

Abstract
AIM
Propylene glycol (PG) is often applied as an excipient in drug formulations. As these formulations may also be used in neonates, the aim of this study was to characterize the pharmacokinetics of propylene glycol, co-administered intravenously with paracetamol (800 mg PG/1000 mg paracetamol) or phenobarbital (700 mg PG/200 mg phenobarbital) in preterm and term neonates.
METHODS
A population pharmacokinetic analysis was performed based on 372 PG plasma concentrations from 62 (pre)term neonates (birth weight (bBW) 630–3980 g, postnatal age (PNA) 1–30 days) using NONMEM 6.2. The model was subsequently used to simulate PG exposure upon administration of paracetamol or phenobarbital in neonates (gestational age 24–40 weeks).
RESULTS
In a one compartment model, birth weight and PNA were both identified as covariates for PG clearance using an allometric function (CLi= 0.0849 × {(bBW/2720)1.69× (PNA/3)0.201}). Volume of distribution scaled allometrically with current bodyweight (Vi= 0.967 × {(BW/2720)1.45}) and was estimated 1.77 times higher when co-administered with phenobarbital compared with paracetamol. By introducing these covariates a large part of the interindividual variability on clearance (65%) as well as on volume of distribution (53%) was explained. The final model shows that for commonly used dosing regimens, the population mean PG peak and trough concentrations range between 33–144 and 28–218 mg l−1 (peak) and 19–109 and 6–112 mg l−1 (trough) for paracetamol and phenobarbital formulations, respectively, depending on birth weight and age of the neonates.
CONCLUSION
A pharmacokinetic model was developed for PG co-administered with paracetamol or phenobarbital in neonates. As such, large variability in PG exposure may be expected in neonates which is dependent on birth weight and PNA.
Keywords: neonates, population pharmacokinetics, propylene glycol, toxicity
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Propylene glycol is commonly used as an excipient in dose forms and is ingested by neonates when administering different drugs.
While propylene glycol is generally considered to be safe, toxic effects like bradycardia, lactic acidosis and convulsions have been reported.
Information on the pharmacokinetics of propylene glycol in neonates is lacking to provide insight into the possible risk of toxicity.
WHAT THIS STUDY ADDS
This study describes the pharmacokinetics of propylene glycol in preterm and term neonates co-administered with paracetamol and phenobarbital.
A pharmacokinetic model was developed which identified birth weight and postnatal age as important covariates for clearance.
The model was used to simulate exposure to propylene glycol co-administered with both drugs.
Introduction
Since a substantial number of drugs have poor solubility or stability, excipients are often needed. Propylene glycol (PG) is a frequently applied cosolvent to increase the solubility and/or stability of several drugs like, for example, phenobarbital, paracetamol, lopinavir, ritonavir or lorazepam, compounds which are also often administered to neonates [1]. Although PG is generally regarded as safe, concentration related toxicity has been reported in the adult, paediatric and neonatal population and may involve bradycardia, depression of the central nervous system, increase in anion gap, lactic acidosis, hepatic dysfunction or kidney injury [1–4].
Little is known about the pharmacokinetics of PG in children. In adults, it has been described that approximately 45% of the administered dose of PG is eliminated through the kidney. The other 55% is metabolized through alcohol dehydrogenase in the liver to lactate and pyruvate and eventually to carbon dioxide and water [5–7]. While the elimination half-life of PG is estimated to be 2–5 h in adults [2, 8], prolonged elimination half-lives of 10.8–30.5 h have been reported in preterm neonates (<1.5 kg) [5, 9]. In particular neonates and infants are therefore potentially at increased risk for toxic effects due to a more pronounced PG exposure [10]. In spite of this, current guidelines on the use of PG in drugs or food are limited and conflicting. Although the Food and Drug Administration (FDA) as well as the European Medicine Agency (EMA) have developed guidelines concerning the safe use of PG, these guidelines vary largely between these agencies. The FDA established an acceptable daily intake of PG of 25 mg kg−1 bodyweight. The EMA proposed a maximum daily dose of 400 mg kg−1 for adults and 200 mg kg−1 for children [11]. This discordance in the different guidelines reflects the lack of information on the safe use of PG in general, and of specific advice for the paediatric and neonatal age ranges in particular.
To date, to our best knowledge, no pharmacokinetic studies on PG have been performed in children nor in the full spectrum of neonates. Only a limited number of paediatric reports, exploring possible toxic effects of PG, are available [3, 12–15]. In this perspective, it is of relevance that the FDA recently warned of serious health problems in premature neonates receiving Kaletra®, which contains a combination of lopinavir and ritonavir dissolved in ethanol (356.3 mg ethanol ml−1) and PG (152.7 mg ml−1). Adverse events such as cardiac, renal and respiratory problems were reported in premature neonates, likely due to a decreased ability to eliminate either ethanol, PG or both [16, 17].
Because of the conflicting guidelines and observations on the (in)tolerabililty to PG in neonates, the aim of this study was to characterize the pharmacokinetics of PG, when co-administered with intravenous paracetamol or phenobarbital in preterm and term neonates.
Methods
Patients
This pharmacokinetic analysis was based on observations collected in 68 (pre)term neonates from a previously published study [1] evaluating short term clinical and biochemical tolerability to PG co-administered with intravenous paracetamol (Paracetamol Sintetica, Mendrisio, Italy) containing 800 mg PG per 1000 mg paracetamol solution or intravenous phenobarbital (Luminal Injektionlösung, Desitin Arzneimittel, Hamburg, Germany) containing 700 mg PG per 200 mg of phenobarbital. The study was conducted at the University Hospitals Leuven (Belgium) at the neonatal intensive care unit following approval by the local ethical board (B-32220084836) and study registration (PARANEO, EUdraCT 2009-011243-39, http://www.clinicaltrials.gov). Neonates were included after informed written parental consent. The decision to prescribe a source of intravenous PG, either paracetamol-PG or phenobarbital-PG, was made by the attending physician and based on the clinical needs. For paracetamol, a loading dose of 20 mg kg−1 was given, followed by a maintenance dose of 5–10 mg kg−1 every 6 h, depending on postmenstrual age [1]. For phenobarbital, a loading dose of 20 mg kg−1 phenobarbital was given, followed by a maintenance dose of 5 mg kg−1 day−1[18]. The number of samples in every individual neonate ranged from 1 to 11 collected between 20 min until 20.5 h after dose administration. Six patients were considered as outliers due to unexplainably high concentrations of PG, likely caused by analytical interferences after visual inspection of the individual chromatographies. The clinical characteristics of the included patients (n= 62) are summarized in Table 1.
Table 1.
Clinical characteristics of the patients, receiving propylene glycol co-administered with paracetamol, phenobarbital or both, presented as median (range)
| Characteristics | Paracetamol | Phenobarbital | Paracetamol + Phenobarbital |
|---|---|---|---|
| Number of patients | 34 | 25 | 3 |
| Gestational age (weeks) | 38 (24–41) | 34 (27–40) | 36 (35–37) |
| Postmenstrual age (weeks) | 38 (25–41) | 34 (28–46) | 36 (35–37) |
| Postnatal age (days) | 3 (1–28) | 2 (1–82) | 3 (2–5) |
| Birth weight (g) | 2990 (630–3820) | 1965 (815–3980) | 2490 (2245–2514) |
| Current bodyweight (g) | 2990 (700–4100) | 1965 (780–3980) | 2435 (2145–2490) |
Birth weight, weight at day of birth; current bodyweight, weight at day of blood sampling.
Analytical assay
PGconcentrations were determined by high performance liquid chromatography with photodiode array detection described by Kulo et al. [19]. The developed accurate, specific, sensitive and rapid method was validated for quantification of PG in low volume neonatal plasma (15–46 mg l−1) and urine (20–175 mg l−1). Samples with concentrations higher than this were re-analyzed after dilution until they fell within the calibration range. The inter-assay and intra-assay precision was between 8.1–14.1% and 2.3–12.7%, respectively, while the lower limit of quantification was 0.25 mg l−1.
Population pharmacokinetic analysis and model evaluation
The population pharmacokinetic analysis was performed using the non-linear mixed effect modeling software NONMEM version 6.2. (Globomax LLC, Hanover, MD, USA). S-Plus, PsN and R were used for visualization and evaluation of the models. Development of the model was performed in four different steps: (i) choice of the structural model, (ii) choice of the statistical sub-model, (iii) covariate analysis and (iv) model evaluation. The descriptive and predictive performance between different models was evaluated by different diagnostic tools [20]. A decrease in objective function (OFV) of 3.9 points or more was considered as a statistically significant difference (P < 0.05 based on X2 distribution) for structural and statistical models while a more stringent P value of 0.005 was used for the evaluation of covariate models. In addition, goodness-of-fit plots, including observed vs. individual predicted, observed vs. population predicted, conditional weighted residuals vs. time and conditional weighted residuals vs. population predicted, were used for diagnostic purposes. Furthermore, the total number of parameters, visual improvement of individual plots, confidence intervals of parameter estimates and correlation matrix were assessed as diagnostic criteria during model development. Finally, ill-conditioning [21] and shrinkage [22], which may occur in paediatric analyses [20], were determined.
Structural model
A one and two compartment model was fitted to the data. The interindividual variability in the pharmacokinetic parameters was assumed to follow a log normal distribution. The value of a particular parameter in an individual i (post hoc value) is given by the following equation:
| (Equation 1) |
in which θTV is the typical value of the parameter and ηi is assumed to be a random variable with mean value zero and variance ω2. The residual variability was best described by a proportional error model. This means for the jth observed concentration of the ith individual the relation (Yij):
| (Equation 2) |
where Cpred is the predicted concentration and εij is a random variable with a mean of zero and a variance of σ2.
Covariate analysis
To visualize potential relationships between covariates and parameter estimates, plots of the individual post hoc parameter estimates and weighted residuals vs. covariates were generated. The following covariates were evaluated: gestational age, postmenstrual age, postnatal age (PNA), birth weight (weight at day of birth) and current bodyweight (weight at day of blood sampling). Potential covariates were implemented into the model using a linear or allometric equation (equation 3).
| (Equation 3) |
In this equation Pi represents the individual parameter estimate of the ith subject, Pp equals the population parameter estimate, Cov is the covariate and k is the exponent which was fixed to 1 for a linear function or estimated for an allometric function.
Covariates were separately implemented into the model and considered statistically significant when the OFV decreased with at least 7.8 points (P value <0.005). When more than one covariate significantly reduced the OFV, the covariate causing the largest drop in OFV was left into the model. Additional covariates had to reduce this OFV further to be retained in the model. Subsequently, the contribution of each covariate was re-evaluated in the backward deletion for which a more stringent P value <0.001 (OFV 10.83 points) was used. To select the final covariate model, the individual and population predicted values were plotted against the most predictive covariate to evaluate whether the individual predicted parameters were equally distributed around the population predicted parameters [20]. The covariate model was further evaluated as discussed previously in the section ‘population pharmacokinetic analysis’. Finally, the results of the model validation procedure (see below) were also considered.
Internal validation
For the internal validation of the final pharmacokinetic model, two different evaluation tools were used. The first method was the bootstrap resampling method to evaluate model precision and stability. The bootstrap analysis was performed in S-plus, version 6.2.1 (Insightful software, Seattle, WA) with NM.SP.interface version 05.03.01 (© by LAP&P Consultants BV, Leiden, the Netherlands) in which 1000 replicates were generated. Parameter estimates obtained in the bootstrap analysis were compared with the parameter estimates of the original dataset.
For the second internal evaluation method, the normalized prediction distribution error method (NPDE) was used, which is a simulation-based diagnostic to determine the accuracy of the model [23, 24]. The observed and simulated concentrations were compared using the NPDE package in R. A histogram of the NPDE distribution and scatterplots showing the NPDE vs. time and vs. predicted concentration were used to evaluate the final model.
Model-based simulations for PG co-administered with paracetamol or phenobarbital
Using the final PK model, simulations were performed in three different patients (birth weight 630 g, 1500 g and 3500 g and gestational age 24, 32 and 40 weeks) with a PNA of 1 and 28 days. The current bodyweight at a PNA of 28 days was 950 g, 1950 g and 4100 g, respectively. These three patients were selected to cover the entire population of the current study in terms of gestational age and body weight. The parameter estimates obtained in the final pharmacokinetic model were used to simulate concentrations of PG after administration of intravenous paracetamol (Paracetamol Sintetica, Mendrisio, Italy: 800 mg PG/1000 mg paracetamol) or intravenous phenobarbital (Luminal Injektionlösung, Desitin Arzneimittel, Hamburg, Germany: 700 mg PG/200 mg phenobarbital) in the dosing regimens applied in this study. For paracetamol, a loading dose of 20 mg kg−1 was given, followed by a maintenance dose of 10 mg kg−1 every 6 h [1]. For phenobarbital, a loading dose of 20 mg kg−1 phenobarbital was given, followed by a maintenance dose of 5 mg kg−1 day−1[18].
Maximally acceptable concentrations of PG in neonates
Different approaches were applied to provide a basis for maximally acceptable concentrations of PG in neonates. First, the exposure to PG upon administration of PG as a result of paracetamol or phenobarbital was compared with concentrations observed in a previously published study in 68 preterm and term neonates in which tolerability of PG was evaluated and no toxic effects were reported [1]. In a second approach, a maximum concentration was defined on basis of the toxic effects related to the osmolar changes. The increase in osmolar gap can be directly linked to PG concentrations by the following relationship [2]: [osmolar gap = concentration of PG (mg dl−1)/7.6] while osmolar gap is considered the first indicator of PG accumulation before PG toxicity appears related to other metabolic disturbances or clinical symptoms [6]. In a study of Yahwak et al. [25] in adults, an increase in osmolar gap of 10 mOsm l−1 was linked to elevated PG concentrations and an increase of 12 mOsm l−1 resulted in clinical changes suggestive of PG toxicity. Furthermore, in studies by Feldman et al. [26] and Giacoia et al. [27], a standard deviation of 8 mOsm l−1 in serum osmolality has been described in neonates. Based on these observations, we considered the maximum allowed PG plasma concentration to remain below 608 mg l−1, which corresponds to a maximum change in osmolar gap of 8 mOsm l−1. The proposed maximum concentration of 608 mg l−1 is in close agreement with previously published results by Wilson et al. [6] in which metabolic abnormalities were reported for concentrations ranging between 580 and 1270 mg l−1[6]. However our proposed maximum concentration of PG of 608 mg l−1 should be viewed with caution since it is only based on findings reported in the literature for adult patients. It is therefore not validated in neonates. Finally, a third possible maximum safe concentration was identified by performing simulations based on the guidelines for PG administration in children established by the EMA (200 mg kg−1 day−1) and the FDA (25 mg kg−1 day−1). To the very best of our knowledge, these guidelines are not supported by observational data. In these simulations 100 mg or 12.5 mg of PG depending on the guidelines by the EMA or FDA, respectively, was administered to three different neonates (bBW 630 g, 1500 g and 3500 g) every 12 h since drugs containing PG are often given in this manner in clinical practice in neonates. It was simulated to be given by a bolus injection over 15 min to illustrate the highest potential exposure to PG.
Results
Patients
The pharmacokinetic analysis was based on 372 observations obtained from 62 neonates. The number of samples taken per neonate ranged between 1–11. Thirty-four neonates received PG by intravenous administration of paracetamol compared with 25 neonates who received phenobarbital while three neonates received a combination of both paracetamol and phenobarbital. Patient characteristics are summarized in Table 1.
Structural pharmacokinetic model
A one compartment model parameterized in terms of clearance and volume of distribution with a proportional error model best described the plasma concentrations of PG.
Covariate analysis
In the systematic covariate analysis, birth weight was found the most important covariate for clearance causing a drop in OFV of 82 points (P < 0.001). Birth weight was best implemented on clearance using an allometric function in which a value of 1.69 was estimated for the exponent. When evaluating other covariates, current weight was found the most important covariate for volume of distribution using an allometric function with an estimated exponent of 1.48 (ΔOFV 48 points, P < 0.001). Furthermore, a significant difference in volume of distribution was seen between neonates receiving phenobarbital and paracetamol. The volume of distribution was estimated to be 1.77 times higher (95% confidence interval 1.35, 2.19) for neonates receiving phenobarbital (ΔOFV 18 points, P < 0.001). Finally, further improvement of the model fit was seen when PNA was introduced on clearance using an allometric function with an estimated exponent of 0.201. This last covariate was responsible for the smallest but still significant drop in the objective function (ΔOFV = 15 points, P < 0.001). All parameter estimates of the final pharmacokinetic model are summarized in Table 2. The diagnostic plots are represented in Figure 1. By introducing these covariates a large part of the interindividual variability on clearance (65%) as well as on volume of distribution (53%) is explained (Table 2). This is reflected by the estimates of interindividual variability in clearance and volume of distribution which were reduced from 0.69 to 0.12 and 0.64 to 0.18, respectively.
Table 2.
Model-based population pharmacokinetic parameter estimates and the values obtained after the bootstrap analysis
| Parameter | Simple model without covariates | Final pharmacokinetic covariate model | Bootstrap final pharmacokinetic model |
|---|---|---|---|
| Value (CV%) | Value (CV%) | Value (CV%) | |
| Fixed effects | |||
| CL (l h−1) = CLp | 0.060 (11.8) | – | – |
| CLp in CL = CLp×(bBW/median)m×(PNA/median)n | – | 0.085 (4.9) | 0.085 (5.24) |
| m | – | 1.69 (10.2) | 1.68 (11.44) |
| n | – | 0.20 (31.9) | 0.20 (37.62) |
| V (l) =Vp | 0.90 (10.2) | – | – |
| Vp in V=Vp×(cBW/median)o×p | – | 0.97 (6.58) | 0.97 (7.05) |
| O | – | 1.45 (10.4) | 1.45 (11.28) |
| P (phenobarbital) | – | 1.77 (12.1) | 1.79 (13.10) |
| Interindividual variability (ω2) | |||
| ω2 (CL) | 0.69 (23.9) | 0.12 (26.3) | 0.11 (30.91) |
| ω2 (V) | 0.64 (23.9) | 0.18 (25.6) | 0.17 (27.99) |
| Residual variability | |||
| σ2 (proportional) | 0.036 (12.1) | 0.036 (11.8) | 0.036 (11.40) |
CL, Clearance; CLp, population value for clearance; V, Volume of distribution; Vp, population value for volume; bBW, bodyweight at birth; cBW, current bodyweight; PNA, postnatal age; m, exponential scaling factor for bodyweight at birth; n, exponential scaling factor for PNA; o, exponential scaling factor for current bodyweight; p, increasing factor in volume of distribution of PG when administered with phenobarbital.
Figure 1.
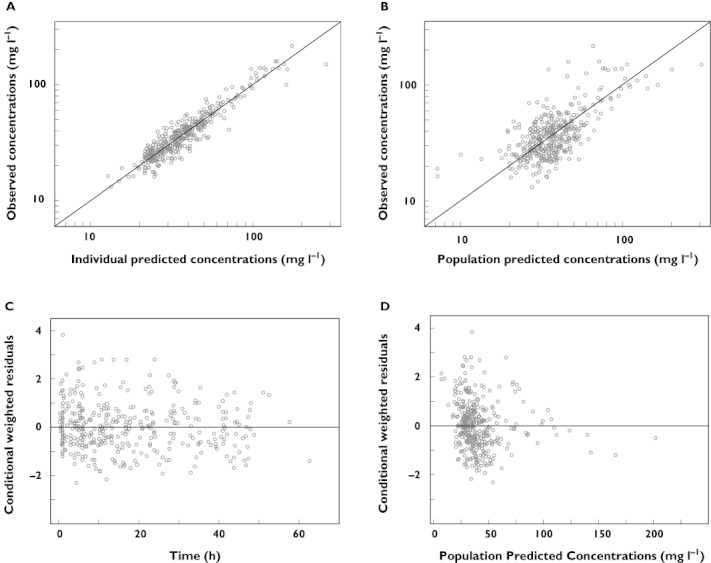
Diagnostic plots for the final pharmacokinetic model. (A) Observed vs. individual predicted concentrations, (B) observed vs. population predicted concentrations, (C) conditional weighted residuals vs. time and (D) conditional weighted residuals vs. population predicted concentrations
Model validation
The values for the parameter estimates obtained during the bootstrap procedure are shown in Table 2. The parameter estimates obtained after bootstrapping were within 8% of the values obtained in the final pharmacokinetic model. Of the total number of runs (n= 1000), 100% were successful and only 34 runs did not have a covariance step.
The results of the NPDE analysis are depicted in Figure 2. The histogram follows the normal distribution indicated by the black solid line (Figure 2A). No trend is seen in the NPDE vs. time (Figure 2B) and the NPDE vs. predicted concentrations (Figure 2C). The plot with the individual predicted parameter estimates and population parameter estimates for clearance and volume of distribution vs. the most predictive covariate, birth weight and current body weight respectively, showed that the individual predicted parameter estimates are randomly scattered around the population parameter estimates (figures not provided). The number of ill-conditioning (8.28) was far below the critical value of 1000 meaning that the final pharmacokinetic model was not over-parameterized. Finally, η-shrinkage expressed as a percentage was identified to be below 20% for clearance (14.8%) and volume of distribution (6.2%).
Figure 2.
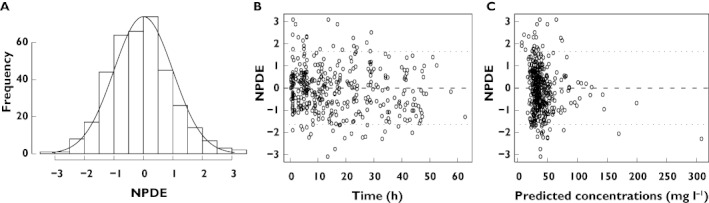
Results of the NPDE analysis. (A) the histogram shows the NPDE distribution, the solid line indicates a normal distribution, (B) NPDE vs. time after first dose and (C) NPDE vs. predicted concentrations
The model-based predicted clearance values for the final pharmacokinetic model vs. birth weight for PNA 1, 7, 14, 21 and 28 days are shown in Figure 3.
Figure 3.
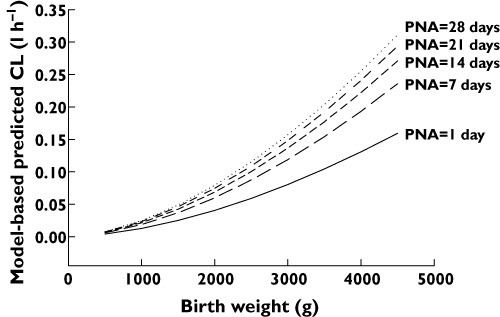
Model-based predicted clearance values of propylene glycol vs. birth weight for postnatal age (PNA) of 0, 7, 14, 21 and 28 days
Model-based simulations for PG co-administered with paracetamol or phenobarbital
Concentration–time profiles of PG after standard dosing regimens of intravenous paracetamol (800 mg PG/1000 mg paracetamol) or phenobarbital (700 mg PG/200 mg phenobarbital) that were used in this study, were simulated in three different neonates (bBW 630 g, 1500 g and 3500 g, respectively) at a PNA of 1 and 28 days (Figure 4). The administered dose of paracetamol, phenobarbital and the corresponding dose of PG are given in Table 3. Figure 4 shows that a population mean value for trough and peak concentrations of PG co-administered with paracetamol for a neonate of 630 g at day 1 were estimated to be 109 and 144 mg l−1, respectively, and for a neonates of 3500 g at day 28 trough and peak concentrations of PG were estimated to be 19 and 33 mg l−1, respectively. The expected population mean peak and trough PG concentrations after administration of phenobarbital varied between 28–218 and 6–112 mg l−1, respectively, depending on birth weight (630 g–3500 g) and PNA (1–28 days) of the neonate (Table 3, Figure 4).
Figure 4.
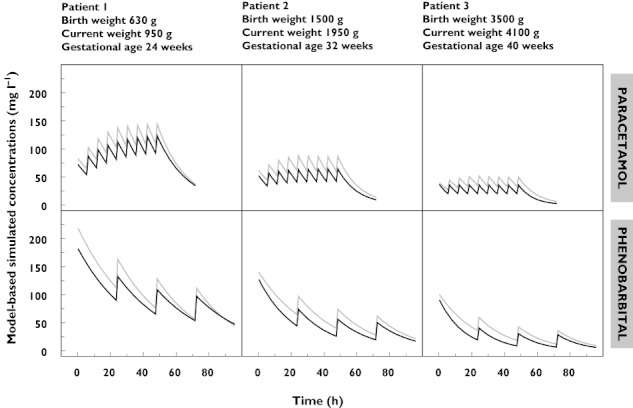
Model-based simulated concentration–time profiles of propylene glycol for three neonates (birth weight 630 g, 1500 g and 3500 g) after administration of paracetamol (800 mg propylene glycol/1000 mg paracetamol, upper panel) and phenobarbital (700 mg propylene glycol/200 mg phenobarbital, lower panel) in doses according to Table 3. The grey lines illustrate the concentration–time profiles for the neonates at birth. The black lines represent the concentration–time profiles at a postnatal age of 28 days (current weight 950 g, 1950 g and 4100 g)
Table 3.
Propylene glycol (PG) dosages when co-administered with paracetamol or phenobarbital in currently used dosages
| Drug | Propylene glycol content | Dosing guideline for drug | Drug-associated daily dose propylene glycol (mg kg−1 day−1) | Reference |
|---|---|---|---|---|
| I.v. paracetamol 10 mg ml−1 | 800 mg PG/1000 mg paracetamol | Loading dose: 20 mg kg−1 | 40 | [1] |
| Maintenance dose: 10 mg kg−1 every 6 h | 16–32 | |||
| I.v. phenobarbital 200 mg ml−1 | 700 mg PG/200 mg phenobarbital | Loading dose: 20 mg kg−1 day−1 | 70 | [18] |
| Maintenance dose: 5 mg kg−1 day−1 | 17.5 |
Discussion
While PG is considered to be safe and inactive, with high concentrations toxic effects like lactic acidosis, bradycardia and convulsions may occur. The risk of PG toxicity is higher in infants and neonates compared with adults since they have a lower metabolic capacity as well as an immature renal function resulting in a lower elimination capacity. The aim of this study was to characterize the pharmacokinetics of PG and its covariates in neonates following intravenous administration.
The pharmacokinetic model developed in this study was based on 372 PG plasma concentrations obtained in 62 preterm and term neonates after administration of paracetamol, phenobarbital or both. Birth weight was found the most important covariate for clearance while an increase in clearance was seen with PNA. The population value for clearance of 0.0849 l h−1 reported here in neonates is very low compared with the clearance value reported in adults which was found to vary between 144–390 ml min−1 1.73 m−2 (8.64–23.4 l h−1 1.73 m−2) [5]. This may indicate that either the alcohol dehydrogenase enzyme pathway or primary renal elimination, or most likely both, are immature during the first month of life. For renal function this has been described before by studying amikacin clearance in neonates, which likely reflects glomerular filtration in neonates [28]. The model-based predicted clearance values of PG vs. birth weight for PNA 1, 7, 14, 21 and 28 are shown in Figure 3. Large differences in clearance values are seen between neonates of 1 kg (0.013 l h−1) and neonates of 4 kg (0.13 l h−1) at day of birth. This 10-fold difference in clearance is still seen 1 month after birth. Furthermore this figure illustrates that during the first 2 weeks of life the largest increase in clearance is observed. These results correspond well with the advice of the FDA to avoid Kaletra®, a PG containing oral solution in premature babies until 14 days after due date, or in full-term babies younger than 14 days PNA [16, 17]. Volume of distribution scaled with current weight and was estimated 1.77 times higher in neonates receiving phenobarbital compared with neonates receiving paracetamol. The volume of distribution of a neonate of 1 kg (0.23 l or 0.40 l) (co-administered with paracetamol or phenobarbital, respectively) was very different compared with a neonate of 4 kg (1.69 l or 3 l). This difference may possibly be explained by the fact that phenobarbital is often given to neonates after perinatal asphyxia which may lead to a change in the pharmacokinetic parameters, e.g. higher volume of distribution. Unfortunately asphyxia could not be investigated as a covariate since no potential indicators (e.g. Apgar score, serum lactate concentration) were identified. The large variability in clearance and volume of distribution as a result of birth weight, PNA and current weight is reflected by the large range in expected peak and trough concentrations that can be expected with commonly used doses of paracetamol and phenobarbital in neonates varying in birth weight between 630 g and 3500 g and between a PNA of 1–28 days (Figure 4). The stability and predictability of the final pharmacokinetic model was demonstrated by the bootstrap (Table 2) as well as the NPDE (Figure 2), which are both advanced validation methods for paediatric pharmacokinetic models.
Although dose-related toxic effects have been reported upon administration of PG, only a limited number of paediatric reports are available in the literature. Glasgow et al. [13] and MacDonald et al. [14] described hyperosmolality and clinical symptoms of PG toxicity in small infants (<1500 g birth weight) due to very high PG exposure (3000 mg kg−1) in multivitamins injections. In retrospective studies of Shehab et al. [3] and Whittaker et al. [15] it was concluded that neonates in the neonatal intensive care unit are indeed exposed to potentially toxic doses of PG due to administration of commonly used drugs (e.g. phenobarbital, lorazepam, phenytoin, paracetamol) cosolved in PG but data on toxicity were not reported. In a study of Chicella et al. [12] a PG containing lorazepam formulation was administered to 11 infants between 1–15 months of age. In this study, there were neither clinical nor laboratory abnormalities observed, but accumulation of PG occurred during continuous infusion of lorazepam. Consequently, PG containing formulations should be used with caution in the paediatric and certainly in the neonatal age range especially when this results in high PG exposure. Based on the literature, the first indicator of a risk for subsequent PG toxicity is PG accumulation and changes in osmolar gap. Accumulation may subsequently result in biochemical changes and eventually toxic effects like, e.g. bradycardia, hepatic or renal injury, depression of the central nervous system.
To provide a basis to interpret the simulated concentrations of PG co-administered with paracetamol or phenobarbital in neonates, different approaches were provided in the methods section. However to identify maximum safe concentrations, more pharmacokinetic and pharmacodynamic studies are needed in neonates, particularly with drugs containing high concentrations of PG. To illustrate this concept, simulations were performed to illustrate the potential exposure of PG co-administered with lorazepam (828 mg PG/2 mg lorazepam). Based on the final pharmacokinetic model of PG co-administered with paracetamol, substantially higher concentrations of PG are obtained depending on the dose of lorazepam. Simulated PG concentrations with lorazepam in a dose of 0.015 mg kg−1 h−1[18] (daily dose of 149 mg kg−1 day−1 of PG) varied between 540 mg l−1 for a neonate of 630 g at day 1 and 123 mg l−1 for a neonate of 3500 g at day 28. With a dose of lorazepam of 0.1 mg kg−1 day−1 as described by Chicella et al. [12], concentrations of PG varying between 798–3563 mg l−1 were obtained, depending on birth weight and PNA. It should be noted that these concentrations are generated under the assumption of linear pharmacokinetics of PG, while higher daily doses of PG were administered to the neonates (149 mg kg−1 day−1 or 996 mg kg−1 day−1) with lorazepam compared with paracetamol or phenobarbital. As a result of the assumption of linear pharmacokinetics, the estimates of the exposure to PG must be considered conservative. In case of non-linearity in pharmacokinetics, even higher exposures are expected. At least, PG accumulation with the lorazepam dosing in neonates is in line with PG accumulation and toxicity described in adults [2, 6].
In conclusion, a pharmacokinetic model with birth weight and PNA as covariates for clearance was developed for PG co-administered with paracetamol or phenobarbital in preterm and term neonates. As such, large variability in exposure of PG may be expected in neonates which are dependent on birth weight and PNA. The model can be used to simulate concentrations of PG co-administered with paracetamol and phenobarbital in neonates. As the exact safe concentrations are still undefined, more studies are needed to characterize the pharmacokinetics of PG in neonates and children.
Acknowledgments
This study was performed within the framework of Top Institute Pharma project number D2-104.
The clinical research of K. Allegaert is supported by the Fund for Scientific Research, Flanders (Belgium) (FWO Vlaanderen) by a Fundamental Clinical Investigatorship (1800209 N). The clinical research of A. Kulo is supported by a JoinEU-SEE scholarship (2009–2010). The authors also would like to thank LAP&P Consultants for their technical support with NONMEM.
Competing Interests
There are no competing interests to declare.
REFERENCES
- 1.Allegaert K, Vanhaesebrouck S, Kulo A, Cosaert K, Verbesselt R, Debeer A, de Hoon J. Prospective assessment of short-term propylene glycol tolerance in neonates. Arch Dis Child. 2010;95:1054–8. doi: 10.1136/adc.2010.190330. [DOI] [PubMed] [Google Scholar]
- 2.Zar T, Graeber C, Perazella MA. Recognition, treatment, and prevention of propylene glycol toxicity. Semin Dial. 2007;20:217–9. doi: 10.1111/j.1525-139X.2007.00280.x. [DOI] [PubMed] [Google Scholar]
- 3.Shehab N, Lewis CL, Streetman DD, Donn SM. Exposure to the pharmaceutical excipients benzyl alcohol and propylene glycol among critically ill neonates. Pediatr Crit Care Med. 2009;10:256–9. doi: 10.1097/PCC.0b013e31819a383c. [DOI] [PubMed] [Google Scholar]
- 4.‘Inactive’ ingredients in pharmaceutical products: update (subject review). American Academy of Pediatrics Committee on Drugs. Pediatrics. 1997;99:268–78. doi: 10.1542/peds.99.2.268. [DOI] [PubMed] [Google Scholar]
- 5.Speth PA, Vree TB, Neilen NF, de Mulder PH, Newell DR, Gore ME, de Pauw BE. Propylene glycol pharmacokinetics and effects after intravenous infusion in humans. Ther Drug Monit. 1987;9:255–8. doi: 10.1097/00007691-198709000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Wilson KC, Reardon C, Theodore AC, Farber HW. Propylene glycol toxicity: a severe iatrogenic illness in ICU patients receiving IV benzodiazepines: a case series and prospective, observational pilot study. Chest. 2005;128:1674–81. doi: 10.1378/chest.128.3.1674. [DOI] [PubMed] [Google Scholar]
- 7.CBG-MEB. Medicinal products: allergy information. Accessed September 9, 2011.
- 8.Yu DK, Elmquist WF, Sawchuk RJ. Pharmacokinetics of propylene glycol in humans during multiple dosing regimens. J Pharm Sci. 1985;74:876–9. doi: 10.1002/jps.2600740815. [DOI] [PubMed] [Google Scholar]
- 9.Fligner CL, Jack R, Twiggs GA, Raisys VA. Hyperosmolality induced by propylene glycol. A complication of silver sulfadiazine therapy. JAMA. 1985;253:1606–9. [PubMed] [Google Scholar]
- 10.Nahata MC. Safety of ‘inert’ additives or excipients in paediatric medicines. Arch Dis Child Fetal Neonatal Ed. 2009;94:F392–3. doi: 10.1136/adc.2009.160192. [DOI] [PubMed] [Google Scholar]
- 11.European Medicines Agency (EMA) Guidelines medicinal products for human use; safety, environment and information. Accessed September 9, 2011.
- 12.Chicella M, Jansen P, Parthiban A, Marlowe KF, Bencsath FA, Krueger KP, Boerth R. Propylene glycol accumulation associated with continuous infusion of lorazepam in pediatric intensive care patients. Crit Care Med. 2002;30:2752–6. doi: 10.1097/00003246-200212000-00021. [DOI] [PubMed] [Google Scholar]
- 13.Glasgow AM, Boeckx RL, Miller MK, MacDonald MG, August GP, Goodman SI. Hyperosmolality in small infants due to propylene glycol. Pediatrics. 1983;72:353–5. [PubMed] [Google Scholar]
- 14.MacDonald MG, Fletcher AB, Johnson EL, Boeckx RL, Getson PR, Miller MK. The potential toxicity to neonates of multivitamin preparations used in parenteral nutrition. JPEN J Parenter Enteral Nutr. 1987;11:169–71. doi: 10.1177/0148607187011002169. [DOI] [PubMed] [Google Scholar]
- 15.Whittaker A, Currie AE, Turner MA, Field DJ, Mulla H, Pandya HC. Toxic additives in medication for preterm infants. Arch Dis Child Fetal Neonatal Ed. 2009;94:F236–40. doi: 10.1136/adc.2008.146035. [DOI] [PubMed] [Google Scholar]
- 16.Food and Drug Administration. Drug safety communication: serious health problems seen in premature babies given Kaletra (lopinavir/ritonavir) oral solution. Accessed September 9, 2011.
- 17.Boxwell D, Cao K, Lewis L, Marcus K, Nikhar B. 2011. Kaletra oral solution toxicity in neonates – lopinavir, ethanol, and/or propylene glycol? 18th Conference on Retroviruses and Opportunistic Infections (2011)
- 18.Yaffe SJ, Aranda JV. Neonatal and Pediatric Pharmacology: Therapeutic Principles in Practice. 4th edn. Philadelphia, PA: Lippincott Williams and Wilkins, Wolters Kluwer; 2010. [Google Scholar]
- 19.Kulo A, Allegaert K, de Hoon J, Verbesselt R. Determination of propylene glycol in low volume plasma and urine samples of neonates by LC with photodiode array detection. Chromatographia. 2011;95:1054–8. [Google Scholar]
- 20.Krekels EH, van Hasselt JG, Tibboel D, Danhof M, Knibbe CA. Systematic evaluation of the descriptive and predictive performance of paediatric morphine population models. Pharm Res. 2011;28:797–811. doi: 10.1007/s11095-010-0333-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montgomery DC, Peck EA. Introduction to Linear Regression Analysis. New York: Wiley; 1982. pp. 301–02. [Google Scholar]
- 22.Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther. 2007;82:17–20. doi: 10.1038/sj.clpt.6100241. [DOI] [PubMed] [Google Scholar]
- 23.Comets E, Brendel K, Mentre F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput Methods Programs Biomed. 2008;90:154–66. doi: 10.1016/j.cmpb.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 24.Brendel K, Comets E, Laffont C, Laveille C, Mentre F. Metrics for external model evaluation with an application to the population pharmacokinetics of gliclazide. Pharm Res. 2006;23:2036–49. doi: 10.1007/s11095-006-9067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yahwak JA, Riker RR, Fraser GL, Subak-Sharpe S. Determination of a lorazepam dose threshold for using the osmol gap to monitor for propylene glycol toxicity. Pharmacotherapy. 2008;28:984–91. doi: 10.1592/phco.28.8.984. [DOI] [PubMed] [Google Scholar]
- 26.Feldman W, Drummond KN. Serum and urine osmolality in normal full-term infants. Can Med Assoc J. 1969;101:73–4. [PMC free article] [PubMed] [Google Scholar]
- 27.Giacoia GP, Miranda R, West KI. Measured vs calculated plasma osmolality in infants with very low birth weights. Am J Dis Child. 1992;146:712–7. doi: 10.1001/archpedi.1992.02160180072020. [DOI] [PubMed] [Google Scholar]
- 28.De Cock RF, Allegaert K, Schreuder MF, Sherwin CM, de Hoog M, van den Anker JN, Danhof M, Knibbe CA. Maturation of the glomerular filtration rate in neonates, as reflected by amikacin clearance. Clin Pharmacokinet. 2012;51:105–17. doi: 10.2165/11595640-000000000-00000. [DOI] [PubMed] [Google Scholar]