

Abstract
AIM
The objective of this study was to determine the extent to which the CYP2C8*3 allele influences pharmacokinetic variability in the drug–drug interaction between gemfibrozil (CYP2C8 inhibitor) and pioglitazone (CYP2C8 substrate).
METHODS
In this randomized, two phase crossover study, 30 healthy Caucasian subjects were enrolled based on CYP2C8*3 genotype (n = 15, CYP2C8*1/*1; n = 15, CYP2C8*3 carriers). Subjects received a single 15 mg dose of pioglitazone or gemfibrozil 600 mg every 12 h for 4 days with a single 15 mg dose of pioglitazone administered on the morning of day 3. A 48 h pharmacokinetic study followed each pioglitazone dose and the study phases were separated by a 14 day washout period.
RESULTS
Gemfibrozil significantly increased mean pioglitazone AUC(0,∞) by 4.3-fold (P < 0.001) and there was interindividual variability in the magnitude of this interaction (range, 1.8- to 12.1-fold). When pioglitazone was administered alone, the mean AUC(0,∞) was 29.7% lower (P= 0.01) in CYP2C8*3 carriers compared with CYP2C8*1 homozygotes. The relative change in pioglitazone plasma exposure following gemfibrozil administration was significantly influenced by CYP2C8 genotype. Specifically, CYP2C8*3 carriers had a 5.2-fold mean increase in pioglitazone AUC(0,∞) compared with a 3.3-fold mean increase in CYP2C8*1 homozygotes (P= 0.02).
CONCLUSION
CYP2C8*3 is associated with decreased pioglitazone plasma exposure in vivo and significantly influences the pharmacokinetic magnitude of the gemfibrozil–pioglitazone drug-drug interaction. Additional studies are needed to evaluate the impact of CYP2C8 genetics on the pharmacokinetics of other CYP2C8-mediated drug–drug interactions.
Keywords: CYP2C8 protein human, drug interaction, gemfibrozil, pharmacogenetics, pharmacokinetics, pioglitazone
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
A drug–drug interaction exists between gemfibrozil (CYP2C8 inhibitor) and pioglitazone (CYP2C8 substrate), whereby gemfibrozil increases pioglitazone plasma exposure. Substantial interindividual variability exists in the pharmacokinetic magnitude of this drug–drug interaction.
CYP2C8*3 is associated with increased metabolism and decreased plasma exposure of pioglitazone.
Polymorphisms in CYP metabolizing enzyme genes, namely CYP2C19 and CYP2D6, have been shown to influence the magnitude of inhibitory drug–drug interactions. However, the extent to which CYP2C8 polymorphisms (e.g. CYP2C8*3) affect the interaction between gemfibrozil and pioglitazone is not known.
WHAT THIS STUDY ADDS
The CYP2C8*3 allele influences pharmacokinetic variability in the drug–drug interaction between gemfibrozil and pioglitazone. CYP2C8*3 carriers experienced a larger relative increase in pioglitazone plasma exposure following gemfibrozil administration than wild-type homozygotes.
Consideration should be given to the contribution of polymorphic CYP2C8 alleles to interindividual variability in the pharmacokinetic magnitude of CYP2C8-mediated drug–drug interactions.
Introduction
Drug–drug interactions involving inhibition or induction complicate the management of cardiometabolic diseases and interindividual variability exists in the pharmacokinetic magnitude of these interactions. There is increasing evidence that genetic variation influences the extent of drug–drug interactions, particularly those involving cytochrome P450 (CYP) metabolizing enzymes [1]. An example of an inhibitory drug–drug interaction that is germane to cardiometabolic pharmacotherapy is the gemfibrozil-mediated CYP2C8 inhibition of pioglitazone metabolism.
CYP2C8 plays an important role in the hepatic metabolism of numerous pharmacologic agents including pioglitazone (thiazolidinedione), repaglinide (meglitinide), cerivastatin (HMG-CoA reductase inhibitor) and paclitaxel (chemotherapeutic agent) [2, 3]. Pioglitazone, a peroxisome proliferator-activated receptor-γ agonist, is indicated for the treatment of type 2 diabetes. It is hepatically metabolized by CYP2C8, and to a lesser extent by CYP3A4, CYP1A2, CYP2C9 and CYP2D6 [4–7]. Gemfibrozil, a fibric acid derivative used in the treatment of hypertriglyceridaemia, potently inhibits CYP2C8 in vitro and in vivo[8–17]. Two clinical studies have shown that gemfibrozil increases pioglitazone plasma exposure approximately 3-fold due to CYP2C8 inhibition [5, 18]. Notably, substantial interindividual variability exists in the magnitude of this interaction, with increases in pioglitazone plasma exposure ranging from 2.3-fold to 6.5-fold [5, 18]. Previous studies have shown that polymorphisms in CYP genes influence the magnitude of CYP-mediated inhibitory drug–drug interactions [1]. For example, the extent of CYP2C19- and CYP2D6-mediated inhibition tends to be greater in extensive vs. poor metabolizers [19–23]. To our knowledge, the impact of CYP2C8 polymorphisms on the drug–drug interaction between gemfibrozil and pioglitazone has not been prospectively evaluated in clinical studies.
CYP2C8*3 is the most commonly studied functional polymorphism in CYP2C8. The CYP2C8*3 allele is comprised of two highly linked nonsynonymous polymorphisms, Arg139Lys and Lys399Arg, in exons 3 and 8, respectively. CYP2C8*3 is common in Caucasians (10% to 23%) but is rare in African and Asian populations [2, 3, 24]. There are conflicting in vitro data regarding the effect of CYP2C8*3 on metabolic activity, with reports of increased, decreased or no change in metabolism [7, 24–30]. In vivo, the consequences of CYP2C8*3 also appear to be substrate-dependent, with increased metabolism of agents such as pioglitazone, rosiglitazone and repaglinide, but decreased metabolism of R-ibuprofen [31–36]. In terms of the clinical pharmacokinetics of pioglitazone, a healthy volunteer study showed that carriers of the CYP2C8*3 allele had lower pioglitazone plasma exposure and a higher rate of metabolite formation than subjects with the CYP2C8*1/*1 genotype [31].
Given the known influence of CYP2C8*3 on pioglitazone pharmacokinetics, the objective of this study was to determine the extent to which CYP2C8*3 influences interindividual pharmacokinetic variability in the drug–drug interaction between gemfibrozil and pioglitazone in healthy volunteers.
Methods
Participants
The study was approved by the Colorado Multiple Institutional Review Board and all subjects provided written informed consent. The study consisted of healthy Caucasian men and women between 21 to 60 years of age. Participants were prospectively screened and stratified according to CYP2C8 genotype as follows: Group 1 =CYP2C8*1/*1 genotype (reference); Group 2 = carriers of at least one copy of the CYP2C8*3 allele (i.e. *1/*3 or *3/*3). Participants were excluded from the study for any of the following: body mass index <18 kg m−2 or ≥35 kg m−2, current or past history of cardiovascular, hepatic, renal, endocrine, gastrointestinal, haematologic, immunologic, or neurologic diseases, history of rhabdomyolysis, active malignancy, self-reported HIV positivity, active drug or alcohol abuse or pregnancy or lactation. Laboratory exclusion criteria included fasting plasma glucose ≥126 mg dl−1, serum potassium >5 mEq l−1 or <3.3 mEq l−1, serum creatinine >1.2 mg dl−1, liver function tests ≥ two times the upper limit of normal, haematocrit <36% in men or <34% in women, platelets <150 × 109 l−1, white blood cell count <4.0 × 109 l−1 or >11.1 × 109 l−1, or any other laboratory abnormalities classified as grade 2 or higher per published grading criteria [37]. Subjects were also excluded for concomitant use of any of the following: antidiabetic medications, statins, fibrates, systemic glucocorticoids and/or any other agent known to inhibit or induce the CYP2C8 and/or CYP3A4 metabolizing enzymes (e.g. trimethoprim, fluvoxamine, rifampicin, grapefruit juice).
Study design
The study was conducted in an open-label, randomized, two phase crossover design. In one phase, subjects received a single 15 mg dose of pioglitazone by mouth at 09.00 h. In the other phase, subjects received 600 mg of gemfibrozil by mouth at 08.00 h and 20.00 h for 4 days, with a single 15 mg dose of pioglitazone administered by mouth on day 3 at 09.00 h. The two phases were separated by a 14 day washout period. An intensive 48 h pharmacokinetic study was conducted after each pioglitazone dose. For the pharmacokinetic studies, subjects were admitted to the University of Colorado Denver Clinical and Translational Research Center (CTRC) Inpatient Unit after an overnight fast. In both phases, subjects received a standardized breakfast (600 calories; 55% carbohydrates, 15% protein and 30% fat) 2 h after pioglitazone ingestion. Subjects also received meals 6, 10 and 24 h after pioglitazone dosing. All meals were caffeine-free and subjects were asked to abstain from caffeine and smoking during the 48 h period. Blood samples (5 ml in ethylene-diaminetetraacetic acid [EDTA]) were collected pre-dose and 1, 2, 3, 4, 5, 7, 9, 12, 18, 24 and 48 h post pioglitazone dosing in both phases. Plasma was harvested within 30 min of each blood draw and stored at −80°C until analytical processing.
Genetic analyses
For DNA collection during the screening process, subjects were asked to swish vigorously 15 ml of Scope® mouthwash (Procter and Gamble, Cincinnati, OH, USA) for 1 min and then expectorate into a sterile collection tube [38]. Genomic DNA was isolated from buccal cells using a commercially available kit (QIAmp DNA Mini Kit, Qiagen, Valencia, CA, USA). The nonsynonymous CYP2C8 polymorphisms, Arg139Lys (rs11572080) and Lys399Arg (rs10509681), were genotyped using PCR-Pyrosequencing analysis (PSQ 96MA, Qiagen, Valencia, CA, USA) according to our previously published method [33]. Automated PSQ 96MA SNP software version 2.0 (Qiagen, Valencia, CA, USA) was used to make genotype determinations. CYP2C8*3 was denoted as the presence of the Lys and Arg alleles at codons 139 and 399, respectively.
Drug concentration analyses
Plasma concentrations of pioglitazone were measured with a validated LC/MS assay. The internal standard was deuterated pioglitazone (pioglitazone-d4; Toronto Research Chemicals, North York, Ontario, Canada). Pioglitazone was extracted from plasma using a liquid–liquid extraction procedure with t-butylmethylether at an acidic pH. Chromatographic separation was conducted on a 2.1 × 50 mm, 5 µm Sunfire C18 column (Waters Corporation, Milford, MA, USA) maintained at 40°C. The mobile phase consisted of 60% ammonium acetate (pH 4.5) : 40% acetonitrile (v/v) and was delivered at a flow rate of 300 µl min−1. The retention time for pioglitazone was 3.5 min. A single quadrupole mass spectrometer (Thermo Fisher MSQ, Thermo Fisher, San Jose, CA, USA) was used in ESI, positive polarity mode. Analytes were detected using single ion monitoring mode, with pioglitazone and pioglitazone-d4 detected at m/z of 357.07 and 361.21, respectively. The needle voltage was set at 2.5 kV and cone voltage at 125 V. Nitrogen was used as the source gas and was maintained at 75 psi. Data acquisition and processing were performed using Xcalibur software, version 1.3 (Thermo Fisher, San Jose, CA, USA). Calculations were based on peak area ratios of analyte to internal standard. Concentrations were interpolated from a linear least squares regression calibration curve, based on 1/concentration weighting. The lower limit of quantification (LLOQ) of pioglitazone was 5 ng ml−1, and the assay was linear over the range of 5 ng ml−1–2000 ng ml−1. Validation data for pioglitazone non-LLOQ samples were within ± 15% for inter- and intra-day accuracy and precision. The LLOQ data were within ± 20% for both accuracy and precision. Possible interference between pioglitazone, gemfibrozil and gemfibrozil 1-O-β-glucuronide was evaluated, but not observed, in this method. Plasma concentrations of gemfibrozil were measured with a validated LC/MS assay as previously described [39]. Deuterated gemfibrozil (gemfibrozil-d6) was used as the internal standard. The LLOQ of gemfibrozil was 0.5 µg ml−1 and the assay was linear over the range of 0.5 to 50 µg ml−1. Inter- and intra-day accuracy and precision were within ± 15% [39]. Gemfibrozil 1-O-β-glucuronide metabolite area ratios were determined from LC/MS chromatograms initially analyzed for gemfibrozil concentrations, which contained a glucuronide peak as a result of in-source dissociation, and were normalized against the internal standard, gemfibrozil-d6. Gemfibrozil 1-O-β-glucuronide concentrations were then determined from the gemfibrozil parent calibration curve, using 1/concentration weighting [39].
Pharmacokinetic analysis
Plasma concentration–time curves of pioglitazone, gemfibrozil and gemfibrozil 1-O-β-glucuronide were generated, and the maximum plasma concentration (Cmax) and time to reach Cmax (tmax) were observed directly from these curves. Pharmacokinetic parameters were estimated by noncompartmental analysis using WinNonlin version 5.2.1 software (Pharsight Corporation, Mountain View, CA, USA). The terminal elimination rate constant (λz) was obtained by regression analysis of the log-linear portion of the concentration–time curves. Pioglitazone area under the plasma concentration–time curve from 0 to infinity (AUC(0,∞)), and gemfibrozil and gemfibrozil 1-O-β-glucuronide AUC from 0 to 10 h (AUC(0,10 h)) were calculated using the log linear trapezoidal rule. Pioglitazone and gemfibrozil half-life (t1/2) were calculated from the following equation, t1/2= ln(2)/λz. Apparent oral clearance (CL/F) was calculated as dose (in mg)/AUC(0,∞). Weight-adjusted oral clearance (CL/F kg−1) was calculated as [dose (mg)/AUC(0,∞)]/subject weight (kg).
Statistical analyses
The primary endpoint was the relative change in pioglitazone AUC(0,∞) between CYP2C8 genotype groups (i.e., *1/*1 vs. *3 carriers). Secondary pharmacokinetic endpoints that were compared between CYP2C8*1 homozygotes and CYP2C8*3 carriers included: (i) other pioglitazone pharmacokinetic parameters (e.g., AUC(0,48 h), CL/F kg−1, t1/2) and (ii) gemfibrozil and gemfibrozil 1-O-β-glucuronide pharmacokinetic parameters.
Baseline demographics were compared between CYP2C8*1 homozygotes and CYP2C8*3 carriers by independent t-tests for continuous data, and Chi-square or Fisher's exact tests for categorical data. Non-normally distributed pharmacokinetic parameters (e.g. AUC and Cmax) were log-transformed prior to analysis. In the entire study cohort, the changes in pioglitazone pharmacokinetic parameters when it was given alone and in the presence of gemfibrozil were assessed with paired t-tests. Pharmacokinetic data, including relative changes, were compared between CYP2C8*1 homozygotes and CYP2C8*3 carriers, using independent Student's t-tests or Mann Whitney U tests (for time data). The relationship between gemfibrozil AUC(0,10 h) and the relative change in pioglitazone AUC(0,∞) was assessed by Pearson correlation coefficients. Statistical analyses were conducted using SPSS version 18.0 software. A P value <0.05 was considered statistically significant.
Results
One hundred forty-two subjects were prospectively genotyped for the CYP2C8*3 allele, and 34 subjects were started on study protocol. Four subjects withdrew after only one phase of the study due to personal reasons. Results are presented for the 30 subjects who completed both intensive pharmacokinetic study visits, i.e. pioglitazone alone and pioglitazone plus gemfibrozil. The study consisted of 21 women and nine men, mean age of 36 ± 11 years and mean weight of 72.7 ± 15.9 kg. Subjects had the following CYP2C8 genotypes: *1/*1 (n = 15); *1/*3 (n = 14) and *3/*3 (n = 1). Baseline demographics did not differ significantly between CYP2C8 genotype groups, and are shown in Table 1.
Table 1.
Baseline demographics (n = 30) by CYP2C8 genotype group
| Demographic variable | CYP2C8 *1/*1 | CYP2C8 *3 carriers | P value |
|---|---|---|---|
| n = 15 | n = 15 | ||
| Male, n (%) | 4 (27%) | 5 (33%) | 0.99 |
| Hispanic ethnicity, n (%) | 0 | 2 (13%) | 0.48 |
| Current smoker, n (%) | 3 (20%) | 1 (7%) | 0.60 |
| Hormonal contraceptive, n (%) | 4 (27%) | 4 (27%) | – |
| Age (years) | 35 ± 9 | 37 ± 12 | 0.45 |
| Weight (kg) | 74.5 ± 15.7 | 71.0 ± 16.4 | 0.56 |
| Body mass index (kg m−2) | 24.9 ± 3.0 | 25.3 ± 4.3 | 0.77 |
Data are presented as number (%) or mean ± SD. P values are for the comparison of baseline demographics between CYP2C8 genotype groups. All subjects in the study classified their race as Caucasian.
Pioglitazone pharmacokinetic parameters in the absence and presence of gemfibrozil in the entire study cohort (n = 30) are shown in Table 2. Gemfibrozil significantly increased mean pioglitazone AUC(0,∞) by 4.3-fold (P < 0.001) and there was substantial interindividual variability in the magnitude of this interaction (range, 1.8- to 12.1-fold). Gemfibrozil also significantly decreased pioglitazone weight-adjusted apparent oral clearance by approximately 70% (P < 0.001) and lengthened the mean t1/2 of pioglitazone by 3-fold (P < 0.001). The mean Cmax of pioglitazone did not significantly change following gemfibrozil administration. The median tmax of pioglitazone was 2.0 h in the absence and presence of gemfibrozil.
Table 2.
Single dose pharmacokinetics of pioglitazone 15 mg in all study participants (n = 30) when pioglitazone was administered alone and in combination with gemfibrozil
| Pioglitazone pharmacokinetic parameter | Pioglitazone alone | Pioglitazone + gemfibrozil | Relative change [(Pioglitazone + gemfibrozil)/pioglitazone alone] | P value |
|---|---|---|---|---|
| Cmax (ng ml−1) | 608 ± 215 | 678 ± 187 | 1.3 (1.0, 1.6) | 0.10 |
| AUC(0,∞) (ng ml−1 h) | 5 770 ± 2 840 | 21 700 ± 8 820 | 4.3 (3.5, 5.1) | <0.001 |
| AUC(0,48 h) (ng ml−1 h) | 5 440 ± 2 620 | 15 300 ± 4 790 | 3.2 (2.7, 3.8) | <0.001 |
| CL/F kg−1 (l h−1 kg−1) | 0.045 ± 0.02 | 0.011 ± 0.005 | 0.29 (0.24, 0.34) | <0.001 |
| V/F (l) | 36.7 ± 19.1 | 24.4 ± 6.2 | 0.80 (0.68, 0.91) | 0.001 |
| t1/2 (h) | 8.1 ± 2.9 | 23.7 ± 10.4 | 3.1 (2.6, 3.6) | <0.001 |
Data are expressed as mean ± SD and mean (95% confidence interval). Cmax= maximum plasma concentration; AUC = area under the plasma concentration–time curve; CL/F kg−1= weight-adjusted apparent oral clearance; V/F= apparent volume of distribution; t1/2= half-life.
Pioglitazone plasma concentration–time curves in the absence and presence of gemfibrozil, by CYP2C8 genotype group, are shown in Figure 1. Pioglitazone AUC(0,∞) in the absence and presence of gemfibrozil for each subject is shown in Figure 2. When pioglitazone was administered alone, mean AUC(0,∞) was 29.7% lower (P= 0.01) and mean weight-adjusted oral clearance was 64.7% higher (P= 0.002) in CYP2C8*3 carriers compared with CYP2C8*1 homozygotes (Table 3). In the presence of gemfibrozil, pioglitazone pharmacokinetic parameters did not differ significantly between CYP2C8 genotype groups (Table 3). However, the mean relative change in pioglitazone pharmacokinetic parameters following gemfibrozil administration was significantly influenced by CYP2C8 genotype (Table 3, Figure 3). Specifically, CYP2C8*3 carriers had a mean 5.2-fold increase in pioglitazone AUC(0,∞) compared with a mean 3.3-fold increase in CYP2C8*1 homozygotes (P= 0.02) following gemfibrozil administration. The relative change in pioglitazone t1/2 was also larger in CYP2C8*3 carriers compared with CYP2C8*1 homozygotes (3.3-fold vs. 2.9-fold), although this difference did not reach statistical significance. The subject with the largest relative increase in pioglitazone AUC(0,∞) (12.1-fold) had the CYP2C8*1/*3 genotype. There was one subject with the CYP2C8*3/*3 genotype in the study cohort and this subject experienced a 7.0-fold increase in pioglitazone AUC(0,∞). Of the 10 subjects with the largest relative increases in pioglitazone AUC(0,∞), eight subjects were CYP2C8*3 carriers.
Figure 1.

Pioglitazone plasma concentration–time curves by CYP2C8 genotype group when pioglitazone was administered alone and in combination with gemfibrozil. CYP2C8*1/*1, pioglitazone alone ( ); CYP2C8*3 carriers, pioglitazone alone (
); CYP2C8*3 carriers, pioglitazone alone ( ); CYP2C8*1/*1, pioglitazone + gemfibrozil (
); CYP2C8*1/*1, pioglitazone + gemfibrozil ( ); CYP2C8*3 carriers, pioglitazone + gemfibrozil (
); CYP2C8*3 carriers, pioglitazone + gemfibrozil ( ). Data are shown as mean ± SEM
). Data are shown as mean ± SEM
Figure 2.
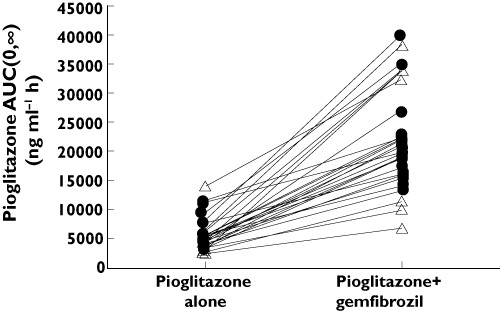
Pioglitazone AUC(0,∞) in the absence and presence of gemfibrozil for each participant. CYP2C8*1/*1 ( ); CYP2C8*3 carriers (
); CYP2C8*3 carriers ( )
)
Table 3.
Pioglitazone pharmacokinetic parameters by CYP2C8 genotype group when pioglitazone was administered alone and in combination with gemfibrozil
| Pharmacokinetic parameter | CYP2C8 *1/*1 | CYP2C8 *3 carriers | P value (between genotype groups) |
|---|---|---|---|
| (n = 15) | (n = 15) | ||
| Cmax (ng ml−1) | |||
| Pioglitazone | 641 ± 171 | 575 ± 253 | 0.22 |
| Pioglitazone + gemfibrozil | 696 ± 164 | 660 ± 212 | 0.51 |
| Mean relative change | 1.1 (0.96, 1.3) | 1.4 (0.88, 2.0) | 0.28 |
| P value (within genotype group) | 0.30 | 0.21 | |
| AUC(0,∞) (ng ml−1 h) | |||
| Pioglitazone | 6 770 ± 2 480 | 4 760 ± 2 900 | 0.01 |
| Pioglitazone + gemfibrozil | 21 100 ± 7 800 | 22 200 ± 9 970 | 0.99 |
| Mean relative change | 3.3 (2.7, 4.0) | 5.2 (3.8, 6.7) | 0.02 |
| P value (within genotype group) | <0.001 | <0.001 | |
| AUC(0,48 h) (ng ml−1 h) | |||
| Pioglitazone | 6 340 ± 2 250 | 4 540 ± 2 730 | 0.01 |
| Pioglitazone + gemfibrozil | 15 800 ± 3 840 | 14 800 ± 5 670 | 0.39 |
| Mean relative change | 2.7 (2.2, 3.2) | 3.7 (2.8, 4.7) | 0.05 |
| P value (within genotype group) | <0.001 | <0.001 | |
| CL/F kg−1 (l h−1 kg−1) | |||
| Pioglitazone | 0.034 ± 0.0097 | 0.056 ± 0.023 | 0.002 |
| Pioglitazone + gemfibrozil | 0.011 ± 0.0026 | 0.012 ± 0.0072 | 0.712 |
| Mean relative change | 0.34 (0.27, 0.42) | 0.23 (0.17, 0.30) | 0.02 |
| P value (within genotype group) | <0.001 | <0.001 | |
| t1/2 (h) | |||
| Pioglitazone | 8.2 ± 3.0 | 8.0 ± 3.0 | 0.85 |
| Pioglitazone + gemfibrozil | 21.7 ± 8.5 | 25.7 ± 12.0 | 0.30 |
| Mean relative change | 2.9 (2.1, 3.6) | 3.3 (2.5, 4.1) | 0.35 |
| P value (within genotype group) | <0.001 | <0.001 |
Data are expressed as mean ± SD, or mean (95% confidence interval) for relative change data. Cmax= maximum plasma concentration; AUC = area under the plasma concentration–time curve; CL/F kg−1= weight-adjusted apparent oral clearance; t1/2= half-life.
Figure 3.
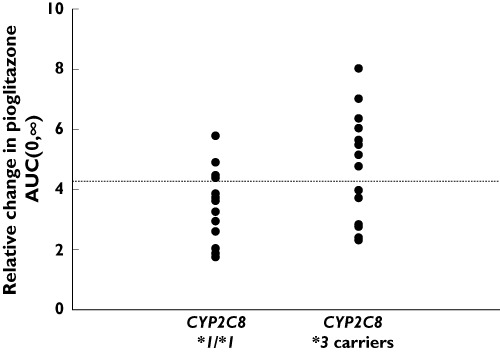
Relative change in pioglitazone AUC(0,∞) by CYP2C8 genotype group. The dashed line represents the mean of the study cohort. CYP2C8*1/*1 (n = 15), CYP2C8*3 carriers (n = 15)
Gemfibrozil and gemfibrozil 1-O-β-glucuronide pharmacokinetic parameters did not differ significantly between CYP2C8 genotype groups (Table 4). Furthermore, gemfibrozil AUC(0,10 h) was not significantly correlated with the relative change in pioglitazone AUC(0,∞) in the entire study cohort (r= 0.04, P= 0.83), nor by CYP2C8 genotype group (CYP2C8*1/*1, r= 0.03, P= 0.91; CYP2C8*3 carriers, r= 0.04, P= 0.90).
Table 4.
Gemfibrozil and gemfibrozil 1-O-β-glucuronide pharmacokinetic parameters by CYP2C8 genotype group
| Pharmacokinetic parameter | CYP2C8 *1/*1 | CYP2C8 *3 carriers | P value |
|---|---|---|---|
| (n = 15) | (n = 15) | ||
| Gemfibrozil | |||
| Cmax (µg ml−1) | 24.9 ± 9.4 | 24.8 ± 9.5 | 0.96 |
| AUC(0,10 h) (µg ml−1 h) | 92.9 ± 37.3 | 94.9 ± 38.6 | 0.94 |
| CL/F kg−1 (l h−1 kg−1) | 0.1 ± 0.03 | 0.1 ± 0.03 | 0.88 |
| t1/2 (h) | 2.0 ± 0.4 | 1.8 ± 0.3 | 0.20 |
| tmax (h) | 2.0 (2.0–3.0) | 2.0 (2.0–4.0) | 0.73 |
| Gemfibrozil 1-O-β-glucuronide | |||
| Cmax (µg ml−1) | 7.5 ± 6.6 | 6.8 ± 1.7 | 0.74 |
| AUC(0,10 h) (µg ml−1 h) | 30.7 ± 12.7 | 32.6 ± 8.6 | 0.45 |
Data are presented as mean ± SD or median (range).
Discussion
Previously, it has been shown that gemfibrozil increases pioglitazone plasma exposure in healthy volunteers, and interindividual variability exists in the magnitude of this interaction [5, 18]. We prospectively set out to determine if the CYP2C8*3 allele influences the extent of this inhibitory drug–drug interaction. Our primary finding was that the relative change in pioglitazone plasma exposure following gemfibrozil administration was significantly influenced by CYP2C8 genotype. Specifically, the relative increase in pioglitazone plasma exposure was greater in CYP2C8*3 carriers compared with CYP2C8*1 homozygotes. These data suggest that a portion of the interindividual variability in the drug-drug interaction between gemfibrozil and pioglitazone may be explained by the CYP2C8*3 allele.
To date, most investigations of the impact of pharmacogenetics on inhibitory drug–drug interactions have been conducted in relation to CYP2D6 and CYP2C19 metabolizing enzymes, with a focus on extensive and poor metabolizers. In these cases, the magnitude of substrate inhibition tends to be greater in genetically-determined extensive metabolizers vs. poor metabolizers because inhibition cannot occur in individuals who lack the metabolizing enzyme [19–23]. However, less is known about the impact of CYP2C8 polymorphisms or increased metabolic activity phenotypes (e.g. ultrarapid metabolizers) on inhibitory drug–drug interactions. To our knowledge, the finding that CYP2C8 genotype significantly influences the magnitude of the interaction between gemfibrozil and pioglitazone has not been reported before.
When pioglitazone was administered alone, we found that its plasma exposure was significantly lower and weight-adjusted apparent oral clearance was significantly higher in carriers of the CYP2C8*3 allele as compared with CYP2C8*1 homozygotes. This finding is consistent with previous clinical reports of increased thiazolidinedione metabolism and decreased plasma exposure in carriers of the CYP2C8*3 allele [31–33]. The observed magnitude of genotype effect was also similar to other clinical studies, with an approximate 25% to 30% lower pioglitazone plasma exposure in CYP2C8*3 carriers compared with wild-type homozygotes [31]. In terms of functional significance, there have been conflicting data regarding the effects of the CYP2C8*3 allele on substrate metabolism, with reports of increased metabolic activity, decreased metabolic activity, and substrate dependency. However, an in vitro study has recently shed more light on this topic [30]. Kaspera and colleagues found that recombinant CYP2C8*3 exhibited higher overall activity than CYP2C8*1 in the presence of cytochrome b5, a redox partner [30]. This finding is thought to be due to greater affinity of CYP2C8*3 for cytochrome b5 and cytochrome P450 reductase [30]. Taking recent in vitro and in vivo data together, it appears that CYP2C8*3 is associated with increased metabolism and decreased plasma concentrations of pioglitazone.
Our pharmacogenetic drug–drug interaction study found a significantly greater relative increase in pioglitazone plasma exposure in CYP2C8*3 carriers (5.2-fold) compared with CYP2C8*1 homozygotes (3.3-fold) following gemfibrozil administration. A few other studies have assessed the role of CYP2C8 polymorphisms on the magnitude of CYP2C8-mediated thiazolidinedione drug–drug interactions. One study showed that trimethoprim, a weak competitive CYP2C8 inhibitor, increased the plasma exposure of pioglitazone by 42% in healthy volunteers [31]. However, the CYP2C8*3 allele did not influence the extent of the interaction. Along the same lines, another healthy volunteer study reported that fluvoxamine, a weak competitive CYP2C8 inhibitor, increased the plasma exposure of rosiglitazone by 21% and the effects were consistent across CYP2C8 genotype groups [40]. The variable genetic findings between studies are not surprising given that different inhibitor-substrate combinations were tested in each of these scenarios. Gemfibrozil is one of the most potent in vivo CYP2C8 inhibitors, primarily due to mechanism-based inhibition of CYP2C8 by its 1-O-β-glucuronide metabolite [13, 15]. As such, gemfibrozil is classified by the Food and Drug Administration as a strong in vivo CYP2C8 inhibitor (i.e. ≥5-fold increase in plasma exposure of CYP2C8 substrates) [41, 42]. In contrast, trimethoprim and fluvoxamine are classified as weak competitive in vivo inhibitors of CYP2C8 (i.e., ≥1.25 but <2-fold increase in plasma exposure of CYP2C8 substrates) [41, 42]. It is reasonable to hypothesize that CYP2C8*3 carriers may be more susceptible to CYP2C8 inhibition by mechanism-based inhibitors, such as gemfibrozil 1-O-β-glucuronide, due to a greater amount of inactivating species produced as a result of the CYP2C8*3 allele. This hypothesis is consistent with data from Tornio and colleagues who showed that the interaction between gemfibrozil and repaglinide (a CYP2C8 substrate) was stronger in CYP2C8 *3 carriers than in non-carriers [14]. In vitro and in vivo studies are needed to elucidate further the impact of genetic polymorphisms on mechanism-based versus competitive inhibitory drug–drug interactions.
Our study highlights several important considerations regarding the impact of pharmacogenetics on the evaluation of inhibitory drug–drug interactions. As previously reviewed by Lee and colleagues, reports of drug–drug interaction data are often limited in scope because interindividual variability in the magnitude of the interaction is not fully explored nor explained [1]. As such, caution must be exerted when extrapolating mean pharmacokinetic drug interaction data to the clinical setting. Importantly, genetic subgroups may exist which are susceptible to differing magnitudes of the interaction. In the case of gemfibrozil-pioglitazone, we observed the mean relative change in pioglitazone plasma exposure to be 4.3-fold, with a range of 1.8-fold to 12.1-fold. Although pioglitazone plasma exposure in the presence of gemfibrozil did not differ significantly between CYP2C8 genotype groups, the magnitude of change in pioglitazone pharmacokinetics was affected by CYP2C8 genotype. With regards to clinical pharmacology and the drug development process, it would seem prudent to routinely interrogate CYP2C8 polymorphisms when gemfibrozil is used as a CYP2C8 inhibitor, or when pioglitazone is used as a CYP2C8 probe drug, in order to characterize comprehensively the contribution of genetics to interindividual variability in the magnitude of potential drug–drug interactions.
There are limitations of our study that deserve to be acknowledged. First, based on previous findings of increased parent pioglitazone concentrations in the presence of gemfibrozil, we only measured parent pioglitazone concentrations in our study [5, 18]. Pioglitazone is metabolized to a number of different active metabolites, namely M-III, and M-IV [4]. A previous study found no significant differences in M-III or M-IV plasma exposure between CYP2C8 genotype groups [31]. However, M-III : parent and M-IV : parent AUC ratios were significantly greater in carriers of CYP2C8*3 allele compared with wild-type homozygotes [31]. Second, only one subject with the CYP2C8*3/*3 genotype was present in our cohort. This subject experienced a 7.0-fold increase in pioglitazone plasma exposure, which was the third highest relative change among all subjects in the study. Additional studies are needed to determine if a CYP2C8*3 gene–dose effect exists during pioglitazone monotherapy and in the setting of inhibitory drug-drug interactions. Third, because we intentionally used a prospective CYP2C8*3 genotype enrichment design, we did not interrogate other polymorphisms in the CYP2C8 gene or other CYP metabolizing enzymes. Future consideration should be given to the effect of other polymorphic alleles (e.g., CYP2C8*4, CYP2C8−271 C>A) or novel CYP2C8 haplotypes on pioglitazone plasma concentrations and drug-drug interactions [28]. Along the same lines, future studies should evaluate the extent to which polymorphisms in UGT2B7, the enzyme that mediates the conversion of gemfibrozil to its 1-O-β-glucuronide metabolite, influence interindividual pharmacokinetic variability in gemfibrozil-mediated drug-drug interactions [43, 44]. Although not evaluated in our CYP2C8 genotype-focused analysis, it is possible that polymorphic UGT2B7 alleles are an additional source of variability in the pioglitazone-gemfibrozil interaction. However, it is unlikely that UGT2B7 polymorphisms confounded our study results given that gemfibrozil and gemfibrozil 1-O-β-glucuronide plasma exposure did not differ significantly between CYP2C8 genotype groups.
In summary, the polymorphic CYP2C8*3 allele influences pharmacokinetic variability in the magnitude of the drug–drug interaction between gemfibrozil and pioglitazone. Additional studies are needed to evaluate the impact of CYP2C8 polymorphisms on other CYP2C8-mediated drug–drug interactions, particularly those involving mechanism-based inhibitors.
Acknowledgments
The authors wish to thank the study volunteers for their participation, Peter Anderson, Pharm.D. for providing laboratory resources and the nursing and administrative staff at the University of Colorado Clinical and Translational Research Center for assisting with the conduct of the study. The study was funded by NIH K23 DK073197 (to CLA) and NIH/NCRR Colorado CTSA UL1 RR025780. Contents are the authors' sole responsibility and do not necessarily represent official NIH views.
Competing Interests
There are no competing interests to declare.
REFERENCES
- 1.Lee LS, Nafziger AN, Bertino JS., Jr Evaluation of inhibitory drug interactions during drug development: genetic polymorphisms must be considered. Clin Pharmacol Ther. 2005;78:1–6. doi: 10.1016/j.clpt.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 2.Totah RA, Rettie AE. Cytochrome P450 2C8: substrates, inhibitors, pharmacogenetics, and clinical relevance. Clin Pharmacol Ther. 2005;77:341–52. doi: 10.1016/j.clpt.2004.12.267. [DOI] [PubMed] [Google Scholar]
- 3.Daily EB, Aquilante CL. Cytochrome P450 2C8 pharmacogenetics: a review of clinical studies. Pharmacogenomics. 2009;10:1489–510. doi: 10.2217/pgs.09.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckland DA, Danhof M. Clinical pharmacokinetics of pioglitazone. Exp Clin Endocrinol Diabetes. 2000;108:S234–42. [Google Scholar]
- 5.Jaakkola T, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics of pioglitazone. Clin Pharmacol Ther. 2005;77:404–14. doi: 10.1016/j.clpt.2004.12.266. [DOI] [PubMed] [Google Scholar]
- 6.Jaakkola T, Laitila J, Neuvonen PJ, Backman JT. Pioglitazone is metabolised by CYP2C8 and CYP3A4 in vitro: potential for interactions with CYP2C8 inhibitors. Basic Clin Pharmacol Toxicol. 2006;99:44–51. doi: 10.1111/j.1742-7843.2006.pto_437.x. [DOI] [PubMed] [Google Scholar]
- 7.Muschler E, Lal J, Jetter A, Rattay A, Zanger U, Zadoyan G, Fuhr U, Kirchheiner J. The role of human CYP2C8 and CYP2C9 variants in pioglitazone metabolism in vitro. Basic Clin Pharmacol Toxicol. 2009;105:374–9. doi: 10.1111/j.1742-7843.2009.00457.x. [DOI] [PubMed] [Google Scholar]
- 8.Wang JS, Neuvonen M, Wen X, Backman JT, Neuvonen PJ. Gemfibrozil inhibits CYP2C8-mediated cerivastatin metabolism in human liver microsomes. Drug Metab Dispos. 2002;30:1352–6. doi: 10.1124/dmd.30.12.1352. [DOI] [PubMed] [Google Scholar]
- 9.Backman JT, Kyrklund C, Neuvonen M, Neuvonen PJ. Gemfibrozil greatly increases plasma concentrations of cerivastatin. Clin Pharmacol Ther. 2002;72:685–91. doi: 10.1067/mcp.2002.128469. [DOI] [PubMed] [Google Scholar]
- 10.Niemi M, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia. 2003;46:347–51. doi: 10.1007/s00125-003-1034-7. [DOI] [PubMed] [Google Scholar]
- 11.Shitara Y, Hirano M, Sato H, Sugiyama Y. Gemfibrozil and its glucuronide inhibit the organic anion transporting polypeptide 2 (OATP2/OATP1B1:SLC21A6)-mediated hepatic uptake and CYP2C8-mediated metabolism of cerivastatin: analysis of the mechanism of the clinically relevant drug-drug interaction between cerivastatin and gemfibrozil. J Pharmacol Exp Ther. 2004;311:228–36. doi: 10.1124/jpet.104.068536. [DOI] [PubMed] [Google Scholar]
- 12.Prueksaritanont T, Richards KM, Qiu Y, Strong-Basalyga K, Miller A, Li C, Eisenhandler R, Carlini EJ. Comparative effects of fibrates on drug metabolizing enzymes in human hepatocytes. Pharm Res. 2005;22:71–8. doi: 10.1007/s11095-004-9011-5. [DOI] [PubMed] [Google Scholar]
- 13.Ogilvie BW, Zhang D, Li W, Rodrigues AD, Gipson AE, Holsapple J, Toren P, Parkinson A. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: implications for drug-drug interactions. Drug Metab Dispos. 2006;34:191–7. doi: 10.1124/dmd.105.007633. [DOI] [PubMed] [Google Scholar]
- 14.Tornio A, Niemi M, Neuvonen M, Laitila J, Kalliokoski A, Neuvonen PJ, Backman JT. The effect of gemfibrozil on repaglinide pharmacokinetics persists for at least 12 h after the dose: evidence for mechanism-based inhibition of CYP2C8 in vivo. Clin Pharmacol Ther. 2008;84:403–11. doi: 10.1038/clpt.2008.34. [DOI] [PubMed] [Google Scholar]
- 15.Baer BR, DeLisle RK, Allen A. Benzylic oxidation of gemfibrozil-1-O-beta-glucuronide by P450 2C8 leads to heme alkylation and irreversible inhibition. Chem Res Toxicol. 2009;22:1298–309. doi: 10.1021/tx900105n. [DOI] [PubMed] [Google Scholar]
- 16.Honkalammi J, Niemi M, Neuvonen PJ, Backman JT. Mechanism-based inactivation of CYP2C8 by gemfibrozil occurs rapidly in humans. Clin Pharmacol Ther. 2011;89:579–86. doi: 10.1038/clpt.2010.358. [DOI] [PubMed] [Google Scholar]
- 17.Honkalammi J, Niemi M, Neuvonen PJ, Backman JT. Dose-dependent interaction between gemfibrozil and repaglinide in humans: strong inhibition of CYP2C8 with subtherapeutic gemfibrozil doses. Drug Metab Dispos. 2011;39:1977–86. doi: 10.1124/dmd.111.040931. [DOI] [PubMed] [Google Scholar]
- 18.Deng LJ, Wang F, Li HD. Effect of gemfibrozil on the pharmacokinetics of pioglitazone. Eur J Clin Pharmacol. 2005;61:831–6. doi: 10.1007/s00228-005-0042-6. [DOI] [PubMed] [Google Scholar]
- 19.Brosen K, Hansen JG, Nielsen KK, Sindrup SH, Gram LF. Inhibition by paroxetine of desipramine metabolism in extensive but not in poor metabolizers of sparteine. Eur J Clin Pharmacol. 1993;44:349–55. doi: 10.1007/BF00316471. [DOI] [PubMed] [Google Scholar]
- 20.Hamelin BA, Bouayad A, Methot J, Jobin J, Desgagnes P, Poirier P, Allaire J, Dumesnil J, Turgeon J. Significant interaction between the nonprescription antihistamine diphenhydramine and the CYP2D6 substrate metoprolol in healthy men with high or low CYP2D6 activity. Clin Pharmacol Ther. 2000;67:466–77. doi: 10.1067/mcp.2000.106464. [DOI] [PubMed] [Google Scholar]
- 21.Ieiri I, Kimura M, Irie S, Urae A, Otsubo K, Ishizaki T. Interaction magnitude, pharmacokinetics and pharmacodynamics of ticlopidine in relation to CYP2C19 genotypic status. Pharmacogenet Genomics. 2005;15:851–9. doi: 10.1097/01213011-200512000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Yasui-Furukori N, Saito M, Uno T, Takahata T, Sugawara K, Tateishi T. Effects of fluvoxamine on lansoprazole pharmacokinetics in relation to CYP2C19 genotypes. J Clin Pharmacol. 2004;44:1223–9. doi: 10.1177/0091270004269015. [DOI] [PubMed] [Google Scholar]
- 23.Yasui-Furukori N, Takahata T, Nakagami T, Yoshiya G, Inoue Y, Kaneko S, Tateishi T. Different inhibitory effect of fluvoxamine on omeprazole metabolism between CYP2C19 genotypes. Br J Clin Pharmacol. 2004;57:487–94. doi: 10.1111/j.1365-2125.2004.02047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bahadur N, Leathart JB, Mutch E, Steimel-Crespi D, Dunn SA, Gilissen R, Houdt JV, Hendrickx J, Mannens G, Bohets H, Williams FM, Armstrong M, Crespi CL, Daly AK. CYP2C8 polymorphisms in Caucasians and their relationship with paclitaxel 6alpha-hydroxylase activity in human liver microsomes. Biochem Pharmacol. 2002;64:1579–89. doi: 10.1016/s0006-2952(02)01354-0. [DOI] [PubMed] [Google Scholar]
- 25.Dai D, Zeldin DC, Blaisdell JA, Chanas B, Coulter SJ, Ghanayem BI, Goldstein JA. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics. 2001;11:597–607. doi: 10.1097/00008571-200110000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Soyama A, Saito Y, Hanioka N, Murayama N, Nakajima O, Katori N, Ishida S, Sai K, Ozawa S, Sawada JI. Non-synonymous single nucleotide alterations found in the CYP2C8 gene result in reduced in vitro paclitaxel metabolism. Biol Pharm Bull. 2001;24:1427–30. doi: 10.1248/bpb.24.1427. [DOI] [PubMed] [Google Scholar]
- 27.Taniguchi R, Kumai T, Matsumoto N, Watanabe M, Kamio K, Suzuki S, Kobayashi S. Utilization of human liver microsomes to explain individual differences in paclitaxel metabolism by CYP2C8 and CYP3A4. J Pharmacol Sci. 2005;97:83–90. doi: 10.1254/jphs.fp0040603. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez-Antona C, Niemi M, Backman JT, Kajosaari LI, Neuvonen PJ, Robledo M, Ingelman-Sundberg M. Characterization of novel CYP2C8 haplotypes and their contribution to paclitaxel and repaglinide metabolism. Pharmacogenomics J. 2008;8:268–77. doi: 10.1038/sj.tpj.6500482. [DOI] [PubMed] [Google Scholar]
- 29.Rowbotham SE, Boddy AV, Redfern CP, Veal GJ, Daly AK. Relevance of nonsynonymous CYP2C8 polymorphisms to 13-cis retinoic acid and paclitaxel hydroxylation. Drug Metab Dispos. 2010;38:1261–6. doi: 10.1124/dmd.109.030866. [DOI] [PubMed] [Google Scholar]
- 30.Kaspera R, Naraharisetti SB, Evangelista EA, Marciante KD, Psaty BM, Totah RA. Drug metabolism by CYP2C8.3 is determined by substrate dependent interactions with cytochrome P450 reductase and cytochrome b5. Biochem Pharmacol. 2011;82:681–91. doi: 10.1016/j.bcp.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tornio A, Niemi M, Neuvonen PJ, Backman JT. Trimethoprim and the CYP2C8*3 allele have opposite effects on the pharmacokinetics of pioglitazone. Drug Metab Dispos. 2008;36:73–80. doi: 10.1124/dmd.107.018010. [DOI] [PubMed] [Google Scholar]
- 32.Kirchheiner J, Thomas S, Bauer S, Tomalik-Scharte D, Hering U, Doroshyenko O, Jetter A, Stehle S, Tsahuridu M, Meineke I, Brockmoller J, Fuhr U. Pharmacokinetics and pharmacodynamics of rosiglitazone in relation to CYP2C8 genotype. Clin Pharmacol Ther. 2006;80:657–67. doi: 10.1016/j.clpt.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 33.Aquilante CL, Bushman LR, Knutsen SD, Burt LE, Rome LC, Kosmiski LA. Influence of SLCO1B1 and CYP2C8 gene polymorphisms on rosiglitazone pharmacokinetics in healthy volunteers. Hum Genomics. 2008;3:7–16. doi: 10.1186/1479-7364-3-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niemi M, Leathart JB, Neuvonen M, Backman JT, Daly AK, Neuvonen PJ. Polymorphism in CYP2C8 is associated with reduced plasma concentrations of repaglinide. Clin Pharmacol Ther. 2003;74:380–7. doi: 10.1016/S0009-9236(03)00228-5. [DOI] [PubMed] [Google Scholar]
- 35.Martinez C, Garcia-Martin E, Blanco G, Gamito FJ, Ladero JM, Agundez JA. The effect of the cytochrome P450 CYP2C8 polymorphism on the disposition of (R)-ibuprofen enantiomer in healthy subjects. Br J Clin Pharmacol. 2005;59:62–9. doi: 10.1111/j.1365-2125.2004.02183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Martin E, Martinez C, Tabares B, Frias J, Agundez JA. Interindividual variability in ibuprofen pharmacokinetics is related to interaction of cytochrome P450 2C8 and 2C9 amino acid polymorphisms. Clin Pharmacol Ther. 2004;76:119–27. doi: 10.1016/j.clpt.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 37.Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events. December 2004. [online]. Available at http://www.niaid.nih.gov/labsandresources/resources/daidsclinrsrch/documents/daidsaegradingtable.pdf (last accessed 1 July 2012)
- 38.Andrisin TE, Humma LM, Johnson JA. Collection of genomic DNA by the noninvasive mouthwash method for use in pharmacogenetic studies. Pharmacotherapy. 2002;22:954–60. doi: 10.1592/phco.22.12.954.33598. [DOI] [PubMed] [Google Scholar]
- 39.Rower JE, Bushman LR, Hammond KP, Kadam RS, Aquilante CL. Validation of an LC/MS method for the determination of gemfibrozil in human plasma and its application to a pharmacokinetic study. Biomed Chromatogr. 2010;24:1300–8. doi: 10.1002/bmc.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pedersen RS, Damkier P, Brosen K. The effects of human CYP2C8 genotype and fluvoxamine on the pharmacokinetics of rosiglitazone in healthy subjects. Br J Clin Pharmacol. 2006;62:682–9. doi: 10.1111/j.1365-2125.2006.02706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.US Food and Drug Administration. Drug Development and Drug Interactions. Available at http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm080499.htm (last accessed 1 July 2012)
- 42.Zhang L, Reynolds KS, Zhao P, Huang SM. Drug interactions evaluation: an integrated part of risk assessment of therapeutics. Toxicol Appl Pharmacol. 2010;243:134–45. doi: 10.1016/j.taap.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 43.Mano Y, Usui T, Kamimura H. The UDP-glucuronosyltransferase 2B7 isozyme is responsible for gemfibrozil glucuronidation in the human liver. Drug Metab Dispos. 2007;35:2040–4. doi: 10.1124/dmd.107.017269. [DOI] [PubMed] [Google Scholar]
- 44.Innocenti F, Liu W, Fackenthal D, Ramirez J, Chen P, Ye X, Wu X, Zhang W, Mirkov S, Das S, Cook E, Jr, Ratain MJ. Single nucleotide polymorphism discovery and functional assessment of variation in the UDP-glucuronosyltransferase 2B7 gene. Pharmacogenet Genomics. 2008;18:683–97. doi: 10.1097/FPC.0b013e3283037fe4. [DOI] [PMC free article] [PubMed] [Google Scholar]