

Abstract
AIMS
This paper describes findings from the first-in-human study for GSK1482160, an orally available allosteric P2X7 receptor modulator. The study aimed to assess the pharmacokinetics (PK), pharmacodynamics (PD), safety and tolerability of the compound in healthy subjects.
METHODS
Escalating single doses of up to 1 g were administered to healthy subjects in a single-blind and placebo-controlled fashion. Safety, tolerability, blood drug concentrations and ex vivo Il-1β production in blood were evaluated.
RESULTS
Drug concentration peaked within 3.5 h of dosing under fasting conditions and declined thereafter with a relatively short half-life of less than 4.5 h. Exposure was proportional to dose with between subject variability of less than 60%. A PK/PD model quantified Il-1β as a function of drug exposure. The model allowed simulation of in vivo pharmacology for various untested dose levels and regimens. Furthermore, the mechanistic model supported the hypothesis that the compound reduces the efficacy of ATP at the P2X7 receptor without affecting its affinity. No major safety or tolerability concerns were identified in this small study (n = 29), except for one case of asymptomatic accelerated idioventricular rhythm at the top dose.
CONCLUSION
The model-based approach maximized analysis power by integrating all biomarker data and revealed mechanistic insight into the pharmacology of P2X7 modulation by GSK1482160. Simulations by this model ultimately led to the discontinuation of the development of this compound. The therapeutic relevance of the P2X7 receptor remains to be tested in patients. The mechanistic-model-based approach can be applied widely to drug development.
Keywords: clinical pharmacology, first-in-human, GSK1482160, Il-1β, P2X7 receptor, population pharmacokinetic-pharmacodynamic modelling
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
P2X7 receptors are involved in the production of pro-inflammatory cytokines, such as Il-1β, by central and peripheral immune cells. Il-1β has been implicated as an important mediator of inflammation. Therefore, the P2X7 receptor is an attractive therapeutic target for inflammatory diseases.
WHAT THIS STUDY ADDS
Findings in pharmacokinetics, pharmacodynamics, safety and tolerability of a P2X7 receptor modulator, GSK1482160, from the first human study for the molecule are described. A pharmacokinetic/pharmacodynamic model for LPS-stimulated ATP-induced Il-1β production in blood allowed the integration of all relevant data and provided in vivo evidence of the nature of the drug–receptor interaction. The model has the potential to be used to simulate future trials.
Introduction
This paper describes the findings from the first-in-human study (FIH) for GSK1482160. The study aimed to evaluate the single dose safety, tolerability, pharmacokinetics (PK) and pharmacokinetic/pharmacodynamic (PK/PD) relationship (in terms of a biomarker) of the molecule within an exposure range defined by pre-clinical findings, which could be used to identify dosing regimens for phase 2 studies.
The purinergic receptor P2X ligand-gated ion channel type 7 (P2X7 receptor [1]) is an adenosine triphosphate (ATP) gated non-selective ion channel which is expressed on surface membranes of cells that are largely, but not exclusively, of haematopoietic origin including macrophages, monocytes and microglia [2, 3]. There are several amino acid changing single nucleotide polymorphisms (SNPs) of the P2X7 receptor gene including the Ala348Thr, Thr357Ser, Gln460Arg and Glu496Ala SNPs that are predicted to alter protein structure or function [4]. Of these, the Glu496Ala (rs3751143) SNP has been associated with loss of P2X7 receptor function [5, 6].
P2X7 receptor is an attractive target for the treatment of inflammatory pain conditions as it plays a pivotal role in the release of pro-inflammatory cytokines including interleukin-1 beta (Il-1β) and interleukin 18 (Il-18) from cells of the immune system in both the periphery and central nervous system (CNS) [3, 7–9]. It has also been investigated as a potential target for neuroprotection [9, 10]. Il-1β has been implicated as an important mediator of inflammatory diseases such as rheumatoid arthritis (RA), since increased systemic levels of Il-1β have been detected in RA patients both in vivo[11] and by ex vivo stimulation of blood [12]. The involvement of the P2X7 receptor in inflammatory and neuropathic pain is supported by findings in knock-out mouse models [13]. Furthermore, there is a therapeutic precedent for targeting Il-1β in RA, as the non-selective Il-1 receptor antagonist anakinra has been shown to attenuate clinical and laboratory markers associated with RA [14]. Therefore, P2X7 receptor modulators may have the potential to reduce the impact of inflammation and pain in conditions such as RA.
Allosteric modulators bind at a site on the receptor topographically distinct from the endogenous agonist, and can modulate the affinity and/or the intrinsic efficacy of the agonist or even directly affect the signalling [15]. GSK1482160, N-{[2-chloro-3-(trifluoromethyl)phenyl]methyl}-1-methyl-5-oxo-L-prolinamide, is an orally available negative allosteric modulator of the P2X7 receptor with excellent in vitro potency (in functional and electrophysiological assays at recombinant and naive P2X7 receptors across multiple species including humans) and cross-target selectivity. In target validation studies, the compound demonstrated efficacy comparable with the gold standards celecoxib and gabapentin in rat models for inflammatory (Freund's complete adjuvant-induced chronic joint pain) and neuropathic pain (chronic constriction injury), respectively. GSK1482160 was shown to have desirable physicochemical properties and an acceptable PK and safety profile in pre-clinical species. The compound was therefore progressed into clinical development for these indications.
Early understanding of the potentially clinically efficacious or pharmacologically active concentration range of a drug, in relation to the exposure limit that is defined by pharmaceutical properties and non-clinical toxicological findings, is important. The clinical development of novel treatments for conditions such as pain is often impaired by the lack of quantifiable markers of direct pharmacological activity that are relevant to the human conditions. Apart from using imaging technologies, it is often not possible to determine confidently and easily PD activities of novel analgesic compounds in the tissues of interest such as the knee joint or spinal cord [16].
To measure the pharmacology of a P2X7 receptor modulator, an assay has been developed to determine Il-1β release following ex vivo stimulation of human blood with lipopolysaccharide (LPS) and ATP. Whilst this methodology has previously been used to characterize the pharmacology of the receptor [17], we sought to understand better the variability of the assay and to optimize the assay conditions in a pilot study prior to its application in the FIH of a new molecule. Based upon the results of this pilot study, the assay was optimized and subsequently incorporated into the design of the FIH for GSK1482160 so that PK/PD relationships could be determined.
Methods
Subjects
Volunteers for both the pilot study and the FIH with GSK1482160 were recruited from a healthy volunteer panel. Both studies were carried out in the GSK Clinical Unit (Addenbrooke's Hospital, Cambridge, UK) in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines and following receipt of regulatory and ethics committee's approval. Subjects provided fully informed written consent for their participation in the study. A separate informed consent procedure was used for participation in the genetic research part of the protocol. Subjects included in the study were non-obese healthy male or female individuals aged between 18–75 years in the pilot study and 18–55 years in the FIH. Females included in FIH were only enrolled if they were deemed as being of non-child-bearing potential. Prescription or non-prescription medications were prohibited during the studies.
Pilot study
This was a test-retest study design involving a single cohort of 10 volunteers tested on two separate occasions, 1 week apart. Whole blood (10 ml) was taken into anticoagulant (sodium citrate) blood tubes by venepuncture of the median cubital vein on the anterior forearm. For each subject, blood was drawn at the same time of day during the consecutive visits. Laboratory staff were blinded to the subject identification and visit information.
Working stock solutions of ATP (20, 10, 5 and 2.5 mm) (Sigma) and LPS 1 mg ml−1 (Sigma) were prepared for each study day. Blood samples (2 ml) were incubated without or with LPS (final concentration 1 µg ml−1) for 2 h at 37°C in a humidified 5% CO2 incubator. Samples were then aliquotted into 96 well plates (80 µl well−1) and ATP solutions (20 µl well−1) added to give final ATP concentrations of 0, 0.5, 1, 2 and 4 mm and incubated further for 30 min. Reactions were stopped with the addition of 150 µl of ice cold plain Roswell Park Memorial Institute (RPMI) 1640 tissue culture medium to each well and incubated for 10 min on ice. Plates were spun at 300 g (1200 rev min−1) for 5 min in a refrigerated (4°C) centrifuge (Beckton Dickinson). Plasma supernatants were aliquoted into fresh 96 well plates (10 µl well−1) and stored at −80°C until required for assay.
Measurement of plasma Il-1β was determined using the BioPlex bead array system (Bio-Rad Laboratories). Samples were diluted 1 in 15 in human serum diluent (Bio-Rad) before analysis. Concentrations were interpolated from standard curves generated within the Bio-Plex software.
Data were analyzed by mixed model analysis of variance (anova). Experimental conditions were treated as fixed effects and subject was treated as a random effect. Sample collection occasion (week) was treated as a fixed effect to assess occasion variation in the assay. Order of the blood sample (i.e. whether the blood was taken at 08.45, 09.00, 09.15, 09.30 or 09.45 h) was also investigated as a fixed effect to see if there was a difference according to how long the blood sample had been kept before stimulating it.
First-in-human study
Study design
This was a single-blind, randomized, placebo-controlled, single dose escalation study in healthy volunteers. Consecutive cohorts of eight subjects were dosed upon four separate occasions within each cohort, with adequate washout periods between occasions. Dose was increased within and between cohorts. Each subject received three active doses, plus placebo which was randomly assigned across the four dosing sessions. Therefore for each dose escalation step, six subjects received the active dose and two subjects received placebo to allow for evaluation of whether adverse events might be attributable to treatment. Upon completion of dose escalation, the effects of co-administration of a high fat and high calorie meal on the PK of GSK1482160 were evaluated in a crossover fashion in eight subjects. It was anticipated that dose escalation would require up to three cohorts of eight subjects. Therefore four cohorts were planned for the entire study.
Dose selection
The study aimed to assess PK, PD, tolerability and safety of GSK1482160 over a range of blood concentrations between the level predicted to cause a minimum anticipated biological effect level (MABEL) and a pre-defined maximum safe level. Accordingly, doses were selected to generate exposure levels at three-fold increments for this intended concentration range.
The MABEL was defined as 10% ex vivo inhibition of Il-1β release in human monocytes following stimulation with LPS/ATP. The concentration of GSK1482160 for this level of inhibition was identified in an in vitro assay. The maximum safe exposure level was based on the no observed adverse effect level (NOAEL) achieved in 28 day toxicology studies. The studies were conducted at 10, 30 and 100 mg kg−1 day−1 doses in the rat and the dog. The NOAEL for the rat study was the highest tested dose of 100 mg kg−1 day−1, which produced a mean AUC of 500 µg ml−1 h and Cmax of 40.8 µg ml−1. In the dog study, lesions in multiple organs were found in animals receiving 100 mg kg−1 day−1 and the 30 mg kg−1 day−1 dose was identified as the NOAEL. The mean AUC of 72.3 µg ml−1 h and Cmax of 15.8 µg ml−1 at the NOAEL, both of which were markedly lower than the corresponding values in the rat study, were therefore used to define the safety exposure limits for the FIH.
GSK1482160 was shown to be metabolically stable in liver microsomes, liver S9 fraction and hepatocytes from multiple species including humans and there was no evidence of metabolic activation in microsomes. There was evidence of hydroxylation in isolated perfused rat livers. In animal species, the majority of drug-related materials was recovered in the urine. For the prediction of human PK, scaling techniques using data derived from rat, dog and monkey indicated that a dose range of 0.3 mg to 1000 mg would produce the exposure range defined above. For this dose range, the AUC was predicted to be in the range of 34 to 113 000 ng ml−1 h and the Cmax was predicted to be in the range of 4 to 12 000 ng ml−1.
The starting dose of 0.3 mg, which targeted the MABEL exposure for a Cmax of 4 ng ml−1, compared with the maximum safe Cmax of 15.8 µg ml−1, was selected according to human PK prediction. Subsequent doses were chosen for three-fold exposure increments following a review by a GSK study team and the investigator of all available in-trial safety, tolerability, PK and PD data. Dose escalation was halted when the observed AUC or Cmax approached the corresponding pre-defined safe level. Although efficacy was observed in animal models at about 50% inhibition of Il-1β release, we considered it necessary to reach exposure in humans that would result in at least 90% inhibition. Exploratory modelling of in vitro data generated in human blood suggested the potential to achieve a sufficient therapeutic window.
Dosing and safety monitoring
During the screening visit, subjects provided a medical history and were assessed for good health by a physician on the basis of physical examination, vital signs, 12-lead electrocardiogram (ECG), ambulatory 24 h continuous cardiac monitoring (Holter ECG) and clinical laboratory tests. During the dosing sessions, subjects in each of the respective cohorts attended the clinical unit the evening prior to dosing and remained in-house for 24 h after dosing. Subjects were required to undergo a physical examination, vital sign and clinical laboratory assessments (haematology, clinical chemistry and urinalysis) prior to being fasted from 22.00 h the evening before dosing. Baseline vital signs and 12-lead ECG were recorded immediately prior to dosing. Subjects were dosed orally with GSK1482160 tablets or matching placebo, and at each new dose level individual subject dosing was staggered at hourly intervals to minimize the number of subjects exposed to the drug in the event of any unexpected serious acute reaction to the drug. Less than half the subjects were administered active treatment on the first day of dosing and the remainder of the cohort was dosed 24 h later following a preliminary review of the safety and tolerability results in the subjects who had already been dosed. Following dosing, subjects were assessed at frequent pre-defined intervals for vital signs and 12-lead ECG for 24 h, whilst Holter ECG and lead II telemetry were continuously monitored for 24 h and 8 h respectively. Clinical laboratory samples for safety purposes were collected at 12 h, 24 h and on day 3 post dose. Food and drinks were only allowed at 6 h and 12 h after dosing with the exception of room temperature water, which was allowed at anytime. All subjects underwent a safety follow-up visit between 7 and 10 days post dose.
PK data collection and analysis
Approximately 2 ml of blood was collected by venepuncture immediately prior to dosing, as well as at 5, 15, 30, 45 min and 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 16 and 24 h post dosing. Triplicates of 20 µl aliquots were applied to FTA Elute filter paper discs on a blood spot card (Whatman grade BFC 180) and allowed to dry for approximately 2 h. GSK1482160 was extracted by addition of methanol and analyzed by high performance liquid chromatography mass spectrometry/mass spectrometry. The quantification range was 1–1000 ng ml−1. Assay bias was within 14% and precision was within 12%. Individual non-compartmental PK parameter values were estimated using software WinNonlin Professional Edition version 5.2 (Pharsight Corporation, Mountain View, CA).
PD data collection
Approximately 2.7 ml blood was collected by venepuncture into Sarstedt Monovette sodium citrate tubes and the samples were kept at room temperature on a roller for a maximum of 90 min before stimulation. Five samples, including a baseline, were collected from each subject and analyzed as described above, at final ATP concentrations of 0, 0.5, 1 and 4 mm. The post dose sampling times were initially 0.25, 1.5, 5 and 8 h. They were modified to 0.5, 1, 4 and 8 h during the study based on interim findings to capture adequately the time course of PD response.
PK/PD analysis
Modelling was conducted to investigate the relationship between GSK1482160 blood concentration, corrected for dilution in the assay, and the LPS/ATP induced Il-1β release. A mechanistic model was applied to provide insight on the nature of the pharmacology at receptor level. Given that GSK1482160 is an allosteric negative modulator of the P2X7 receptor, the measured Il-1β concentrations were considered as a sum of pre-treatment baseline level, ATP-induced release that was independent of the P2X7 receptor and a response which was subject to P2X7 receptor modulation (equation 1).
| (1) |
where [Il-1β] is the total Il-1β concentration (ng ml−1), BASE is the pre-treatment Il-1β concentration (ng ml−1), SLOPE is the slope of the ATP-induced Il-1β release that is non-specific to P2X7 receptor [(ng ml−1)/mm], RESP is the Il-1β release by ATP (mm) that is subject to GSK1482160 modulation (ng ml−1) via the P2X7 receptor and [ATP] is the ATP concentration (mm).
Variations of a general model for allosteric modulation (equation 2) [14] were evaluated. In this case, ATP was the agonist and GSK1482160 the allosteric modulator. It can be shown that fixing certain parameters in this model to their null-hypothesis values reflects special cases of the interaction.
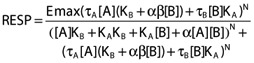 |
(2) |
where Emax is maximal possible response, [A] and [B] are the ligand concentrations for the agonist (ATP) and the modulator (GSK1482160) respectively, τA and τB are the operational measures of efficacy for the respective ligands, KA and KB are the receptor affinities for the respective ligands, α is the binding cooperativity factor describing the allosteric effect of the modulator on the agonist affinity and vice versa, β is the activation cooperativity factor which quantifies the allosteric effect of the modulator on the signalling efficacy of the agonist and N is a slope factor.
Blood concentration of GSK1482160 was treated as the independent variable, Il-1β concentration as the dependent variable and ATP concentration as a continuous covariate. A direct PK/PD correlation was applied to observed drug and Il-1β concentrations. Data from all subjects at all doses including placebo were fitted simultaneously using software NONMEM (version VI, ICON Solutions, 2008). In addition to the population mean values of the model parameters, between subject variability was estimated for model parameters when supported by the data. Model testing and selection was based on graphical diagnostics, residual variability, estimation precision and difference in objection functions (which approximates Chi-square distribution) between nested models.
PGx sample collection and analysis
Pre-dose venous blood was collected into a 7.5 ml vacutainer tube containing K2EDTA anticoagulant. The nucleic acids were isolated via a modified salting-out precipitation method using the Gentra PureGene kit. Genotyping of Glu496Ala (rs3751143) was performed using a TaqMan® single nucleotide polymorphism assay according to the manufacturer's specifications. Purified genomic DNA (20 ng) was used for each TaqMan® reaction. Genotyping assay details can be provided upon request.
Results
Subject characteristics
In the pilot study, subjects consisted of nine female and one male Caucasian healthy volunteers (mean age 49 years, range 33–70 years). Nine of the 10 subjects enrolled completed both sessions of the pilot study with one subject withdrawing for personal reasons. In the FIH study, 29 subjects with an average age of 38.0 years (range 19–55years) were randomized and of these 28 were male and one was female. Twenty-seven of the subjects were White, one was of mixed race and one was Asian. Three of the randomized subjects were withdrawn, one for an SAE, one for an AE and one at the investigator's discretion. The safety and PK/PD populations comprised all 29 subjects. The PK population comprised 27 subjects (having quantifiable concentration data); and valid genotyping data was generated for 26 subjects (PGx population).
Pilot study
Elevated levels of Il-1β were detected following stimulation with 1 µg ml−1 LPS and various concentrations of ATP (Figure 1). While there was no statistically significant difference between LPS 1 µg ml−1 (without ATP) and RPMI, LPS caused statistically significantly greater Il-1β production than RPMI in the presence of ATP. There is a clear dose–response in Il-1β release with larger differences being observed as the ATP concentration increased (Table 1). The standard deviations of the assay in the presence of ATP translated to 9–26% within subjects and 17–33% between subjects. The comparison for Il-1β release between week 1 and week 2 at 0.5 mm ATP was not statistically significant. However, for all other concentrations of ATP, small (<30%) but significant reductions in week 2 levels when compared with week 1 were detected (data not shown).
Figure 1.
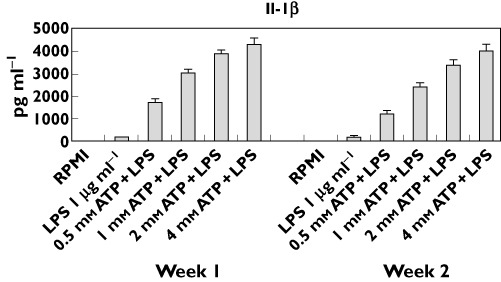
ATP caused Il-1β production after ex vivo stimulation of whole blood by LPS
Table 1.
LPS (1 µg ml−1) stimulated Il-1β release compared with reference (RPMI)
| ATP (mm ) | Test (pg ml−1) | Reference (pg ml−1) | Difference (95% CI)* (pg ml−1) | Between subject SD (pg ml−1) |
|---|---|---|---|---|
| 0 | 192.46 | 3.33 | 189.13 (−62.95, 441.20) | 136.5 |
| 0.5 | 1497.36 | 3.33 | 1494.03 (1241.95, 1746.11) | 493.0 |
| 1.0 | 2769.76 | 3.33 | 2766.43 (2514.35, 3018.51) | 472.0 |
| 2.0 | 3666.82 | 3.33 | 3663.49 (3411.41, 3915.57) | 652.5 |
| 4.0 | 4195.24 | 3.33 | 4191.90 (3939.82, 4443.98) | 835.0 |
P < 0.05 when the 95% CI excludes 0. Within subject SD = 391.5 pg ml−1.
First-in-human study
Three cohorts of eight subjects were dosed. Treatment allocation and sequence are shown in Table 2.
Table 2.
Treatment allocation in the first-in-human study
| Dose (mg) | |||
|---|---|---|---|
| Cohort 1 (n = 8) | Placebo, 0.3, 1, 3, 10 | ||
| Cohort 2 (n = 8) | Placebo, 30, 100, 300, 600 | ||
| Cohort 3 (n = 8) | Placebo, 1000, 300 (fed), 300 (fasting) | ||
In cohorts 1 and 2, each subject underwent four dosing events, when they received three of the four active doses in increasing order plus placebo. The occasion when the placebo was administered was randomized. In cohort 3, each subject underwent three dosing events. They were randomized to first receive 1000 mg (n = 6) or placebo (n = 2), and then receive 300 mg doses in fed and fasting condition in a crossover fashion.
Safety and tolerability
Dose escalation continued until the planned maximum exposures were achieved in line with the pre-defined PK stopping criteria of AUC 72 µg ml−1 h and Cmax 15.8 µg ml−1. A total of 81 AEs were reported by 20/29 subjects (69%) in this study. Most were of mild or moderate intensity, with the most common being headache (Table 3). Two subjects were withdrawn from the study due to adverse events. Following review of the 24 h Holter ECGs, a serious adverse event of a single asymptomatic run of accelerated idioventricular rhythm was observed in a 41-year-old male at 1 h 25 min following dosing with 1000 mg GSK1482160. He was then withdrawn from the study and hospitalized for further continuous cardiac monitoring. There was no further evidence of arrhythmic disturbance. A follow-up cardiology consultation concluded that the subject had no identifiable structural cardiac pathology or cardiovascular risk factors based upon echocardiogram, 12-lead ECG and clinical laboratory assessments. This event was considered by the investigator to be related to the study drug. In addition, a second subject reported AEs of pain and headache of moderate intensity following dosing with 3 mg GSK1482160. The following day, the same subject also reported body pains, cough, nasal congestion and dry throat and was then withdrawn from the study. The AEs in this second subject was considered to be un-related to the study drug. There were no patterns or trends for changes in any of the clinical laboratory parameters, vital signs or 12-lead ECG parameters in relation to GSK1482160 dose.
Table 3.
Summary of adverse events in the FIH study
| Dose (mg) | 0.3 | 1 | 3 | 10 | 30 | 100 | 300 | 600 | 1000 | 300 Fasting | 300 Fed | 0 (Placebo) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of subjects | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 8 | 8 | 17 |
| Number of subjects (%) reporting AEs | 3 (50) | 4 (67) | 3 (50) | 4 (67) | 2 (33) | 3 (50) | 1 (17) | 2 (33) | 2 (33) | 3 (38) | 4 (50) | 6 (35) |
| Number of subjects reporting AEs that were reported by more than one subject (%) | ||||||||||||
| Headache | 2 (33) | 2 (33) | 1 (17) | 2 (33) | 0 | 1 (17) | 1 (17) | 0 | 1 (17) | 1 (13) | 1 (13) | 3 (18) |
| Application site rash | 0 | 0 | 2 (33) | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 0 | 0 |
| Fatigue | 0 | 2 (33) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Nausea | 0 | 2 (33) | 1 (17) | 0 | 1 (17) | 1 (17) | 0 | 0 | 0 | 0 | 0 | 0 |
| Cough | 0 | 2 (33) | 1 (17) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (6) |
PK profile
The median blood concentration–time profile is illustrated in Figure 2 and blood PK parameters are summarized in Table 4.
Figure 2.
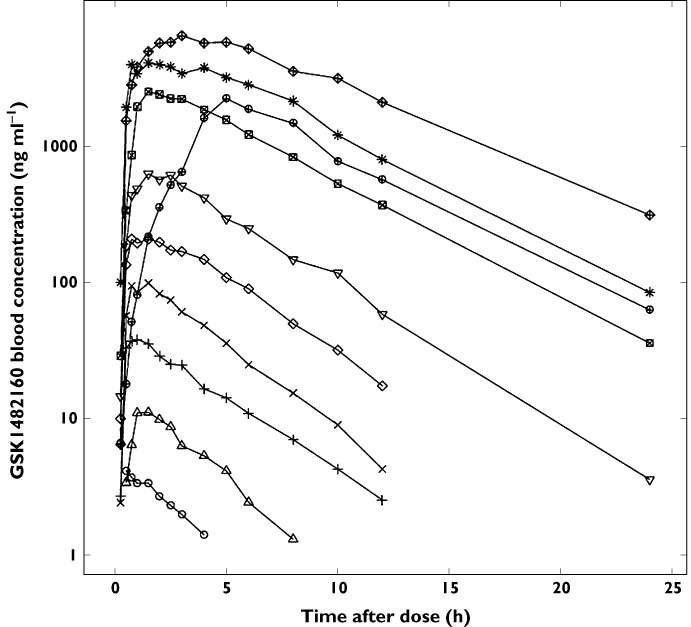
Median blood concentration of GSK1482160 after a single oral dose.  , 0.3 mg;
, 0.3 mg;  , 1 mg;
, 1 mg;  , 3 mg;
, 3 mg;  , 10 mg;
, 10 mg;  , 30 mg;
, 30 mg;  , 100 mg;
, 100 mg;  , 300 mg;
, 300 mg;  , 300 mg-fed;
, 300 mg-fed;  , 600 mg;
, 600 mg;  , 1000 mg
, 1000 mg
Table 4.
Blood exposure [geometric mean (CV%)] to GSK1482160
| Treatment | n | AUC(0,∞) (ng ml−1 h) | Cmax (ng ml−1) |
|---|---|---|---|
| 0.3 mg | 6 | 15.3 (32.5) | 4.9 (39.4) |
| 1 mg | 5 | 57.4 (38.4) | 15.3 (10.6) |
| 3 mg | 5 | 171 (40.9) | 46.7 (24.7) |
| 10 mg | 5 | 467 (34.9) | 131 (17.5) |
| 30 mg | 6 | 1 270 (18.5) | 268 (22.1) |
| 100 mg | 6 | 3 750 (12.5) | 737 (32.5) |
| 300 mg | 6 | 16 500 (21.3) | 2780 (18.0) |
| 600 mg | 6 | 34 000 (13.5) | 4820 (20.3) |
| 1000 mg | 6 | 64 900 (30.4) | 7100 (39.6) |
| 300 mg Fasting | 8 | 18 100 (29.2) | 2600 (31.0) |
| 300 mg Fed | 8 | 19 700 (57.8) | 2520 (52.5) |
Both AUC(0,∞) and Cmax appeared proportional to dose. Within the dose range tested, each milligram dose generated a median AUC(0,∞) of 57 ng ml−1 h and Cmax of 9 ng ml−1. A power model was used to assess dose proportionality for these parameters. The linearized slope (adjusted mean, 90% confidence interval) for AUC(0,∞) (1.04, 1.01, 1.07) and Cmax (0.908, 0.879, 0.938) were both close to unity, confirming that the exposure measures were overall proportional to dose within the tested range. Between subject variability was low to moderate at any given dose level, 13–58% for AUC(0,∞) and 11–53% for Cmax. Cmax was observed relatively early with geometric mean from 0.6 h at 0.3 mg to 3.5 h at 1000 mg, occurring slightly later at higher doses. Half-life was relatively short and independent of dose. Its geometric mean at any given dose ranged from 2.2–4.5 h.
Food delayed Cmax at 300 mg slightly from 2.5 h to 5 h. The food effect on AUC(0,∞) and Cmax was assessed using an analysis of variance (anova) model. The fed-to-fasted ratio (geometric mean, 90% confidence interval) for AUC(0,∞) (1.00, 0.860, 1.16) and Cmax (0.97, 0.756, 1.25) showed that a high fat meal did not alter either exposure parameter appreciably.
PK/PD relationship
Graphical assessment showed that GSK1482160 had no efficacy in the absence of ATP (Figure 3, ATP concentration = 0 nm) and therefore τB was fixed to 0. The possibilities that allosteric modulation could affect the affinity of ATP (α ≠ 1), the efficacy of ATP (expressed by β) or both were tested. The attempt to estimate both parameters resulted in a β close to zero, suggesting that full suppression of the efficacy of ATP was possible. This was supported by graphical inspection of the data. Fixing β to zero resulted in a small effect of α = 2, with several parameters poorly estimated, suggesting that the main effect was on efficacy. To improve estimation precision, α and β were fixed to 1 and 0, respectively. Allowing for an additional non-specific ATP effect (SLOPE) on Il-1β concentration improved the fitting.
Figure 3.
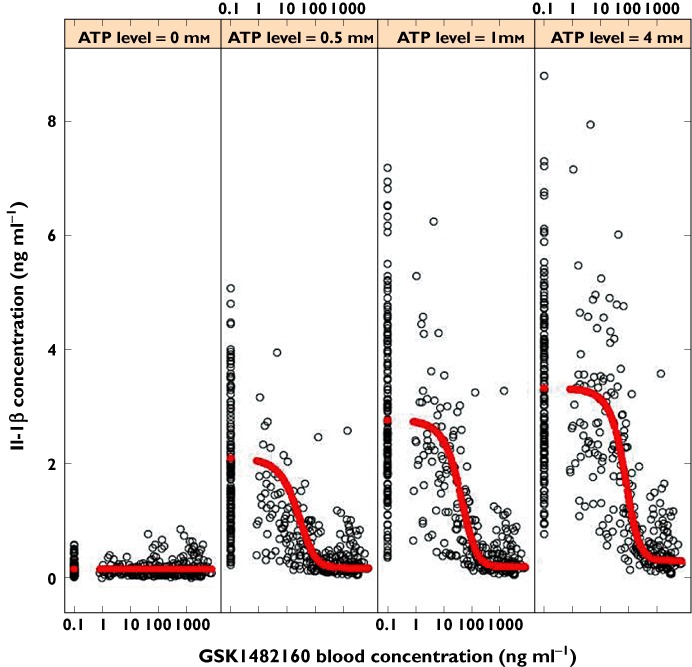
Observed (black) and model predicted (red) Il-1β concentration as a function of GSK1482160 blood concentration in FIH
Applying proportional between subject variability (BSV) on baseline, slope and efficacy parameters for ATP (τA and Emax) improved the fit. Because affinity is the feature of a chemical interaction for which large BSV was unlikely, BSV for KA and KB were not tested. In addition to a proportional residual error, an additive residual error was justified by the reduction of objective function. The final model selected fitted the data significantly better than the alternative models explored, as judged by the objective function.
The final PK/PD model for Il-1β (ng ml−1) therefore took the form of equation 3. Parameter estimates are shown in Table 5.
Table 5.
Summary of final PK/PD model parameters
| Parameter | Estimate (CV%) | 95% CI | BSV or residual variability (%) |
|---|---|---|---|
| Fixed effect parameter | |||
| BASE [ng ml−1] | 0.158 (11%) | (0.124, 0.192) | |
| KA[mm] | 1.17 (24%) | (0.61, 1.73) | |
| τA | 3.9 (23%) | (2.1, 5.7) | |
| N | 2.19 (12%) | (1.65, 2.73) | |
| Emax [ng ml−1] | 3.28 (8%) | (2.75, 3.81) | |
| KB[ng ml−1] | 32.2 (20%) | (19.1, 45.3) | |
| τB | 0 Fixed | ||
| α | 1 Fixed | ||
| β | 0 Fixed | ||
| SLOPE [(ng ml−1)/mm] | 0.037 (28%) | (0.016, 0.057) | |
| Random effect parameter | |||
| Variance BSV IL0 | 0.364 (23%) | (44, 73) | 60 |
| Variance BSVτA | 0.784 (59%) | (0, 131) | 89 |
| Variance BSV Emax | 0.0763 (19%) | (22, 32) | 28 |
| Variance BSV SLOPE | 0.584 (63%) | (0, 115) | 76 |
| Variance proportional residual | 0.141 (13%) | (32, 42) | 38 |
| Additive residual [ng ml−1] | 1.66 × 10−3 (42%) | (2.64 × 10−4, 3.06 × 10−3) |
The 95% CI for the variance parameters are the 95% CI for the BSV or Residual variability (%). See equations 1 and 2 for explanation for other abbreviations. BSV, between subject variability; CI, confidence interval; CV, coefficient of variation.
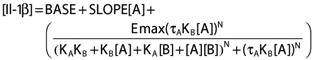 |
(3) |
The model fit the data well (Figure 3). The lack of clear bias was supported by graphical diagnostics. The baseline Il-1β concentration in absence of ATP was estimated to be 0.16 ng ml−1. This parameter was associated with a considerable between subject variability of 60% CV. There appeared to be a P2X7 receptor independent release of Il-1β, which was linearly related to ATP concentration by a SLOPE of 0.037 [(ng ml−1)/mm]. This means that under the experimental conditions, for an ATP concentration of 1 mm, 0.037 ng ml−1 Il-1β release could not be antagonized by GSK1482160. The between subject variability for this slope appeared large though it was poorly estimated.
The drug response was adequately described by the allosteric modulation model. The pharmacological effect of ATP on the P2X7 receptor was characterized by an affinity (KA) of 1.2 mm, an operational measure of efficacy (τA) of 3.9 and a maximal response (Emax) of 3.3 ng ml−1 Il-1β. Between subject variability in the Emax was a moderate 28% and the data were insufficient for reliable estimation of the variability in τA. The pharmacological effect of GSK1482160 on the P2X7 receptor was characterized by an affinity (KB) of 32 ng ml−1 and a lack of efficacy (τB = 0). While ATP and GSK1482160 did not affect each other's binding to the P2X7 receptor (α = 1), binding of GSK1482160 to the receptor abolishes the effect of the bound ATP. As such, the receptor-ATP-GSK1482160 binding complex had no residual efficacy (β = 0).
Visual inspection suggested a lack of marked effect on model parameters, whose values could be estimated for individuals, by several covariates (genotype, age or body weight). However, the ranges of these covariates were narrow and the sample size was small.
PGx
Among the FIH study subjects who provided genetic data (n = 26), the minor allele frequency of Glu496Ala was 17%. There was only one subject who carried two copies of the minor allele, while seven subjects carried one copy of the minor allele.
Pre-dose Il-1β release levels measured from lymphocytes ex vivo are presented in Figure 4 by ATP concentration and rs3751143 (Glu496Ala) genotype. There was an indication that the rs3751143 polymorphism influences Il-1β release levels, and that P2X7 receptor function increased with the number of RS3751143 T alleles (encoding for glutamine at amino acid position 496) carried. The difference in Il-1β release levels at 0.5 mm ATP, between subjects in the GT and TT genotype groups, was statistically significant (P= 0.049).
Figure 4.
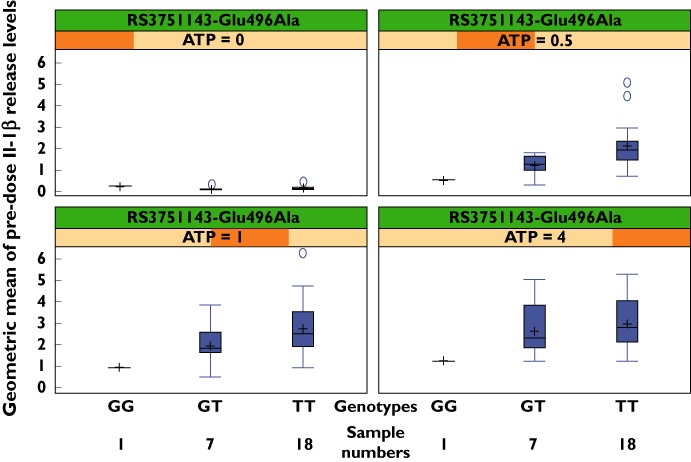
Pre-dose Il-1β release concentrations (ng ml−1) for Glu496Ala genotype in FIH of GSK1482160
Discussion
Blood exposures of GSK1482160 in humans were consistent with the predictions based on pre-clinical data. Following single oral doses over the range 0.3–1000 mg, the Cmax and AUC of the compound were moderately variable and proportional to dose. The time course of the drug concentration appeared mono-exponential, with relatively early Cmax and short half-life. Findings from this study suggest that full suppression of ATP induced Il-1β release via the P2X7 receptor after a single oral dose is well tolerated in healthy volunteers. One subject was withdrawn from the trial due to an asymptomatic run of accelerated idioventricular rhythm. There had been no preclinical observations of cardiac toxicity.
The Il-1β data were adequately fitted by a mechanistic model describing the nature of the allosteric interaction between ATP and GSK1482160. Besides characterizing the pharmacology of ATP at the P2X7 receptor level, the analysis provided an estimate of the affinity of GSK1482160 to the P2X7 receptor, which had not been possible to measure in vitro. The model also indicated that GSK1482160 itself had no efficacy for LPS -stimulated Il-1β release in the absence of ATP. The compound reduced the efficacy of ATP at the P2X7 receptor without affecting its affinity. An effect of ATP on Il-1β release that was not subject to GSK1482160 modulation was also quantified. However, this effect may be either truly physiological or an artefact of the assay.
The between subject variability shown in the pilot study probably reflected physiological variation and contributed to the variability in some of the PK–PD model parameters. Whilst responses to a number of different concentrations of ATP were explored, it is not clear which concentration, if any, is most physiologically relevant. Regardless, the highest concentration of 4 mm ATP administered represents a robust stimulus which may be relevant to the inhibition of Il-1β release in target tissues where the local concentrations of mediators may be high.
Consistent with previous findings, Il-1β release was lower in subjects carrying one or more copies of the G allele of rs3751143 which encodes alanine at amino acid position 496 [5]. The implications of this are not clear but one possibility is that P2X7 receptor modulators may not demonstrate the same level of efficacy in subjects carrying one or more copies of this particular allele.
In recent years there has been a notable lack of success in the field of analgesic drug discovery with compounds of novel mechanisms, including antagonists to NK1, glycine or adenosine A1 receptors, sodium channel blockers, anti-NGF (nerve growth factor) treatments and compounds targeting the endocannabinoid system. Whilst some of these failures may be attributable to the lack of relevance of the targeted mechanism in the pre-clinical models to the human pain conditions, there are cases when PK and/or toxicological limitations of the compound exposure have precluded sufficient target engagement in humans to test fully the mechanism.
To avoid some of the assumptions associated with pre-clinical models of nociception applied to the prediction of therapeutically relevant exposure in humans, the use of PD biomarkers in humans presents a useful strategy. The utility of the Il-1β release assay as a biomarker of PD activity of a P2X7 receptor modulator is supported by evidence of P2X7 receptors being primary mediators of Il-1β release in humans. Therefore, the assay provided an opportunity to measure directly in vivo target pharmacology, which needs to be sufficient to test the mechanism of action in future patient trials. One limitation of this biomarker is that the PD activity determined systemically might not necessarily reflect the situation in the tissue of interest which may be in a peripheral compartment such as the knee joint or the CNS. However, GSK1482160 readily crosses the blood brain barrier and is not a substrate for the known active efflux transporters. Free drug concentration is therefore expected to be the same in the entire distribution space at equilibrium. Assuming that the free concentration drives the interaction with the receptor, the ex vivo human blood assay should reflect the pharmacology at other sites at steady-state. A further consideration is that the PD effects for GSK1482160 were measured by ex vivo stimulation of blood with LPS and ATP, instead of by circulating endogenous levels of Il-1β, because the latter are low and variable, thus making it difficult to quantify. Finally, PD response in patients, which may be different from that in healthy subjects due to alterations in both P2X7 receptor expression and physiological concentrations of ATP, remains to be investigated.
A model-based approach was applied to the early clinical development of this compound. Such an approach enhances the efficiency of drug development by virtue of its capacity for enabling the quantitative interpretation of data in their entirety, as well as its versatility in scenario testing by simulation. We modelled the FIH PK/PD data from all subjects, at all dose levels and for all ATP concentrations simultaneously, thus maximizing the power for inference. Concentration–response modelling for the biomarker Il-1β informed a number of key decisions. Initially, a preliminary PK/PD model was developed based on in vitro Il-1β data. This model was used to assess the feasibility that adequate pharmacological engagement of the target could be achieved using human exposures of GSK1482160 that remain below the maximum toxicology NOAEL with sufficient safety margin. The model was then used in conjunction with human PK parameters predicted using pre-clinical data, to choose a range of doses for the FIH to explore safety, tolerability, PK and PD for the exposure range between minimal pharmacology and maximum safe level. The PK and PD data subsequently collected during the single dose FIH were subjected to population PK/PD modelling. The next step would be to develop a PK model, with the objective that the PK and PK/PD models could be used in combination for scenario testing, by precise simulation, to help select a steady-state dose regimen that would have the desirable pharmacology profile for future repeat dose trials. Preliminary simulations of various dose regimens using the PK/PD model concluded that it was not possible to achieve the level of pharmacology (>90% inhibition of Il-1β release throughout entire dosing interval) that was considered necessary to test the P2X7 mechanism adequately, while maintaining a sufficient safety margin with respect to the steady-state NOAEL. As such, these simulations informed a decision to terminate the development of GSK1482160 for a chronic inflammatory pain indication. Interestingly, Stock and colleagues have recently reported the results of an exploratory efficacy study with the selective P2X7 receptor antagonist, CP-224 535, in patients with RA [18]. That compound failed to achieve separation from placebo over a 12 week period despite the achievement of plasma concentrations that were 25 times the estimated IC90 for inhibition of Il-1 release ex vivo. The therapeutic relevance of P2X7 receptor modulation for rheumatoid arthritis or other inflammatory conditions remains to be established.
Acknowledgments
We gratefully acknowledge the subjects who participated in the studies. We also thank John Haselden, John Tonkyn, Nadia Garman, Yujay Ramakrishnan, Shilina Roman, Bill Davis, Joanna Ward, Kimberley Stephens and Astrid Yeo for their contribution to study design (JH) and conduct (JT, NG, YR), Il-1? sample preparation and assay (SR, BD, JW), statistical analysis (KS) and genetic evaluation (AY).
Competing Interests
The work described in this paper was funded by GlaxoSmithKline R&D. All authors were employees of GlaxoSmithKline R&D at the time of the study. BL has occasionally received fees for consulting from GSK. PS-S, LH, OD, JR and CC hold shares in the company.
REFERENCES
- 1.Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (5th edn.) 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–67. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- 3.Collo G, Neidhart S, Kawashima E, Kosco-Vilbois M, North RA, Buell G. Tissue distribution of the P2X7 receptor. Neuropharmacology. 1997;36:1277–83. doi: 10.1016/s0028-3908(97)00140-8. [DOI] [PubMed] [Google Scholar]
- 4.Lucae S, Salyakina D, Barden N, Harvey M, Gagné B, Labbé M, Binder EB, Uhr M, Paez-Pereda M, Sillaber I, Ising M, Brückl T, Lieb R, Holsboer F, Müller-Myhsok B. P2RX7, a gene coding for a purinergic ligand-gated ion channel, is associated with major depressive disorder. Hum Mol Genet. 2006;15:2438–45. doi: 10.1093/hmg/ddl166. [DOI] [PubMed] [Google Scholar]
- 5.Sluyter R, Shemon A, Wiley J. Glu496 to Ala polymorphism in the P2X7 receptor impairs ATP-induced IL1β release from human monocytes. J Immunol. 2004;172:3399–405. doi: 10.4049/jimmunol.172.6.3399. [DOI] [PubMed] [Google Scholar]
- 6.Gu BJ, Zhang W, Worthington RA, Sluyter R, Dao-Ung P, Petrou S, Barden JA, Wiley JS. A Glu-496 to Ala polymorphism leads to loss of function of the human P2X7 receptor. J Biol Chem. 2001;276:11135–42. doi: 10.1074/jbc.M010353200. [DOI] [PubMed] [Google Scholar]
- 7.Sim JA, Young MT, Sung H-Y, North RA, Surprenant A. Reanalysis of P2X7 receptor expression in rodent brain. J Neurosci. 2004;24:6307–14. doi: 10.1523/JNEUROSCI.1469-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Virgilio FD. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877–83. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 9.Engel T, Gomez-Villafuertes R, Tanaka K, Mesuret G, Sanz-Rodriguez A, Garcia-Huerta P, Miras-Portugal MT, Henshall DC, Diaz-Hernandez M. Seizure suppression and neuroprotection by targeting the purinergic P2X7 receptor during status epilepticus in mice. FASEB J. 2011;26:1616–28. doi: 10.1096/fj.11-196089. [DOI] [PubMed] [Google Scholar]
- 10.Arbeloa J, Pérez-Samartín A, Gottlieb M, Matute C. P2X7 receptor blockade prevents ATP excitotoxicity in neurons and reduces brain damage after ischemia. Neurobiol Dis. 2011;45:954–61. doi: 10.1016/j.nbd.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Altomonte L, Zoli A, Mirone L, Scolieri P, Magaró M. Serum levels of interleukin-1β, tumour necrosis factor-α and interleukin-2 in rheumatoid arthritis. Correlation with disease activity. Clin Rheumatol. 1992;11:202–5. doi: 10.1007/BF02207957. [DOI] [PubMed] [Google Scholar]
- 12.Al-Shukaili A, Al-Kaabi J, Hassan B. A comparative study of interleukin-1beta production and P2X7 expression after ATP stimulation by peripheral blood mononuclear cells isolated from rheumatoid arthritis patients and normal healthy controls. Inflammation. 2008;31:84–90. doi: 10.1007/s10753-007-9052-0. [DOI] [PubMed] [Google Scholar]
- 13.Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, Egerton J, Murfin M, Richardson J, Peck WL, Grahames CB, Casula MA, Yiangou Y, Birch R, Anand P, Buell GN. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain. 2005;114:386–96. doi: 10.1016/j.pain.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Cohen S, Hurd E, Cush J, Schiff M, Weinblatt ME, Moreland LW, Kremer J, Bear MB, Rich WJ, McCabe D. Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate. Arthritis Rheum. 2002;46:614–24. doi: 10.1002/art.10141. [DOI] [PubMed] [Google Scholar]
- 15.Conn P, Christopoulos A, Lindsley C. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nature Reviews Drug Discovery. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matthews PM, Rabiner I, Gunn R. Non-invasive imaging in experimental medicine for drug development. Curr Opin Pharmacol. 2011;11:501–7. doi: 10.1016/j.coph.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 17.Roman S, Cusdin FS, Fonfria E, Goodwin JA, Reeves J, Lappin SC, Chambers L, Walter DS, Clay WC, Michel AD. Cloning and pharmacological characterization of the dog P2X7 receptor. Br J Pharmacol. 2009;158:1513–26. doi: 10.1111/j.1476-5381.2009.00425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stock TC, Bloom BJ, Wei N, Ishaq S, Park W, Wang X, Gupta P, Mebus CA. Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. J Rheumatol. 2012;39:720–727. doi: 10.3899/jrheum.110874. DOI: 10.3899/jrheum.110874 ( http://www.jrheum.org/content/early/2012/02/28/jrheum.110874) [DOI] [PubMed] [Google Scholar]