

Abstract
AIM
To assess the safety, tolerability, pharmacodynamics and pharmacokinetics in healthy subjects of a novel, highly selective, sigma-1 receptor antagonist (S1RA).
METHODS
Three randomized, double-blind, placebo-controlled trials evaluated single oral doses (5–500 mg, study 101; 500–800 mg, study 106) and multiple doses (50–400 mg once daily for 8 days, study 102) of S1RA. Safety and tolerability were assessed by adverse event reporting, clinical laboratory, physical examinations, vital signs and electrocardiography, including Holter monitoring. Pharmacodynamic assessments included computerized cognitive testing. Plasma samples were analyzed using validated HPLC-MS/MS methods.
RESULTS
One hundred and seventy-five subjects were enrolled. Single and multiple doses were safe and well tolerated, with no serious adverse events. The most common side effects were headache and dizziness. The highest single doses were associated with some mild to moderate transient CNS effects. The maximum tolerated dose was not reached. There were no clinically significant changes in the electrocardiogram (ECG), 24 h Holter monitoring, or in vital signs and laboratory assessments. Subjective CNS pharmacodynamics evaluations showed no relevant differences vs. placebo. Cognitive testing showed no effects on visual memory, executive function, attention or somnolence, while revealing some transient slowing of response for simple reaction time and choice reaction time at 2 h following the administration of higher doses. A fast absorption, rapid distribution and slow elimination were observed (tmax 0.75–2.0 h, t1/2 compatible with once a day administration) and steady-state was reached. No gender differences were observed.
CONCLUSIONS
S1RA exhibited an acceptable safety, tolerability, pharmacodynamic and pharmacokinetic profile in healthy subjects over the dose range studied.
Keywords: pain, pharmacodynamics, pharmacokinetics, S1RA (E-52862), safety, tolerability
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
A novel and highly selective sigma-1 receptor antagonist (S1RA) provides a new approach for pain management. S1RA has shown activity in animal models of neuropathic pain and potentiation of opioid analgesia.
WHAT THIS STUDY ADDS
Phase I studies of single and multiple oral dose administration show that S1RA is safe and well tolerated by healthy subjects. S1RA is rapidly absorbed and its rate and extent of exposure increases with dose.
The safety, tolerability, pharmacokinetic and pharmacodynamic profiles of S1RA support its further phase II development in different pain indications.
Introduction
The sigma-1 receptor (S1R) is a 223 amino acid protein which shares no sequence homology with any known mammalian proteins [1, 2]. The S1R is specifically located at the interface of the endoplasmic reticulum (ER) and the mitochondrion where it regulates ER-mitochondrion signalling and ER-nucleus crosstalk, modulating the function of a variety of ion channels and receptors [1–3]. As a molecular chaperone, S1R and its ligands have no effect upon ion channels and receptors under normal physiological conditions, but in the presence of disease or dysfunction, the assistance of S1R chaperones may be demanded by changes in the activity of specific ion channels and receptors (and/or other cellular signalling mediators), resulting in the potential for S1R-based pharmacotherapy to become a beneficial and selective therapeutic intervention [1].
The potential of S1R targeting in pain has been an area of particular interest. S1Rs have been shown to modulate opioid receptor-mediated antinociception [4–6], with their actions being mainly supraspinal [7]. Most recently, a direct physical and functional association of S1R with µ-opioid receptors has been reported [8]. Interestingly, S1R also plays a role in nociception in the absence of opioids as demonstrated using S1R knockout mice [9, 10] and in pharmacological studies [11, 12] where attenuated pain hypersensitivity was found in sensitizing conditions (i.e. capsaicin sensitization and nerve injury).
A novel chemical entity, highly selective sigma-1 receptor antagonist (S1RA), has been identified and found to have high affinity for human sigma-1 receptors (Ki = 17 nm) [13, 14]. S1RA or E-52862, which corresponds to 4-[2-[[5-methyl-1-(2-naphthalenyl)-1H-pyrazol-3-yl]oxy]ethyl] morpholine, is active by the oral route, crosses the blood–brain barrier and binds to S1Rs in the brain [14]. S1RA has been shown to inhibit dose-dependently formalin-induced nociception, capsaicin-induced mechanical hypersensitivity and nerve injury-induced mechanical and thermal hypersensitivity in preclinical studies [14]. A significant correlation between the extent of central nervous system (CNS) S1R occupancy and the degree of antinociceptive effect exerted by S1RA was found, with no pharmacological tolerance to S1RA's antiallodynic and antihyperalgesic effects developed following repeated administration to nerve-injured mice [14]. S1RA has also been shown to enhance the analgesic effect of morphine, while reducing its rewarding effect [15]. Also, no findings that preclude further development of S1RA have been found in safety pharmacology and toxicity studies (including 13 week repeat dose toxicity studies in rats, dogs and monkeys).
The purpose of this article is to present the results of early investigations in healthy subjects on safety, tolerability, pharmacodynamics and pharmacokinetics of S1RA.
Methods
Study design
Three phase I studies (Figure 1A–C) were conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines: studies 101 and 102 were performed at the Centre d'Investigació de Medicaments, Institut de Recerca de l'Hospital de la Santa Creu i Sant Pau, Barcelona, Spain and study 106 was carried out at Richmond Pharmacology Ltd (RPL), Croydon University Hospital, London, UK. Study protocols and informed consent forms were approved by independent ethics committees: the Ethics Committee for Clinical Research of the Hospital de la Santa Creu i Sant Pau for studies 101 and 102 (reference numbers 08/064 and 09/028, respectively) and the Yorkshire Independent Research Ethics Committee for study 106 (reference number 10/IEC07/9). All subjects gave written informed consent prior to initial screening. The studies were registered with EudraCT numbers: 2008-000751-94 (study 101), 2009-009424-37 (study 102) and 2010-020343-13 (study 106).
Figure 1.
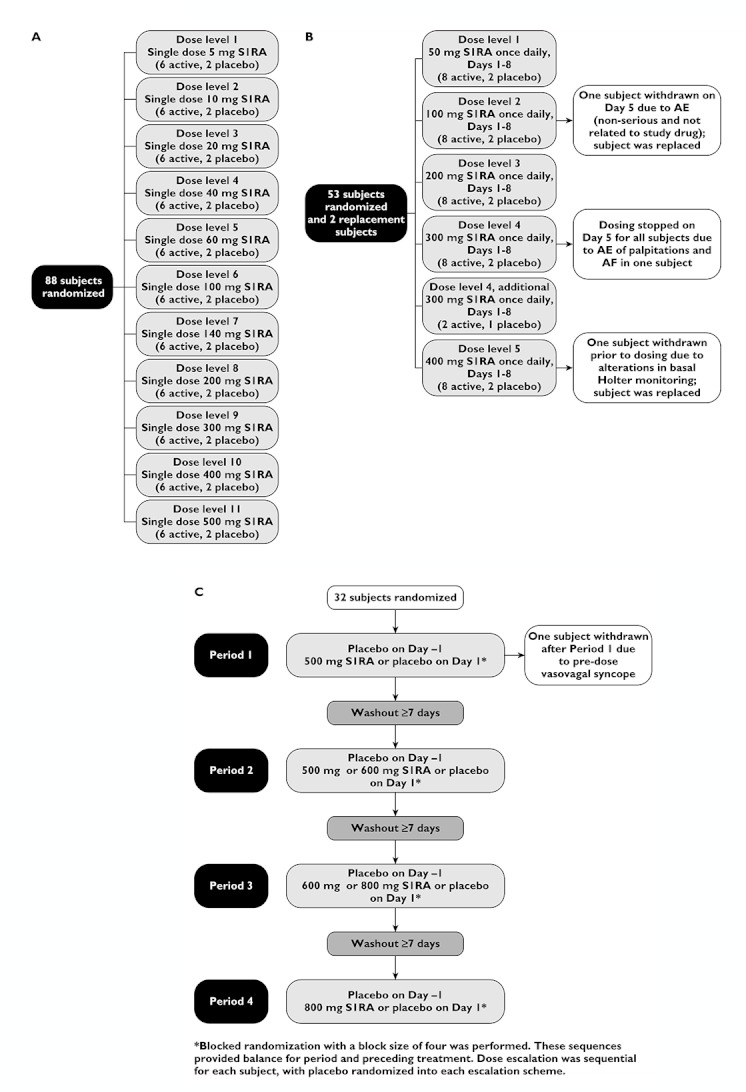
(A) Design and subject allocation scheme for study 101. (B) Design and subject allocation scheme for study 102. (C) Design and subject allocation scheme for study 106
All studies were single-centre, randomized, double-blind and placebo-controlled. Study 101, the first-in-man study, was a parallel, dose-escalation study with 11 consecutive cohorts of healthy subjects receiving single doses of S1RA (5–500 mg) or placebo (Figure 1A). Dose escalation was planned to stop if the minimum intolerated dose (MID) was reached, in order to determine the maximum tolerated dose (MTD). Study 102 was a parallel, multiple dose-escalation study in which subsequent cohorts of healthy subjects received oral doses of S1RA (50–400 mg) or placebo daily for 8 days (Figure 1B). Study 106 was a four-way crossover study of single doses of S1RA (500–800 mg) or placebo in healthy subjects, with the multiple crossover design allowing sequential dose escalation in the same subjects (Figure 1C). No formal sample size calculations were performed for these three phase I trials but the number of subjects was considered to be adequate to answer the respective study objectives, based on empirical criteria.
Inclusion/exclusion criteria
All subjects studied were healthy adults more than 18 years old. Only male subjects up to 45 years were eligible for study 101, while male and female subjects up to 45 years were eligible for study 102 and up to 35 years for study 106.
Women taking part were of non-childbearing potential, or agreed to use medically acceptable methods of contraception. Subjects were required to have a body weight within the normal range and not to have smoked for ≥6 months before study (101, 102) or ≥3 months before the study (106). Subjects participating in study 106 were also required to be of Caucasian origin and to have a triplicate 12-lead ECG and a 24 h 5-lead Holter ECG without clinically relevant abnormalities.
Exclusion criteria included: abnormal ECG results, including QTc interval >450 ms or ECG changes which might interfere with the interpretation of QTc interval changes, history of serious adverse reactions or hypersensitivity to any drug, abnormal physical findings of clinical significance, history of drug or alcohol abuse, clinically significant abnormal laboratory values or existence of any surgical or medical condition which might interfere with the distribution, metabolism or excretion of S1RA. In addition, for study 106, subjects were excluded if they met any of the following criteria: history of clinically significant syncope, family history of sudden death, family history of premature cardiovascular death, clinically significant history or family history of congenital long QT syndrome or a history of clinically significant arrhythmias or ischemic heart disease.
Study drug administration
Each S1RA or placebo dose was administered orally in fasting conditions. All study medication was supplied in pre-packaged bottles labelled with individual subject numbers to maintain blinding. In studies 101 and 102, all study medications were presented as active pharmaceutical ingredient (API) in capsules, whereas in study 106 S1RA was formulated as capsules containing immediate release pellets. Bioequivalence of both formulations was demonstrated in a previous study. Subject randomization was performed using a computer-generated list, using either SPSS (SPSS Inc. Chicago, IL) or SAS™ (SAS Institute Inc., Cary, NC) by an independent statistician.
In each of the 11 groups in study 101, six subjects were randomized to receive active drug and two subjects to receive placebo. Subjects were resident in the facility from the afternoon of day −1 until 36 h after dosing.
In each of the five dose levels in study 102, eight subjects were randomized to receive active drug and two subjects to receive placebo. Subjects were resident in the facility from the afternoon of day −1 until approximately 32 h after administration of the last dose of study medication (day 9).
In study 106, all subjects received a single dose of placebo on day −1 of each period in order to establish a single-blinded baseline for ECG recordings. On day 1 of each period, subjects received a single dose study medication, either S1RA or placebo. Subjects were resident in the facility from day −2 of period 1 until the morning of day 5 of period 4.
Safety and tolerability assessments and analyses
In all studies, the following assessments of safety and tolerability were made: adverse events recording following questioning and spontaneous reporting; physical examinations, 12-lead ECG evaluations, vital signs measurements in the sitting position (systolic and diastolic blood pressure, heart and respiratory rate and axillary body temperature) and laboratory safety tests (haematology and blood coagulation, biochemistry, urinalysis).
In study 102, participants at the 300 mg and 400 mg dose levels underwent three channel continuous Holter monitoring from 10 min pre dose on day 1 to 24 h post administration of the last dose. In study 106, thorough cardiac monitoring via telemetry, triplicate 12-lead ECGs and 24 h Holter ECG was performed during each treatment period from 1 h pre dose on day −1 and day 1 until 24 h post dose. In addition, blood pressure and heart rate were measured in both the supine and the standing position on day −1 to day 5 in each treatment period, and on day −2 of treatment period 1.
All subjects randomized into the studies and receiving at least one dose of study medication were included for safety and tolerability analysis. The Medical Dictionary for Regulatory Affairs (MedDRA) was used to code the adverse events of the trials. Descriptive statistics were calculated for all variables collected at the screening phase (demographic characteristics, ECG evaluations, vital signs and laboratory safety tests). During the treatment period, descriptive statistics were calculated for safety evaluations (ECG evaluations, vital signs and laboratory safety tests). Analyses of QT interval changes were also made. All statistical analyses were performed using SAS™.
Pharmacodynamic assessments and analyses
All pharmacodynamic analyses included all subjects who received at least one dose of study drug and had at least one complete set of evaluable parameters, following the intent-to-treat principle.
In order to explore any potential CNS activity of S1RA, various assessments were conducted. In each study, VAS were used to evaluate subjective effects, mainly somnolence and potential abuse liability related to the drug, and were completed prior to dosing and at time points up to 48 h post dose. In studies 101 and 102, subjects completed the Addiction Research Center Inventory (ARCI) [16], a true/false questionnaire based on experienced drug users' reports of drug effects, and measuring sedation, euphoria, dysphoria, intellectual efficiency and amphetamine effects, before and after dosing. In addition, subjects in study 101 were asked to complete an end-of-study (EOS) questionnaire about drug-liking and drug-identification at 24 h after drug administration, and in study 102, the self-assessment scale for the evaluation of sleep and awakening quality (SSA) [17], a self-rating questionnaire that allows subjective evaluation of sleep and awakening quality, was completed prior to dosing on days 1–7 and 24 h post administration of the last dose.
In study 106, cognitive testing using a computerized battery (CogState Clinical Trials Software, CogState Ltd, Warminster, UK) was performed on day −1 and day 1, at 2 h and 3 h post dose, and on day 2 at 24 h post dose. The battery included a Groton maze learning task assessing executive function, a one card learning task measuring working memory and learning, a detection task measuring simple reaction time and psychomotor function, an identification test measuring choice reaction time and a sustained vigilance test (repeat of the detection task).
In studies 101 and 102, descriptive statistics were performed for sedation, euphoria, somatic-dysphoric and amphetamine effects measured by subscales of the ARCI and for the VAS, measuring drowsiness, liking, high, good effects, bad effects, sedation, nausea and salivation. Descriptive statistics were also performed for the EOS question about drug liking in study 101, and the quality of sleep status, the quality of awakening status and the somatic symptoms measured by the SSA in study 102.
In study 106, a linear mixed model was used to compare the treatment effect on cognitive tests and VAS. The model had treatment, sequence, period, gender, time and time by treatment interaction as fixed effects, baseline value as covariate, and subject as random effect. All statistical analyses were performed using SAS™ and a two-tailed 0.05 level of significance.
Blood sampling and laboratory analyses
Blood samples for pharmacokinetic evaluation in plasma were collected into tubes containing K2-EDTA prior to dosing and at various time points after dosing up to 48 h, 60 h and 96 h in studies 101, 102 and 106 respectively.
The quantification of S1RA was performed using a validated HPLC/MS/MS method (API4000 AB Sciex, Foster City, CA, USA), which involved the extraction of S1RA and the deuterium-labelled internal standard from plasma samples (100 µl) [18]. The lower limit of quantification was 2 ng ml−1 in study 101 with a between-day precision and accuracy of 5.9% (CV) and 108.3% (nominal value), respectively, and 20 ng ml−1 in studies 102 and 106 with a between day precision and accuracy of 7.7% (CV) and 91.6% (nominal value) respectively. The calibration curve was linear up to 6000 ng ml−1.
Pharmacokinetic evaluations and analyses
The population for pharmacokinetic analysis consisted of all subjects who had received at least one dose of study medication and had an evaluable pharmacokinetic profile. Non-compartmental pharmacokinetic analysis was performed using appropriate software (WinNonlin, Pharsight Corporation, Mountain View, CA, and SAS™). The following pharmacokinetic parameters were obtained: maximum plasma concentration (Cmax), time to maximum plasma concentration (tmax), elimination half-life (t1/2), area under the concentration–time curve from time zero to different time points (AUC(0,∞), AUC(0,24 h), AUC(0,τ)), minimum concentration after dose interval (Cmin) and accumulation ratio (R).
For single and multiple dose studies, descriptive statistical analyses for pharmacokinetic parameters were performed using SAS™. An analysis of variance (anova) was performed for Cmax and AUC(0,24 h) for male and female subjects of study 106 and for the last three values of Cmin at days 6, 7 and 8 to assess whether steady-state had been reached.
For the dose range studied (5–800 mg), a power model using individual subject data was carried out to assess the dose proportionality which assumed the following relationship: PK = α × Doseβ, where PK is the pharmacokinetic parameter, in this case, Cmax or AUC. β can be obtained by natural log transforming PK and dose, followed by linear regression on ln(PK) = ln(α) + β × ln(Dose).When β is not statistically significantly different from unity (i.e. the 90% confidence interval includes 1), dose proportionality can be established [19–21].
Results
Demographics and baseline characteristics
A total of 175 healthy subjects (134 men and 41 women) were randomized in the three studies (Table 1), with 162 completing as planned. In study 101, 88 male subjects were randomized (66 to active treatment and 22 to placebo) and all completed the study (Figure 1A). In study 102, 55 subjects were randomized, of whom 54 (28 male and 26 female) received at least one dose of study medication (43 active treatment and 11 placebo) and 43 completed the study. Eleven subjects did not complete the study (two due to adverse events and nine due to the discontinuation of a dose cohort) (Figure 1B).
Table 1.
Demographics and baseline characteristics (mean (SD)) of subjects in studies 101, 102 and 106
| Dose (mg) | n | Age (years) | Body weight (kg) | Height (cm) | Body mass index (kg m–2) |
|---|---|---|---|---|---|
| Study 101 | |||||
| 5 | 8 | 28.0 (6.5) | 69.5 (5.3) | 172.0 (8.3) | 23.4 (2.0) |
| 10 | 8 | 25.6 (5.4) | 73.2 (9.1) | 177.4 (7.9) | 23.3 (2.2) |
| 20 | 8 | 24.0 (3.2) | 65.5 (6.7) | 172.5 (5.1) | 22.0 (1.6) |
| 40 | 8 | 30.8 (6.11) | 70.9 (7.3) | 177.0 (5.9) | 22.6 (1.9) |
| 60 | 8 | 25.2 (2.4) | 73.9 (10.1) | 178.9 (6.0) | 23.0 (2.3) |
| 100 | 8 | 24.6 (5.3) | 70.7 (9.1) | 174.4 (8.8) | 23.2 (2.0) |
| 140 | 8 | 27.3 (3.5) | 75.2 (5.7) | 174.3 (7.5) | 24.8 (1.4) |
| 200 | 8 | 27.5 (5.9) | 72.0 (9.8) | 172.4 (6.8) | 24.1 (1.7) |
| 300 | 8 | 29.3 (7.8) | 70.5 (6.3) | 171.4 (8.0) | 24.1 (1.8) |
| 400 | 8 | 28.6 (5.9) | 70.4 (8.6) | 174.3 (5.4) | 23.1 (2.3) |
| 500 | 8 | 24.5 (3.3) | 68.3 (7.5) | 175.0 (6.4) | 22.2 (1.7) |
| Study 102 | |||||
| 50 | 10 | 31.8 (5.8) | 66.1 (11.8) | 167.7 (15.0) | 23.4 (1.7) |
| 100 | 11 | 27.7 (4.1) | 63.5 (9.9) | 166.2 (7.7) | 22.9 (2.6) |
| 200 | 10 | 27.4 (6.5) | 65.2 (11.3) | 169.7 (10.6) | 22.5 (2.2) |
| 300 (subgroup 1) | 5 | 30.5 (7.8) | 67.7 (9.2) | 167.0 (14.1) | 24.2 (0.8) |
| 300 (subgroup 2) | 5 | 28.8 (5.7) | 65.9 (11.7) | 167.8 (7.9) | 23.2 (2.2) |
| 300 (additional subgroup) | 3 | 33.2 (10.3) | 66.2 (11.3) | 166.0 (13.5) | 24.0 (1.9) |
| 400 | 10 | 28.5 (6.7) | 69.0 (11.7) | 171.0 (9.6) | 23.4 (2.1) |
| Study 106 | |||||
| 500–800 | 32 | 24.2 (4.7) | 67.4 (9.7) | 173.09 (9.8) | 22.4 (1.6) |
In study 106, 32 subjects (17 male and 15 female) were randomized and 31 completed all study periods. One subject was withdrawn from the study after the first period (S1RA 500 mg) due to an adverse event recorded before active study medication was administered (Figure 1C).
Demographic and baseline characteristics of study subjects are shown in Table 1.
Safety and tolerability
There were no serious adverse events reported in any of the three studies. Few (46) treatment-emergent adverse events (TEAEs), i.e. events after dosing with either placebo or active treatment, occurred in studies 101 and 102. As would be anticipated, a greater number of TEAEs (137) were reported after administration of higher single doses of S1RA in study 106.
Study 101
In study 101, 12 adverse events (two with placebo and 10 with S1RA at any dose) occurred in eight subjects, two with placebo (9.1%) and six with active treatment (9.1%). Seven of them experienced one event and one subject receiving 200 mg S1RA experienced five events: headache, pruritus, swollen lip, skin rash and pruritic skin rash. All were mild or moderate in intensity and resolved without sequelae. The single most reported symptom was headache (four subjects receiving S1RA and one subject receiving placebo). All adverse events were considered by investigators to be possibly related to study drug, except for an event of elevated triglycerides in the blood in a subject who received 100 mg S1RA which was considered unlikely to be related to study drug. One subject in the placebo group also exhibited an increase in triglycerides and these elevations were without clinical relevance.
Study 102
In study 102, 34 adverse events occurred (30 with active treatment and four with placebo) in 15 subjects (12 receiving S1RA, i.e. 27.9% and three receiving placebo i.e. 27.3%). The most common was headache (nine episodes, all with active treatment), followed by dizziness (four out of six episodes with S1RA). Relationship with study drug was classified as possible in 28 events, not related in three events and unlikely in three events. One subject receiving 100 mg S1RA withdrew on day 6 due to a non-serious, non-related adverse event (dysmenorrhoea) of severe intensity, and one subject receiving 300 mg S1RA withdrew on day 4 due to paroxysmal atrial fibrillation (AF), which was assessed as non-serious, possibly related to study drug and of moderate intensity. This subject experienced two episodes of paroxysmal AF (detected on ECG and then palpitations without other signs or symptoms) which resolved spontaneously without treatment and without sequelae. In this subject, plasma concentrations of S1RA were slightly lower than the mean concentration for the group. Three days after stopping administration of S1RA, the subject experienced the second AF event which also reverted spontaneously. He was suffering from acute emotional stress (which is a potential alternative explanation for the occurrence of the event) when he experienced the two events and reported palpitations several months prior to entering the study, also related to emotional stress. Follow-up ECG, echocardiogram and Holter monitoring were normal.
For prudence, and considering that S1RA is a new chemical entity, dosing of the entire 300 mg dose cohort (10 subjects) was stopped after the third (subgroup 1) or the fourth (subgroup 2) dose administration (Table 1). Based on the known safety profile of the drug and the acceptable tolerability demonstrated in all the rest of the healthy subjects previously tested, it was decided to continue with the 300 mg administration, but with a new and smaller group of healthy subjects (additional subgroup). The Ethics Committee approved a protocol amendment for the continuation of the trial with 24 h Holter monitoring throughout the 8 day repeated administration period. Subjects in the new 300 mg cohort presented a good safety and tolerability profile and no cardiac adverse events were reported. Subsequently, the pre-planned group of 400 mg was begun, also with 24 h Holter monitoring, which was continued during the 8-day multiple dosing period. No cardiac adverse events were reported.
Study 106
In study 106, a total of 137 TEAEs were reported in 25 subjects. Adverse events reported by at least 5% of subjects in association with any treatment are shown in Table 2. TEAEs are divided into those occurring during the ‘pre dose’ time interval (from dosing time on day −1, i.e. placebo administration, to dosing time on day 1) and the ‘post dose’ time interval (from dosing time on day 1 to dosing time on day −1 of the next period).
Table 2.
Treatment-emergent adverse events occurring in ≥5% of subjects receiving any study treatment in study 106*
| Number of subjects | n= 32 | n= 31 | n= 32 | n= 31 | n= 31 |
|---|---|---|---|---|---|
| MedDRA System Organ Class and Preferred Term | Pre dose | Placebo | 500 mg S1RA | 600 mg S1RA | 800 mg S1RA |
| Any system | 21 (11, 34.4%) | 7 (6, 19.4%) | 17 (12, 37.5%) | 42 (17, 54.8%) | 50 (15, 48.4%) |
| Nervous system disorders | 11 (8, 25.0%) | 3 (3, 9.7%) | 8 (6, 18.8%) | 13 (10, 32.3%) | 17 (10, 32.3%) |
| Dizziness postural | 6 (4, 12.5%) | 1 (1, 3.2%) | 3 (3, 9.4%) | 4 (4, 12.9%) | 2 (2, 6.5%) |
| Headache | 0 | 2 (2, 6.5%) | 1 (1, 3.1%) | 5 (5, 16.1%) | 6 (6, 19.4%) |
| Dizziness | 2 (2, 6.3%) | 0 | 1 (1, 3.1%) | 0 | 6 (6, 19.4%) |
| Syncope | 1 (1, 3.1%) | 0 | 2 (2, 6.3%) | 0 | 0 |
| Disturbance in attention | 0 | 0 | 0 | 2 (2, 6.5%) | 0 |
| Psychiatric disorders | 0 | 0 | 4 (3, 9.4%) | 14 (5, 16.1%) | 14 (8, 25.8%) |
| Euphoric mood | 0 | 0 | 0 | 5 (5, 16.1%) | 1 (1, 3.2%) |
| Dissociation | 0 | 0 | 1 (1, 3.1%) | 2 (2, 6.5%) | 2 (2, 6.5%) |
| Thinking abnormal | 0 | 0 | 2 (2, 6.3%) | 0 | 3 (3, 9.7%) |
| Hallucination, visual | 0 | 0 | 0 | 2 (2, 6.5%) | 1 (1, 3.2%) |
| Paranoia | 0 | 0 | 0 | 2 (2, 6.5%) | 1 (1, 3.2%) |
| Abnormal dreams | 0 | 0 | 0 | 0 | 2 (2, 6.5%) |
| Restlessness | 0 | 0 | 0 | 2 (2, 6.5%) | 0 |
| Gastrointestinal disorders | 5 (3, 9.4%) | 0 | 0 | 5 (5, 16.1%) | 5 (4, 12.9%) |
| Nausea | 2 (2, 6.3%) | 0 | 0 | 3 (3, 9.7%) | 4 (4, 12.9%) |
| Cardiac disorders | 1 (1, 3.1%) | 0 | 0 | 3 (3, 9.7%) | 3 (3, 9.7%) |
| Palpitations | 0 | 0 | 0 | 2 (2, 6.5%) | 1 (1, 3.2%) |
| Sinus tachycardia | 0 | 0 | 0 | 1 (1, 3.2%) | 2 (2, 6.5%) |
| Vascular disorders | 1 (1, 3.1%) | 0 | 2 (2, 6.3%) | 2 (2, 6.5%) | 1 (1, 3.2%) |
| Orthostatic hypotension | 1 (1, 3.1%) | 0 | 2 (2, 6.3%) | 2 (2, 6.5%) | 0 |
Number of AEs (subjects, % subjects with AE). Percentages in parentheses within table are calculated by dividing the number of subjects with at least one adverse event by the number of subjects receiving that study treatment.
Two adverse events were rated as severe (both episodes of syncope following 500 mg S1RA), and the rest were mild or moderate in intensity. Fifteen were considered by the investigator to be unrelated to treatment. One subject was withdrawn from the study due to telemetry findings of AF coinciding with an episode of vasovagal syncope during cannulation observed before study medication was administered.
Following the 500 mg dose, the most common adverse event was postural dizziness (three subjects), and two subjects had an episode of syncope judged to be severe. These episodes were considered by the investigator to be most likely due to a drop in blood pressure rather than related to the study drug. This alternative explanation is supported by the fact that the study included frequent blood sampling, the subjects were often required to stand up for blood pressure assessments, the weather was unusually warm and the subjects' liquid intake was limited as per protocol. For the subsequent treatment groups (600 mg and 800 mg), sudden standing for blood pressure assessment was not permitted and no further episodes of syncope were observed at the higher dose levels. Four TEAEs reported in three subjects were classified in the psychiatric disorders MedDRA SOC (System Organ Class) (Table 2).
Following the 600 mg dose, the most frequently reported adverse events were headache and euphoric mood (five episodes of each) and there were three events of nausea. Several other psychiatric adverse events were reported (Table 2). After the 800 mg dose, the most common adverse events were headache and dizziness (six episodes of each). In addition, eight subjects experienced a total of 14 psychiatric adverse events at this dose level, including three cases of abnormal thinking and two cases each of abnormal dreams and dissociation. Seven of the psychiatric and three of the CNS adverse events were reported retrospectively at a later stage, that is, there were no clinical indications of these events during the day of dosing and no obvious concerns during monitoring by health care professionals. All subjects performed and completed their tasks including their cognitive function tests.
Cardiovascular safety
As discussed above, one subject in study 102 receiving 300 mg S1RA developed AF on day 4 of dosing and was withdrawn from the study. One subject in study 106 developed AF prior to dosing (during cannulation) and was withdrawn from the study.
Two subjects experienced an episode of sinus tachycardia during study 106. One of the subjects experienced asymptomatic episodes of tachycardia 1 h after receiving 600 mg S1RA (up to 140 beats min–1) and 25 min after 800 mg S1RA (up to 150 beats min–1). The other subject experienced asymptomatic sinus tachycardia with a maximum heart rate of 174 beats min–1 approximately 5 h after receiving 800 mg S1RA. Both episodes were observed through telemetric monitoring. Subsequent examinations by a consultant cardiologist were normal for both subjects. Another subject was noted to have a brief, asymptomatic episode of non-sustained ventricular tachycardia on Holter monitoring prior to dosing. This subject was also seen by a cardiologist, who found no evidence of underlying cardiac disease.
There were no clinically relevant changes in QTc interval observed during the three studies (data not shown). No other clinically relevant changes were observed in any other parameters of the 12-lead ECG, or in the 24 h Holter monitoring, in any subjects participating in the three studies (data not shown).
Other safety results
Blood chemistry, haematology and urinalysis results were not clinically relevant at baseline and remained normal throughout the studies. Overall, supine blood pressure and pulse rates were normal at baseline and there were no clinically relevant changes throughout the studies. Orthostatic hypotension was noted in two subjects receiving 500 mg S1RA when standing for blood pressure measurements (at 2 h 5 min and at 4 h 15 min post dose, respectively). Three other subjects at this dose level reported dizziness on standing at 2–3 h post dose and two experienced episodes of syncope (at 2 h 15 min and at 3 h 10 min post dose, respectively). As discussed above, sudden standing for blood pressure measurement was not permitted at higher dose levels and no further episodes of syncope were observed. No clinically significant findings in physical examination were observed during the studies.
Pharmacodynamics
In studies 101 and 102, no consistent pattern of CNS effects was demonstrated in any pharmacodynamic evaluations, including SSA, ARCI or the EOS questionnaire about drug-liking and drug-identification.
All VAS evaluated in studies 101 and 102 showed no effect at any of the tested doses, even at the highest ones (500 mg single dose and 400 mg multiple dose). In study 106, VAS assessments generally showed a dose-related trend with no significant effect at 500 mg, while the 800 mg dose exhibited the greatest effect (Figure 2). At 2 h post dose, subjects receiving 800 mg S1RA were rated as being more drowsy, dreamy and excited, and less proficient, energetic, quick-witted, clear-headed and relaxed, compared with subjects receiving placebo. After 24 h, ratings were similar for all treatment groups.
Figure 2.
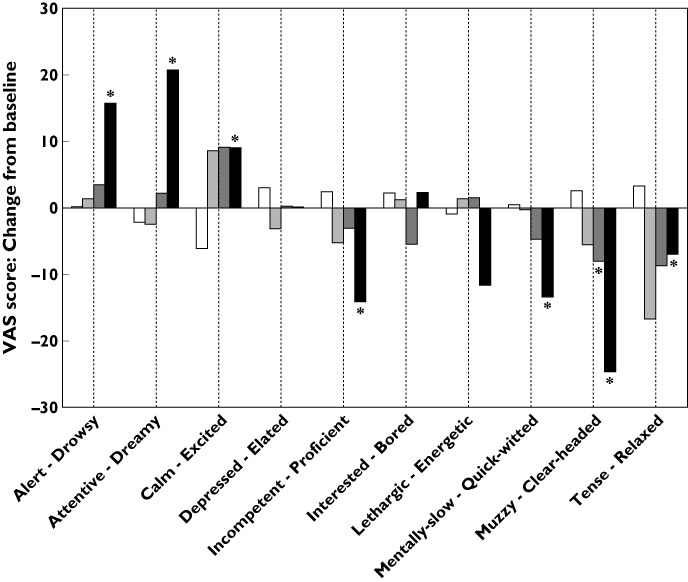
VAS scores for study 106. The figure shows mean values of difference from baseline scores at 2 h post-dose for the 10 measured VAS. The VAS pairs of adjectives are shown so the first one corresponds to low scores. An asterisk indicates a significant difference vs. placebo in the linear mixed model analysis, adjusted for the baseline value. Baseline values were similar for all groups and in the range of 40 to 70 for all scales, except for ‘calm-excited’, which ranged from 25 to 35. □, Placebo;  , 500 mg;
, 500 mg;  , 600 mg,
, 600 mg,  , 800 mg
, 800 mg
In study 106, some slowing of response for simple reaction time (detection task) and choice reaction time (identification task) was observed 2 h following the administration of S1RA, compared with placebo (data not shown). This recovered by 24 h post dose. Also on computerized cognitive testing, there was no effect on visual memory, executive function, attention or somnolence at any assessment.
Pharmacokinetics
Pharmacokinetic parameters of single and multiple doses of S1RA obtained in the single dose studies (101 and 106) and the multiple dose study (102) are summarized in Table 3 and Table 4, respectively. Also, pharmacokinetic parameters for male and female subjects in study 106 are shown in Table 5.
Table 3.
Pharmacokinetic parameters of single doses of S1RA in studies 101 and 106*
| Dose (mg) | Cmax (µg ml–1) | tmax (h) | t1/2 (h) | AUC(0,24 h) (µg ml–1 h) | AUC(0,∞) (µg ml–1h) | Cmax/Dose (µg ml–1)/g | AUC(0,24 h)/Dose (µg ml–1 h)/g |
|---|---|---|---|---|---|---|---|
| Study 101 | |||||||
| 5 | 0.07 (0.02) | 0.8 (0.6–0.8) | 19.7 (4.7) | 0.24 (0.06) | 0.35 (0.10) | 13.9 (4.0) | 48.9 (11.1) |
| 10 | 0.11 (0.05) | 1.0 (0.8–1.2) | 15.7 (2.5) | 0.53 (0.36) | 0.77 (0.45) | 10.7 (4.9) | 52.6 (36.0) |
| 20 | 0.25 (0.07) | 0.8 (0.8–0.9) | 15.7 (6.2) | 0.95 (0.27) | 1.24 (0.31) | 12.6 (3.7) | 47.7 (13.3) |
| 40 | 0.51 (0.19) | 0.8 (0.8–0.9) | 15.6 (2.8) | 2.19 (0.80) | 3.09 (1.36) | 12.8 (4.7) | 54.9 (20.1) |
| 60 | 0.62 (0.13) | 0.8 (0.6–1.2) | 17.0 (3.0) | 3.11 (0.77) | 4.30 (1.20) | 10.4 (2.2) | 51.9 (12.8) |
| 100 | 1.22 (0.45) | 0.9 (0.8–1.2) | 14.8 (4.7) | 7.92 (3.98) | 8.70 (2.37) | 12.2 (4.4) | 79.2 (39.8) |
| 140 | 1.43 (0.26) | 0.8 (0.8–0.9) | 17.1 (6.3) | 9.05 (2.90) | 13.98 (4.84) | 10.2 (1.9) | 64.6 (20.7) |
| 200 | 2.51 (1.05) | 0.9 (0.8–1.5) | 18.4 (3.4) | 15.10 (3.95) | 23.20 (7.88) | 12.5 (5.3) | 75.5 (19.7) |
| 300 | 3.44 (0.47) | 0.8 (0.6–1.4) | 19.1 (4.8) | 22.96 (2.99) | 35.96 (5.62) | 11.4 (1.6) | 76.5 (10.0) |
| 400 | 3.11 (0.79) | 1.2 (1.0–1.3) | 15.9 (3.1) | 21.25 (8.09) | 39.99 (10.12) | 7.8 (2.0) | 53.1 (20.2) |
| 500 | 4.72 (1.82) | 1.3 (0.9–1.8) | 15.1 (4.6) | 30.78 (6.61) | 46.69 (17.37) | 9.4 (3.6) | 61.6 (13.2) |
| Study 106 | |||||||
| 500 | 3.94 (0.79) | 1.5 (1.4–2.0) | 31.9 (8.0) | 36.90 (6.46) | 85.75 (19.06) | 7.9 (1.6) | 73.8 (12.9) |
| 600 | 4.36 (0.71) | 2.0 (1.5–2.0) | 35.5 (6.8) | 44.98 (7.15) | 112.07 (24.07) | 8.7 (1.4) | 90.0 (14.3) |
| 800 | 5.58 (1.18) | 2.0 (1.5–2.5) | 41.5 (7.1) | 56.98 (8.50) | 155.91 (28.83) | 11.2 (2.4) | 114.0 (17.0) |
Mean (SD). tmax values are expressed as median (25th percentile–75th percentile).
Table 4.
Pharmacokinetic parameters of multiple doses of S1RA in study 102*
| Dose (mg) | Day | Cmax (µg ml–1) | tmax (h) | AUC(0,τ) (µg ml–1 h) | Cmax/Dose (µg ml–1)/g | AUC(0,τ)/Dose (µg ml–1 h)/g | Cmin (µg ml–1) | R Cmax | R AUC(0,τ) |
|---|---|---|---|---|---|---|---|---|---|
| 50 | 1 | 0.67 (0.15) | 0.8 (0.8–0.8) | 3.63 (1.95) | 13.5 (3.0) | 72.7 (38.9) | – | – | – |
| 100 | 1 | 1.36 (0.39) | 0.8 (0.8–1.0) | 7.56 (1.29) | 13.6 (3.9) | 75.6 (12.9) | – | – | – |
| 200 | 1 | 3.12 (0.96) | 1.0 (0.9–1.0) | 23.85 (6.88) | 15.6 (4.8) | 119.3 (34.4) | – | – | – |
| 300 | 1 | 4.41 (1.07) | 1.0 (0.8–1.8) | 27.38 (6.33) | 14.7 (3.6) | 91.3 (21.1) | – | – | – |
| 400 | 1 | 3.64 (0.98) | 1.0 (0.9–1.5) | 27.97 (5.53) | 9.1 (2.4) | 69.9 (13.8) | – | – | – |
| 50 | 8 | 0.99 (0.46) | 0.8 (0.7–0.8) | 6.50 (4.17) | 19.9 (9.2) | 130.0 (83.5) | 0.14 (0.13) | 1.5 | 1.8 |
| 100 | 8 | 2.04 (0.39) | 0.8 (0.5–0.8) | 17.34 (5.38) | 20.4 (3.9) | 173.4 (53.8) | 0.48 (0.23) | 1.6 | 2.3 |
| 200 | 8 | 3.86 (0.85) | 1.0 (0.8–1.3) | 44.23 (11.59) | 19.3 (4.2) | 221.2 (57.9) | 1.28 (0.44) | 1.3 | 1.9 |
| 300 | 8 | 4.29 (0.86) | 1.4 (1.1–1.7) | 72.67 (23.78) | 14.3 (2.9) | 242.2 (79.3) | 2.63 (1.17) | 0.9 | 2.4 |
| 400 | 8 | 5.42 (0.64) | 0.9 (0.8–1.3) | 64.80 (8.80) | 13.5 (1.6) | 162.0 (22.0) | 1.95 (0.41) | 1.6 | 2.4 |
Mean (SD). tmax values are expressed as median (25th percentile–75th percentile).
Table 5.
Pharmacokinetic parameters of male and female subjects in study 106*
| Dose (mg) | Gender | Cmax (µg ml–1) | tmax (h) | t1/2 (h) | AUC(0,24 h) (µg ml–1 h) |
|---|---|---|---|---|---|
| 500 | Male | 3.84 (3.46, 4.22) | 1.5 (1.5–2.0) | 30.4 (8.9) | 37.41 (34.04, 40.78) |
| 600 | 4.35 (3.99, 4.71) | 1.5 (1.5–2.0) | 34.6 (7.7) | 46.46 (42.36, 50.56) | |
| 800 | 5.51 (4.83, 6.19) | 1.8 (1.4–2.0) | 40.2 (6.2) | 57.81 (52.97, 62.66) | |
| 500 | Female | 4.05 (3.57, 4.53) | 1.5 (1.3–2.0) | 35.8 (6.6) | 36.32 (32.71, 39.94) |
| 600 | 4.36 (3.94, 4.79) | 2.0 (1.5–2.0) | 37.7 (4.9) | 43.40 (39.85, 46.94) | |
| 800 | 5.66 (5.05, 6.26) | 2.0 (1.5–3.0) | NC | 56.09 (51.64, 60.54) |
Cmax and AUC(0,24 h) values are expressed as mean (95 % CI), tmax values are expressed as median (25th percentile–75th percentile) and t1/2 values are expressed as mean (SD). NC, not calculated.
Single dose studies
After single oral dose administration, fast absorption, rapid distribution and slow terminal elimination were observed (Figure 3). The rate and extent of exposure to S1RA increased with dose in both studies (Figure 4). Slight differences in the mean exposure at 500 mg among subjects from study 101 and study 106 were observed (Table 3).
Figure 3.
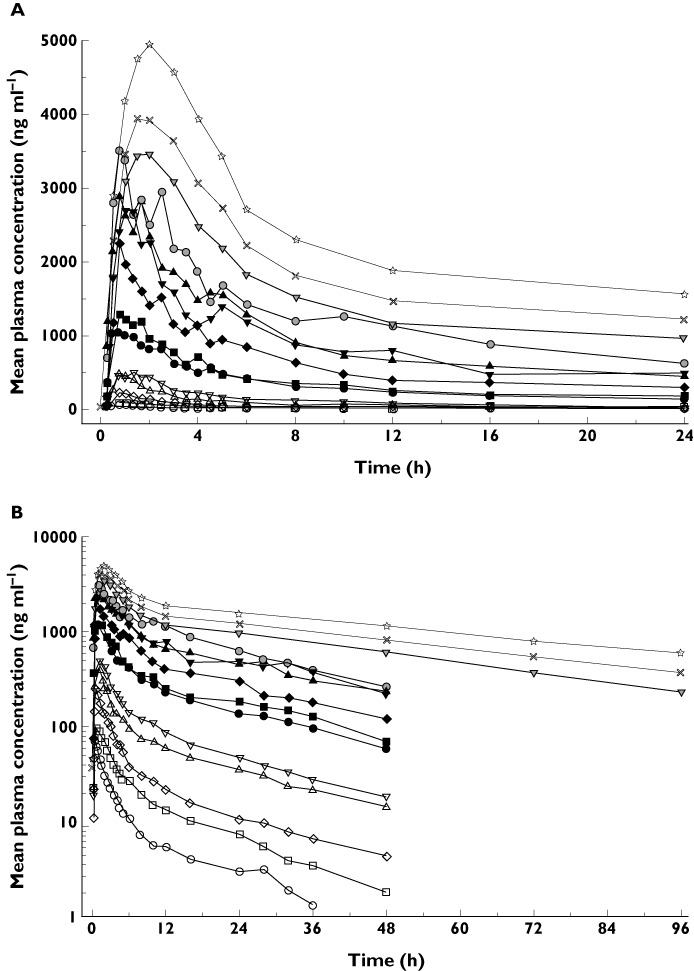
(A) Mean plasma concentration of S1RA over a 24 h period after single oral doses of 5–800 mg (linear scale). (B) Mean plasma concentration of S1RA over time after single oral doses of 5–800 mg (logarithmic scale). Study 101:  , 5 mg;
, 5 mg;  , 10 mg;
, 10 mg;  , 20 mg;
, 20 mg;  , 40 mg;
, 40 mg;  , 60 mg;
, 60 mg;  , 100 mg;
, 100 mg;  , 140 mg;
, 140 mg;  , 200 mg;
, 200 mg;  , 300 mg;
, 300 mg;  , 400 mg;
, 400 mg;  , 500 mg. Study 106:
, 500 mg. Study 106:  , 500 mg;
, 500 mg;  , 600 mg;
, 600 mg;  , 800 mg
, 800 mg
Figure 4.
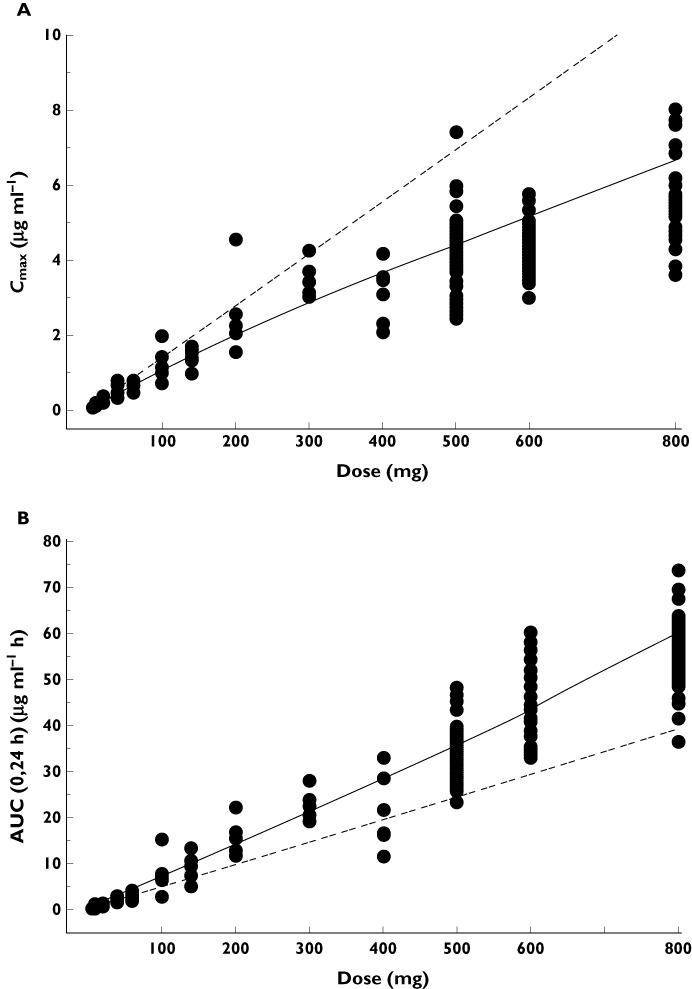
(A) Individual Cmax values of S1RA over the dose range following a single administration. (B) Individual AUC(0,24 h) values of S1RA over the dose range following a single administration.  , Power model;
, Power model;  , Linearity
, Linearity
The mean terminal monoexponential phase half-life for S1RA ranged from 14.8 to 19.7 h in the lower single dose study (5–500 mg, study 101) and 31.9 to 41.5 h in the higher single dose study (500–800 mg, study 106).
The S1RA maximum plasma concentration (Cmax) increased with increasing dose over the dose range 5–800 mg although this increase appeared to be less than the proportionate dose increment (Table 3 and Figure 4A) and the Cmax value at the highest dose level (800 mg) was approximately 49.8% lower than the value predicted from a linear relationship. The power model analysis concluded that Cmax proportionality was maintained in the dose range from 5–300 mg. The extent of systemic exposure of humans to S1RA characterized by AUC(0,24 h) increased proportionately with increasing dose in the range 5–60 mg, according to the power model analysis. However, at higher doses, the AUC(0,24 h) increase was greater than the proportionate dose increment and at 800 mg AUC(0,24 h) was approximately 45.8% higher than the value predicted from a linear relationship (Table 3 and Figure 4B).
Median tmax appeared to increase with increasing dose of S1RA (Table 3 and Figure 5), being 2 h for 600 mg and 800 mg, compared with 0.75–1.5 h for doses of 5–500 mg.
Figure 5.
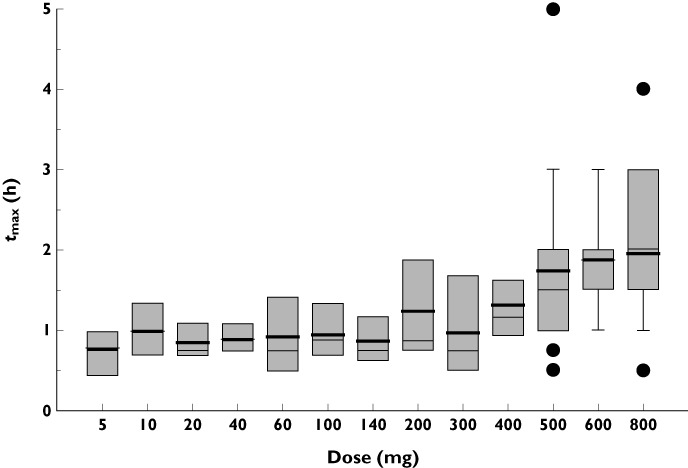
Whisker box plot of tmax values of S1RA over the dose range following a single administration
The evaluation of demographic covariates on S1RA pharmacokinetics showed that the rate and extent of systemic exposure of humans to S1RA was not affected by gender (P > 0.05) at doses of 500–800 mg (Table 5). Also, there was a weak negative correlation between final drug exposure parameter (AUC(0,24 h)/Dose) and total body weight (r = 0.262). On the other hand, the narrow range of ages (from 18 to 45 years) precluded a robust examination of the influence of this covariate on the pharmacokinetics of S1RA.
Multiple dose study
Following the multiple oral administration of S1RA for 8 days, fast absorption, rapid distribution and slow terminal elimination of S1RA were also observed (Figure 6). Median tmax did not change after multiple oral administration of S1RA for 8 days.
Figure 6.
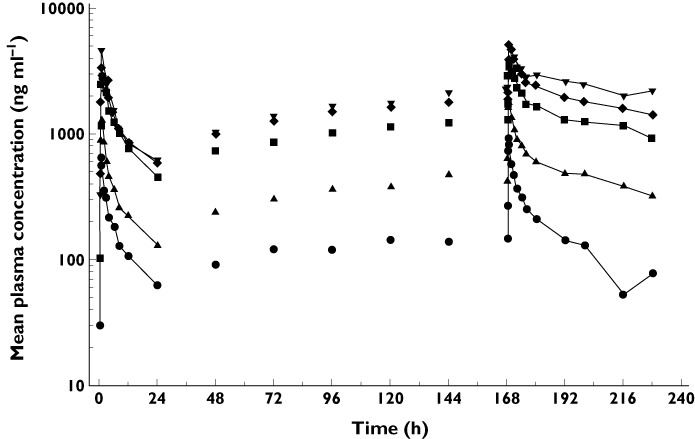
Mean plasma concentration of S1RA over time after multiple oral doses of 50–400 mg once daily for 8 days (logarithmic scale).  , 50 mg;
, 50 mg;  , 100 mg;
, 100 mg;  , 200 mg;
, 200 mg;  , 300 mg;
, 300 mg;  , 400 mg
, 400 mg
The rate and extent of systemic exposure of humans to S1RA in terms of Cmax and AUC(0,τ) increased proportionately with increasing dose over the dose range 50–300 mg (Table 4). At the 400 mg dose, the Cmax value was approximately 31.8% lower, and the AUC(0,τ) value was approximately 24.7% higher, than the value predicted from a linear relationship.
To evaluate whether steady-state had been reached, a statistical analysis of the Cmin values obtained at days 6, 7 and 8 was performed. The results showed that there were no significant differences in the Cmin values at the administered doses (P > 0.05). Therefore, it can be proposed that steady-state had been reached after 8 days of administration of S1RA once a day.
A theoretical assessment of time-dependent pharmacokinetics was also conducted based on the comparison of the expected accumulation factor and the observed accumulation factor calculated as R = AUC(0,τ) last day : AUC(0,τ) first day (Table 4). Based on the expected (2.7 for a half-life in the range of 31.9 to 41.5 h obtained in the higher single dose study) and observed accumulations at various S1RA doses (1.8–2.4), it seems that S1RA does not exhibit time-dependent pharmacokinetics.
Discussion
The new chemical entity S1RA has demonstrated an acceptable safety and tolerability profile in healthy male and female subjects. The MTD of S1RA was not reached following the administration of single doses of up to 800 mg or multiple doses of up to 400 mg daily for 8 days.
These three studies enrolled a total of 175 subjects, providing a robust evidence base. The most common adverse events were headache and dizziness, with the majority of adverse events reported being of mild or moderate intensity. Importantly, no serious adverse events occurred in any of the three studies.
As might be expected, increased numbers of adverse events were reported with higher single doses of S1RA, with the majority being classified as nervous system or psychiatric disorders, as would be anticipated for a centrally acting drug. Some of these adverse events were reported retrospectively. Importantly, there were no clinical indications of these events during the day of dosing, and all cognitive testing and monitoring was performed without incident, suggesting the clinical relevance of these events is limited. While there was some degree of slowing of simple reaction time and choice reaction time on cognitive testing performed 2 h after administration of S1RA (500, 600 or 800 mg), S1RA had no effect on visual memory, executive function, attention or somnolence and all cognitive tests were normal 24 h post dose. Although no concerns about nervous or psychiatric disorders were raised, it will be important to continue to evaluate the impact of S1RA in the CNS.
Among all studies, three subjects experienced cardiac rhythm adverse events while on treatment with S1RA. There were two episodes of AF in the same subject, only one of which occurred after dosing (300 mg S1RA), and was assessed as being possibly related to study drug. This event, occurring on the fourth day of dosing in a subject experiencing acute emotional stress, and who reported a prior episode of palpitations in association with stress months before, resolved spontaneously without sequelae. Two subjects experienced asymptomatic episodes of sinus tachycardia shortly after receiving 600 and 800 mg S1RA. Subsequent examinations by a consultant cardiologist were normal. It should also be noted that three subjects not receiving active treatment experienced arrhythmic events (one episode of AF and two episodes of non-sustained ventricular tachycardia).
ECG monitoring conducted in the three studies indicated that administration of S1RA at doses of up to 800 mg once daily was not associated with prolongation of the QTc interval, and there were no significant changes in terms of rhythm, conduction and ECG variations. Furthermore, other collected cardiac safety data did not indicate any trend or results of clinical relevance. Hence, in the current studies S1RA was not associated with significant cardiac side effects and this will continue to be reviewed in future studies.
The pharmacokinetic profile of S1RA in humans allows once daily dosing. Absorption was rapid and the rate and extent of exposure increased with dose. At higher doses the extent of exposure increased in a greater than dose-proportional manner and the rate of exposure at a lower than dose-proportional manner. Median tmax values were also extended at higher doses. This finding together with the decrease of Cmax with dose could probably be due to a saturation of the absorption process at high dose levels.
The mean terminal half-life ranged from 14.8 to 41.5 h. The longer value observed in study 106 compared with study 101 might be a reflection of a multicompartment drug disposition that could not be properly estimated in study 101 due to the sampling difference between studies (last post dose sample at 48 h and 96 h for 101 and 106, respectively). The presence of this peripheral compartment, which leads to a longer terminal half-life but with relatively low drug concentrations, is considered to have minimal impact based on drug accumulation upon multiple dosing (once a day) since this terminal half-life may only represent a small fraction of the total clearance of a drug.
The slight difference in the mean exposure at 500 mg observed among subjects from study 101 and study 106 might be attributable to the inherent pharmacokinetic variability, the low number of subjects in study 101 with respect to study 106 (n = 6 and 32, respectively) and the identification of the slow peripheral compartment with a longer mean terminal half-life in study 106 due to the extension of the blood collecting period.
Although the studied population was relatively homogenous, the influence of several demographic covariates on S1RA pharmacokinetics was investigated and it was concluded that the pharmacokinetic profile of S1RA does not appear to be affected by gender and weight.
The pharmacokinetic/pharmacodynamic relationship based on plasma concentrations of S1RA associated with analgesic activity in preclinical pharmacological models of pain suggests that the estimated therapeutic dose range for S1RA in humans should be around 100–400 mg in terms of Cmax and 20–150 mg in terms of AUC. Both estimations are well within the dose range already administered to humans that is associated with an acceptable safety and tolerability profile.
Many currently available treatments for pain are associated with troublesome side effects, and/or limited efficacy, and/or require frequent dosing and/or titration. Therefore, pain management is not satisfactorily resolved. In this context, the identification of new mechanisms of action and the development of treatments strategies is necessary. S1RA provides a novel approach with a new mechanism of action which is complementary to those of other drugs used in pain management, and also supports potential combination usage with other analgesic compounds. Taken together with preclinical data, these phase I results support the further investigation of S1RA in clinical studies to evaluate its efficacy, safety and tolerability in patients suffering from pain of various aetiologies, which are currently underway.
In conclusion, S1RA is well tolerated in healthy male and female subjects at single doses of up to 800 mg and multiple doses of up to 400 mg once daily for 8 days, with no dose-limiting toxicities or serious adverse events observed. S1RA is rapidly absorbed with rate and extent of exposure increasing with increasing dose and t1/2 compatible with once a day administration. The safety, tolerability, pharmacokinetic and pharmacodynamic profiles of S1RA demonstrated in these three phase I studies support its further development.
Acknowledgments
The authors would like to acknowledge the following individuals from ESTEVE for their contributions to these studies: Gemma Casadevall, Imma Cervera, Núria Cubel, Lluís Soler, Jaume Tomas (Pharmaceutical Innovation); Adelaida Morte, Roser Vives, Esther Ortiz, Soledad Casals, Sebastián Videla, Alberto Puyada (Clinical Investigation); Daniel Zamanillo, José Miguel Vela (Drug Discovery and Preclinical Development); Albert González (R&D Portfolio Management); Mercedes Encabo, Anna Cabot, Ramón Farrán, Zhengguo Xu (ADME and Bioanalysis); Fina Camacho (Quality Assurance); and Neus Gascón (Drug Safety and Pharmacovigilance). The authors also thank the Principal Investigator and their team at Richmond Pharmacology and the Centre d'Investigació de Medicaments, Institut de Recerca de l'Hospital de la Santa Creu i Sant Pau for their participation in the phase 1 studies described in this report and Complete Medical Communications for their assistance in the writing and editing of this publication.
Competing Interests
This research and publication were funded by ESTEVE (sponsor of the studies and owner of the compound) and partially financed with a grant from the Centro para el Desarrollo Tecnológico Industrial (CDTI) of the Spanish Ministerio de Ciencia e Innovación. All authors are employees of ESTEVE and all authors agree with the manuscript as written. The authors declare that they have no other real or potential conflicts of interest.
REFERENCES
- 1.Su TP, Hayashi T, Maurice T, Buch S, Ruoho AE. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol Sci. 2010;31:557–66. doi: 10.1016/j.tips.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cobos EJ, Entrena JM, Nieto FR, Cendán CM, Del Pozo E. Pharmacology and therapeutic potential of sigma(1) receptor ligands. Curr Neuropharmacol. 2008;6:344–66. doi: 10.2174/157015908787386113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maurice T, Su TP. The pharmacology of sigma-1 receptors. Pharmacol Ther. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chien CC, Pasternak GW. Functional antagonism of morphine analgesia by (+)-pentazocine: evidence for an anti-opioid system. Eur J Pharmacol. 1993;250:R7–R8. doi: 10.1016/0014-2999(93)90650-7. [DOI] [PubMed] [Google Scholar]
- 5.Chien CC, Pasternak GW. Selective antagonism of opioid analgesia by a sigma system. J Pharmacol Exp Ther. 1994;271:1583–90. [PubMed] [Google Scholar]
- 6.Chien CC, Pasternak GW. Sigma antagonists potentiate opioid analgesia in rats. Neurosci Lett. 1995;190:137–9. doi: 10.1016/0304-3940(95)11504-p. [DOI] [PubMed] [Google Scholar]
- 7.Mei J, Pasternak GW. Sigma 1 receptor modulation of opioid analgesia in the mouse. J Pharmacol Exp Ther. 2002;300:1070–4. doi: 10.1124/jpet.300.3.1070. [DOI] [PubMed] [Google Scholar]
- 8.Kim FJ, Kovalyshyn I, Burgman M, Neilan C, Chien CC, Pasternak GW. Sigma-1 receptor modulation of G-protein-coupled receptor signaling: potentiation of opioid transduction independent from receptor binding. Mol Pharmacol. 2010;77:695–710. doi: 10.1124/mol.109.057083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De la Puente B, Nadal X, Portillo-Salido E, Sánchez-Arroyos R, Ovalle S, Palacios G, Muro A, Romero L, Entrena JM, Baeyens JM, López-García JA, Maldonado R, Zamanillo D, Vela JM. Sigma-1 receptors regulate activity-induced spinal sensitization and neuropathic pain after peripheral nerve injury. Pain. 2009;145:294–303. doi: 10.1016/j.pain.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Entrena JM, Cobos EJ, Nieto FR, Cendán CM, Gris G, Del Pozo E, Zamanillo D, Baeyens JM. Sigma-1 receptors are essential for capsaicin-induced mechanical hypersensitivity: studies with selective sigma-1 ligands and sigma-1 knockout mice. Pain. 2009;143:252–61. doi: 10.1016/j.pain.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 11.Entrena JM, Cobos EJ, Nieto FR, Cendán CM, Baeyens JM, Del Pozo E. Antagonism by haloperidol and its metabolites of mechanical hypersensitivity induced by intraplantar capsaicin in mice: role of sigma-1 receptors. Psychopharmacology. 2009;205:21–33. doi: 10.1007/s00213-009-1513-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rho DH, Kim HW, Yoon SY, Seo HS, Kwon YB, Kim KW, Han HJ, Beitz AJ, Na HS, Lee JH. Intrathecal injection of the sigma-1 receptor antagonist BD1047 blocks both mechanical allodynia and increases in spinal NR1 expression during the induction phase of rodent neuropathic pain. Anesthesiology. 2008;109:879–89. doi: 10.1097/ALN.0b013e3181895a83. [DOI] [PubMed] [Google Scholar]
- 13.Díaz JL, Zamanillo D, Corbera J, Baeyens JM, Maldonado R, Pericàs MA, Vela JM, Torrens A. Selective sigma-1 (sigma1) receptor antagonists: emerging target for the treatment of neuropathic pain. Cent Nerv Syst Agents. Med Chem. 2009;9:172–83. doi: 10.2174/1871524910909030172. [DOI] [PubMed] [Google Scholar]
- 14.Romero L, Zamanillo D, Nadal X, Sánchez-Arroyos R, Rivera-Arconada I, Dordal A, Montero A, Muro A, Bura A, Segalés C, Laloya M, Hernández E, Portillo-Salido E, Escriche M, Codony X, Encina G, Burgueño J, Merlos M, Baeyens J, Giraldo J, López-García JA, Maldonado R, Plata-Salamán CR, Vela JM. Pharmacological properties of S1RA, a new sigma-1 receptor antagonist that inhibits neuropathic pain and activity-induced spinal sensitization. Br J Pharmacol. 2012 doi: 10.1111/j.1476-5381.2012.01942.x. Accepted Article, doi: 10.1111/j.1476-5381.2012.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vidal A, Touriño C, Romero L, Baeyens JM, Zamanillo D, Maldonado R, Vela JM. Potentiation of morphine analgesia but inhibition of the rewarding effect of morphine following co-administration of a new selective sigma-1 receptor antagonist (S1RA) Eur J Pain. 2009;13(Suppl. 1):S104. [Google Scholar]
- 16.Arasteh K, Poudevida S, Farre M, Roset PN, Cami J. Response patterns of the Spanish version of the 49-item short form of the Addiction Research Center Inventory after the use of sedatives, stimulants, and opioids. Drug Alcohol Depend. 1999;55:117–25. doi: 10.1016/s0376-8716(98)00185-9. [DOI] [PubMed] [Google Scholar]
- 17.Saletu B, Wessely P, Grünberger J, Schultes M. Erste klinische Erfahrungen mit einem neuen schlafanstoßenden Benzodiazepin, Cinolazepam, mittels eines Selbstbeurteilungsbogens für Schlaf- und Aufwachqualität (SSA) Neuropsychiatrie. 1987;1:169–76. [Google Scholar]
- 18.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research. Center for Veterinary Medicine. 2001. Guidance for industry: bioanalytical method validation. U.S. Food and Drug Administration, Center for Drug Evaluation and Research, Washington D.C. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107.pdf (last accessed 30 January 2012)
- 19.Smith B, Vandenhende F, DeSante K, Farid NA, Welch PA, Callaghan JT, Forgue ST. Confidence interval criteria for assessment of dose proportionality. Pharm Res. 2000;17:1278–83. doi: 10.1023/a:1026451721686. [DOI] [PubMed] [Google Scholar]
- 20.Smith B. Assessment of dose proportionality. In: Bonate P, Howard D, editors. Pharmacokinetics in Drug Development: Clinical Study Design and Analysis. Arlington, VA: AAPS Press; 2004. pp. 363–82. [Google Scholar]
- 21.Patterson ST, Smith B. Analysis of human pharmacokinetic data. In: Dmitrienko A, Chuang-Stein C, D'Agostino R, editors. Pharmaceutical Statistics Using SAS – A Practical Guide. Cary NC: SAS Press; 2008. pp. 197–211. [Google Scholar]