong et al. [135] are based on identified insurance claims for gastrointestinal event related treatment. Also, caution should be taken when interpreting across country cost comparisons.
Figure 7.
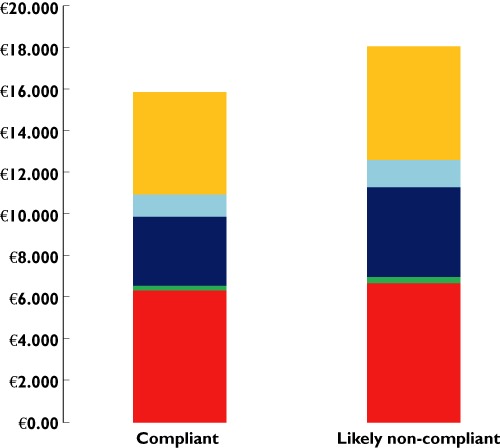
Health care costs (per patient) in a 1 year follow up period: Compliant vs. likely non-compliant chronic opioid users. Increases in the utilization of inpatient care and prescription costs are the main cost drivers for the difference in costs (70% of the savings) between adherent and non-adherent patients. Data adapted from Leider et al. and converted to € using the 2008 average exchange rate [134].  , Prescription medicines;
, Prescription medicines;  , Other medical;
, Other medical;  , Inpatient;
, Inpatient;  , Emergency;
, Emergency;  , Ambulatory
, Ambulatory
Discontinuation
A systematic review by Noble et al. [136] comprising a total of 2473 patients from 11 trials with a variety of painful indications, showed that the overall rate of discontinuation from analgesics was 22.9%. The discontinuation rate changed between weak (11.4%) and strong opioids (34.1%). Broekmanset et al. [137]. found that only 38% of chronic non-malignant pain patients were compliant with their prescribed course of pain treatment. These finding are supported by the findings by Leider et al. [134] suggesting that only 21% of patients, across various medical conditions, were compliant with prescribed treatment of pain. The level of discontinuation and non-compliance due to side effects by prescribing opioids for long term use, have been shown to lead to increased health care costs beyond the cost of managing the side effects, by carrying over additional cost to other parts of the health care system and by the cost associated with switching medicines [134, 138, 139]. As some formulations such as oxycodone and naloxone combinations, supplement of opioid therapy with parenteral methylnaltrexone and tapentadol, have been shown to have less side effects on the gastrointestinal tract compared with traditional opioids [140–142], the use of such drugs may reduce costs related to side effects and may guide recommendation of opioid use from an economic point of view.
Figure 7 above shows the increased costs among likely non-compliant patient compared with compliant chronic opioid users.
A key factor for ensuring a high level of compliance with a prescribed course of treatment is to create a balance between treatment effect and the side effect profile of the different pharmacological treatments individualized for each patient. Hence, restricting a physician's armamentarium in pain management will reduce the ability to tailor a planned course of treatment. On the other hand, ensuring a personalized approach that balances treatment effect, tolerability and convenience in alignment with the individual patient's need will likely imply a larger overall saving in health care costs as well as reducing production loss. Hence, access to a wide range of treatment options, provided at the discretion of the physician in dialogue with the patient is likely to ensure an effective allocation of health care resources.
Conclusion
In clinical practice strong opioids are effective agents in the management of moderate to severe pain. Although most opioids exert their main effect on the μ receptor, there are obviously differences between classes in their metabolism and mechanisms of action. On an individual level there is also marked variation between drugs in response and the opioid dose required. In this review we have highlighted that opioids differ with respect to:
absorption, distribution, metabolism and elimination
experimental pain
clinical efficacy and side effects
effect in relation to other diseases such as impaired renal and liver function
meta-analysis when parallel design and pooled estimates are used
costs with major differences between countries
The data presented in this review also support the practice of switching to an alternative opioid in an endeavour to improve the balance between opioid analgesia and side effects if a patient does not achieve an acceptable clinical outcome on one strong opioid. The experimental and clinical data presented support the need for access to opioids within different pharmacological classes in the clinical setting. Naturally, health economic considerations should also be taken into consideration, but restricting a physician's armamentarium in pain management will reduce the ability to tailor a planned course of treatment for the individual patients. This may lead to more pain and suffering and increase costs for society in the long run due to sick leave and early retirement. Due to the major individual variability in the response to opioids, future studies should focus on search for biomarkers that can predict the effect and side effect profile of opioid analgesics in the individual patient and in this way optimize individualized pain treatment.
Competing Interests
R. C. D. J. is currently an employee of Johnson and Johnson and previous employee of Novo Nordisk. R. C. D. J. has shares in both Johnson and Johnson and Novo Nordisk A/S. There are no other competing interests to declare.
REFERENCES
- 1.International Narcotics Control Board. Narcotic Drugs [online] Available at http://www.incb.org/incb/en/narcotic-drugs-technical-report_2011.html (last accessed 29 May 2012)
- 2.Dale O, Moksnes K, Kaasa S. European Palliative Care Research Collaborative pain guidelines: opioid switching to improve analgesia or reduce side effects. A systematic review. Palliat Med. 2011;25:494–503. doi: 10.1177/0269216310384902. [DOI] [PubMed] [Google Scholar]
- 3.Chou R, Fanciullo GJ, Fine PG, Adler JA, Ballantyne JC, Davies P, Donovan MI, Fishbain DA, Foley KM, Fudin J, Gilson AM, Kelter A, Mauskop A, O'Connor PG, Passik SD, Pasternak GW, Portenoy RK, Rich BA, Roberts RG, Todd KH, Miaskowski C. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain. 2009;10:113–30. doi: 10.1016/j.jpain.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caraceni A, Hanks G, Kaasa S, Bennett MI, Brunelli C, Cherny N, Dale O, De Conno F, Fallon M, Hanna M, Haugen DF, Juhl G, King S, Klepstad P, Laugsand EA, Maltoni M, Mercadante S, Nabal M, Pigni A, Radbruch L, Reid C, Sjogren P, Stone PC, Tassinari D, Zeppetella G. Use of opioid analgesics in the treatment of cancer pain: evidence-based recommendations from the EAPC. Lancet Oncol. 2012;13:e58–68. doi: 10.1016/S1470-2045(12)70040-2. [DOI] [PubMed] [Google Scholar]
- 5.Gaynes BN, Warden D, Trivedi MH, Wisniewski SR, Fava M, Rush AJ. What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Psychiatr Serv. 2009;60:1439–45. doi: 10.1176/ps.2009.60.11.1439. [DOI] [PubMed] [Google Scholar]
- 6.Christrup LL, Foster D, Popper LD, Troen T, Upton R. Pharmacokinetics, efficacy, and tolerability of fentanyl following intranasal versus intravenous administration in adults undergoing third-molar extraction: a randomized, double-blind, double-dummy, two-way, crossover study. Clin Ther. 2008;30:469–81. doi: 10.1016/j.clinthera.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 7.Joshi GP. Sufentanil for chronic pain management. Fut Neurol. 2010;5:791–6. [Google Scholar]
- 8.Strain EC, Walsh SL, Preston KL, Liebson IA, Bigelow GE. The effects of buprenorphine in buprenorphine-maintained volunteers. Psychopharmacology (Berl) 1997;129:329–38. doi: 10.1007/s002130050199. [DOI] [PubMed] [Google Scholar]
- 9.Poklis A. Fentanyl: a review for clinical and analytical toxicologists. J Toxicol Clin Toxicol. 1995;33:439–47. doi: 10.3109/15563659509013752. [DOI] [PubMed] [Google Scholar]
- 10.Streisand JB, Varvel JR, Stanski DR, Le ML, Ashburn MA, Hague BI, Tarver SD, Stanley TH. Absorption and bioavailability of oral transmucosal fentanyl citrate. Anesthesiology. 1991;75:223–9. doi: 10.1097/00000542-199108000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Kharasch ED, Hoffer C, Altuntas TG, Whittington D. Quinidine as a probe for the role of p-glycoprotein in the intestinal absorption and clinical effects of fentanyl. J Clin Pharmacol. 2004;44:224–33. doi: 10.1177/0091270003262075. [DOI] [PubMed] [Google Scholar]
- 12.Somogyi AA, Barratt DT, Coller JK. Pharmacogenetics of opioids. Clin Pharmacol Ther. 2007;81:429–44. doi: 10.1038/sj.clpt.6100095. [DOI] [PubMed] [Google Scholar]
- 13.Tzvetkov MV, Saadatmand AR, Lotsch J, Tegeder I, Stingl JC, Brockmoller J. Genetically polymorphic OCT1: another piece in the puzzle of the variable pharmacokinetics and pharmacodynamics of the opioidergic drug tramadol. Clin Pharmacol Ther. 2011;90:143–50. doi: 10.1038/clpt.2011.56. [DOI] [PubMed] [Google Scholar]
- 14.Thorn M, Finnstrom N, Lundgren S, Rane A, Loof L. Cytochromes P450 and MDR1 mRNA expression along the human gastrointestinal tract. Br J Clin Pharmacol. 2005;60:54–60. doi: 10.1111/j.1365-2125.2005.02389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nies AT, Koepsell H, Damme K, Schwab M. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol. 2011;201:105–67. doi: 10.1007/978-3-642-14541-4_3. [DOI] [PubMed] [Google Scholar]
- 16.Wandel C, Kim R, Wood M, Wood A. Interaction of morphine, fentanyl, sufentanil, alfentanil, and loperamide with the efflux drug transporter P-glycoprotein. Anesthesiology. 2002;96:913–20. doi: 10.1097/00000542-200204000-00019. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez M, Ortega I, Soengas I, Suarez E, Lukas JC, Calvo R. Effect of P-glycoprotein inhibition on methadone analgesia and brain distribution in the rat. J Pharm Pharmacol. 2004;56:367–74. doi: 10.1211/0022357022782. [DOI] [PubMed] [Google Scholar]
- 18.Park HJ, Shinn HK, Ryu SH, Lee HS, Park CS, Kang JH. Genetic polymorphisms in the ABCB1 gene and the effects of fentanyl in Koreans. Clin Pharmacol Ther. 2007;81:539–46. doi: 10.1038/sj.clpt.6100046. [DOI] [PubMed] [Google Scholar]
- 19.Bostrom E, Simonsson US, Hammarlund-Udenaes M. Oxycodone pharmacokinetics and pharmacodynamics in the rat in the presence of the P-glycoprotein inhibitor PSC833. J Pharm Sci. 2005;94:1060–6. doi: 10.1002/jps.20327. [DOI] [PubMed] [Google Scholar]
- 20.Bostrom E, Simonsson US, Hammarlund-Udenaes M. In vivo blood-brain barrier transport of oxycodone in the rat: indications for active influx and implications for pharmacokinetics/pharmacodynamics. Drug Metab Dispos. 2006;34:1624–31. doi: 10.1124/dmd.106.009746. [DOI] [PubMed] [Google Scholar]
- 21.Wang D, Guo Y, Wrighton SA, Cooke GE, Sadee W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J. 2011;11:274–86. doi: 10.1038/tpj.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guitton J, Buronfosse T, Desage M, Lepape A, Brazier JL, Beaune P. Possible involvement of multiple cytochrome P450s in fentanyl and sufentanil metabolism as opposed to alfentanil. Biochem Pharmacol. 1997;53:1613–9. doi: 10.1016/s0006-2952(96)00893-3. [DOI] [PubMed] [Google Scholar]
- 23.Labroo RB, Paine MF, Thummel KE, Kharasch ED. Fentanyl metabolism by human hepatic and intestinal cytochrome P450 3A4: implications for interindividual variability in disposition, efficacy, and drug interactions. Drug Metab Dispos. 1997;25:1072–80. [PubMed] [Google Scholar]
- 24.Dayer P, Desmeules J, Leemann T, Striberni R. Bioactivation of the narcotic drug codeine in human liver is mediated by the polymorphic monooxygenase catalyzing debrisoquine 4-hydroxylation (cytochrome P-450 dbl/bufI) Biochem Biophys Res Commun. 1988;152:411–6. doi: 10.1016/s0006-291x(88)80729-0. [DOI] [PubMed] [Google Scholar]
- 25.Lalovic B, Phillips B, Risler LL, Howald W, Shen DD. Quantitative contribution of CYP2D6 and CYP3A to oxycodone metabolism in human liver and intestinal microsomes. Drug Metab Dispos. 2004;32:447–54. doi: 10.1124/dmd.32.4.447. [DOI] [PubMed] [Google Scholar]
- 26.Mikus G, Weiss J. Influence of CYP2D6 genetics on opioid, kinetics, metabolism and response. Curr Pharmacogen. 2005;3:43–52. [Google Scholar]
- 27.Kirchheiner J, Schmidt H, Tzvetkov M, Keulen JT, Lotsch J, Roots I, Brockmoller J. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J. 2007;7:257–65. doi: 10.1038/sj.tpj.6500406. [DOI] [PubMed] [Google Scholar]
- 28.Madadi P, Ross CJ, Hayden MR, Carleton BC, Gaedigk A, Leeder JS, Koren G. Pharmacogenetics of neonatal opioid toxicity following maternal use of codeine during breastfeeding: a case-control study. Clin Pharmacol Ther. 2009;85:31–5. doi: 10.1038/clpt.2008.157. [DOI] [PubMed] [Google Scholar]
- 29.Samer CF, Daali Y, Wagner M, Hopfgartner G, Eap CB, Rebsamen MC, Rossier MF, Hochstrasser D, Dayer P, Desmeules JA. Genetic polymorphisms and drug interactions modulating CYP2D6 and CYP3A activities have a major effect on oxycodone analgesic efficacy and safety. Br J Pharmacol. 2010;160:919–30. doi: 10.1111/j.1476-5381.2010.00709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andreassen TN, Eftedal I, Klepstad P, Davies A, Bjordal K, Lundstrom S, Kaasa S, Dale O. Do CYP2D6 genotypes reflect oxycodone requirements for cancer patients treated for cancer pain? A cross-sectional multicentre study. Eur J Clin Pharmacol. 2012;68:55–64. doi: 10.1007/s00228-011-1093-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Subrahmanyam V, Renwick AB, Walters DG, Young PJ, Price RJ, Tonelli AP, Lake BG. Identification of cytochrome P-450 isoforms responsible for cis-tramadol metabolism in human liver microsomes. Drug Metab Dispos. 2001;29:1146–55. [PubMed] [Google Scholar]
- 32.Lotsch J, Skarke C, Schmidt H, Rohrbacher M, Hofmann U, Schwab M, Geisslinger G. Evidence for morphine-independent central nervous opioid effects after administration of codeine: contribution of other codeine metabolites. Clin Pharmacol Ther. 2006;79:35–48. doi: 10.1016/j.clpt.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 33.Romberg R, Olofsen E, Sarton E, den Hartigh J, Taschner PE, Dahan A. Pharmacokinetic-pharmacodynamic modeling of morphine-6-glucuronide-induced analgesia in healthy volunteers: absence of sex differences. Anesthesiology. 2004;100:120–33. doi: 10.1097/00000542-200401000-00021. [DOI] [PubMed] [Google Scholar]
- 34.Bhasker CR, McKinnon W, Stone A, Lo AC, Kubota T, Ishizaki T, Miners JO. Genetic polymorphism of UDP-glucuronosyltransferase 2B7 (UGT2B7) at amino acid 268: ethnic diversity of alleles and potential clinical significance. Pharmacogenetics. 2000;10:679–85. doi: 10.1097/00008571-200011000-00002. [DOI] [PubMed] [Google Scholar]
- 35.Coffman BL, King CD, Rios GR, Tephly TR. The glucuronidation of opioids, other xenobiotics, and androgens by human UGT2B7Y268 and UGT2B7H268. Drug Metab Dispos. 1998;26:73–7. [PubMed] [Google Scholar]
- 36.Holthe M, Klepstad P, Zahlsen K, Borchgrevink PC, Hagen L, Dale O, Kaasa S, Krokan HE, Skorpen F. Morphine glucuronide-to-morphine plasma ratios are unaffected by the UGT2B7 H268Y and UGT1A1*28 polymorphisms in cancer patients on chronic morphine therapy. Eur J Clin Pharmacol. 2002;58:353–6. doi: 10.1007/s00228-002-0490-1. [DOI] [PubMed] [Google Scholar]
- 37.Holthe M, Rakvag TN, Klepstad P, Idle JR, Kaasa S, Krokan HE, Skorpen F. Sequence variations in the UDP-glucuronosyltransferase 2B7 (UGT2B7) gene: identification of 10 novel single nucleotide polymorphisms (SNPs) and analysis of their relevance to morphine glucuronidation in cancer patients. Pharmacogenomics J. 2003;3:17–26. doi: 10.1038/sj.tpj.6500139. [DOI] [PubMed] [Google Scholar]
- 38.Sawyer MB, Innocenti F, Das S, Cheng C, Ramirez J, Pantle-Fisher FH, Wright C, Badner J, Pei D, Boyett JM, Cook E, Jr, Ratain MJ. A pharmacogenetic study of uridine diphosphate-glucuronosyltransferase 2B7 in patients receiving morphine. Clin Pharmacol Ther. 2003;73:566–74. doi: 10.1016/S0009-9236(03)00053-5. [DOI] [PubMed] [Google Scholar]
- 39.Gerber JG, Rhodes RJ, Gal J. Stereoselective metabolism of methadone N-demethylation by cytochrome P4502B6 and 2C19. Chirality. 2004;16:36–44. doi: 10.1002/chir.10303. [DOI] [PubMed] [Google Scholar]
- 40.Galvan A, Skorpen F, Klepstad P, Knudsen AK, Fladvad T, Falvella FS, Pigni A, Brunelli C, Caraceni A, Kaasa S, Dragani TA. Multiple loci modulate opioid therapy response for cancer pain. Clin Cancer Res. 2011;17:4581–7. doi: 10.1158/1078-0432.CCR-10-3028. [DOI] [PubMed] [Google Scholar]
- 41.Klepstad P, Fladvad T, Skorpen F, Bjordal K, Caraceni A, Dale O, Davies A, Kloke M, Lundstrom S, Maltoni M, Radbruch L, Sabatowski R, Sigurdardottir V, Strasser F, Fayers PM, Kaasa S. Influence from genetic variability on opioid use for cancer pain: a European genetic association study of 2294 cancer pain patients. Pain. 2011;152:1139–45. doi: 10.1016/j.pain.2011.01.040. [DOI] [PubMed] [Google Scholar]
- 42.Laugsand EA, Fladvad T, Skorpen F, Maltoni M, Kaasa S, Fayers P, Klepstad P. Clinical and genetic factors associated with nausea and vomiting in cancer patients receiving opioids. Eur J Cancer. 2011;47:1682–91. doi: 10.1016/j.ejca.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 43.Osborne R, Joel S, Grebenik K, Trew D, Slevin M. The pharmacokinetics of morphine and morphine glucuronides in kidney failure. Clin Pharmacol Ther. 1993;54:158–67. doi: 10.1038/clpt.1993.127. [DOI] [PubMed] [Google Scholar]
- 44.Coller JK, Christrup LL, Somogyi AA. Role of active metabolites in the use of opioids. Eur J Clin Pharmacol. 2009;65:121–39. doi: 10.1007/s00228-008-0570-y. [DOI] [PubMed] [Google Scholar]
- 45.Duttaroy A, Yoburn BC. The effect of intrinsic efficacy on opioid tolerance. Anesthesiology. 1995;82:1226–36. doi: 10.1097/00000542-199505000-00018. [DOI] [PubMed] [Google Scholar]
- 46.Gourlay G. Different opioids – same actions? In: Kalso E, McQuay HJ, Wiesenfeld-Hallin Z, editors. Opioid Sensitivity of Chronic Noncancer Pain. Vol. 14. Seattle: JASP Press; 1999. pp. 97–116. Progess in Pain Research and Management. [Google Scholar]
- 47.Kanaan M, Daali Y, Dayer P, Desmeules J. Uptake/efflux transport of tramadol enantiomers and O-desmethyl-tramadol: focus on P-glycoprotein. Basic Clin Pharmacol Toxicol. 2009;105:199–206. doi: 10.1111/j.1742-7843.2009.00428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keith DE, Anton B, Murray SR, Zaki PA, Chu PC, Lissin DV, Monteillet-Agius G, Stewart PL, Evans CJ, von ZM. mu-opioid receptor internalization: opiate drugs have differential effects on a conserved endocytic mechanism in vitro and in the mammalian brain. Mol Pharmacol. 1998;53:377–84. [PubMed] [Google Scholar]
- 49.Kumar P, Sunkaraneni S, Sirohi S, Dighe SV, Walker EA, Yoburn BC. Hydromorphone efficacy and treatment protocol impact on tolerance and mu-opioid receptor regulation. Eur J Pharmacol. 2008;597:39–45. doi: 10.1016/j.ejphar.2008.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lemberg KK, Siiskonen AO, Kontinen VK, Yli-Kauhaluoma JT, Kalso EA. Pharmacological characterization of noroxymorphone as a new opioid for spinal analgesia. Anesth Analg. 2008;106:463–70. doi: 10.1213/ane.0b013e3181605a15. [DOI] [PubMed] [Google Scholar]
- 51.Leppert W, Luczak J. The role of tramadol in cancer pain treatment – a review. Support Care Cancer. 2005;13:5–17. doi: 10.1007/s00520-004-0720-4. [DOI] [PubMed] [Google Scholar]
- 52.Lotsch J. Opioid metabolites. J Pain Symptom Manage. 2005;29(5 Suppl.):S10–24. doi: 10.1016/j.jpainsymman.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 53.McRae A, Sonne S. Opioid equivalency: a review. J Pharm Pract. 1998;11:394–405. [Google Scholar]
- 54.Medzihradsky F, Emmerson PJ, Mousigian CA. Lipophilicity of opioids determined by a novel micromethod. J Pharmacol Toxicol Methods. 1992;27:67–9. doi: 10.1016/1056-8719(92)90023-t. [DOI] [PubMed] [Google Scholar]
- 55.Nielsen CK, Ross FB, Lotfipour S, Saini KS, Edwards SR, Smith MT. Oxycodone and morphine have distinctly different pharmacological profiles: radioligand binding and behavioural studies in two rat models of neuropathic pain. Pain. 2007;132:289–300. doi: 10.1016/j.pain.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 56.Ross FB, Smith MT. The intrinsic antinociceptive effects of oxycodone appear to be kappa-opioid receptor mediated. Pain. 1997;73:151–7. doi: 10.1016/S0304-3959(97)00093-6. [DOI] [PubMed] [Google Scholar]
- 57.Tzschentke TM, Christoph T, Kogel B, Schiene K, Hennies HH, Englberger W, Haurand M, Jahnel U, Cremers TI, Friderichs E, De VJ. (–)-(1R,2R)-3-(3-dimethylamino-1-ethyl-2-methyl-propyl)-phenol hydrochloride (tapentadol HCl): a novel mu-opioid receptor agonist/norepinephrine reuptake inhibitor with broad-spectrum analgesic properties. J Pharmacol Exp Ther. 2007;323:265–76. doi: 10.1124/jpet.107.126052. [DOI] [PubMed] [Google Scholar]
- 58.Vallejo R, Barkin RL, Wang VC. Pharmacology of opioids in the treatment of chronic pain syndromes. Pain Phys. 2011;14:E343–60. [PubMed] [Google Scholar]
- 59.Xie R, Hammarlund-Udenaes M. Blood-brain barrier equilibration of codeine in rats studied with microdialysis. Pharm Res. 1998;15:570–5. doi: 10.1023/a:1011929910782. [DOI] [PubMed] [Google Scholar]
- 60.Zuo Z. The role of opioid receptor internalization and beta-arrestins in the development of opioid tolerance. Anesth Analg. 2005;101:728–34. doi: 10.1213/01.ANE.0000160588.32007.AD. [DOI] [PubMed] [Google Scholar]
- 61.Avdeef A, Barrett DA, Shaw PN, Knaggs RD, Davis SS. Octanol-, chloroform-, and propylene glycol dipelargonat-water partitioning of morphine-6-glucuronide and other related opiates. J Med Chem. 1996;39:4377–81. doi: 10.1021/jm960073m. [DOI] [PubMed] [Google Scholar]
- 62.Leander JD. Buprenorphine has potent kappa opioid receptor antagonist activity. Neuropharmacology. 1987;26:1445–7. doi: 10.1016/0028-3908(87)90112-2. [DOI] [PubMed] [Google Scholar]
- 63.Andresen T, Staahl C, Oksche A, Mansikka H, rendt-Nielsen L, Drewes AM. Effect of transdermal opioids in experimentally induced superficial, deep and hyperalgesic pain. Br J Pharmacol. 2011;164:934–45. doi: 10.1111/j.1476-5381.2010.01180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ebert B, Thorkildsen C, Andersen S, Christrup LL, Hjeds H. Opioid analgesics as noncompetitive N-methyl-D-aspartate (NMDA) antagonists. Biochem Pharmacol. 1998;56:553–9. doi: 10.1016/s0006-2952(98)00088-4. [DOI] [PubMed] [Google Scholar]
- 65.Volpe DA, Mahon Tobin GA, Mellon RD, Katki AG, Parker RJ, Colatsky T, Kropp TJ, Verbois SL. Uniform assessment and ranking of opioid mu receptor binding constants for selected opioid drugs. Regul Toxicol Pharmacol. 2011;59:385–90. doi: 10.1016/j.yrtph.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 66.Codd EE, Shank RP, Schupsky JJ, Raffa RB. Serotonin and norepinephrine uptake inhibiting activity of centrally acting analgesics: structural determinants and role in antinociception. J Pharmacol Exp Ther. 1995;274:1263–70. [PubMed] [Google Scholar]
- 67.Tzschentke TM, Jahnel U, Kogel B, Christoph T, Englberger W, De VJ, Schiene K, Okamoto A, Upmalis D, Weber H, Lange C, Stegmann JU, Kleinert R. Tapentadol hydrochloride: a next-generation, centrally acting analgesic with two mechanisms of action in a single molecule. Drugs Today (Barc) 2009;45:483–96. doi: 10.1358/dot.2009.45.7.1395291. [DOI] [PubMed] [Google Scholar]
- 68.Pasternak GW. Molecular biology of opioid analgesia. J Pain Symptom Manage. 2005;29(5 Suppl.):S2–9. doi: 10.1016/j.jpainsymman.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 69.Bodnar RJ. Endogenous opiates and behavior: 2009. Peptides. 2010;31:2325–59. doi: 10.1016/j.peptides.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 70.Chizh BA, Priestley T, Rowbotham M, Schaffler K. Predicting therapeutic efficacy – experimental pain in human subjects. Brain Res Rev. 2009;60:243–54. doi: 10.1016/j.brainresrev.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 71.Dolgin E. Animalgesic effects. Nat Med. 2010;16:1237–40. doi: 10.1038/nm1110-1237. [DOI] [PubMed] [Google Scholar]
- 72.Staahl C, Olesen AE, Andresen T, rendt-Nielsen L, Drewes AM. Assessing analgesic actions of opioids by experimental pain models in healthy volunteers – an updated review. Br J Clin Pharmacol. 2009;68:149–68. doi: 10.1111/j.1365-2125.2009.03456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kalso E, Edwards JE, Moore RA, McQuay HJ. Opioids in chronic non-cancer pain: systematic review of efficacy and safety. Pain. 2004;112:372–80. doi: 10.1016/j.pain.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 74.Arendt-Nielsen L, Curatolo M, Drewes A. Human experimental pain models in drug development: translational pain research. Curr Opin Investig Drugs. 2007;8:41–53. [PubMed] [Google Scholar]
- 75.Bingel U, Tracey I. Imaging CNS modulation of pain in humans. Physiology (Bethesda) 2008;23:371–80. doi: 10.1152/physiol.00024.2008. [DOI] [PubMed] [Google Scholar]
- 76.Drewes AM, Gregersen H, rendt-Nielsen L. Experimental pain in gastroenterology: a reappraisal of human studies. Scand J Gastroenterol. 2003;38:1115–30. doi: 10.1080/00365520310004399. [DOI] [PubMed] [Google Scholar]
- 77.Curatolo M, Petersen-Felix S, Gerber A, rendt-Nielsen L. Remifentanil inhibits muscular more than cutaneous pain in humans. Br J Anaesth. 2000;85:529–32. doi: 10.1093/bja/85.4.529. [DOI] [PubMed] [Google Scholar]
- 78.Staahl C, Christrup LL, Andersen SD, rendt-Nielsen L, Drewes AM. A comparative study of oxycodone and morphine in a multi-modal, tissue-differentiated experimental pain model. Pain. 2006;123:28–36. doi: 10.1016/j.pain.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 79.Mogil JS, Ritchie J, Smith SB, Strasburg K, Kaplan L, Wallace MR, Romberg RR, Bijl H, Sarton EY, Fillingim RB, Dahan A. Melanocortin-1 receptor gene variants affect pain and mu-opioid analgesia in mice and humans. J Med Genet. 2005;42:583–7. doi: 10.1136/jmg.2004.027698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eisenach JC, Carpenter R, Curry R. Analgesia from a peripherally active kappa-opioid receptor agonist in patients with chronic pancreatitis. Pain. 2003;101:89–95. doi: 10.1016/s0304-3959(02)00259-2. [DOI] [PubMed] [Google Scholar]
- 81.Arendt-Nielsen L, Olesen AE, Staahl C, Menzaghi F, Kell S, Wong GY, Drewes AM. Analgesic efficacy of peripheral kappa-opioid receptor agonist CR665 compared to oxycodone in a multi-modal, multi-tissue experimental human pain model: selective effect on visceral pain. Anesthesiology. 2009;111:616–24. doi: 10.1097/ALN.0b013e3181af6356. [DOI] [PubMed] [Google Scholar]
- 82.Staahl C, Upton R, Foster DJ, Christrup LL, Kristensen K, Hansen SH, Arendt-Nielsen L, Drewes AM. Pharmacokinetic-pharmacodynamic modeling of morphine and oxycodone concentrations and analgesic effect in a multimodal experimental pain model. J Clin Pharmacol. 2008;48:619–31. doi: 10.1177/0091270008314465. [DOI] [PubMed] [Google Scholar]
- 83.Sengupta JN, Snider A, Su X, Gebhart GF. Effects of kappa opioids in the inflamed rat colon. Pain. 1999;79:175–85. doi: 10.1016/s0304-3959(98)00175-4. [DOI] [PubMed] [Google Scholar]
- 84.Staahl C, Dimcevski G, Andersen SD, Thorsgaard N, Christrup LL, Arendt-Nielsen L, Drewes AM. Differential effect of opioids in patients with chronic pancreatitis: an experimental pain study. Scand J Gastroenterol. 2007;42:383–90. doi: 10.1080/00365520601014414. [DOI] [PubMed] [Google Scholar]
- 85.Bruera E, Belzile M, Pituskin E, Fainsinger R, Darke A, Harsanyi Z, Babul N, Ford I. Randomized, double-blind, cross-over trial comparing safety and efficacy of oral controlled-release oxycodone with controlled-release morphine in patients with cancer pain. J Clin Oncol. 1998;16:3222–9. doi: 10.1200/JCO.1998.16.10.3222. [DOI] [PubMed] [Google Scholar]
- 86.Koppert W, Ihmsen H, Korber N, Wehrfritz A, Sittl R, Schmelz M, Schuttler J. Different profiles of buprenorphine-induced analgesia and antihyperalgesia in a human pain model. Pain. 2005;118:15–22. doi: 10.1016/j.pain.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 87.Olesen AE, Staahl C, Arendt-Nielsen L, Drewes AM. Different effects of morphine and oxycodone in experimentally evoked hyperalgesia: a human translational study. Br J Clin Pharmacol. 2010;70:189–200. doi: 10.1111/j.1365-2125.2010.03700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koppert W, Dern SK, Sittl R, Albrecht S, Schuttler J, Schmelz M. A new model of electrically evoked pain and hyperalgesia in human skin: the effects of intravenous alfentanil, S(+)-ketamine, and lidocaine. Anesthesiology. 2001;95:395–402. doi: 10.1097/00000542-200108000-00022. [DOI] [PubMed] [Google Scholar]
- 89.Brainin-Mattos J, Smith ND, Malkmus S, Rew Y, Goodman M, Taulane J, Yaksh TL. Cancer-related bone pain is attenuated by a systemically available delta-opioid receptor agonist. Pain. 2006;122:174–81. doi: 10.1016/j.pain.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 90.Delaney A, Fleetwood-Walker SM, Colvin LA, Fallon M. Translational medicine: cancer pain mechanisms and management. Br J Anaesth. 2008;101:87–94. doi: 10.1093/bja/aen100. [DOI] [PubMed] [Google Scholar]
- 91.Andresen T, Upton RN, Foster DJ, Christrup LL, Arendt-Nielsen L, Drewes AM. Pharmacokinetic/pharmacodynamic relationships of transdermal buprenorphine and fentanyl in experimental human pain models. Basic Clin Pharmacol Toxicol. 2011;108:274–84. doi: 10.1111/j.1742-7843.2010.00649.x. [DOI] [PubMed] [Google Scholar]
- 92.Kent DM, Rothwell PM, Ioannidis JP, Altman DG, Hayward RA. Assessing and reporting heterogeneity in treatment effects in clinical trials: a proposal. Trials. 2010;11:85. doi: 10.1186/1745-6215-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rothwell PM. Can overall results of clinical trials be applied to all patients? Lancet. 1995;345:1616–9. doi: 10.1016/s0140-6736(95)90120-5. [DOI] [PubMed] [Google Scholar]
- 94.Wiffen PJ, McQuay HJ. Oral morphine for cancer pain. Cochrane Database Syst Rev. 2007;(4) doi: 10.1002/14651858.CD003868.pub2. CD003868. [DOI] [PubMed] [Google Scholar]
- 95.Tassinari D, Sartori S, Tamburini E, Scarpi E, Raffaeli W, Tombesi P, Maltoni M. Adverse effects of transdermal opiates treating moderate-severe cancer pain in comparison to long-acting morphine: a meta-analysis and systematic review of the literature. J Palliat Med. 2008;11:492–501. doi: 10.1089/jpm.2007.0200. [DOI] [PubMed] [Google Scholar]
- 96.Bekkering GE, Soares-Weiser K, Reid K, Kessels AG, Dahan A, Treede RD, Kleijnen J. Can morphine still be considered to be the standard for treating chronic pain? A systematic review including pair-wise and network meta-analyses. Curr Med Res Opin. 2011;27:1477–91. doi: 10.1185/03007995.2011.586332. [DOI] [PubMed] [Google Scholar]
- 97.Dahan A, Olofsen E, Sigtermans M, Noppers I, Niesters M, Aarts L, Bauer M, Sarton E. Population pharmacokinetic-pharmacodynamic modeling of ketamine-induced pain relief of chronic pain. Eur J Pain. 2011;15:258–67. doi: 10.1016/j.ejpain.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 98.Cullberg M, Eriksson UG, Wahlander K, Eriksson H, Schulman S, Karlsson MO. Pharmacokinetics of ximelagatran and relationship to clinical response in acute deep vein thrombosis. Clin Pharmacol Ther. 2005;77:279–90. doi: 10.1016/j.clpt.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 99.Yassen A, Olofsen E, Kan J, Dahan A, Danhof M. Pharmacokinetic-pharmacodynamic modeling of the effectiveness and safety of buprenorphine and fentanyl in rats. Pharm Res. 2008;25:183–93. doi: 10.1007/s11095-007-9440-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dahan A, Romberg R, Teppema L, Sarton E, Bijl H, Olofsen E. Simultaneous measurement and integrated analysis of analgesia and respiration after an intravenous morphine infusion. Anesthesiology. 2004;101:1201–9. doi: 10.1097/00000542-200411000-00021. [DOI] [PubMed] [Google Scholar]
- 101.Hanks GW, Conno F, Cherny N, Hanna M, Kalso E, McQuay HJ, Mercadante S, Meynadier J, Poulain P, Ripamonti C, Radbruch L, Casas JR, Sawe J, Twycross RG, Ventafridda V. Morphine and alternative opioids in cancer pain: the EAPC recommendations. Br J Cancer. 2001;84:587–93. doi: 10.1054/bjoc.2001.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Klepstad P, Kaasa S, Cherny N, Hanks G, De Conno F. Pain and pain treatments in European palliative care units. A cross sectional survey from the European Association for Palliative Care Research Network. Palliat Med. 2005;19:477–84. doi: 10.1191/0269216305pm1054oa. [DOI] [PubMed] [Google Scholar]
- 103.Pergolizzi J, Alegre C, Blake D, Alen JC, Caporali R, Casser HR, Correa-Illanes G, Fernandes P, Galilea E, Jany R, Jones A, Mejjad O, Morovic-Vergles J, Oteo AA, Radrigan Araya FJ, Simoes ME, Uomo G. Current considerations for the treatment of severe chronic pain: the potential for tapentadol. Pain Pract. 2011;2(12):290–306. doi: 10.1111/j.1533-2500.2011.00487.x. [DOI] [PubMed] [Google Scholar]
- 104.Stone P, Minton O. European Palliative Care Research collaborative pain guidelines. Central side-effects management: what is the evidence to support best practice in the management of sedation, cognitive impairment and myoclonus? Palliat Med. 2011;25:431–41. doi: 10.1177/0269216310380763. [DOI] [PubMed] [Google Scholar]
- 105.Laugsand EA, Kaasa S, Klepstad P. Management of opioid-induced nausea and vomiting in cancer patients: systematic review and evidence-based recommendations. Palliat Med. 2011;25:442–53. doi: 10.1177/0269216311404273. [DOI] [PubMed] [Google Scholar]
- 106.Ahmedzai S, Brooks D. Transdermal fentanyl versus sustained-release oral morphine in cancer pain: preference, efficacy, and quality of life. The TTS-Fentanyl Comparative Trial Group. J Pain Symptom Manage. 1997;13:254–61. doi: 10.1016/s0885-3924(97)00082-1. [DOI] [PubMed] [Google Scholar]
- 107.Daniell HW. Hypogonadism in men consuming sustained-action oral opioids. J Pain. 2002;3:377–84. doi: 10.1054/jpai.2002.126790. [DOI] [PubMed] [Google Scholar]
- 108.Eisenstein TK, Rahim RT, Feng P, Thingalaya NK, Meissler JJ. Effects of opioid tolerance and withdrawal on the immune system. J Neuroimmune Pharmacol. 2006;1:237–49. doi: 10.1007/s11481-006-9019-1. [DOI] [PubMed] [Google Scholar]
- 109.Mercadante S, Bruera E. Opioid switching: a systematic and critical review. Cancer Treat Rev. 2006;32:304–15. doi: 10.1016/j.ctrv.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 110.Mercadante S, Ferrera P, Villari P, Casuccio A, Intravaia G, Mangione S. Frequency, indications, outcomes, and predictive factors of opioid switching in an acute palliative care unit. J Pain Symptom Manage. 2009;37:632–41. doi: 10.1016/j.jpainsymman.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 111.Mercadante S, Ferrera P, Villari P, Adile C, Casuccio A. Switching from oxycodone to methadone in advanced cancer patients. Support Care Cancer. 2012;20:191–4. doi: 10.1007/s00520-011-1259-9. [DOI] [PubMed] [Google Scholar]
- 112.Kanbayashi Y, Hosokawa T, Okamoto K, Fujimoto S, Konishi H, Otsuji E, Yoshikawa T, Takagi T, Miki T, Taniwaki M. Factors predicting requirement of high-dose transdermal fentanyl in opioid switching from oral morphine or oxycodone in patients with cancer pain. Clin J Pain. 2011;27:664–7. doi: 10.1097/AJP.0b013e3182168fed. [DOI] [PubMed] [Google Scholar]
- 113.Fillingim RB, King CD, Ribeiro-Dasilva MC, Rahim-Williams B, Riley JL., III Sex, gender, and pain: a review of recent clinical and experimental findings. J Pain. 2009;10:447–85. doi: 10.1016/j.jpain.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Macintyre PE, Jarvis DA. Age is the best predictor of postoperative morphine requirements. Pain. 1996;64:357–64. doi: 10.1016/0304-3959(95)00128-X. [DOI] [PubMed] [Google Scholar]
- 115.Niesters M, Dahan A, Kest B, Zacny J, Stijnen T, Aarts L, Sarton E. Do sex differences exist in opioid analgesia? A systematic review and meta-analysis of human experimental and clinical studies. Pain. 2010;151:61–8. doi: 10.1016/j.pain.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 116.Knudsen AK, Brunelli C, Kaasa S, Apolone G, Corli O, Montanari M, Fainsinger R, Aass N, Fayers P, Caraceni A, Klepstad P. Which variables are associated with pain intensity and treatment response in advanced cancer patients? – Implications for a future classification system for cancer pain. Eur J Pain. 2011;15:320–7. doi: 10.1016/j.ejpain.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 117.McQuay HJ, Carroll D, Faura CC, Gavaghan DJ, Hand CW, Moore RA. Oral morphine in cancer pain: influences on morphine and metabolite concentration. Clin Pharmacol Ther. 1990;48:236–44. doi: 10.1038/clpt.1990.145. [DOI] [PubMed] [Google Scholar]
- 118.Quigley C. Opioid switching to improve pain relief and drug tolerability. Cochrane Database Syst Rev. 2004;(3) doi: 10.1002/14651858.CD004847. CD004847. [DOI] [PubMed] [Google Scholar]
- 119.Mercadante S, Casuccio A, Fulfaro F, Groff L, Boffi R, Villari P, Gebbia V, Ripamonti C. Switching from morphine to methadone to improve analgesia and tolerability in cancer patients: a prospective study. J Clin Oncol. 2001;19:2898–904. doi: 10.1200/JCO.2001.19.11.2898. [DOI] [PubMed] [Google Scholar]
- 120.Riley J, Ross JR, Rutter D, Wells AU, Goller K, du Bois R, Welsh K. No pain relief from morphine? Individual variation in sensitivity to morphine and the need to switch to an alternative opioid in cancer patients. Support Care Cancer. 2006;14:56–64. doi: 10.1007/s00520-005-0843-2. [DOI] [PubMed] [Google Scholar]
- 121.Slatkin NE. Opioid switching and rotation in primary care: implementation and clinical utility. Curr Med Res Opin. 2009;25:2133–50. doi: 10.1185/03007990903120158. [DOI] [PubMed] [Google Scholar]
- 122.Pasternak GW. Incomplete cross tolerance and multiple mu opioid peptide receptors. Trends Pharmacol Sci. 2001;22:67–70. doi: 10.1016/s0165-6147(00)01616-3. [DOI] [PubMed] [Google Scholar]
- 123.Nielsen CK, Ross FB, Smith MT. Incomplete, asymmetric, and route-dependent cross-tolerance between oxycodone and morphine in the Dark Agouti rat. J Pharmacol Exp Ther. 2000;295:91–9. [PubMed] [Google Scholar]
- 124.Pasternak GW. Molecular insights into mu opioid pharmacology: from the clinic to the bench. Clin J Pain. 2010;26(Suppl. 10):S3–9. doi: 10.1097/AJP.0b013e3181c49d2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Knotkova H, Fine PG, Portenoy RK. Opioid rotation: the science and the limitations of the equianalgesic dose table. J Pain Symptom Manage. 2009;38:426–39. doi: 10.1016/j.jpainsymman.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 126.Fine PG, Portenoy RK. Establishing ‘best practices’ for opioid rotation: conclusions of an expert panel. J Pain Symptom Manage. 2009;38:418–25. doi: 10.1016/j.jpainsymman.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mercadante S, Caraceni A. Conversion ratios for opioid switching in the treatment of cancer pain: a systematic review. Palliat Med. 2011;25:504–15. doi: 10.1177/0269216311406577. [DOI] [PubMed] [Google Scholar]
- 128.Portenoy RK. Treatment of cancer pain. Lancet. 2011;377:2236–47. doi: 10.1016/S0140-6736(11)60236-5. [DOI] [PubMed] [Google Scholar]
- 129.NIH 1998. National Institute of Health.New Directions in Pain Research. 1998. Available at http://grants.nih.gov/grants/guide/pa-files/PA-98-102.html (last accessed 29 May 2012)
- 130.Gustavsson A, Bjorkman J, Ljungcrantz C, Rhodin A, Rivano-Fischer M, Sjolund KF, Mannheimer C. Socio-economic burden of patients with a diagnosis related to chronic pain – Register data of 840,000 Swedish patients. Eur J Pain. 2011;16:289–99. doi: 10.1016/j.ejpain.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 131.Ritzwoller DP, Crounse L, Shetterly S, Rublee D. The association of comorbidities, utilization and costs for patients identified with low back pain. BMC Musculoskelet Disord. 2006;7:72. doi: 10.1186/1471-2474-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Berger A, Dukes EM, Oster G. Clinical characteristics and economic costs of patients with painful neuropathic disorders. J Pain. 2004;5:143–9. doi: 10.1016/j.jpain.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 133.Benyamin R, Trescot AM, Datta S, Buenaventura R, Adlaka R, Sehgal N, Glaser SE, Vallejo R. Opioid complications and side effects. Pain Phys. 2008;2(Suppl.):S105–20. [PubMed] [Google Scholar]
- 134.Leider HL, Dhaliwal J, Davis EJ, Kulakodlu M, Buikema AR. Healthcare costs and nonadherence among chronic opioid users. Am J Manag Care. 2011;17:32–40. [PubMed] [Google Scholar]
- 135.Kwong WJ, Diels J, Kavanagh S. Costs of gastrointestinal events after outpatient opioid treatment for non-cancer pain. Ann Pharmacother. 2010;44:630–40. doi: 10.1345/aph.1M520. [DOI] [PubMed] [Google Scholar]
- 136.Noble M, Treadwell JR, Tregear SJ, Coates VH, Wiffen PJ, Akafomo C, Schoelles KM. Long-term opioid management for chronic noncancer pain. Cochrane Database Syst Rev. 2010;(1) doi: 10.1002/14651858.CD006605.pub2. CD006605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Broekmans S, Dobbels F, Milisen K, Morlion B, Vanderschueren S. Determinants of medication underuse and medication overuse in patients with chronic non-malignant pain: a multicenter study. Int J Nurs Stud. 2010;47:1408–17. doi: 10.1016/j.ijnurstu.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 138.Neighbors DM, Bell TJ, Wilson J, Dodd SL. Economic evaluation of the fentanyl transdermal system for the treatment of chronic moderate to severe pain. J Pain Symptom Manage. 2001;21:129–43. doi: 10.1016/s0885-3924(00)00247-5. [DOI] [PubMed] [Google Scholar]
- 139.Radbruch L, Sabatowski R, Petzke F, Brunsch-Radbruch A, Grond S, Lehmann KA. Transdermal fentanyl for the management of cancer pain: a survey of 1005 patients. Palliat Med. 2001;15:309–21. doi: 10.1191/026921601678320296. [DOI] [PubMed] [Google Scholar]
- 140.Vondrackova D, Leyendecker P, Meissner W, Hopp M, Szombati I, Hermanns K, Ruckes C, Weber S, Grothe B, Fleischer W, Reimer K. Analgesic efficacy and safety of oxycodone in combination with naloxone as prolonged release tablets in patients with moderate to severe chronic pain. J Pain. 2008;9:1144–54. doi: 10.1016/j.jpain.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 141.Buynak R, Shapiro DY, Okamoto A, Van Hove I, Rauschkolb C, Steup A, Lange B, Lange C, Etropolski M. Efficacy and safety of tapentadol extended release for the management of chronic low back pain: results of a prospective, randomized, double-blind, placebo- and active-controlled Phase III study. Expert Opin Pharmacother. 2010;11:1787–804. doi: 10.1517/14656566.2010.497720. [DOI] [PubMed] [Google Scholar]
- 142.Thomas J, Karver S, Cooney GA, Chamberlain BH, Watt CK, Slatkin NE, Stambler N, Kremer AB, Israel RJ. Methylnaltrexone for opioid-induced constipation in advanced illness. N Engl J Med. 2008;358:2332–43. doi: 10.1056/NEJMoa0707377. [DOI] [PubMed] [Google Scholar]
- 143.Annemans L. Pharmacoeconomic impact of adverse events of long-term opioid treatment for the management of persistent pain. Clin Drug Investig. 2011;31:73–86. doi: 10.1007/BF03256935. [DOI] [PubMed] [Google Scholar]