

Abstract
Aim
SRT2104 is a novel, first-in-class, highly selective small molecule activator of the NAD + dependent deacetylase SIRT1. SRT2104 was dosed to healthy male and female volunteers in a series of phase 1 clinical studies that were designed to elucidate tolerability and pharmacokinetics associated with oral dosing to aid in dose selection for subsequent clinical trials.
Methods
In the first-in-human study, there was both a single dose phase and 7 day repeat dose phase. Doses used ranged from 0.03 to 3.0 g. A radioactive microtracer study was subsequently conducted to determine systemic clearance, bioavailability and preliminary metabolism, and a crossover study was conducted to determine the effect of gender, formulation and feeding state on SRT2104 pharmacokinetics.
Results
SRT2104 was well tolerated in all of these studies, with no serious adverse reactions observed. SRT2104 displayed a dose-dependent, but sub-proportional increase in exposure following single dose and repeated dose administration. Accumulation of three-fold or less occurs after 7 days of repeat dosing. The mean bioavailability was circa 14% and the mean clearance was circa 400 ml min−1. Although there were no substantial effects on exposure resulting from gender or formulation differences, a notable food effect was observed, manifested as up to four-fold increase in exposure parameters.
Conclusions
In the absence of an optimized formulation of SRT2104, the food effect can be used to maximize exposure in future clinical studies. Combined with the good tolerability of all doses demonstrated in these studies, the favourable selectivity profile of SRT2104 allows for the use of this SIRT1 modulator for target validation in the clinic.
Keywords: activator, microtracer, pharmacokinetics, SIRT1, sirtuin, SRT2104
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT?
Resveratrol is a putative small molecule activator of SIRT1, but also possesses significant off-target activity and has been found to be poorly bioavailable. Validation of SIRT1 as a clinical target requires a probe with acceptable pharmacokinetics and selectivity,and is critical to advancing the understanding of SIRT1 biology in humans.
WHAT THIS STUDY ADDS?
This manuscript provides a comprehensive understanding of SRT2104 pharmacokinetics in humans and will serve as the foundation for future clinical studies. As the field of SIRT1 science matures, the data shown in this manuscript provide a critical component for determining appropriate doses of SRT2104 to evaluate SIRT1 activation in a clinical setting.
Introduction
The sirtuin class of deacetylases plays important regulatory roles in numerous physiological pathways with examples in all three sub-cellular compartments: nuclear, cytosolic and mitochondria [1, 2]. Of the seven mammalian sirtuin isoforms, the biology of SIRT1 is the most well characterized and its drug discovery platform is also the most advanced [3]. Studies that involve SIRT1 over-expression in mammalian cellular models [4, 5] as well as those that involve the use of transgenic SIRT1 over-expressing mice have demonstrated significant SIRT1-dependent improvements to pathological phenotypes [6–8]. Furthermore, the efficacy induced by small molecule SIRT1 activators in preclinical models of metabolic, inflammatory, cardiovascular and neurodegenerative disease [9–13] suggests that pharmacological activation of SIRT1 may be beneficial for human health. For example, small molecule SIRT1 activators have shown efficacy in ob/ob and diet-induced obesity (DIO) mouse models [9] with significant effects on weight control, glucose homeostasis and insulin. Moreover, notable observations in additional animal models have included decreased production of inflammatory markers, reduced steatosis, and increased exercise endurance and muscle function, a convergence of effects that promote improved overall health [14–17]. There have been contradictory data published in the literature that link sirtuins to cancer, with both beneficial effects of increased expression [18] and inhibition or reduced expression [19, 20] of SIRT1 as cancer therapy strategies.
Although the full extent of the biological activity of SIRT1 has yet to be determined, the extraordinary breadth of the pharmacological potential presented by SIRT1 modulation can be more fully appreciated by considering that it has more than 70 known sub-cellular protein substrates, prominent examples of which include p53, PGC1α, FOXO, ACS1 and p65-NFκB, as well as a host of other nuclear and cytosolic proteins with established roles in numerous disease states [21–26] that generally are responsive to periods of cellular stress. Acetylation and deacetylation cycles associated with post translational modification of proteins have been implicated in critical control of cellular processes. Beyond serving as an on/off functional switch, deacetylation can alter protein stability [27], protein–protein interactions [28] and sub-cellular localization [29], thereby controlling downstream biological effects at multiple levels. Furthermore, the unique enzymatic reaction carried out by sirtuins involves NAD+ as a co-substrate, coupling cellular energy status to the regulatory mechanisms. A relatively comprehensive expression analysis of sirtuins in human tissues indicates a high level of SIRT1 expression in skeletal muscle, brain, kidney, thymus and reproductive tissues [30, 31]. Classical drug discovery has predominantly targeted protein classes such as GPCRs, kinases, nuclear receptors, ion channels and enzymes with a narrow, specific function. Their upstream modulation of several protein classes at once makes sirtuin enzymes unique targets for drug development. As such, pharmacological activation of sirtuins has emerged as an approach with great potential for the discovery of therapeutics in multiple disease indications.
The first small scale screen [32] for SIRT1 activators identified a narrow hit series comprised mostly of polyphenols. The most prominent and well-known hit was resveratrol, a stilbene abundantly available from sources such as grape skin, peanuts and roots of Polygonum cuspidatum. Resveratrol has been investigated in several exploratory studies in man [33, 34] although its lack of selectivity for SIRT1, significant metabolic liabilities and extremely low inherent bioavailability [35, 36] limit its use as a drug targeting SIRT1. Furthermore, some recent studies have proposed alternate mechanisms for the pharmacology of resveratrol [37] as well as its potential impact on SIRT1 expression levels [38, 39], assertions that are difficult to refute unambiguously. In order to address some of these issues, a search was undertaken for compounds with a high degree of selectivity for SIRT1, and that had improved potency, physiochemical pharmacokinetic properties and no chemical relationship to resveratrol led to SRT2104, a first-in-class activator of SIRT1. Following pharmacology and toxicology assessments, a series of phase 1 clinical studies in normal healthy volunteers were designed to characterize the pharmacokinetics of SRT2104, and to assess whether there were any deleterious effects of dosing a SIRT1 activator in humans. The overall goal of these studies was to identify appropriate doses of SRT2104 for use in future clinical trials designed to probe its safety and efficacy.
Methods
Study designs
SRT2104 was evaluated in four phase 1 clinical studies designed to characterize its safety, tolerability and pharmacokinetics. All of the protocols were approved by a local medical ethics committee and all studies were conducted at Simbec Research Ltd (Wales, UK) following Good Clinical Practice (GCP) guidelines. The protocols for the first-in-human (FIH) study, the food/gender effect study and the second food effect study were approved by South East Wales Research Ethics Committee (Cardiff, UK) and the protocol for the bioavailability study was approved by Independent Ethics Committee, Plymouth (Devon, UK). All subjects were deemed eligible based on medical history, physical examination and standard clinical laboratory parameters. Subjects gave written consent to participate. The FIH study was a randomized, double-blind, placebo-controlled, dose escalation study in which seven cohorts were given single doses of 0.03 g, 0.1 g, 0.25 g, 0.5 g, 1.0 g, 2.0 g or 3.0 g of SRT2104 as an oral suspension [1% hypromellose acetate succinate (HPMC-AS) in water] followed by a 7 day repeat administration of the same dose after a 1 week washout period. The starting dose for this study was selected to be more than 100-fold lower than the no observable adverse effect level (NOAEL) of the most sensitive toxicological species (female rats), which achieved an exposure of 33 μg ml−1 h following 28 days of consecutive dosing at 1.0 g kg−1. Each dose escalation in this study occurred following a review of safety parameters (including physical examination findings, vital signs, ECG studies, adverse events and laboratory values) and pharmacokinetic data by the study director, the sponsor and an independent medical monitor. The second study conducted was a single dose bioavailability study to estimate the relative contribution of systemic clearance and drug absorption to the SRT2104 pharmacokinetic (PK) profile in humans. The study design for the bioavailability study called for subjects to be given a single 0.25 g dose of SRT2104 as an oral suspension (1% HPMC-AS in water) followed at or near tmax (2.75 h after oral dosing) by a 15 min intravenous (i.v.) infusion containing 100 μg of 14C-SRT2104. This microtracer [40] design allowed simultaneous determination of absolute bioavailability and i.v. pharmacokinetic parameters together with preliminary information on the human metabolism of SRT2104. The third study (food/gender effect study) was a four-way crossover study where two cohorts of subjects (male and female) were given 0.5 g of SRT2104 as either a suspension or capsule formulation, in both the fed and fasted states. Subjects in the fed state were administered SRT2104 30 min following a standardized meal consisting of 602 kcal (18.3% protein, 30.4% fat and 45.3% carbohydrates). The final study (second food effect study) was a single dose and repeat dose study in male volunteers to assess the food effect at the 2.0 g dose. This study was conducted to provide assurance that the magnitude of the food effect observed at the 0.5 g dose would be similar at 2.0 g, and that exposure in future studies at this dose could be accurately predicted if administered in the fed state. A single cohort of subjects was given one dose of 2.0 g SRT2104 as a capsule in the fed state. Following a 1 week washout period, a 2.0 g dose of SRT2104 was administered for 7 consecutive days in the fed state to the same individuals, utilizing the standardized meal described above.
Clinical assessments
Clinical safety was evaluated throughout the studies by monitoring vital signs, physical examinations, 12-lead electrocardiograms (ECGs), continuous cardiac telemetry, biochemistry, hematology, coagulation and urinalyses, as well as recording treatment emergent adverse events.
Bioanalysis and pharmacokinetics
For all four clinical trials, plasma samples were obtained for pharmacokinetic analysis, pre dose and at 0.25 h, 0.5 h, 1 h, 2 h, 4 h, 8 h, 12 h and 24 h after dosing. Studies subsequent to the FIH clinical trial contained additional pharmacokinetic time points. For the bioavailability study, additional post dose time points included 3 h, 5 h, 6 h, 10 h, 18 h, 48 h and 72 h. Both the food/gender effect study and the second food effect study contained additional post dose time points including 0.75 h, 1.5 h, 2.5 h, 3 h, 5 h, 6 h, 10 h, 15 h, 36 h (bioavailability study only), 48 h, 72 h and 168 h (second food effect study only). In studies which contained a multiple dose component, pre dose samples were taken on all days in which study drug was administered, and time points listed above were taken on day 1 and day 7 of the dosing period. For the 100 μg 14C-SRT2104 i.v. infusion, plasma samples were taken before the start of the infusion, at 5 min and 10 min during infusion and at the end of infusion. Post infusion plasma samples were taken at 5 min, 10 min, 20 min, 30 min and 45 min as well as 1 h, 2 h, 3 h, 4 h, 6 h, 8 h, 10 h, 12 h, 15 h, 21 h, 45 h and 69 h. All blood samples were collected in vacuum tubes containing lithium heparin and were separated by centrifugation at 1500 g with a temperature of 4°C for 10 min. SRT2104 plasma concentrations were determined by a validated liquid chromatography method with tandem mass spectrometry (LC-MS/MS) using d8-SRT2104 as a stable label, internal standard. Assay performance was evaluated using four quality control (QC) concentrations: 0.5 ng ml−1, 2.1 ng ml−1, 42.5 ng ml−1 and 425 ng ml−1. Intra- and inter-batch QC samples showed accuracy ranging from 90.8% to 103.2% and the coefficient of variation did not exceed 8.0%. The upper limit of quantification was 500 ng ml−1 and the lower limit of quantification was 0.5 ng ml−1. Samples with concentrations outside of the quantitation limits were reported as below the limit of quantitation (BLQ) if less than 0.5 ng ml−1 and were diluted and reanalyzed if above 500 ng ml−1. Dilution integrity up to 50 μg ml−1 was demonstrated during method validation. Quantification of 14C-SRT2104 was performed by utilizing accelerator mass spectrometry (AMS) which traces low abundance 14C for high isotopic specificity, but detects carbon atoms with mass spectrometry for high sensitivity [41]. The lower limit of quantification for 14C-SRT2104 was 6.4 pg ml−1.
Preliminary metabolism
Plasma and urine samples from the radiolabelled microtracer study (bioavailability study) were analyzed by AMS, liquid chromatography-mass spectrometry (LC-MS) and nuclear magnetic resonance spectroscopy (NMR) to characterize and quantify the metabolites present.
Two plasma pools representative of the AUC(0,3.25 h) and AUC(4.25,21.25 h) after the start of the i.v. infusion for all subjects together with a pool representative of the AUC(0,24 h) after the oral dose were prepared using plasma volumes in proportion to the nominal time interval between individual samples, performed according to the method described by Hop et al. [42]. Pooled urine was prepared by combining proportional volumes (circa 1.5% of total volume) from the 0−24 h collections from each volunteer to supply a representative 0–24 h pool across all subjects. The AUC(0,3.25 h) and AUC(4.25,21.25 h) representative plasma pools together with the urine pool were assayed for radioactivity using AMS to provide metabolite profiles for the i.v. dose route. The plasma pool representative of the AUC(0,24 h) after oral dosing and urine pool were analyzed by LC-MS and fractionated by semi-preparative high performance liquid chromatography (HPLC) prior to proton NMR to provide structural information on the metabolites. In addition, relative concentrations of SRT2104 and the major plasma and urine metabolites after oral dosing were estimated from the proton NMR data by integration of a suitable proton in a manner similar to that described by Dear et al. [43]. Absolute levels of observed drug-related material in urine, following oral administration, were also estimated by comparison of the proton NMR integrals with a standard of known concentration.
Data evaluation and statistics
Standard non-compartmental pharmacokinetic parameters, including AUC(0,t), AUC(0,τ), Cmax, and tmax were derived from the concentration–time data for all oral doses. Additional parameters, including clearance (CL) and steady-state volume of distribution (Vss) were derived from the concentration−time data for the i.v. dose.
Results
Study population, tolerability and safety
In the four phase 1 pharmacokinetics studies described, a total of 55 male and 10 female subjects received at least one dose of SRT2104 via oral administration. Eight subjects also received a 100 μg i.v. dose of SRT2104 labelled with 14C. Adverse events (AEs) that occurred in three or more individuals are shown in Table 1. AEs reported in three or more subjects receiving SRT2104 in these studies included headache (18 subjects, 28%), flatulence (nine subjects, 14%), infusion site pain (five subjects; 8%), nausea (five subjects, 8%), diarrhoea (four subjects, 6%) and nasopharyngitis (four subjects, 6%). One severe AE was reported (headache), and the remaining AEs were of mild to moderate severity and self-limiting. There was no dose relationship to the observed AEs. There were no clinically relevant changes in laboratory parameters or ECG recordings observed in any of the trials. In general, the incidence of AEs was comparable between subjects receiving SRT2104 and those receiving placebo.
Table 1.
Adverse events occurring in three or more subjects
| *Preferred term | Placebo | 0.03 g | 0.10 g | 0.25 g | 0.50 g | 1.0 g | 2.0 g* | 3.0 g | All SRT2104 |
|---|---|---|---|---|---|---|---|---|---|
| (n = 16) | (n = 4) | (n = 4) | (n = 12) | (n = 24) | (n = 5) | (n = 12) | (n = 4) | (n = 65) | |
| Headache | 1 (6%) | 0 | 1 (25%) | 0 | 8 (33%) | 1 (20%) | 6 (50%) † | 2 (50%) | 18 (28%) |
| Flatulence | 3 (19%) | 2 (50%) | 0 | 0 | 3 (13%) | 2 (40%) | 2 (17%) | 0 | 9 (14%) |
| Infusion site pain | 0 | 0 | 0 | 5 (42%) | 0 | 0 | 0 | 0 | 5 (8%) |
| Nausea | 1 (6%) | 0 | 1 (25%) | 0 | 2 (8%) | 0 | 1 (8%) | 1 (25%) | 5 (8%) |
| Diarrhoea | 0 | 3 (75%) | 0 | 0 | 1 (4%) | 0 | 0 | 0 | 4 (6%) |
| Nasopharyngitis | 0 | 0 | 0 | 0 | 2 (8%) | 0 | 1 (8%) | 1 (25%) | 4 (6%) |
MedDRA. Expressed as count (% of total).
The only AEs occurring in three or more subjects in the fed state were headaches.
Headache occurred in three subjects in the fasted state and three in the fed state.
Pharmacokinetics
Following a single dose, SRT2104 concentrations exhibited a monophasic rise (Figure 1), with median tmax occurring 1–3 h post dose. Concentrations then exhibited a gradual decline with a half-life of 8–24 h. Median tmax after 7 days of consecutive dosing was similar at 1 to 2 h post dose, while mean half-life ranged from 12–20 h. For both single and repeat dose administration, tmax and half-life showed dose independence. The pharmacokinetic parameters measured indicate that SRT2104 exhibits a dose-dependent increase in Cmax and AUC(0,t) following single doses (Figure 2, Panel 1) and daily doses for 7 days. However, the increase in exposure was less than proportional to dose. Accumulation of SRT2104 was observed on day 7 as compared with day 1. The average accumulation ratio (RA) ranged from 1.5 to 2.0 over the dose range 0.03−2.0 g, which is consistent with the observed half-life of up to 24 h. There was no obvious relationship between dose and RA. Overall, high inter-subject variability in the plasma concentration vs. time profiles and derived pharmacokinetic parameters was observed. The percent coefficient of variation of geometric mean AUC(0,τ) from the FIH study ranged from 57%–143%. Pharmacokinetic parameters for the repeat dose phase are shown in Table 2 (see Table S1 for that of the single dose).
Figure 1.
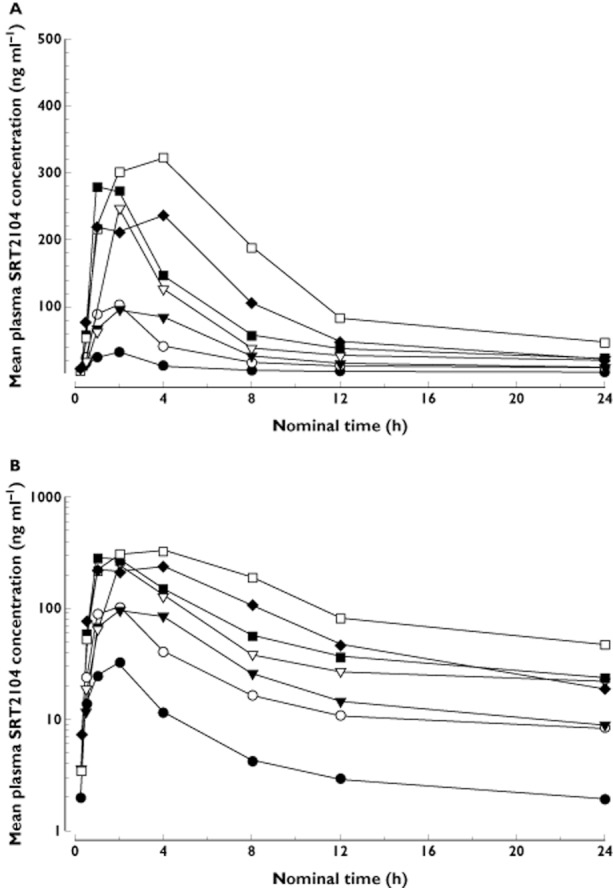
Mean plasma concentration–time profile of SRT2104 following single oral dose as a suspension on linear (A) and log (B) scale (FIH study). •, 0.3 g; ○, 0.1 g; ▾, 0.25 g; ▿, 0.5 g; ▪, 1.0 g; □, 2.0 g; ♦, 3.0 g
Figure 2.
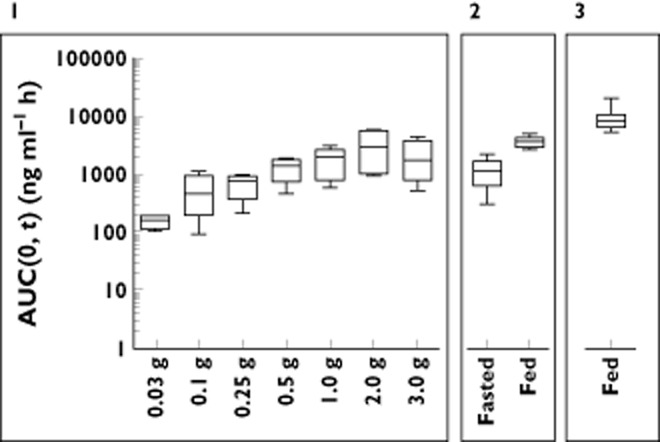
Exposure in terms of AUC(0,t) for single doses in the fasted state (Panel 1, FIH study), 0.5 g single doses in the fed and fasted state (Panel 2, food/gender effect study) and 2.0 g single dose in the fed state (Panel 3, second food effect study)
Table 2.
Summary of pharmacokinetic parameters for SRT2104 after oral administration to healthy human volunteers for 7 consecutive days (FIH and the second food effect study)
| Fasted | Fed | |||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | 0.03 g | 0.1 g | 0.25 g | 0.5 g | 1.0 g | 2.0 g | 3.0 g | 2.0 g |
| n | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 8 |
| Cmax (ng ml−1) | 50.1 (40) | 127.3 (77) | 218.7 (81) | 293.3 (37) | 452.7 (55) | 536.8 (55) | 261.2 (78) | 1873.6 (28) |
| tmax (h) | 1.5 (1.0–2.0) | 1.5 (1.0–4.0) | 2.0 (2.0–8.0) | 1.0 (1.0–4.0) | 1.5 (1.0–4.0) | 2.0 (1.0–4.0) | 2.0 (1.0–2.0) | 3.0 (2.5–5.0) |
| t1/2 (h) | 12.7 (20) | 12.1 (33) | 21.6† (79) | 20.0 (26) | 17.7* (2) | 14.2† (27) | 14.6 (31) | 25.4 (23) |
| AUC(0,τ) (ng ml−1 h) | 247 (57) | 602 (171) | 1191 (143) | 1878 (59) | 3087 (82) | 3665 (86) | 1573 (132) | 13 690 (23) |
| RA§ | 1.6 | 1.5 | 1.9 | 1.6 | 2.1 | 1.5 | 1.0 | 1.5 |
| dnAUC¶ | 8.2 | 6.0 | 4.8 | 3.8 | 3.1 | 1.8 | 0.5 | 6.8 |
Data are mean (%CV), except tmax which is median (range) and AUC which is geometric mean (%CV).
n = 2.
n = 3.
RA = AUC(0),τ)/AUC(0,t).
Dose normalized AUC (AUC(ng ml-1 h)/Dose (mg)) expressed as mean.
Absolute oral bioavailability was calculated using a 100 μg i.v. 14C-labelled microdose as a reference (Table 3), administered concurrently with a 0.25 g oral dose of a suspension in the fasted state. SRT2104 was found to be 13.8% bioavailable (2–27%). The mean clearance of 14C-SRT2104 was 404 ml min−1 and ranged from 287−493 ml min−1.
Table 3.
Mean ± SD values of pharmacokinetic parameters for SRT2104 (Bioavailability study)
| 0.25 g SRT2104 Oral dose (n = 8) | 100 μg 14C-SRT2104 15 min i.v. infusion (n = 8) | ||
|---|---|---|---|
| Parameter | SRT2104 | Total 14C | 14C-SRT2104 |
| Cmax (ng ml−1) | 132 ± 90.2 | 1.42 ± 0.239 | 1.23 ± 0.282 |
| tmax (h) | 2.00 (1.00–3.03) * | 0.267 (0.250–0.333) * | 0.250 (0.167–0.333) * |
| AUC(0,t) (ng ml−1 h) | 1219 ± 1057 | 7.98 ± 1.80 | 2.95 ± 0.757 |
| AUC(0,∞) (ng ml−1 h) | 1192 ± 1211† | 8.90 ± 2.50§ | 3.41 ± 0.835§ |
| t1/2 (h) | 25.5 ± 6.45† | 28.0 ± 3.93§ | 23.7 ± 9.37§ |
| CL (ml min−1) | – | – | 404 ± 92.6§ |
| Vss (l) | – | – | 428 ± 152§ |
| Fabs (%) | 13.8 ± 10.6 (n = 5) ¶ | – | – |
Median (range).
n = 7.
n = 6.
n = 5.
The dose-normalized AUC(0,t) (AUC in ng ml−1 h divided by dose in mg) from the single ascending dose study ranged from 8.2−0.5 and decreased with increasing dose, demonstrating limited absorption at higher doses. In this study, SRT2104 was administered as an oral suspension. The food/gender effect study compared exposures from suspension formulation with those from powder in capsule formulation in both the fed and fasted states (Table S2). In the fasted state, dose normalized AUC(0,t) of the suspension and capsule formulations was 2.8 and 2.1, respectively, and in the fed state was 12.1 and 10.7 respectively. Although a slight decrease in exposure was observed from the drug-in-capsule presentation, the effect was not sufficient to preclude use of capsules in future studies. The effect of dose administration in the fed state, however, was dramatic (Figure 2, Panel 2). An approximate four-fold increase in AUC(0,t) was observed in individuals receiving 0.5 g of SRT2104 immediately following a meal, regardless of formulation. A food effect similar in magnitude was also observed following a 2.0 g dose (Figure 3, Panel 3), with a dose normalized AUC(0,t) in that study of 4.4 compared with 1.2 in the fasted state.
Figure 3.
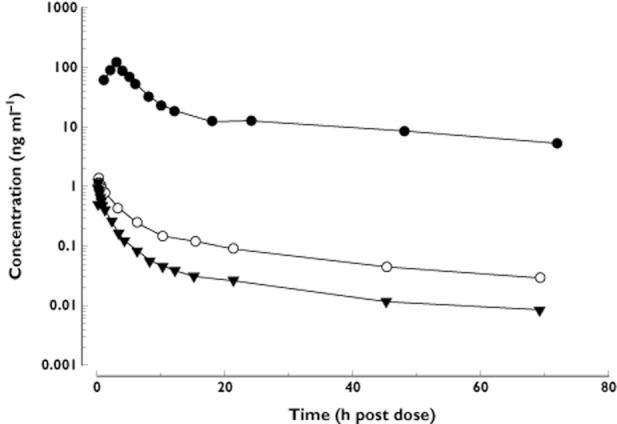
Plasma concentration–time curves for SRT2104 co-administered as an oral dose (0.25 g) and an i.v. microdose (100 μg). (Bioavailability study). •, 0.25 g oral dose; ○, 100 μg i.v. dose – total radioactivity; ▾, 100 μg i.v. dose – SRT2104
In these studies, the data were consistent with gender having minimal impact in overall exposure to SRT2104. In the fasted state, the AUC ratio of females to males is 2.2 for the suspension formulation and 2.4 for capsule administrations. However, this apparent increase in exposure for females reduces to 1.1 and 1.2 in the fed state for suspension and capsule formulations, respectively.
Further to the pharmacokinetic parameters, some insights into metabolism were obtained from the microtracer study. Mean AUC(0,t) for total radioactivity was approximately 2.5 times greater than the corresponding AUC for parent 14C-SRT2104 as shown in Figure 3, indicating the presence of drug metabolites. The parallel half-lives for total radioactivity and parent 14C-SRT2104 after i.v. dosing indicated that radiolabelled metabolites of 14C-SRT2104 are likely to be formation rate-limited and thus are not expected to accumulate more than parent drug.
Approximately 30 metabolites were identified by LC-MS and proton NMR in plasma and urine using the high concentrations of drug-related material present after oral dosing, with unequivocal structures proposed for more than 10 of these metabolites. The peaks in the urine and plasma radio profiles, constructed from the AMS data, were identified with the aid of drug-related material derived from the oral dose and as far as could be assessed, the routes of metabolism for SRT2104 after oral and i.v. administration were qualitatively similar, involving both phase 1 and phase 2 mechanisms. The major drug-related component in plasma was unchanged SRT2104 following both dosing routes with nine metabolites identified. The major metabolite accounted for 20–30% of drug-related material.
Urinary excretion of drug-related material was a minor route of elimination, accounting for approximately 2% and 13% of the oral and i.v. doses, respectively, with drug-related material eliminated primarily as metabolites. Unchanged SRT2104 was minor and accounted for less than 2% of the urinary elimination following both dosing routes.
Discussion
Pharmacological activation of the human SIRT1 enzyme provides a novel therapeutic strategy with potential in a variety of indications including metabolic, inflammation and neurodegenerative diseases. Currently, there exists no validated biomarker to assess directly SIRT1 activation in humans. As such, the dose selected for human efficacy studies will be the dose that achieves the highest exposure whilst maintaining an excellent tolerability profile. The objective of this approach is to maximize the chance of observing a beneficial clinical signal while demonstrating the safety of dosing a SIRT1 activator. The phase I clinical plan reflects the demands of this approach: four separate trials to elucidate safety/tolerability, dose linearity and exposure after single and 7 day repeat dosing as well as formulation/gender/food effect, i.v. PK parameters and absolute bioavailability.
The initial dose escalation study indicated that SRT2104 was poorly absorbed following oral dosing as a suspension. Although some accumulation was observed from 1 to 7 days, there was concern that the drug concentrations achieved might be insufficient to generate a pharmacological response. At that time, it was not known whether the poor exposure was due primarily to poor absorption, high first pass metabolism or a combination of the two. It should be noted, however, that while SRT2104 demonstrates greater than 20 mg ml−1 solubility in simulated gastric fluid (SGF), the solubility in simulated intestinal fluid (SIF) drops to less than 2 μg ml−1 (unpublished data). As such, one explanation for the exposure variability in humans is that it is associated with the variability of gastric emptying times in the fasted state. Regardless, it was assumed that the poor solubility profile in intestinal fluid adversely affected the absorption of SRT2104, and that an optimized formulation, particularly one that improved the intestinal fluid solubility or that was retained for an extended period in the stomach (thereby maximizing drug dissolution in the acidic environment and releasing into the duodenum the dissolved drug over an extended period), might be a viable way to increase exposure and reduce variability. Furthermore, SRT2104 is currently characterized as Biopharmaceutics Classification System (BCS) II, suggesting that solubility, not permeability, is the greatest barrier to absorption.
It was important to determine to what extent clearance of SRT2104 prevented exposure. In the absence of a preclinical toxicology package supporting i.v. administration of SRT2104, an i.v. microdose of 100 μg SRT2104 fortified with 14C-SRT2104 was used. A common criticism of microdose studies is that due to the very small dose, transporters and enzymes involved in the disposition of the drug, which might otherwise be saturated at a pharmacological dose, can exert an influence on the pharmacokinetic parameters. Sirtris employed a microtracer design whereby subjects were administered a 0.25 g oral dose of SRT2104 followed by the microtracer at approximately tmax, thus minimizing the potential for bias resulting from the small i.v. dose. As shown in Table 3, SRT2104 exhibits low to moderate clearance, suggesting that improving the presentation of the drug through formulation would not be made irrelevant by clearance. Further need for an improved formulation was identified when a substantial food effect was observed following a single dose of 0.5 g SRT2104. Indeed, an approximately four fold increase in AUC(0,t) and Cmax was observed following a meal. Thus, further investigation into the causes underlying this observation could lead to an enhanced and improved formulation.
The goal of this phase 1 programme was to determine a dosing strategy to maximize exposure and it has been demonstrated that the food effect and repeat dose accumulation can contribute to such a strategy. The FIH study showed that maximal exposure was observed at the 2.0 g dose and that a further increase in dose provided no additional exposure. SRT2104 was well tolerated after dosing 2.0 g of SRT2104 in the fed state for 7 days, suggesting that this could be an appropriate dosing strategy for further early stage studies. Furthermore, the absence of a substantial gender effect suggests that this can be appropriately used in a mixed gender population. Although there was some evidence of increased exposure in females in the fasted state, this may have been primarily due to variability, which was substantially higher for males in the fasted state in this study (one male subject showed no meaningful exposure after being dosed with SRT2104 in the fasted state). Regardless, the variability and sample size prevents definitive conclusions with regard to gender effect on exposure, but the data suggests that any gender differences would be minimal. Finally, SRT2104 in a capsule does not greatly impact exposure (compared with a suspension formulation), allowing a more facile drug product to be utilized in larger studies.
In general, the use of a 2.0 g dose for target validation in the clinic presents a risk that pharmacological effects resulting from interaction with targets other than SIRT1 could confound any SIRT1-specific efficacy signals that might be observed. In the absence of a valid biomarker for SIRT1 activation, one approach to discharge the liability of a large dose with respect to target engagement in vivo is to evaluate extensively the selectivity of SRT2104. Indeed, SRT2104 displays minimal non-SIRT1 related activity as assessed by an in vitro profiling study including over 180 targets representing kinases, GPCRs, nuclear recptors, ion channels, enzymes and transporters (unpublished results). In vitro characterization of this favourable selectivity profile, along with its safety and tolerability profile in healthy volunteers, suggested the evaluation of high doses of SRT2104 in exploratory clinical trials in patient populations is acceptable.
The clinical development for SRT2104 will include studies designed to demonstrate that administration of this compound can exert a pharmacological effect in humans and help to validate SIRT1 as a target of therapeutic value. Increased SIRT1 activity by overexpression of both the gene and protein has been reported to have beneficial effects in inflammation and metabolic disease [46–49]. Attenuation of the immune response can be evaluated through the measurement of cytokines and other pro-inflammatory markers, as well as by clinical assessment of symptoms known to be associated with acute LPS administration (fever, headache, generalized malaise, and myalgia) [51–54], exercise tolerance and disuse atrophy [55], all of which could be assessed in a series of small, focused exploratory medicine studies.
The current formulation of SRT2104 is extremely minimal and does not circumvent the inherently poor physical properties of the molecule. Of particular concern is the low bioavailability and the significant inter-subject variability in exposure. Although there appears to be opportunity for formulation optimization, this may be a significant challenge should a high dose be required. In addition, reliance on a food effect to maximize exposure, while useful in exploratory studies, would present some challenges later in development since patients would be required to take SRT2104 with food. In fact, some patient populations (type II diabetics, ulcerative colitis patients) may be unable to comply with instructions to take SRT2104 following a standardized meal based on dietary restrictions. Despite these issues, the remarkable pharmacology associated with SIRT1 modulation in preclinical models makes assessment of this target in humans of paramount importance. Indeed, SRT2104 is the first, highly selective small molecule activator of SIRT1 to progress to human subjects. Demonstration of pharmacological activity in humans, and correlation of this activity with preclinical efficacy data, will provide support for the tractability of SIRT1 as a drug target. Despite some of the challenges highlighted above, SRT2104 is well-tolerated and has limited non-SIRT1 activity, making it a useful molecule for interrogating SIRT1 activation in the clinic during early stage development. Future clinical studies, with comprehensive pharmacokinetic assessments, may allow for detection of a clinical signal as well as associated pharmacodynamic drivers, thereby allowing for a better understanding of the biological impact of selected SIRT1 activation in a variety of human diseases.
Acknowledgments
The bioanalysis was performed by Simbec, Ltd, UK under contract by the sponsor, with special thanks to Tracy Thomas. Quotient Clinical Ltd and Dr Lloyd Stevens, in particular, assisted with the bioavailability study under contract by the sponsor. The quantification of the 14C-SRT2104 was performed by Vitalea Inc (Davis, CA) under contract by the sponsor. In addition, the authors are grateful to Rachel D. Collier, Steve Corless, Clive Felgate, Sian Hilton, Andy Roberts, Rita Tailor, Graeme Young and Claire Beaumont for the metabolite identification.
Competing Interests
SRT2104 is currently under development by GlaxoSmithKline. All authors of this paper are currently or have been employed by GlaxoSmithKline and may own shares in the company.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1
Summary of pharmacokinetic parameters for SRT2104 after oral administration to healthy human volunteers (FIH and second food effect study)
Table S2
Summary of pharmacokinetic parameters for SRT2104 after oral administration of a 0.5 g dose to healthy human volunteers (Food/gender effect study)
References
- 1.Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins – novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov. 2008;7:841–853. doi: 10.1038/nrd2665. [DOI] [PubMed] [Google Scholar]
- 3.Blum CA, Ellis JL, Loh C, Ng PY, Perni RB, Stein RL. SIRT1 modulation as a novel approach to the treatment of diseases of aging. J Med Chem. 2010;54:417–432. doi: 10.1021/jm100861p. [DOI] [PubMed] [Google Scholar]
- 4.Lee JH, Song MY, Song EK, Kim EK, Moon WS, Han MK, Park JW, Kwon KB, Park BH. Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes. 2009;58:344–351. doi: 10.2337/db07-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo W, Qian L, Zhang J, Zhang W, Morrison A, Hayes P, Wilson S, Chen T, Zhao J. Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mTOR signaling. J Neurosci Res. 2011;89:1723–1736. doi: 10.1002/jnr.22725. [DOI] [PubMed] [Google Scholar]
- 6.Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L, Gu W, Accili D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang QJ, Wang Z, Chen HZ, Zhou S, Zheng W, Liu G, Wei YS, Cai H, Liu DP, Liang CC. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res. 2008;80:191–199. doi: 10.1093/cvr/cvn224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshizaki T, Milne JC, Imamura T, Schenk S, Sonoda N, Babendure JL, Lu JC, Smith JJ, Jirousek MR, Olefsky JM. SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Mol Cell Biol. 2009;29:1363–1374. doi: 10.1128/MCB.00705-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamazaki Y, Usui I, Kanatani Y, Matsuya Y, Tsuneyama K, Fujisaka S, Bukhari A, Suzuki H, Senda S, Imanishi S, Hirata K, Ishiki M, Hayashi R, Urakaze M, Nemoto H, Kobayashi M, Tobe K. Treatment with SRT1720, a SIRT1 activator, ameliorates fatty liver with reduced expression of lipogenic enzymes in MSG mice. Am J Physiol Endocrinol Metab. 2009;297:E1179–1186. doi: 10.1152/ajpendo.90997.2008. [DOI] [PubMed] [Google Scholar]
- 12.Shindler KS, Ventura E, Rex TS, Elliott P, Rostami A. SIRT1 activation confers neuroprotection in experimental optic neuritis. Invest Ophthalmol Vis Sci. 2007;48:3602–3609. doi: 10.1167/iovs.07-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gracia-Sancho J, Villarreal G, Zhang Y, Garcia-Cardena G. Activation of SIRT1 by resveratrol induces KLF2 expression conferring an endothelial vasoprotective phenotype. Cardiovasc Res. 2010;85:514–519. doi: 10.1093/cvr/cvp337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008;8:347–358. doi: 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 15.Smith JJ, Kenney RD, Gagne DJ, Frushour BP, Ladd W, Galonek HL, Israelian K, Song J, Razvadauskaite G, Lynch AV, Carney DP, Johnson RJ, Lavu S, Iffland A, Elliott PJ, Lambert PD, Elliston KO, Jirousek MR, Milne JC, Boss O. Small molecule activators of SIRT1 replicate signaling pathways triggered by calorie restriction in vivo. BMC Syst Biol. 2009;3:31. doi: 10.1186/1752-0509-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshizaki T, Schenk S, Imamura T, Babendure JL, Sonoda N, Bae EJ, Oh DY, Lu M, Milne JC, Westphal C, Bandyopadhyay G, Olefsky JM. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am J Physiol Endocrinol Metab. 2010;298:E419–428. doi: 10.1152/ajpendo.00417.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minor RK, Baur JA, Gomes AP, Ward TM, Csiszar A, Mercken EM, Abdelmohsen K, Shin Y-K, Canto C, Scheibye-Knudsen M, Krawczyk M, Irusta PM, Martin-Montalvo A, Hubbard BP, Zhang Y, Lehrmann E, White AA, Price NL, Swindell WR, Pearson KJ, Becker KG, Bohr VA, Gorospe M, Egan JM, Talan MI, Auwerx J, Westphal CH, Ellis JL, Ungvari Z, Vlasuk GP, Elliott PJ, Sinclair DA, de Cabo R. SRT1720 improves survival and healthspan of obese mice. Sci Rep. 2011;1:70. doi: 10.1038/srep00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, Campbell J, Bhimavarapu A, Luikenhuis S, de Cabo R, Fuchs C, Hahn WC, Guarente LP, Sinclair DA. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE. 2008;3:e2020. doi: 10.1371/journal.pone.0002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SH, Watkins DN, Herman JG, Baylin SB. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006;2:e40. doi: 10.1371/journal.pgen.0020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kabra N, Li Z, Chen L, Li B, Zhang X, Wang C, Yeatman T, Coppola D, Chen J. SirT1 is an inhibitor of proliferation and tumor formation in colon cancer. J Biol Chem. 2009;284:18210–18217. doi: 10.1074/jbc.M109.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 22.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 23.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 24.Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A. 2006;103:10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daitoku H, Sakamaki J, Fukamizu A. Regulation of FoxO transcription factors by acetylation and protein-protein interactions. Biochim Biophys Acta. 2011;1813:1954–1960. doi: 10.1016/j.bbamcr.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 28.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 29.Li L, Wang L, Wang Z, Ho Y, McDonald T, Holyoake TL, Chen W, Bhatia R. Activation of p53 by SIRT1 Inhibition Enhances Elimination of CML Leukemia Stem Cells in Combination with Imatinib. Cancer Cell. 2012;21:266–281. doi: 10.1016/j.ccr.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 31.Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 33.Timmers S, Konings E, Bilet L, Houtkooper Riekelt H, van de Weijer T, Goossens Gijs H, Hoeks J, van der Krieken S, Ryu D, Kersten S, Moonen-Kornips E, Hesselink Matthijs KC, Kunz I, Schrauwen-Hinderling Vera B, Blaak EE, Auwerx J, Schrauwen P. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011;14:612–622. doi: 10.1016/j.cmet.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smoliga JM, Baur JA, Hausenblas HA. Resveratrol and health – A comprehensive review of human clinical trials. Mol Nutr Food Res. 2011;55:1129–1141. doi: 10.1002/mnfr.201100143. [DOI] [PubMed] [Google Scholar]
- 35.Walle T, Hsieh F, DeLegge MH, Oatis JE, Walle UK. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab Dispos. 2004;32:1377–1382. doi: 10.1124/dmd.104.000885. [DOI] [PubMed] [Google Scholar]
- 36.Boocock DJ, Faust GE, Patel KR, Schinas AM, Brown VA, Ducharme MP, Booth TD, Crowell JA, Perloff M, Gescher AJ, Steward WP, Brenner DE. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol Biomarkers Prev. 2007;16:1246–1252. doi: 10.1158/1055-9965.EPI-07-0022. [DOI] [PubMed] [Google Scholar]
- 37.Park SJ, Ahmad F, Philp A, Baar K, Williams T, Luo H, Ke H, Rehmann H, Taussig R, Brown AL, Kim MK, Beaven MA, Burgin AB, Manganiello V, Chung JH. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell. 2012;148:421–433. doi: 10.1016/j.cell.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Franco JGO, de Moura EG, Koury JC, Trotta PA, Cordeiro A, Souza LL, Almeida NADS, Lima NLDS, Pazos-Moura CC, Lisboa PCC, Passos MCF. Resveratrol reduces lipid peroxidation and increases sirtuin 1 expression in adult animals programed by neonatal protein restriction. J Endocrinol. 2010;207:319–328. doi: 10.1677/JOE-10-0124. [DOI] [PubMed] [Google Scholar]
- 39.Costa Cdos S, Rohden F, Hammes TO, Margis R, Bortolotto JW, Padoin AV, Mottin CC, Guaragna RM. Resveratrol upregulated SIRT1, FOXO1, and adiponectin and downregulated PPARgamma1-3 mRNA expression in human visceral adipocytes. Obes Surg. 2011;21:356–361. doi: 10.1007/s11695-010-0251-7. [DOI] [PubMed] [Google Scholar]
- 40.Lappin G, Stevens L. Biomedical accelerator mass spectrometry: recent applications in metabolism and pharmacokinetics. Expert Opin Drug Metab Toxicol. 2008;4:1021–1033. doi: 10.1517/17425255.4.8.1021. [DOI] [PubMed] [Google Scholar]
- 41.Lappin G, Garner RC. Current perspectives of 14C-isotope measurement in biomedical accelerator mass spectrometry. Anal Bioanal Chem. 2004;378:356–364. doi: 10.1007/s00216-003-2348-5. [DOI] [PubMed] [Google Scholar]
- 42.Hop CE, Wang Z, Chen Q, Kwei G. Plasma-pooling methods to increase throughput for in vivo pharmacokinetic screening. J Pharm Sci. 1998;87:901–903. doi: 10.1021/js970486q. [DOI] [PubMed] [Google Scholar]
- 43.Dear GJ, Roberts AD, Beaumont C, North SE. Evaluation of preparative high performance liquid chromatography and cryoprobe-nuclear magnetic resonance spectroscopy for the early quantitative estimation of drug metabolites in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;876:182–190. doi: 10.1016/j.jchromb.2008.10.040. [DOI] [PubMed] [Google Scholar]
- 44.Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, Steele AD, Crowe H, Marmor S, Luo J, Gu W, Guarente L. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007;6:759–767. doi: 10.1111/j.1474-9726.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- 46.de Jonge E, Dekkers PE, Creasey AA, Hack CE, Paulson SK, Karim A, Kesecioglu J, Levi M, van Deventer SJ, van der Poll T. Tissue factor pathway inhibitor does not influence inflammatory pathways during human endotoxemia. J Infect Dis. 2001;183:1815–1818. doi: 10.1086/320723. [DOI] [PubMed] [Google Scholar]
- 47.de Jonge E, Dekkers PE, Creasey AA, Hack CE, Paulson SK, Karim A, Kesecioglu J, Levi M, van Deventer SJ, van Der Poll T. Tissue factor pathway inhibitor dose-dependently inhibits coagulation activation without influencing the fibrinolytic and cytokine response during human endotoxemia. Blood. 2000;95:1124–1129. [PubMed] [Google Scholar]
- 48.Dekkers PE, Juffermans NP, ten Hove T, de Jonge E, van Deventer SJ, van der Poll T. Endotoxin down-regulates monocyte and granulocyte interleukin-6 receptors without influencing gp130 expression in humans. J Infect Dis. 2000;181:1055–1061. doi: 10.1086/315356. [DOI] [PubMed] [Google Scholar]
- 49.Dekkers PE, Lauw FN, ten Hove T, te Velde AA, Lumley P, Becherer D, van Deventer SJ, van der Poll T. The effect of a metalloproteinase inhibitor (GI5402) on tumor necrosis factor-alpha (TNF-alpha) and TNF-alpha receptors during human endotoxemia. Blood. 1999;94:2252–2258. [PubMed] [Google Scholar]
- 50.Van der Poll T. Endotoxemia in healthy subjects as a human model of inflammation. In: Marshall JC, Cohen CJ, editors. The Immune Response in the Critically Ill. Berlin: Springer-Verlag; 1999. pp. 335–357. [Google Scholar]
- 51.Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 52.Heilbronn LK, Civitarese AE, Bogacka I, Smith SR, Hulver M, Ravussin E. Glucose tolerance and skeletal muscle gene expression in response to alternate day fasting. Obes Res. 2005;13:574–581. doi: 10.1038/oby.2005.61. [DOI] [PubMed] [Google Scholar]
- 53.Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- 54.Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.