

Abstract
Aim: Genetic factors are important for outcome after traumatic brain injury (TBI), although exact knowledge of relevant genes/pathways is still lacking. We here used an unbiased approach to define differentially activated pathways between the inbred DA and PVG rat strains. The results prompted us to study further if a naturally occurring genetic variation in glutathione-S-transferase alpha 4 (Gsta4) affects the outcome after TBI. Results: Survival of neurons after experimental TBI is increased in PVG compared to the DA strain. Global expression profiling analysis shows the glutathione metabolism pathway to be the most regulated between the strains, with increased Gsta4 in PVG among top regulated transcripts. A congenic strain (R5) with a PVG genomic insert containing the Gsta4 gene on DA background displays a reversal of the strain pattern for Gsta4 expression and increased survival of neurons compared to DA. Gsta4 is known to effectively reduce 4-hydroxynonenal (4-HNE), a noxious by-product of lipid peroxidation. Immunostaining of 4-HNE was evident in both rat and human TBI. Intracerebral injection of 4-HNE resulted in neurodegeneration with increased levels of a marker for nerve injury in cerebrospinal fluid of DA compared to R5. Innovation: These findings provide strong support for the notion that the inherent capability of coping with increased 4-HNE after TBI affects outcome in terms of nerve cell loss. Conclusion: A naturally occurring variation in Gsta4 expression in rats affects neurodegeneration after TBI. Further studies are needed to explore if genetic variability in Gsta4 can be associated to outcome also in human TBI. Antioxid. Redox Signal. 18, 784–794.
Introduction
Traumatic brain injury (TBI) is an acute condition where immediate actions are required in order to stabilize vital functions and reduce the risk of secondary insults that can be devastating for the prognosis. Current intensive care routines have improved outcome considerably. Still, however, it is evident that tissue reactions induced by the initial injury with ongoing loss of nerve cells continue for days or even weeks after the initial injury. For this reason, major research efforts have been made to understand the pathophysiological mechanisms of TBI better, and based on this knowledge, to develop therapies that limit loss of nerve cells and improve prognosis. A great obstacle to this effort has been the wide clinical spectrum of TBI regarding severity, age, gender, type of injury, and co-morbidity. This may be the main reason why a number of clinical studies have failed to reproduce a beneficial effect in spite of positive outcomes in standardized experimental models of TBI (24). Furthermore, it is now recognized that even when all of the above prognostic factors are taken into consideration, individuals can respond differently to a similar injury, presumably at least in part due to genetic differences (20).
Innovation.
Gsta4 has by far the highest detoxifying capability of the highly toxic product 4-HNE. Lipid peroxidation is one of the most significant pathophysiological processes in TBI. A naturally occurring genetic variability in Gsta4 is here identified to affect expression and protein levels of the enzyme, which is located in neurons and upregulated in these cells upon injury. A congenic strain with higher expression of Gsta4 displays less nerve cell loss in the hippocampus after TBI, which is the first such congenic strain effect ever to be reported in a TBI model. These findings encourage further studies of the role of polymorphism in human Gsta4 in neurodegenerative diseases and open new perspectives for therapies targeting 4-HNE in TBI.
Indeed, a number of studies have found evidence that polymorphisms in the apolipoprotein E (APOE) gene affect outcome of TBI, with a more unfavorable outcome for individuals carrying the e4 allele of the APOE gene (49). Apart from APOE, a smaller number of association studies have suggested a possible genetic influence on TBI outcome for polymorphisms in the tumor protein 53, interleukin-1β, CACNA1A, dopamine receptor D2, and poly(ADO-ribose) polymerase 1 genes (26). However, all these studies have been conducted with a very limited number of patients, leaving a high risk for false positive findings. From other conditions, we now know that in order to unravel the genetic basis of complex traits, cohorts consisting of many thousand patients are needed to achieve the necessary statistical power to pinpoint genetic influences (36).
Experimental studies conducted in models of TBI are valuable tools for studying the impact of naturally occurring genetic polymorphisms on TBI outcome and thereby revealing possible candidate genes. This approach, by using genetic dissection of complex traits, has been particularly successful in autoimmune diseases such as multiple sclerosis and rheumatoid arthritis, where discovery of important information about underlying genetic regulation has led to increased knowledge of disease pathophysiology and treatment response (15, 31). The impact of genetic heterogeneity has been much less studied in the context of TBI. However, it has been demonstrated that TBI outcome differs across different rodent strains (34, 45), and we recently reproduced this finding by showing considerable differences in TBI outcome in the two inbred strains: dark agouti (DA) and piebald virol glaxo (PVGav1) (2). These two strains have previously been extensively studied in autoimmune models such as experimental allergic encephalomyelitis (EAE), a model of multiple sclerosis (MS), and experimental arthritis, where the DA strain is susceptible while the PVGav1 is resistant (8, 19). We have also demonstrated differences in the response to a standardized peripheral nerve lesion with regard to survival of axotomized nerve cells and local glial activation (8, 19, 44).
In this study, we used microarrays in order to determine genetically regulated pathways between the two strains DA and PVGav1. The most significant differentially regulated pathway was glutathione metabolism. Among the strain-regulated genes in this pathway was glutathione-S-transferase alpha 4 (Gsta4), which plays an important role for detoxification of a toxic product of lipid peroxidation; 4-hydroxy-t-2,3-nonenal (4-HNE). We then used a congenic strain, recently demonstrated to have a phenotype related to axotomy-induced nerve cell loss (42), to investigate if the naturally occurring variation in Gsta4 expression has a significant effect on 4-HNE metabolism and neurodegeneration in experimental TBI with relevance for human TBI.
Results
Differential regulation of the glutathione metabolism pathway between DA and PVGav1
The pericontusional area of 5 DA and 5 PVGav1 rats were analyzed with microarrays in order to provide unbiased information on genetically regulated pathways one day after experimental brain injury. The pericontusional area was collected using a sample corer (Fine Science Tools, Heidelberg, Germany) resulting in a 5×5×6 mm piece of tissue consisting of the contusion core, the pericontusional cortex, the underlying hippocampus, and part of the thalamus. The experimental setup that was used to dissect genetic differences is shown in Figure 1. The molecular pathway showing the largest differences between the two strains was the glutathione metabolism pathway and included the transcripts Gsta1 (5.26E-06; 8.50), Gsta4 (3.94E-04;−1.7), Gstm2 (8.65E-04; −1.50), Gstm4 (2.29E-07; −2.93), and PRDX6 (2.63E-03; −1.56) (Fig. 2). The p value and fold change DA/PVGav1 are shown in the parentheses for each transcript, respectively. All transcripts display higher expression in the PVGav1 strain except from Gsta1. Independent confirmation of differences in transcriptional regulation of Gsta1 and Gsta4 was done with RT-PCR. Gsta1 displayed a similar expression pattern, as in the microarray data, with lower levels in PVGav1 compared to DA (data not shown). RT-PCR for Gsta4 included also the congenic strain R5 and showed that the PVGav1 strain displayed higher Gsta4 levels than the DA strain and similar Gsta4 levels to the R5 congenic (Fig. 3A). Microarray analysis of the 35 genes that are included in the genetic region differing between DA and R5 are shown in Table 1. Only the Gsta4 gene displayed both a significant p value and above threshold fold change in the analysis. Protein quantification with Western blot confirmed that the observed differential Gsta4 expression pattern results in corresponding differences at the protein level (Fig. 3B).
FIG. 1.

Experimental set up used to dissect the effect of genetic variability in the Gsta4 gene. Experimental traumatic brain injury was performed in the two inbred rat strains DA and PVGav1 and their genome is shown in black or white color, respectively. The genome of the R5 congenic rat has all genes from the DA strain illustrated in black color except for a fragment with 35 genes, including Gsta4 (illustrated as a small white segment at chromosome 8) from the PVGav1 strain. (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
FIG. 2.

Differentially expressed genes between DA and PVGav1 after TBI. Partek Express (Partek, St Louis, MO) was used to carry out statistical analyses of microarray data files. A t-test and a fold change value were used to examine the significance and degree of the differences between DA and PVGav1 for every expressed target. Transcripts with a p<0.005 and a ≥1.4-fold change were uploaded for analysis with the Ingenuity Pathway Analysis software in order to identify top canonical pathways affected by strain. IPA identified the canonical pathways that were most significant in the data set in 2 different ways: 1) By using Fisher's exact test to calculate the probability (p value) that the association between the genes in the data set and the canonical pathway is explained by chance alone; 2) By calculating the ratio of the number of genes from the data set that map to the pathway divided by the total number of genes that map to the canonical pathway. The bars represent −log p value and the percentage of genes present in the data set compared to the total number of genes present in each selected pathway in the IPA data base. The yellow line represents the ratio of affected genes to the total number of genes in a pathway. Canonical pathways above the threshold (horizontal line) are shown. (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
FIG. 3.

Genetic differences in Gsta4 expression after TBI. Gsta4 transcriptional levels were analyzed by quantitative PCR in the pericontusional area one day after TBI. The DA strain had lower expression of Gsta4 compared to the PVGav1 strain. The congenic R5 strain with the DA genomic background but with the Gsta4 haplotype from PVGav1 had similar levels to the PVGav1 (A). The protein level of Gsta4 was measured with Western blot using beta-actin as reference protein. Data show that the difference in expression is reflected also at the protein level. (B). Statistical analyses were made using one-way ANOVA with a Bonferroni post-hoc test (*p<0.05, **p<0.01, ***p<0.0001).
Table 1.
Gene Names of the 35 Genes Differing in the Genetic Region Between DA and R5
| RGD_ID | Symbol | Name | P value | Fold change |
|---|---|---|---|---|
| 1549773 | Lrrc1 | leucine rich repeat containing 1 | 0.194461 | 1.08761 |
| 1560540 | Klhl31 | kelch-like 31 (Drosophila) | 0.0977909 | 1.12532 |
| 1593347 | LOC691935 | similar to 60S ribosomal protein L27a | * | * |
| 619868 | Gclc | glutamate-cysteine ligase catalytic subunit | 0.208953 | 1.0748 |
| 620583 | Elovl5 | ELOVL family member 5 elongation of long chain fatty acids | 0.022354 | −1.1182 |
| 61852 | Gcm1 | glial cells missing homolog 1 (Drosophila) | 0.805276 | 1.01952 |
| 1593343 | LOC691940 | similar to Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | * | * |
| 1310374 | Fbxo9 | f-box protein 9 | 0.030816 | −1.14769 |
| 71050 | Ick | intestinal cell kinase | 0.319976 | 1.1242 |
| 1306318 | Rpl12-ps1 | ribosomal protein L12 pseudogene 1 | * | * |
| 1309970 | Gsta4 | glutathione S-transferase alpha 4 | 0.000393941 | -1.70008 |
| 1593189 | LOC494499 | LOC494499 protein | † | † |
| 1592003 | LOC367116 | similar to glyceraldehyde-3-phosphate dehydrogenase | * | * |
| 2754 | Gsta2 | glutathione S-transferase alpha 2 | 0.567174 | −1.04368 |
| 1305838 | Dppa5 | developmental pluripotency associated 5 | † | † |
| 1311103 | RGD1311103 | similar to RIKEN cDNA 2410146L05 | * | * |
| 1308830 | Mto1 | mitochondrial translation optimization 1 homolog (S. cerevisiae) | 0.123685 | 1.2266 |
| 67387 | Eef1a1 | eukaryotic translation elongation factor 1 alpha 1 | 0.00580026 | 1.07236 |
| 1311388 | Slc17a5 | solute carrier family 17 (anion/sugar transporter) member 5 | 0.510577 | −1.05204 |
| 1311287 | Cd109 | CD109 molecule | 0.798576 | 1.02068 |
| 1562651 | LOC367117 | similar to RIKEN cDNA 2900055D03 | 0.855682 | −1.0256 |
| 1592002 | LOC367118 | similar to nuclease sensitive element binding protein 1 | * | * |
| 2324749 | LOC100363141 | dynein light chain 1-like | 0.397462 | 1.06638 |
| 1585750 | LOC682240 | similar to 40S ribosomal protein S3a | * | * |
| 2374 | Col12a1 | collagen type XII alpha 1 | 0.121998 | 1.21117 |
| 1587147 | LOC679536 | hypothetical protein LOC679536 | † | † |
| 2323401 | LOC100363239 | Sec61 gamma subunit-like | † | † |
| 2324602 | LOC100363289 | LRRGT00022-like | † | † |
| 68365 | Cox7a2 | cytochrome c oxidase subunit VIIa polypeptide 2 | 0.31234 | 1.13129 |
| 1303315 | Tmem30a | transmembrane protein 30A | 0.161074 | 1.11615 |
| 628597 | Filip1 | filamin A interacting protein 1 | 0.132794 | −1.16512 |
| 1585967 | LOC679622 | hypothetical protein LOC679622 | * | * |
| 1304565 | Senp6 | SUMO1/sentrin specific peptidase 6 | 0.0806125 | 1.16435 |
| 1560646 | Myo6 | myosin VI | 0.703506 | 1.034 |
| 620499 | Impg1 | interphotoreceptor matrix proteoglycan 1 | 0.27957 | 1.09185 |
P values and fold change DA/PVGav1 as obtained by microarray analysis are shown. Significant differences p<0.01 or fold change ≥1.4 depicted in bold.
, pseudogene; †, not analyzed in microarrays.
Cellular localization of Gsta4 and 4-HNE
The glutathione metabolism pathway plays a major role in neutralizing oxidative stress products in several conditions (21, 33, 47). Of the different enzymes in this pathway, Gsta4 has the by far most efficient catalytic activity to reduce 4-HNE by conjugating it to glutathione, hence reducing its toxic activity and thereby preventing formation of adducts to other proteins (4). Immunohistological staining of Gsta4 showed labeling mainly in neurons on both the ipsi- and contralateral sides (Fig. 4A and 4B). A semiquantitative assessment of Gsta4 labeling intensity on the ipsi versus the contralateral side suggested an upregulation in neurons after injury in the PVGav1 and R5 strains, but not in DA, which is consistent with previous findings after peripheral nerve injury (42). Immunostaining with two different antibodies recognizing 4-HNE-conjugates showed an essentially similar pattern, with strong staining of blood cell infiltrates in the contusion, a diffuse staining in the pericontusional area, and also a more distinct cellular staining of both glia and neurons. Neuronal cytoplasmic staining of 4-HNE was seen in degenerating cells in the pericontusional cortex and the hippocampal CA1, CA2/CA3, dentate gyrus, and hilus areas at both 1 and 6 days after TBI (Fig. 4C–4F). No discernible qualitative differences in cellular levels of 4-HNE were evident between the different strains.
FIG. 4.

Gsta4 and 4-HNE-protein adduct immunolocalization. (A, B) Immunohistochemical labeling for Gsta4 in the hilus of a R5 rat at one day after traumatic brain injury on the contralateral and ipsilateral hilus, dentate gyrus, and part of CA3. (C, D) Immunohistochemical labeling of 4-HNE-protein adducts in the lesion and the surrounding cortex and hippocampus of a DA rat at one day after TBI with (D) showing a picture of hilus in higher magnification. (E, F) Confocal micrograph in the same DA rat of fluorescent triple immunolabeling with DAPI, the neuronal marker NeuN, and 4-HNE-protein adducts in the cortex (E) and hilus (F). Neuronal cells both at the contralateral (A) and ipsilateral (B) site of the injury express Gsta4 with a stronger labeling on the ipsilateral site. In addition, a strong labeling for 4-HNE-protein adducts is present in the blood derived infiltrating cells in the injury, as well as a diffuse labeling in the surrounding cortex and the hippocampus (C). Positive staining of a neuron in the hilus is indicated (D, arrow head). Cytoplasmic staining for 4-HNE-protein adducts is present in both neurons (arrow heads) and glia (arrows) in the cortex (E) and the hilus (F). Scale bars are 100 μm in A, B, E and F; 1 cm in C, and 15 μm in D. (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
Analysis of 4-HNE-conjugate levels in cerebrospinal fluid
The detection of 4-HNE-conjugates using ELISA based techniques has been reported previously (7, 33). We therefore attempted to determine CSF 4-HNE-conjugate levels in rat and human cerebrospinal fluid (CSF) after TBI using two different commercial ELISA-kits, of which one gave a positive signal. The analysis was conducted in a clinical material composed of patients with TBI and a control group of patients with other neurological diseases (OND). However, further testing showed that this signal was derived from unspecific binding of the secondary antibody to immunoglobulins (data not shown). In an ELISA set up in the lab using the two different antibodies used for immunohistochemistry, a signal in CSF spiked with 4-HNE down to a concentration of 0.5 μM could be detected. However, no specific signal was present in human CSF as well as rat CSF after TBI or 4-HNE-injection. It is therefore unlikely that 4-HNE-conjugates exist in micromolar concentration in CSF as previously has been reported (38, 39).
The PVGav1 Gsta4 haplotype reduces loss of neurons in hippocampus after TBI
The localization of Gsta4 and 4-HNE in degenerating neurons in the hippocampus prompted us to investigate the effect of the Gsta4 haplotype for neuronal cell loss. Cell numbers in the hilus region were determined with stereology-based cell counts 30 days after TBI in the DA, PVGav1, and R5 strains. All three strains had equal numbers of neurons in sham-operated controls, but lower numbers of neurons after TBI. However, while DA displayed a 47.6% reduction in neuron numbers, the PVGav1 and R5 strain had lost only 24.6% and 29.8% of the neurons, respectively. This corresponds to a more than one-third reduction of neuronal loss in the PVGav1 and R5 strain compared to DA (Fig. 5).
FIG. 5.

Surviving neuronal cells in the hilus 30 days after TBI. Design-based stereology was used to count the neurons in the hilus that had survived TBI 30 days after the injury. The estimated total number of neurons is shown for the DA (black boxes), PVGav1 (red triangles), and R5 (black triangles) rat. Sham operated rats were used as controls. Comparison between the groups were evaluated by one-way ANOVA with a Bonferroni post-hoc test (***p<0.001). (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
Intraparenchymal injection of 4-HNE induces upregulation of Gsta4 and apoptosis of neurons
The biological cascade that follows TBI includes many toxic molecules apart from 4-HNE. In order to test the effect of 4-HNE more specifically, intraparenchymal injections were performed in DA and R5. Gsta4 labeling was upregulated in the area surrounding the site of injection, revealing a feedback mechanism where neurons upregulate Gsta4 in response to increased 4-HNE levels (Fig. 6A). A blinded semiquantitative assessment of Gsta4 protein staining revealed stronger Gsta4 upregulation in the R5 strain compared to the DA strain (Fig. 6B–6E). In addition, CSF levels of neurofilament light, a marker for axonal injury, were lower in R5 compared to DA, further supporting the in vivo role of Gsta4 to detoxify 4-HNE (Fig. 6F). A strong staining for 4-HNE-conjugates was seen in the area of injection, with both glia and neurons displaying labeling, without discernible qualitative differences between strains (Fig. 6G–6J). Further, TUNEL-staining showed apoptotic cell bodies after injection and co-localization of 4-HNE protein adducts in apoptotic neurons (Fig. 6K–6L).
FIG. 6.

Gsta4 expression, 4-HNE detoxification, and apoptotic cell death following intracerebral 4-HNE injection. (A–E) Gsta4 immunohistochemical staining, 24 h after intraparenchymal injection of 4-HNE showing expression of Gsta4 in the site ipsilateral to the injection versus the contralateral site and differences between the DA and R5 strain particularly in the CA1 area. (F) CSF levels of neurofilament light as assessed with ELISA, and (G–J) imunohistochemical detection of 4-HNE-protein adducts in the ipsilateral versus the contralateral site. (K, L) Double immunofluorescent labeling for apoptotic cell death as assessed with TUNEL (green) and 4-HNE-protein adducts (red). Note the upregulation of Gsta4 around the injection site (A) and that neurons of the CA1 area display more Gsta4 staining for the R5 strain compared to the DA (B–E). Also, note in (F) that CSF neurofilament light levels are elevated in the DA strain compared to the R5. Furthermore, in (G–I) note that 4-HNE has diffused in the tissue surrounding the injection to form adducts which are present both in glia and neurons. Finally, (K) shows that injection of 4-HNE results in apoptotic cell death around the injection site and (L) shows a neuron of the CA1 area with an apoptotic fragmented nucleus and the presence of 4-HNE-Michael adducts in the cytoplasm. Scale bars are 1 mm in (A); 250 μm in (K), 100 μm in (B), (D), (G), and (I); 15 μm in (C), (E), (H), (J), and (L). Comparison between the two groups were evaluated by the Student's t test (*p<0.05). (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
4-HNE-conjugates are present in neurons in human TBI
A biopsy was taken from a 76-year-old patient who presented with a Glasgow Coma Scale of 9 after head trauma. Radiological evaluation revealed an expansive left contusional hemorrhage and the patient was treated surgically at 15 hours after trauma. Immunolabeling of human pericontusional tissue with antibodies for the neuronal marker NeuN and two different antibodies for 4-HNE, revealed the presence of 4-HNE-conjugates in neurons also after human TBI (Fig. 7).
FIG. 7.

Presence of 4-HNE-protein adducts in neurons in human TBI. (A, B) Triple immunolabeling of human pericontusional tissue, 15 h after trauma, with DAPI, the neuronal marker NeuN, and 4-HNE-protein adducts. Note the presence of 4-HNE-protein adducts in the cytoplasm of neurons in human pericontusional tissue. Two areas (A, B) in the tissue are shown with the second (B) in higher magnification. Scale bars are 50 μm in (A) and 12.5 μm in (B). (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
Discussion
In this study we provide evidence that naturally occurring differences in Gsta4 regulate outcome in experimental TBI. Microarray data was obtained in the two inbred rat strains DA and PVGav1 as a first step to genetically dissect differences in pathway activation after experimental TBI. These strains have previously been extensively studied in autoimmune disease models such as EAE, a model of MS, which has provided both information on genetically regulated pathways as well as specific genes of importance for disease pathogenesis (5, 13, 29, 31, 44). Furthermore, the strains differ both in inflammatory response and neuronal survival in a standardized peripheral nerve injury model (19, 22). Recently, we demonstrated that these two strains also differ in their inflammatory response to experimental TBI, as well as outcome in terms of lesion volume (2). In the present study, we find that the most significantly genetically regulated pathway between DA and PVGav1 after TBI is the glutathione metabolism, with Gsta4 being one of the most differentially regulated transcripts. Glutathione metabolism has long been known to constitute a molecular system that protects tissues by detoxifying toxic molecules produced by oxidative stress and lipid peroxidation (37). In addition to this, oxidative stress and lipid peroxidation is one of the most extensively studied pathogenic processes known to contribute to the secondary damage after TBI. After a primary insult on the brain tissue, free radicals are formed and react with membrane phospholipids in a chain reaction disrupting their function and producing toxic aldehydes such as 4-HNE. 4-HNE reacts with the amino acids lysine, histidine, and cysteine in cellular proteins, which modify their conformation and disrupt important cellular enzymatic functions, making 4-HNE a highly toxic molecule (16). 4-HNE has been shown to be increased early after TBI and antioxidant therapies acting on upstream processes relevant for 4-HNE generation display a neuroprotective effect (11, 14, 18, 40). Once generated by peroxidation of lipids, a physiologic cellular system composed of glutathione (GSH) and a family of enzymes, glutathione-S-transferases, acts as a defense system to limit the deleterious effects of 4-HNE by conjugating it to glutathione, thus, making it unable to react with tissue proteins. Gsta4 has the by far highest catalytic activity for this reaction (4). This prompted us to investigate the role of the here identified naturally occurring genetic variation in Gsta4 expression between the inbred DA and PVGav1 strains for oxidative damage in TBI. This was done by the use of a congenic strain that contains a small genetic fragment including the Gsta4 gene from PVGav1 on DA background, with significantly increased Gsta4 levels. The finding that PVG alleles confer higher expression of Gsta4, which is translated into higher protein levels, while the sequence of the translated protein is identical between the strains (unpublished data) suggests that higher catalytic activity in PVG is a direct consequence of increased protein levels rather than providing higher catalytic activity in the enzyme per se.
We find 4-HNE immunoreactivity both in the pericontusional area and the hippocampus, including neurons, after experimental TBI, suggesting that it may contribute to neuronal cell loss after TBI. Indeed, when 4-HNE is injected directly into the brain, apoptotic nerve cells are present at one day after the injection both in the area of the injection but also in the hippocampus. We have previously performed an unbiased complex trait mapping of motor neuron loss after ventral root avulsion in the DA and PVGav1 strains, where a significant linkage was identified on chromosome 8 (19, 43). Also a whole genome expression QTL mapping in a F2(DAxPVGav1) intercross combined with fine mapping using congenic strains in the ventral root avulsion model identified one single transcript being highly strain regulated; Gsta4 (42). Further, we have found that the DA vs PVGav1 strain difference is equally strong on the protein level (42). We therefore decided to test the smallest of our congenic strain, R5, which contains 35 protein coding genes including Gsta4, in the TBI model. We find that the R5 strain displays a complete reversal of the strain pattern regarding Gsta4 (i.e., the level of expression is similar to the donor PVGav1 strain and significantly higher than the recipient DA strain). Furthermore, the levels of neurofilament light protein, a marker for neurodegeneration (35), were lower in the CSF of the R5 strain compared to DA after injection of 4-HNE, which provides in vivo evidence for a nonredundant importance of naturally occurring genetic variability in Gsta4 for detoxification of 4-HNE. However, the most significant finding is that neuronal cell death, as quantified stereologically in the hilus region, is similar between the congenic R5 and PVGav1 strains and significantly reduced compared to DA. Thus, the difference between the two parental strains in large part can be explained by variation in the R5 fragment, where Gsta4 is the only differentially regulated transcript. This provides strong support for the notion that higher expression of Gsta4 results in a more efficient detoxification of 4-HNE, in turn reducing nerve cell death. This chain of events are supported by findings in previous studies, where it was shown that 4-HNE induces degeneration of a variety of nerve cell populations ranging from hippocampal to motoneurons both in vitro and using experimental in vivo models (3, 17, 25, 27, 48).
Levels of 4-HNE in CSF have been reported previously in the chronic neurodegenerative disorders Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS) (38, 39, 41). In the studies involving AD and PD subjects, the CSF concentration is reported to be approximately 0.5 μM in controls and further elevated in patients. The ALS studies show levels of only around 600 pg/ml in controls, corresponding to approximately 5 nM (41). Here we could not detect 4-HNE-conjugates in CSF using ELISA, although the assays had sensitivity down to low micromolar levels. Western blot identified a number of bands in 4-HNE-incubated CSF, but not in unmanipulated samples. Thus, there are presently only a limited number of studies on 4-HNE-conjugate levels in CSF, which have been conducted by two different groups showing very divergent results. Our results indicate that soluble 4-HNE-conjugates are not present at micromolar levels in CSF, which suggests that most HNE-conjugates remain captured in the tissue rather than being dissolved into the CSF. This notion is supported by the finding of immunostaining for 4-HNE-conjugates in contused human brain tissue.
Glutathione-S-transferase polymorphisms have been the subject of prior studies, identifying variability due to ethnic origin, but also to be associated to susceptibility for certain cancers (9). Thus, certain Gsta4 polymorphisms increase the risk to develop melanoma and hepatocellular and lung cancer (1, 28, 32). In CNS diseases there are some indications that a Gstm1 null and a Gstm3 polymorphism increase the risk for PD and AD, respectively (23, 30). The effect of genetic variability in Gsta4 has not been studied, except for a single study in 50 patients with PD. Not surprisingly, given the low number of included patients, the study did not reveal any association (6). In order to determine any possible relevance for neurodegeneration in humans, genotyping of much larger patient materials with available information on clinical follow up data that reflects the rate of neurodegeneration is required.
Taken together, these findings underscore both the importance of genetic diversity for outcome of TBI and also more specifically the role of the Gsta4—4-HNE detoxifying process. The former, if confirmed also for human TBI, could partially explain the failure of antioxidant therapy studies in human TBI. These results encourage studies on any possible genetic influence of polymorphism in Gsta4 for outcome in human TBI as well as on therapeutic approaches that targets the 4-HNE pathway, such as, for example, histidine analogues or hydrazine derivatives that scavenge 4-HNE and rescue cells from 4-HNE toxicity (12, 16, 46).
Materials and Methods
Animals
The DA (RT1av1) strain was originally provided by Professor Hans Hedrich (Medizinische Hochschule, Hannover, Germany), while the MHC congenic PVG-RT1av1 (hereafter called PVGav1) strain was obtained from Harlan UK Ltd (Blackthorn, UK). The DA.PVGav1-Vra1R5 (RNO8: D8Rat24-D8Got132 [82.2–88.6 Mb]) (hereafter called R5) congenic was bred in-house as described previously (43). The genome of the R5 congenic has all genes from the DA strain except 35 genes, including Gsta4, from the PVGav1 strain. All animals were bred in our in-house breeding facility with 12 h light/dark cycles and fed standard rodent chow and water ad libitum.
Experimental brain injury and 4-HNE injections
Experimental traumatic brain contusion was performed in 60 male animals weighing approximately 230–300 grams, at an age of 10–14 weeks, under deep isoflurane anesthesia using the weight drop injury model as described previously (2). In brief, the rats were placed in a stereotactic frame, and a 2 mm craniotomy was drilled 3 mm posterior and 2.3 mm lateral to the bregma. A standardized parietal contusion was made by letting a 24 gram weight fall onto a rod with a flat end diameter of 1.8 mm from a height of 7 cm in contused rats, allowed to compress the tissue a maximum of 3 mm. For 4-HNE injections the same procedure as for TBI was followed except at this time the craniotomy was smaller, with slow injection of 1 μl 64 mM 4-HNE (Cayman Chemical) during 2 min at the same coordinates and at 3 mm depth from the surface. This concentration was chosen since it has been shown to induce axonal and oligodendrocyte damage after intracerebral injection (27). All experiments in this study were approved by the local ethical committee for animal experimentation.
Array hybridization and qRT-PCR and Western blot analysis
Animal sacrifice, tissue preparation and preparation of mRNA was done as previously reported (2). The microarray analysis was performed at the microarray core facility of Karolinska Institutet using Affymetrix Rat gene 1.0 ST Array chips (Affymetrix, Santa Clara, CA) as described before (42). The microarray data is available in MIAME-compliant (minimal information about a microarray experiments) format at the ArrayExpress Database (http://www.ebi.ac.uk/arrayexpress) under accession code E-MTAB-795. Preparation of cDNA and qRT-PCR was done as described before (2). Gapdh and Hprt were used as housekeeping genes. The sequences of the used primers are: Gsta1 (Fw: CTGTGCGGTGGCTATTGG, Rev: TTCATAATGGCTCTGGTCTGC), Gsta4 (Fw: CAGGAGTCATGGAAGTCAAAC, Rev: TTCTCATATTGTTCTCTCGTCTC), Gapdh (Fw: TCAACTACATGGTCTACATGTTCCAG, Rev: TCCCATTCTCAGCCTTGACTG) and Hprt (Fw: CTCATGGACTGATTATGGACAGGAC, Rev: GCAGGTCAGCAAAGAACT TATAGCC).
For Western blot analysis, protein homogenate from the same region as used for PCR was prepared and analyzed as previously decribed (42). Briefly, Gsta4 was labeled with a rabbit anti-rat Gsta4 antibody (1:2000) and a secondary HRP-conjugated antibody (donkey anti‐rabbit IgG‐HRP conjugated; 1:2000; Amersham NA9340V). ECL solution (Amersham) was applied and membranes exposed to Chemiluminescence film (Amersham HyperfilmTM ECL). Intensity of bands were measured with Gel DocTM XR+/Image LabTM 3.0(BioRad) and normalized against β-actin (Sigma Monoclonal β-Actin antibody Clone AC-15;1: 25 000).
4-HNE and neurofilament light analysis of cerebrospinal fluid after traumatic brain injury
Rat cerebrospinal fluid (CSF) was obtained directly after sacrifice from the cerebellomedullary cistern. The rat was placed in a stereotaxic frame and shaved over the occipital crest. A puncture through the occipital foramen magnum was made and about 100 μl of CSF was slowly drawn. The CSF was stored at −70°C until further processing.
The human CSF samples were obtained from 27 patients with severe TBI while being treated at the intensive care unit (ICU), and from 37 patients with other neurological diseases (OND) that were used as controls. CSF collection used in this study was approved by the regional ethical review board in Stockholm.
To assess the concentration of 4-HNE-protein adducts (μg/ml) in the CSF, we used two different commercial enzyme-linked immunosorbent assay (OxiSelect HNE-His Adduct ELISA, Cell Biolabs, INC, San Diego and HNE ELISA, Cusabio, Hubei Province 430223, P.R.China). These ELISAs were performed according to the manufacturer's instructions. Third, we set up a sandwich ELISA, using mouse monoclonal anti HNE (Abcam) as capture antibody, and rabbit polyclonal anti-HNE (Abcam, 1:6000) as detection antibody. Samples were incubated overnight and a biotinylated anti-rabbit antibody was used together with ABC-kit (Vector labs) for signal enhancement. As standard, control CSF incubated with HNE (Cayman, Burlingame, CA) was used in a dilution series with un-incubated CSF rendering a standard with linear detection in the range of 32–0.5 μM.
Levels of neurofilament light were quantified using NF-Light Neurofilament ELISA RUO kit (Umandiagnostics, Umeå, Sweden) according to the manufacturer's instructions.
Immunohistochemistry and immunofluorescence
Animals were sacrificed with CO2 and perfused first with 100 ml cold PBS containing heparin and after that with 250 ml parafolmaldehyde. The brains were postfixed at the same fixative followed by 15% sucrose and then stored at −70°C. Tissue preparation, immunohistochemistry, and double labeling immunofluorescense was performed as described previously (2). Brain tissue from the pericontusional area was obtained from a 76-year-old female patient who was surgically treated for a left contusional hemorrhage. The biopsy was snap-frozen in liquid nitrogen, stored at −70°C and postfixed in paraformaldehyde. The biopsy used in this study was approved by the regional ethical review board in Stockholm.
The primary antibodies used were rabbit polyclonal anti-HNE Michael adducts (Merck Chemicals Ltd., Nottingham, UK), mouse monoclonal anti HNE-histidine adducts (Abcam), rabbit polyclonal anti-rat Gsta4 (42), mouse monoclonal and rabbit polyclonal anti-NeuN (Millipore, MA). Apoptotic cell death was visualized using TUNEL. The sections were washed in PBS and permeabilized for 5 min in a 2:1 mixture of ethanol and acetic acid at −20°C. The TUNEL reaction was carried out with the "In situ cell death detection kit, fluorescein" (Boehringer Mannheim, Bromma, Sweden) at 37°C for 30 min.
Stereological assessment of neurodegeneration
We determined the number of neurons in the hilus 30 days after experimental TBI using a design based stereological approach. The hippocampi of the side ipsilateral to the injury, of 7 DA, 6 PVGav1, 7 DA.PVGav1R5, and 2 sham animals from each strain were dissected and straightened manually in order to eliminate its natural curvature. They were then embedded in 5% agar and sectioned perpendicularly to the longest axis at 65 μm thickness on a Vibratome 3,000 (Vibratome, St Louis, MO). Every 15th section (ssf=1/15) was chosen from each animal and was stained with thionin with the first section corresponding to a random number. Neuronal cell counting was performed using a light microscope modified for stereology with a microcator, motorized stage, fast digital camera, and the newCAST software (Visiopharm, Hoersholm, Denmark) as reported previously (42). The border of the hilus was outlined at 10× magnification and cell counting was performed using a Zeiss 40× oil objective (FLUAR, NA 1.30). The sampling frame had an area of 4000 μm2 and the step length was 140 μm×140 μm resulting in an area sampling fraction (asf ) equal to 0.20. A z axis distribution was obtained for 2 animals in order to find the range for the dissector height in which the cell nucleoli densities were similar (unpublished data). This range could be defined to be from 6 μm to 28 μm from the top of the section (dissector height; h(dis)=22 μm). Neurons that fulfilled the sampling criteria were counted (ΣQ-) and their total numbers (Ntotal) were estimated for the whole hilus segment using the equation:
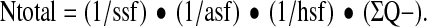 |
Here hsf is the height sampling fraction (hsf=h(dis)/t(Q−), where t(Q−) is the Q-weighted section thickness (10).
Statistical and microarray analyses
All analyses were performed and data illustrated using the software GraphPad Prism 5.0. Data are presented as mean±SD. Partek® Express, version 6.11, copyright 2012 (Partek, St Louis, MO) was used to carry out statistical analyses of microarray data files and Ingenuity Pathway Analysis software (IPA 9.0, http://www.ingenuity.com), a web-based bioinformatics tool, was used to identify functional networks and pathways.
Abbreviations Used
- 4-HNE
4-hydroxy-t-2,3-nonenal
- APOE
apolipoprotein E
- CACNA1A
calcium channel, voltage-dependent, P/Q type, alpha 1A subunit
- CNS
central nervous system
- DA
dark agouti
- DRD2
dopamine receptor D2
- Gsta1, a4
glutathione S transferase alpha1, alpha 4
- Gstm1, m2, m3, m4
glutathione S transferase mu1, mu2, mu3, mu4
- Il1b
interleukin-1 beta
- NeuN
neuronal nuclei
- PARP-1
poly [ADP-ribose] polymerase 1
- PRDX6
peroxiredoxin-6
- PVG
piebald virol glaxo
- QTL
quantitative trait locus
- TP53
tumor protein 53
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- S100b
S100 calcium binding protein B
Acknowledgments
This study was supported by the 6th Framework Program of the European Union, NeuroproMiSe, LSHM-CT-2005-018637, EURATools, LSHG-CT-2005019015, and the 7th Framework Program of the European Union, EURATrans, HEALTH-F4-2010-241504, by the Swedish Research Council, the Swedish Brain Foundation and the Swedish Association of Persons with Neurological Disabilities. Centre for Stochastic Geometry and Advanced Bioimaging is supported by Villum Foundation.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Abel EL. Angel JM. Riggs PK. Langfield L. Lo HH. Person MD. Awasthi YC. Wang LE. Strom SS. Wei Q. DiGiovanni J. Evidence that Gsta4 modifies susceptibility to skin tumor development in mice and humans. J Natl Cancer Inst. 2010;102:1663–1675. doi: 10.1093/jnci/djq392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al Nimer F. Beyeen AD. Lindblom R. Strom M. Aeinehband S. Lidman O. Piehl F. Both MHC and non-MHC genes regulate inflammation and T-cell response after traumatic brain injury. Brain Behav Immun. 2011;25:981–990. doi: 10.1016/j.bbi.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 3.Arakawa M. Ishimura A. Arai Y. Kawabe K. Suzuki S. Ishige K. Ito Y. N-Acetylcysteine and ebselen but not nifedipine protected cerebellar granule neurons against 4-hydroxynonenal-induced neuronal death. Neurosci Res. 2007;57:220–229. doi: 10.1016/j.neures.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 4.Balogh LM. Atkins WM. Interactions of glutathione transferases with 4-hydroxynonenal. Drug Metab Rev. 2011;43:165–178. doi: 10.3109/03602532.2011.558092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beyeen AD. Adzemovic MZ. Ockinger J. Stridh P. Becanovic K. Laaksonen H. Lassmann H. Harris RA. Hillert J. Alfredsson L. Celius EG. Harbo HF. Kockum I. Jagodic M. Olsson T. IL-22RA2 associates with multiple sclerosis and macrophage effector mechanisms in experimental neuroinflammation. J Immunol. 2010;185:6883–6890. doi: 10.4049/jimmunol.1001392. [DOI] [PubMed] [Google Scholar]
- 6.Coppede F. Armani C. Bidia DD. Petrozzi L. Bonuccelli U. Migliore L. Molecular implications of the human glutathione transferase A-4 gene (hGSTA4) polymorphisms in neurodegenerative diseases. Mutat Res. 2005;579:107–114. doi: 10.1016/j.mrfmmm.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 7.Costantini D. Ferrari C. Pasquaretta C. Cavallone E. Carere C. von Hardenberg A. Reale D. Interplay between plasma oxidative status, cortisol and coping styles in wild alpine marmots, Marmota marmota. J Exp Biol. 2012;215:374–383. doi: 10.1242/jeb.062034. [DOI] [PubMed] [Google Scholar]
- 8.Dahlman I. Lorentzen JC. de Graaf KL. Stefferl A. Linington C. Luthman H. Olsson T. Quantitative trait loci disposing for both experimental arthritis and encephalomyelitis in the DA rat; Impact on severity of myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis and antibody isotype pattern. Eur J Immunol. 1998;28:2188–2196. doi: 10.1002/(SICI)1521-4141(199807)28:07<2188::AID-IMMU2188>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 9.Di Pietro G. Magno LA. Rios-Santos F. Glutathione S-transferases: An overview in cancer research. Expert Opin Drug Metab Toxicol. 2010;6:153–170. doi: 10.1517/17425250903427980. [DOI] [PubMed] [Google Scholar]
- 10.Dorph-Petersen KA. Nyengaard JR. Gundersen HJ. Tissue shrinkage and unbiased stereological estimation of particle number and size. J Microsc. 2001;204:232–246. doi: 10.1046/j.1365-2818.2001.00958.x. [DOI] [PubMed] [Google Scholar]
- 11.Ferguson S. Mouzon B. Kayihan G. Wood M. Poon F. Doore S. Mathura V. Humphrey J. O'Steen B. Hayes R. Roses A. Mullan M. Crawford F. Apolipoprotein E genotype and oxidative stress response to traumatic brain injury. Neuroscience. 2010;168:811–819. doi: 10.1016/j.neuroscience.2010.01.031. [DOI] [PubMed] [Google Scholar]
- 12.Galvani S. Coatrieux C. Elbaz M. Grazide MH. Thiers JC. Parini A. Uchida K. Kamar N. Rostaing L. Baltas M. Salvayre R. Negre-Salvayre A. Carbonyl scavenger and antiatherogenic effects of hydrazine derivatives. Free Radic Biol Med. 2008;45:1457–1467. doi: 10.1016/j.freeradbiomed.2008.08.026. [DOI] [PubMed] [Google Scholar]
- 13.Gillett A. Marta M. Jin T. Tuncel J. Leclerc P. Nohra R. Lange S. Holmdahl R. Olsson T. Harris RA. Jagodic M. TNF production in macrophages is genetically determined and regulates inflammatory disease in rats. J Immunol. 2010;185:442–450. doi: 10.4049/jimmunol.0904101. [DOI] [PubMed] [Google Scholar]
- 14.Gilmer LK. Ansari MA. Roberts KN. Scheff SW. Age-related mitochondrial changes after traumatic brain injury. J Neurotrauma. 2010;27:939–950. doi: 10.1089/neu.2009.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gulko PS. Contribution of genetic studies in rodent models of autoimmune arthritis to understanding and treatment of rheumatoid arthritis. Genes Immun. 2007;8:523–531. doi: 10.1038/sj.gene.6364419. [DOI] [PubMed] [Google Scholar]
- 16.Hall ED. Vaishnav RA. Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics. 2010;7:51–61. doi: 10.1016/j.nurt.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang JJ. Lee SJ. Kim TY. Cho JH. Koh JY. Zinc and 4-hydroxy-2-nonenal mediate lysosomal membrane permeabilization induced by H2O2 in cultured hippocampal neurons. J Neurosci. 2008;28:3114–3122. doi: 10.1523/JNEUROSCI.0199-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itoh T. Satou T. Nishida S. Tsubaki M. Imano M. Hashimoto S. Ito H. Edaravone protects against apoptotic neuronal cell death and improves cerebral function after traumatic brain injury in rats. Neurochem Res. 2010;35:348–355. doi: 10.1007/s11064-009-0061-2. [DOI] [PubMed] [Google Scholar]
- 19.Lidman O. Swanberg M. Horvath L. Broman KW. Olsson T. Piehl F. Discrete gene loci regulate neurodegeneration, lymphocyte infiltration, and major histocompatibility complex class II expression in the CNS. J Neurosci. 2003;23:9817–9823. doi: 10.1523/JNEUROSCI.23-30-09817.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lingsma HF. Roozenbeek B. Steyerberg EW. Murray GD. Maas AI. Early prognosis in traumatic brain injury: From prophecies to predictions. Lancet Neurol. 2010;9:543–554. doi: 10.1016/S1474-4422(10)70065-X. [DOI] [PubMed] [Google Scholar]
- 21.Liu H. Wang H. Shenvi S. Hagen TM. Liu RM. Glutathione metabolism during aging and in Alzheimer disease. Ann NY Acad Sci. 2004;1019:346–349. doi: 10.1196/annals.1297.059. [DOI] [PubMed] [Google Scholar]
- 22.Lundberg C. Lidman O. Holmdahl R. Olsson T. Piehl F. Neurodegeneration and glial activation patterns after mechanical nerve injury are differentially regulated by non-MHC genes in congenic inbred rat strains. J Comp Neurol. 2001;431:75–87. [PubMed] [Google Scholar]
- 23.Maes OC. Schipper HM. Chong G. Chertkow HM. Wang E. A GSTM3 polymorphism associated with an etiopathogenetic mechanism in Alzheimer disease. Neurobiol Aging. 2010;31:34–45. doi: 10.1016/j.neurobiolaging.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Marklund N. Bakshi A. Castelbuono DJ. Conte V. McIntosh TK. Evaluation of pharmacological treatment strategies in traumatic brain injury. Curr Pharm Des. 2006;12:1645–1680. doi: 10.2174/138161206776843340. [DOI] [PubMed] [Google Scholar]
- 25.Matsuda S. Umeda M. Uchida H. Kato H. Araki T. Alterations of oxidative stress markers and apoptosis markers in the striatum after transient focal cerebral ischemia in rats. J Neural Transm. 2009;116:395–404. doi: 10.1007/s00702-009-0194-0. [DOI] [PubMed] [Google Scholar]
- 26.McAllister TW. Genetic factors modulating outcome after neurotrauma. PM R. 2010;2:S241–252. doi: 10.1016/j.pmrj.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 27.McCracken E. Valeriani V. Simpson C. Jover T. McCulloch J. Dewar D. The lipid peroxidation by-product 4-hydroxynonenal is toxic to axons and oligodendrocytes. J Cereb Blood Flow Metab. 2000;20:1529–1536. doi: 10.1097/00004647-200011000-00002. [DOI] [PubMed] [Google Scholar]
- 28.McGlynn KA. Hunter K. LeVoyer T. Roush J. Wise P. Michielli RA. Shen FM. Evans AA. London WT. Buetow KH. Susceptibility to aflatoxin B1-related primary hepatocellular carcinoma in mice and humans. Cancer Res. 2003;63:4594–4601. [PubMed] [Google Scholar]
- 29.Ockinger J. Stridh P. Beyeen AD. Lundmark F. Seddighzadeh M. Oturai A. Sorensen PS. Lorentzen AR. Celius EG. Leppa V. Koivisto K. Tienari PJ. Alfredsson L. Padyukov L. Hillert J. Kockum I. Jagodic M. Olsson T. Genetic variants of CC chemokine genes in experimental autoimmune encephalomyelitis, multiple sclerosis and rheumatoid arthritis. Genes Immun. 2010;11:142–154. doi: 10.1038/gene.2009.82. [DOI] [PubMed] [Google Scholar]
- 30.Perez-Pastene C. Graumann R. Diaz-Grez F. Miranda M. Venegas P. Godoy OT. Layson L. Villagra R. Matamala JM. Herrera L. Segura-Aguilar J. Association of GST M1 null polymorphism with Parkinson's disease in a Chilean population with a strong Amerindian genetic component. Neurosci Lett. 2007;418:181–185. doi: 10.1016/j.neulet.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 31.Piehl F. Olsson T. Inflammation and susceptibility to neurodegeneration: The use of unbiased genetics to decipher critical regulatory pathways. Neuroscience. 2009;158:1143–1150. doi: 10.1016/j.neuroscience.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 32.Qian J. Jing J. Jin G. Wang H. Wang Y. Liu H. Li R. Fan W. An Y. Sun W. Ma H. Miao R. Hu Z. Jin L. Wei Q. Shen H. Huang W. Lu D. Association between polymorphisms in the GSTA4 gene and risk of lung cancer: A case-control study in a Southeastern Chinese population. Mol Carcinog. 2009;48:253–259. doi: 10.1002/mc.20478. [DOI] [PubMed] [Google Scholar]
- 33.Radu RA. Hu J. Yuan Q. Welch DL. Makshanoff J. Lloyd M. McMullen S. Travis GH. Bok D. Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J Biol Chem. 2011;286:18593–18601. doi: 10.1074/jbc.M110.191866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reid WM. Rolfe A. Register D. Levasseur JE. Churn SB. Sun D. Strain-related differences after experimental traumatic brain injury in rats. J Neurotrauma. 2010;27:1243–1253. doi: 10.1089/neu.2010.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salzer J. Svenningsson A. Sundstrom P. Neurofilament light as a prognostic marker in multiple sclerosis. Mult Scler. 2010;16:287–292. doi: 10.1177/1352458509359725. [DOI] [PubMed] [Google Scholar]
- 36.Sawcer S. Bayes factors in complex genetics. Eur J Hum Genet. 2010;18:746–750. doi: 10.1038/ejhg.2010.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schulz JB. Lindenau J. Seyfried J. Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- 38.Selley ML. (E)-4-hydroxy-2-nonenal may be involved in the pathogenesis of Parkinson's disease. Free Radic Biol Med. 1998;25:169–174. doi: 10.1016/s0891-5849(98)00021-5. [DOI] [PubMed] [Google Scholar]
- 39.Selley ML. Close DR. Stern SE. The effect of increased concentrations of homocysteine on the concentration of (E)-4-hydroxy-2-nonenal in the plasma and cerebrospinal fluid of patients with Alzheimer's disease. Neurobiol Aging. 2002;23:383–388. doi: 10.1016/s0197-4580(01)00327-x. [DOI] [PubMed] [Google Scholar]
- 40.Shao C. Roberts KN. Markesbery WR. Scheff SW. Lovell MA. Oxidative stress in head trauma in aging. Free Radic Biol Med. 2006;41:77–85. doi: 10.1016/j.freeradbiomed.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 41.Simpson EP. Henry YK. Henkel JS. Smith RG. Appel SH. Increased lipid peroxidation in sera of ALS patients: A potential biomarker of disease burden. Neurology. 2004;62:1758–1765. doi: 10.1212/wnl.62.10.1758. [DOI] [PubMed] [Google Scholar]
- 42.Strom M. Al Nimer F. Lindblom R. Nyengaard JR. Piehl F. Naturally occurring genetic variability in expression of Gsta4 is associated with differential survival of axotomized rat motoneurons. Neuromol Med. 2012;14:15–29. doi: 10.1007/s12017-011-8164-8. [DOI] [PubMed] [Google Scholar]
- 43.Swanberg M. Harnesk K. Strom M. Diez M. Lidman O. Piehl F. Fine mapping of gene regions regulating neurodegeneration. PLoS One. 2009;4:e5906. doi: 10.1371/journal.pone.0005906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swanberg M. Lidman O. Padyukov L. Eriksson P. Akesson E. Jagodic M. Lobell A. Khademi M. Borjesson O. Lindgren CM. Lundman P. Brookes AJ. Kere J. Luthman H. Alfredsson L. Hillert J. Klareskog L. Hamsten A. Piehl F. Olsson T. MHC2TA is associated with differential MHC molecule expression and susceptibility to rheumatoid arthritis, multiple sclerosis and myocardial infarction. Nat Genet. 2005;37:486–494. doi: 10.1038/ng1544. [DOI] [PubMed] [Google Scholar]
- 45.Tan AA. Quigley A. Smith DC. Hoane MR. Strain differences in response to traumatic brain injury in Long-Evans compared to Sprague-Dawley rats. J Neurotrauma. 2009;26:539–548. doi: 10.1089/neu.2008.0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang SC. Arumugam TV. Cutler RG. Jo DG. Magnus T. Chan SL. Mughal MR. Telljohann RS. Nassar M. Ouyang X. Calderan A. Ruzza P. Guiotto A. Mattson MP. Neuroprotective actions of a histidine analogue in models of ischemic stroke. J Neurochem. 2007;101:729–736. doi: 10.1111/j.1471-4159.2006.04412.x. [DOI] [PubMed] [Google Scholar]
- 47.Townsend DM. Tew KD. Tapiero H. The importance of glutathione in human disease. Biomed Pharmacother. 2003;57:145–155. doi: 10.1016/s0753-3322(03)00043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yao X. Effect of zinc exposure on HNE and GLT-1 in spinal cord culture. Neurotoxicology. 2009;30:121–126. doi: 10.1016/j.neuro.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 49.Zhou W. Xu D. Peng X. Zhang Q. Jia J. Crutcher KA. Meta-analysis of APOE4 allele and outcome after traumatic brain injury. J Neurotrauma. 2008;25:279–290. doi: 10.1089/neu.2007.0489. [DOI] [PubMed] [Google Scholar]