

Abstract
AIM
To evaluate the single dose pharmacokinetics, pharmacodynamics, and preliminary tolerability of the γ-secretase inhibitor BMS-708163 (avagacestat) in young and elderly men and women.
METHODS
All subjects received double-blinded administration of a single 50 mg dose of avagacestat in capsule form or matching placebo. Main evaluations included pharmacokinetics, safety, plasma amyloid-β (Aβ)1–40 concentratios and exploration of Notch biomarkers.
RESULTS
Avagacestat 50 mg capsule was well tolerated and rapidly absorbed among young and elderly subjects, with a median tmax between 1 and 2 h post dose and an average half-life between 41 and 71 h. In general, subjects aged 75 years or more had higher AUC(0,∞) values than those aged less than 75 years. An exploratory analysis of Aβ1–40 serum concentrations showed a pattern of decreasing concentrations over the first 4–6 h followed by a rise above baseline that was maintained until the end of the assessment period. Adverse events were generally mild, occurring more frequently in elderly subjects, with no observed difference between subjects receiving avagacestat and placebo. No dose limiting gastrointestinal effects of avagacestat were observed and exploratory biomarkers of Notch inhibition did not change significantly.
CONCLUSIONS
The favourable safety profile and pharmacokinetic effects of avagacestat in this study support its continued development, especially in the target population of elderly subjects with mild cognitive impairment or Alzheimer's disease.
Keywords: γ-secretase inhibitor, avagacestat, Aβ1–40, Notch biomarkers, pharmacodynamics, pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Under homeostatic conditions, sequential cleavage of amyloid precursor protein by β-site cleaving enzyme and γ-secretase forms the common Aβ isoforms, Aβ1–38, Aβ1–40, and Aβ1–42.
However, in the pathological Alzheimer's disease (AD) state, Aβ clearance is decreased nearly 30%. As such, γ-secretase is a promising drug target for AD therapy, as a decrease in its enzymatic activity has the potential to reduce Aβ levels and modify disease.
In contrast, current treatments for AD provide only short term symptomatic relief.
WHAT THIS STUDY ADDS
Unique among previous studies of γ-secretase inhibitors (GSIs), such as semagacestat and begacestat, our results showed that avagacestat, an oral GSI in clinical development for the disease-modifying treatment of AD, decreased Aβ1–40 plasma concentrations and was well-tolerated in both young and elderly subjects, with adverse events being predominantly mild to moderate in severity.
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder that affects 1 in 10 people over the age of 65 years and is characterized by a progressive decline in cognition, behaviour and activities of daily living [1]. Although the aetiology of AD is still a topic of active investigation, the preponderance of evidence suggests that deposition of amyloid-β (Aβ) in the brain is an early, initiating event in AD pathogenesis [2, 3]. Under homeostatic conditions, sequential cleavage of amyloid precursor protein (APP) by β-site cleaving enzyme and γ-secretase forms the common Aβ isoforms, Aβ1–38, Aβ1–40, and Aβ1–42. In the pathological AD disease state, Aβ clearance is decreased about 30% [4]. Thus, γ-secretase is a promising drug target for AD therapy, since a decrease in its activity has the potential to reduce Aβ concentrations and thereby modify disease. This is in contrast to current treatments that only provide short term symptomatic relief [5, 6].
Inhibition of γ-secretase also affects other substrates. Among these substrates, the Notch family of transmembrane receptors is most notable. The activity of γ-secretase on these transmembrane receptors results in the liberation of the intracellular domain of the Notch receptor, enabling it to reach the nucleus and regulate gene transcription [7]. Studies of γ-secretase inhibitors (GSIs) in animal models have shown that alterations in lymphocyte subsets and increased differentiation of intestinal goblet cells are among the Notch-dependent effects of γ-secretase inhibition. These changes can potentially be followed using decreased expression of Notch target genes such as dual specificity phosphatase 6 (DUSP6), hairy and enhancer of split 1 (HES1) [8], baculoviral inhibitor of apoptosis repeat-containing 3 (BIRC3) or increased goblet cell-specific proteins such as trefoil factor family 3 (TFF3) protein [9] as surrogates for Notch toxicity.
BMS-708163 (avagacestat) is an oral GSI designed for selective inhibition of Aβ synthesis. In vitro findings demonstrated up to 190-fold more selectivity for inhibiting APP cleavage (reducing Aβ40 than for inhibiting the cleavage of human Notch proteins) [10], and compared with semagacestat, avagacestat is 15-fold to 32-fold more selective for Aβ synthesis than for Notch processing [11]. Moreover, preclinical studies of avagacestat demonstrated reductions in plasma, brain and cerebrospinal fluid (CSF) Aβ in rats and dogs.
In this manuscript, we report the results of a double-blinded, placebo-controlled, single 50 mg dose study to assess the pharmacokinetics (PK), pharmacodynamics (PD) and safety and tolerability of avagacestat in young and elderly men and women. Study objectives also included assessment of the effect of age on PK profiles and the effects of avagacestat on cortisol concentrations, thyroid-stimulating hormone (TSH), free triiodothyronine (T3), free thyroxine (T4), lymphocyte subsets and key electrocardiogram (ECG) measurements. The same capsule dose strength across age groups was used to make these assessments. The 50 mg dose is within the linear range and is thought to be a potentially viable efficacious dose based upon data from solution dosing [12].
Methods
Subjects
Subjects included males aged 18–59 years and males and females aged 60 years or older (considered elderly for this study). Subjects were screened and eligible to enrol if they were deemed healthy, based on medical history, physical examination, 12-lead ECG, clinical laboratory evaluations, rectal examination and stool evaluation. Elderly subjects with age-related disorders such as hypertension, diabetes and dyslipidaemia were permitted to enrol, provided their condition was being treated with a stable dose of medication for at least 3 months. Subjects were excluded if they had organ system disease, surgery within 4 months of study drug administration or a history of gastrointestinal (GI) surgery that would interfere with drug absorption.
In groups aged 60 years or more, subjects with a diagnosis of mild cognitive impairment (MCI; a diagnosis of amnestic MCI according to Petersen criteria [13] and a Mini-Mental State Examination [MMSE] score of 24–30, inclusive) were allowed to enrol. In the age group ≥75 years of age, subjects with a diagnosis of AD (AD; a diagnosis of probable AD by Diagnostic and Statistical Manual of Mental Disorders, 4th edition [DSM-IV] and National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association [NINCDS-ADRDA] criteria) [14] with mild-to-moderate disease severity were allowed to enrol. However, due to recruitment issues, healthy subjects within these age groups were also allowed to enrol.
Study design
This study was conducted at three centres in the United States. Subjects entered the clinic on day −3 and were allowed to leave on day 7 (Figure 1). A total of 36 subjects were randomized to four dose panels in this study: 1 male subjects aged 18–45 years, 2 male subjects aged 46–59 years, 3 male and female subjects, aged 60–74 years, normal-healthy or subjects with mild cognitive impairment (MCI) and 4 male and female subjects aged 75 years or more, normal healthy or subjects with MCI or AD. On the morning of day 0, subjects in each panel received double-blinded administration of a single oral dose of avagacestat 50 mg in capsule form (n= 6 each for panels 1, 2, and 3 and n= 9 for panel 4) or matching placebo (n= 2 each for panels 1, 2, and 3 and n= 3 for panel 4). Subjects were allowed to leave the clinic on day 7 and were scheduled for follow-up visits to assess safety on days 14 and 28, after which they were discharged from the study.
Figure 1.
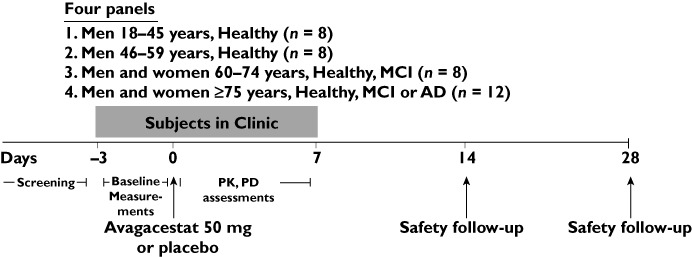
Schematic diagram of the study design. AD, Alzheimer's disease; MCI, mild cognitive impairment; PK, pharmacokinetics; PD, pharmacodynamics
The study was conducted in accordance with the principles of the Declaration of Helsinki and good clinical practice as defined by the International Conference on Harmonization. All centres obtained approval from their corresponding Institutional Review Board before beginning the study and all subjects gave written informed consent.
Analytical methods
Plasma samples were analyzed for concentration of avagacestat by a validated assay using liquid chromatography with tandem mass spectrometry (LC-MS/MS) [15]. The standard curves, which ranged from 0.1 to 100 ng ml−1 for avagacestat, were fitted to a linear regression model. The intra-assay and inter-assay precision was ≤10.5% and ≤3.8%, respectively, while the mean % deviation was ±2.2%.
PK and PD assessments
For avagacestat PK assessments, blood was obtained by an indwelling catheter or by direct venipuncture pre and post dose on day 1 (14 samples collected at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 10, 12 and 18 h), days 2 and 3 (collected at 24, 36, 48 and 60 h), and days 4–7 (collected at 72, 96, 120 and 144 h). Plasma was obtained and concentrations assayed by a validated LC-MS/MS method [15]. Blood plasma samples for assessment of Aβ1–40 concentrations were collected prior to administration of avagacestat (once on day −1 and twice on day 1 [30 min before and immediately prior to drug administration]) and up to 72 h (1, 2, 3, 4, 5, 6, 8, 12, 18, 24, 48 and 72 h) after dosing. Aβ1–40 was extracted by solid phase/high performance liquid chromatography of ethylenediaminetetraacetic acid-containing plasma and was measured using a sandwich electrochemiluminescence assay (Meso Scale Discovery, Gaithersburg, MD). At the time this study was conducted, the plasma assay for Aβ1–42 was not well developed and was therefore not characterized in this study. However, it is expected that the results for Aβ1–40 are reflective of what would be observed for Aβ1–42[16].
Safety assessments
Physical examination, vital signs, 12-lead ECG and laboratory evaluations were performed prior to dosing and at regular intervals post dosing. Measurement of 24 h urinary cortisol, salivary cortisol, and adrenocorticotrophic hormone stimulation tests were performed prior to dosing and at regular intervals post dosing. Thyroid hormone concentrations (TSH, free T3, and free T4) were measured from blood samples taken at screening and on days 1, 2, 4 and 7. Whole blood samples were obtained at screening and on days 7, 14 and 28 for immunophenotyping using flow cytometry with Tritest™ reagents or Multitest™ IMK Kits (Becton Dickinson, Franklin Lakes, NJ). Stool evaluations, including frequency (number of defaecations per day), Bristol Stool Scale [17] and tests for haemoccult blood, were performed at screening and for every stool produced while in the clinic.
PD measurement of exploratory Notch biomarkers
Blood samples were taken on day 1 immediately prior to dosing and at 4, 8 and 12 h post dose, then on the mornings of days 2, 4 and 7. Serum TFF3 was measured using a novel enzyme-linked immunosorbent assay method. Whole blood samples were collected in PAXgene® Blood RNA tubes and RNA extracted with the Blood RNA Molecular Diagnostics (MDx) Kit (Qiagen, Valencia, CA, USA) and reverse-transcribed to cDNA using the iScript™ cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). cDNA was then used for real time polymerase chain reaction (RT-PCR) with TaqMan® primers for BIRC3, DUSP6, and HES1 on the ABI Prism® 7900HT machine (Applied Biosystems, Carlsbad, CA). Moreover, for the RT-PCR analysis, the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene was designated as the experimental housekeeping control gene and its levels of expression were assessed to determine the effect of avagacestat treatment.
Statistical analyses
All statistical analyses were carried out using SAS/STAT® Version 8.2 (SAS Institute Inc., Cary, NC). All subjects who received study drug were included in the safety data set. Analyses were performed by dose group. Subjects receiving placebo were pooled into two groups by age (those aged less than 60 years and those aged 60 years or more), regardless of dose group or gender.
Pharmacokinetics
The single dose PK parameters were derived from plasma concentration vs. time data utilizing noncompartmental methods [18, 19] employing Kinetica (EP Version 2.6.1; Thermo Electron Corporation, Philadelphia, PA, USA). The terminal log linear phase of the plasma concentration vs. time curve was identified by least-squares linear regression of data points that yielded a minimum mean square error. The area under the plasma concentration–time curve from 0 to infinity (AUC(0,∞)) was determined by a combination of trapezoidal and log-trapezoidal methods plus the extrapolated area. The extrapolated area was determined by dividing the observed concentration at the time of the last nonzero plasma concentration by the slope (λz) of the terminal log-linear phase. The elimination half-life (t1/2) of the terminal log linear phase was calculated as ln2 divided by the absolute value of λz. The peak plasma concentration, Cmax, and the time at which Cmax occurred, tmax, were obtained from the observed data. Scatter plots of avagacestat Cmax and AUC(0,∞) were constructed to explore exposure differences among genders, disease states and age categories.
The PD effect of the 50 mg dose of avagacestat was assessed in three age groups (ages 46–59 years, 60–74 years, and 75 years or more). For plasma Aβ1–40 and TFF3, a repeated measurement, linear mixed effects model with fixed dose and time effects was fitted to the log-transformed percentage of baseline (%BL) values. The log-transformed BL (mean of log-transformed BLs, if multiple pre-treatment time points existed) was included in the model to adjust for the BL effect. The resulting estimates of means, adjusted for BL effect, and confidence intervals were then exponentiated back to the original scale (i.e. geometric mean of %BL) before reporting. For HES1, BIRC3 and DUSP6 gene expression levels, the GAPDH-normalized change from BL values (ΔΔCt values [20]) were modelled using a linear mixed effect model similar to the approaches considered for Aβ1–40 and TFF3. The results of gene expression were also exponentiated (with base = 2) back to %BL scale.
All drug and molecular target nomenclature conforms to the British Journal of Pharmacology's Guide to Receptors and Channels [21].
Results
Subject demographics
A total of 27 subjects received avagacestat 50 mg and nine subjects received placebo. Panels with subjects aged 18–59 years were all male, while panels with elderly subjects aged 60 years or more were both male and female. Overall, subject demographic characteristics were well-matched within subgroups except for a notably greater proportion of females than males in the age group with subjects aged 75 years or more (78% females) (Table 1).
Table 1.
Demographic characteristics
| Characteristic | Placebo | Avagacestat | Placebo | Avagacestat | ||
|---|---|---|---|---|---|---|
| 18–59 years (n= 4) | 18–45 years (n= 6) | 46–59 years (n= 6) | 60+ years (n= 5) | 60–74 years (n= 6) | 75+ years (n= 9) | |
| Age (years) mean (SD) | 36.5 (11.7) | 33.7 (6.4) | 51.0 (3.4) | 74.2 (9.5) | 63.2 (3.4) | 80.2 (3.4) |
| Male, n (%) | 4 (100) | 6 (100) | 6 (100) | 3 (60) | 4 (67) | 2 (22) |
| Race, n (%) | ||||||
| White | 3 (75) | 2 (33) | 2 (33) | 3 (60) | 3 (50) | 6 (67) |
| Black | 0 | 4 (67) | 4 (67) | 1 (20) | 0 | 2 (22) |
| Asian | 0 | 0 | 0 | 0 | 2 (33) | 1 (11) |
| Other | 1 (25) | 0 | 0 | 1 (20) | 1 (17) | 0 |
| MCI | 2 (33) | 3 (33) | ||||
| AD | 1 (20) | 2 (22) | ||||
| Weight (kg), mean (SD) | 82.1 (18.8) | 85.1 (13.9) | 83.1 (12.3) | 74.8 (13.7) | 71.6 (9.6) | 70.3 (10.3) |
| BMI (kg m−2), mean (SD) | 25.9 (3.9) | 25.8 (3.2) | 26.3 (3.4) | 25.8 (2.7) | 24.8 (3.6) | 27.2 (3.5) |
BMI, body mass index; SD, standard deviation; MCI, mild cognitive impairment; AD, Alzheimer's disease.
PK of avagacestat
Selected summary statistics for avagacestat PK parameters are presented in Table 2.
Table 2.
Summary statistics of PK parameters for avagacestat (50 mg in capsule formulation)
| Age group (years) | Cmax, ng ml−1 GM (% CV) | AUC(0,∞) ng ml−1 h GM (% CV) | tmax, h median (min, max) | t1/2, h mean (SD) |
|---|---|---|---|---|
| 18–45, (n= 6) | 149 (43) | 1202 (41) | 1.5 (1.0, 2.0) | 41.5 (10.9) |
| 46–59, (n= 6) | 251 (27) | 1693 (27) | 1.0 (1.0, 2.1) | 44.7 (11.6) |
| 60–74, (n= 6) | 175 (19) | 1701 (28) | 1.5 (1.0, 2.1) | 46.0 (14.3) |
| ≥75, (n= 9) | 224 (34) | 2688 (40) | 1.5 (1.0, 2.0) | 70.7 (19.5) |
AUC(0,∞), area under the concentration curve from time 0 extrapolated to infinite time; CV, coefficient of variation; Cmax, maximum observed concentration; GM, geometric mean; SD, standard deviation; tmax, time of maximum observed plasma concentration; t1/2, elimination half-life.
Concentration–time profiles are provided in Figure 2 and the scatter plots of avagacestat Cmax and AUC(0,∞)values vs. age as a continuous variable are shown in Figure 3. From these plots, several observations can be made: (i) there was no apparent association between age and avagacestat Cmax, (ii) the ranges of AUC(0,∞) values were similar in the younger groups (ages 18–45, 46–59 and 60–74 years) and (iii) the four females (two healthy and two MCI) in the age group of 75 years or more had the highest values of avagacestat AUC(0,∞) in the study. The remaining subjects in the age group of 75 years or more (two males and three females) had individual AUC(0,∞) values comparable with the younger subjects.
Figure 2.
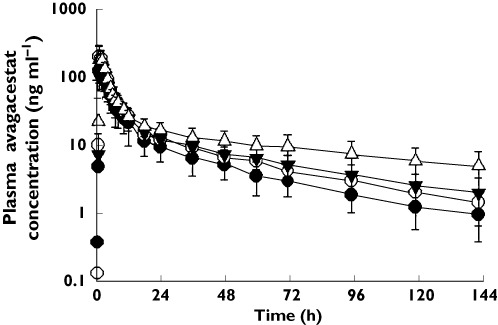
Mean (±SD) plasma concentration–time profiles of healthy young males aged 18–45 years, healthy or MCI males aged 46–59 years, healthy or MCI males aged 60–74 years, and healthy, MCI or AD male and female subjects aged 75 years or older after oral administration of a single 50 mg avagacestat dose.  , healthy young males 18–45 years;
, healthy young males 18–45 years;  , males 46–59 years;
, males 46–59 years;  , males and females 60–74 years healthy or MCI;
, males and females 60–74 years healthy or MCI;  , males and females 75 years and older healthy, MCI or AD
, males and females 75 years and older healthy, MCI or AD
Figure 3.
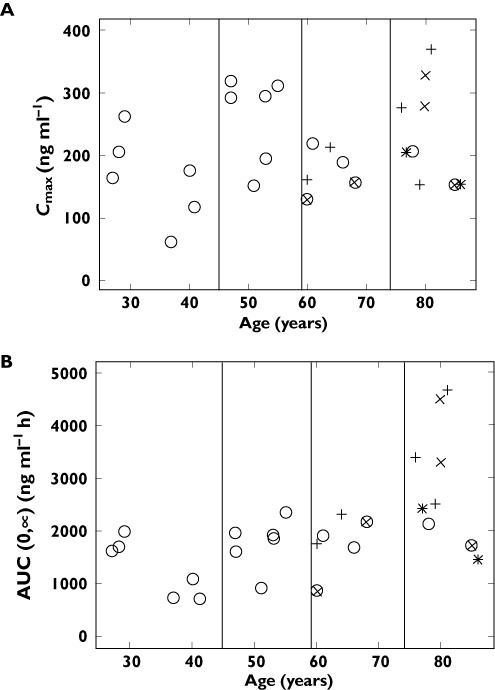
Scatter plots of avagacestat A) Cmax and B) AUC(0,∞) vs. age as a continuous variable. (○ male healthy, × male MCI, + female healthy, × female MCI, * female AD). AD, Alzheimer's disease; AUC, area under the curve; MCI, mild cognitive impairment
PD of Aβ1–40
Following treatment with avagacestat 50 mg, subjects in all three age groups experienced a short period (approximately 4–6 h) of plasma Aβ1–40 reduction which then surpassed the BL concentration and remained elevated until the end of the 72 h monitoring period. The mean reduction in plasma Aβ1–40 concentrations was up to 70–75% of their BL concentrations (Figure 4). Plasma % BL values for the five subjects with MCI and the two subjects with AD in this study were within the range of values of healthy subjects in their corresponding age categories.
Figure 4.
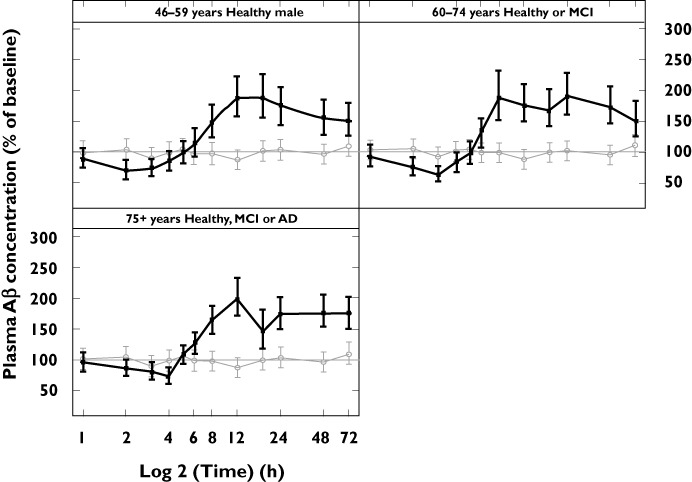
Plots of mean % of BL plasma Aβ1–40 concentrations (with 95% CI) vs. time for the healthy male group (aged 46–59 years) and two elderly groups (aged 60–74 years and 75 years or more). Aβ, amyloid-beta; CI, confidence interval.  light line placebo;
light line placebo;  dark line active
dark line active
Safety and tolerability of avagacestat
A total of 24 adverse events (AEs) occurred in 17 of 36 subjects in this study, 12 subjects (44.4%) receiving avagacestat and five subjects (55.6%) receiving placebo. AEs for the subjects reported in this study are summarized in Table 3. Most AEs (79% in the complete study) were of mild intensity. The most common AEs in the study were dizziness (n= 12 subjects [10.3%]) and headache (n= 6 [5.2%]). The number of AEs was increased in elderly subjects relative to other age groups, but AE frequency was similar between elderly subjects receiving placebo and those receiving avagacestat. Three serious AEs, all deemed unrelated to study medication, occurred during the study: haemorrhagic stroke in a male aged 77 years who had received one dose of placebo, Merkel's carcinoma in a female aged 80 years diagnosed 27 days after taking avagacestat (however, her skin lesion existed for about 1 year prior to the study) and adhesive small bowel obstruction in a female aged 74 years during the screening period prior to randomization to treatment. There were no deaths in this study. Two AEs led to study discontinuation. One was the serious AE of haemorrhagic stroke and the other was a mild-intensity AE of occult blood positive stool that lasted 3 days and resolved without treatment.
Table 3.
Adverse events with avagacestat or placebo
| Events, n | Placebo | Avagacestat | Placebo | Avagacestat | Total placebo (n= 9) | Total Avagacestat (n= 27) | ||
|---|---|---|---|---|---|---|---|---|
| 18–59 years (n= 4) | 18–45 years (n= 6) | 46–59 years (n= 6) | ≥60 years (n= 5) | 60–74 years (n= 6) | ≥75 years (n= 9) | |||
| Total subjects with AEs | 1 | 2 | 1 | 4 | 3 | 6 | 5 | 12 |
| Total AEs | 2 | 2 | 1 | 9 | 4 | 6 | 11 | 13 |
| System organ class preferred term | ||||||||
| General disorders and administration site conditions | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Gastrointestinal disorders | ||||||||
| Constipation | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Infections and infestations | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 |
| Investigations (increases in ALT, AST, blood CPK and LDH, and transaminases); occult blood positive | 2 | 2 | 0 | 0 | 0 | 0 | 2 | 2 |
| Muscoskeletal and connective tissue disorders | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Nervous system disorders | ||||||||
| Dizziness | 0 | 0 | 1 | 2 | 1 | 0 | 2 | 2 |
| Headache | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Haemorrhagic stroke | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| Paresthesia | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| Renal and urinary disorders | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 |
| Skin and subcutaneous tissue disorders | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| Unspecified | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Vascular disorders | 0 | 0 | 0 | 2 | 2 | 0 | 2 | 2 |
AEs, adverse events; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CPK, creatine phosphokinase; LDH, lactate dehydrogenase.
GI AEs, stool evaluations and exploratory biomarkers of Notch toxicity
Because of the mechanism of action of avagacestat, GI AEs were considered of clinical interest. Diarrhoea occurred in a female aged 64 years who received placebo, and constipation occurred in a female aged 76 years who received avagacestat. The events were unlikely to be related to study medication and resolved without treatment. Stool evaluations indicated that avagacestat had no notable effect on stool frequency or consistency. Occult blood positive stools were seen in two young subjects who received avagacestat 50 mg. A male aged 37 years experienced positive stools for 3 days and discontinued from the study, though the AE was considered mild and unlikely to be related to study drug. In addition, a male aged 40 years experienced one mild AE of positive haemocult stool that was considered unrelated to study drug.
To examine potential Notch inhibition by avagacestat, subjects were monitored for decreases in RNA levels of Notch target genes BIRC3, DUSP6, HES1 and for increases in TFF3 protein (Figures 5A–D). There was no apparent change in these biomarkers in avagacestat-treated subjects relative to placebo.
Figure 5.
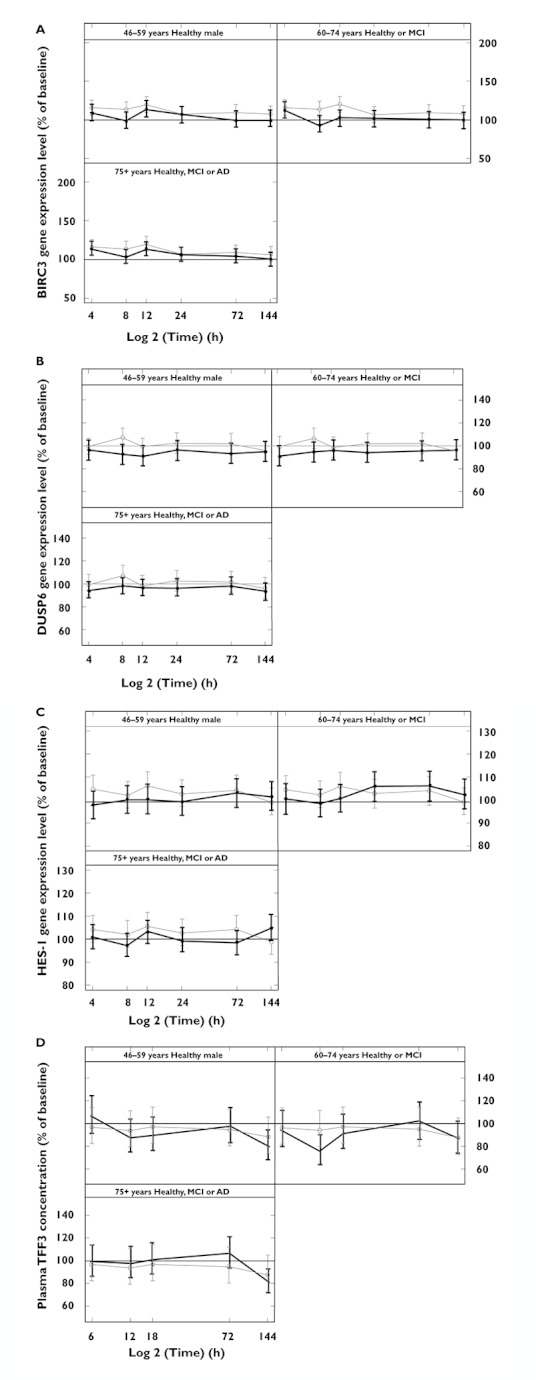
Plots of mean % (with 95% CI) of BL of exploratory biomarkers of Notch toxicity: (A) BIRC3, (B) DUSP6, (C) HES1 and (D) TFF3 vs. time for the healthy male group (aged 46–59 years) and two elderly groups (aged 60–74 years and 75 years or more). AD, Alzheimer's disease; BIRC3, baculoviral IAP repeat-containing protein 3; DUSP6, dual specificity phosphatase 6; HES1, hair and enhancer of split 1; MCI, mild cognitive impairment; TFF3, trefoil factor 3.  light line placebo;
light line placebo;  dark line active
dark line active
ECGs and vital signs
Two male subjects (aged 60 and 66 years) had orthostatic hypotension, both AEs were considered to be related to avagacestat. No clinically relevant effects of avagacestat were observed for ECG parameters. The lone clinically relevant ECG abnormality (a ventricular extrasystole in a Black male aged 35 years) was experienced after receiving placebo.
Laboratory parameters, cortisol, thyroid function and lymphocytes
There were 53 laboratory marked abnormalities (MAs), pre-defined levels greater than the normal concentration range, that occurred during the study. The most common MAs were elevated urine white blood cells (seven [53.8%] avagacestat, three [75%] placebo) and elevated triglycerides (seven [33%] avagacestat, none placebo). No clinically relevant trends were observed for the incidence of any MA. Administration of avagacestat did not cause any clinically relevant changes in salivary or 24 h cortisol concentrations, cortisol after adrenocorticotrophic hormone stimulation, TSH, T3 or T4 concentrations. Avagacestat did not have any clinically relevant effect on leucocyte counts, lymphocyte counts, B cells, T cells, natural killer cells, monocytes, neutrophils, eosinophils or basophils.
Discussion
Single dose avagacestat was well-tolerated in this study by both young and elderly subjects, some of whom had diagnoses of MCI or AD. Avagacestat was rapidly absorbed, with a median tmax between 1 and 2 h post dose and its average half-life was between 41 and 71 h. While sampling out to 144 h is more than three half-lives in the younger subjects, it is only two half-lives in the elderly. However, as can be seen from Figure 2, the terminal phase is very linear and the authors believe this is still a good estimate of half-life in these subjects. The PK profile of avagacestat indicated somewhat higher AUC(0,∞) values for subjects aged 75 years or more compared with those aged less than 75 years. This, however, may be due to the four females in the age group of 75 years or more (two healthy and two MCI) who had the highest values of avagacestat AUC(0,∞) in the study. It is possible that the elderly females had a smaller volume of distribution (perhaps due to less fat depot, for example) which could yield higher peak and overall exposures than that of males. However, the ranges of Cmax and AUC(0,∞) values were similar in the younger groups (aged 18–45, 46–59 and 60–74 years). Also, although the numbers are small, Figure 3 suggests that subjects diagnosed with MCI or AD did not have exposures different from several of the normal healthy subjects, as the Cmax values in these subjects were similar to healthy subjects who were aged less than 60 years and the subjects with AD aged 75 years or more had exposures that were comparable with two of the elderly healthy female subjects in the same age group.
In the 46–59 years, 60–74 years, and 75 years or more age categories, an exploratory analysis of Aβ1–40 concentrations in serum showed a pattern of decreasing concentrations over the first 4–6 h followed by a rise above BL that was maintained until the end of the 72 h assessment period. This in vivo profile of plasma Aβ kinetics mirrors that of many GSIs observed in vitro and in animal models [22]. Studies in animal models have also shown no increase in the concentrations of CSF Aβ, as avagacestat decreased brain and CSF Aβ concentrations in a clinically relevant manner (CF Albright et al., unpublished data). AEs were generally mild and occurred more frequently in elderly subjects, though there was no difference in this age group between subjects on avagacestat and those on placebo. No dose-limiting GI effects of avagacestat were seen in this study. There was no age-related effect on GI AEs and avagacestat had no effect on stool frequency or consistency. Exploratory biomarkers of Notch inhibition did not change significantly, suggesting that single dose avagacestat 50 mg has no effect on Notch.
Because this study was small and short term, and because only a single dose of avagacestat was administered, these findings should be interpreted with caution. In addition, the elderly subject population studied included healthy subjects and those having active AD. Despite these caveats, the favourable safety profile and PK effects of avagacestat in this trial support its further study, particularly in the target population of elderly patients with MCI or AD.
Acknowledgments
The authors acknowledge with deep appreciation all the volunteers and investigative teams (Bristol-Myers Squibb Clinical Pharmacology Unit, Hamilton, NJ, Seaview Research, Inc., Miami, FL and California Clinical Trials Medical Center, Glendale, CA) who participated in this clinical trial. They recognize the efforts of the principal investigators and their clinical staff. Medical writing and editorial support was provided by StemScientific and funded by Bristol-Myers Squibb.
Competing Interests
All authors were employees of Bristol-Myers Squibb, sponsor of the study, at the time when this work was done. The following authors own in excess of $10 000 of company stock: GT, S-P H, RC, LC, RB, CS and CA. Furthermore, the following authors are inventors on patents related to the subject matter: OW and CA. All authors report no other conflicts of interest, including consultancies, honoraria and paid expert testimony.
REFERENCES
- 1.Alzheimer's Association. 2010. Alzheimer's disease facts and figures. Available at http://www.alz.org/documents_custom/report_alzfactsfigures2010.pdf (last accessed 6 February 2012)
- 2.Morris JC, Price AL. Pathologic correlates of nondemented aging, mild cognitive impairment, and early stage Alzheimer's disease. J Mol Neurosci. 2001;17:101–18. doi: 10.1385/jmn:17:2:101. [DOI] [PubMed] [Google Scholar]
- 3.Kinkingnéhun S, Sarazin M, Lehéricy S, Guichart-Gomez E, Hergueta T, Dubois B. VBM anticipates the rate of progression of Alzheimer disease: a 3-year longitudinal study. Neurology. 2008;70:2201–11. doi: 10.1212/01.wnl.0000303960.01039.43. [DOI] [PubMed] [Google Scholar]
- 4.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogers SL, Doody RS, Pratt RD, Ieni JR. Long-term efficacy and safety of donepezil in the treatment of Alzheimer's disease: final analysis of a US multicentre open-label study. Eur Neuropsychopharmacol. 2000;10:195–203. doi: 10.1016/s0924-977x(00)00067-5. [DOI] [PubMed] [Google Scholar]
- 6.Rafii MS, Aisen PS. Recent developments in Alzheimer's disease therapeutics. BMC Med. 2009;7:7. doi: 10.1186/1741-7015-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.LaVoie MJ, Selkoe DJ. The Notch ligands, Jagged and Delta, are sequentially processed by alpha-secretase and presenilin/gamma-secretase and release signaling fragments. J Biol Chem. 2003;278:34427–37. doi: 10.1074/jbc.M302659200. [DOI] [PubMed] [Google Scholar]
- 8.Weerkamp F, Luis TC, Naber BA, Koster EE, Jeannotte L, van Dongen JJ, Staal FJ. Identification of Notch target genes in uncommitted T-cell progenitors: no direct induction of a T-cell specific gene program. Leukemia. 2006;20:1967–77. doi: 10.1038/sj.leu.2404396. [DOI] [PubMed] [Google Scholar]
- 9.Taupin D, Podolsky DK. Trefoil factors: initiators of mucosal healing. Nat Rev Mol Cell Biol. 2003;4:721–32. doi: 10.1038/nrm1203. [DOI] [PubMed] [Google Scholar]
- 10.Meredith J, Jr, Albright CF, Dockens RC, Olson RE, Lentz KA, Wang J-S, Denton RR, Pilcher G, Zaczek R, Macor JE, Houston J, Wong O, Gu H, Berman RM, Tong G. 2011. BMS-708163, a notch-sparing GSI, decreases central Aβ in rats, dogs, and humans with a therapeutic margin relative to notch toxicity. Presented at the 10th International Conference on Alzheimer's and Parkinson's Diseases. 9–13 March 2011, Barcelona, Spain.
- 11.Martone RL, Zhou H, Atchison K, Comery T, Xu JZ, Huang X, Gong X, Jin M, Kreft A, Harrison B, Mayer SC, Aschmies S, Gonzales C, Zaleska MM, Riddell DR, Wagner E, Lu P, Sun SC, Sonnenberg-Reines J, Oganesian A, Adkins K, Leach MW, Clarke DW, Huryn D, Abou-Gharbia M, Magolda R, Bard J, Frick G, Raje S, Forlow SB, Balliet C, Burczynski ME, Reinhart PH, Wan HI, Pangalos MN, Jacobsen JS. Begacestat (GSI-953): a novel, selective thiophene sulphonamide inhibitor of amyloid precursor protein gamma-secretase for the treatment of Alzheimer's disease. J Pharmacol Exp Ther. 2009;331:598–608. doi: 10.1124/jpet.109.152975. [DOI] [PubMed] [Google Scholar]
- 12.Tong G, Wang J-S, Sverdlov O, Huang S-P, Slemmon R, Croop R, Castaneda L, Gu H, Wong O, Li H, Berman RM, Smith C, Albright CF, Dockens R. Multicenter, randomized, double-blinded, placebo-controlled single ascending-dose study of the oral gamma-secretase inhibitor BMS-708163 (avagacestat): tolerability profile, pharmacokinetics, and pharmacodynamics. Manuscript in press. Clin Ther. 2012;34:654–67. doi: 10.1016/j.clinthera.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 13.Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56:303–8. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 14.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 15.Gu H, Deng Y, Wang J, Aubry AF, Arnold ME. Development and validation of sensitive and selective LC-MS/MS methods for the determination of BMS-708163, a gamma-secretase inhibitor, in plasma and cerebrospinal fluid using deprotonated or formate adduct ions as precursor ions. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:2319–26. doi: 10.1016/j.jchromb.2010.06.041. [DOI] [PubMed] [Google Scholar]
- 16.Oh ES, Mielke MM, Rosenberg PB, Jain A, Fedarko NS, Lyketsos CG, Mehta PD. Comparison of conventional ELISA with electrochemiluminescence technology for detection of amyloid-β in plasma. J Alzheimers Dis. 2010;21:769–73. doi: 10.3233/JAD-2010-100456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewis SJ, Heaton KW. Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol. 1997;32:920–4. doi: 10.3109/00365529709011203. [DOI] [PubMed] [Google Scholar]
- 18.Gibaldi M, Perrier D. In: Noncompartmental Analysis Based on Statistical Moment Theory, in Pharmacokinetics. 2nd edn. New York: Marcel Dekker; 1982. pp. 409–17. [Google Scholar]
- 19.Riegelman S, Collier P. The application of statistical moment theory to the evaluation of in vivo dissolution time and absorption time. J Pharmacokinet Biopharm. 1980;8:509–34. doi: 10.1007/BF01059549. [DOI] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 21.Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burton CR, Meredith JE, Barten DM, Goldstein ME, Krause CM, Kieras CJ, Sisk L, Iben LG, Polson C, Thompson MW, Lin XA, Corsa J, Fiedler T, Pierdomenico M, Cao Y, Roach AH, Cantone JL, Ford MJ, Drexler DM, Olson RE, Yang MG, Bergstrom CP, McElhone KE, Bronson JJ, Macor JE, Blat Y, Grafstrom RH, Stern AM, Seiffert DA, Zaczek R, Albright CF, Toyn JH. The amyloid-beta rise and gamma-secretase inhibitor potency depend on the level of substrate expression. J Biol Chem. 2008;283:22992–3003. doi: 10.1074/jbc.M804175200. [DOI] [PubMed] [Google Scholar]