

Abstract
Aim: The glutathione-dependent AdhC-EstD formaldehyde detoxification system is found in eukaryotes and prokaryotes. It is established that it confers protection against formaldehyde that is produced from environmental sources or methanol metabolism. Thus, its presence in the human host-adapted bacterial pathogen Neisseria meningitidis is intriguing. This work defined the biological function of this system in the meningococcus using phenotypic analyses of mutants linked to biochemical and structural characterization of purified enzymes. Results: We demonstrated that mutants in the adhC and/or estD were sensitive to killing by formaldehyde. Inactivation of adhC and/or estD also led to a loss of viability in biofilm communities, even in the absence of exogenous formaldehyde. Detailed biochemical and structural analyses of the esterase component demonstrated that S-formylglutathione was the only biologically relevant substrate for EstD. We further showed that an absolutely conserved cysteine residue was covalently modified by S-glutathionylation. This leads to inactivation of EstD. Innovation: The results provide several conceptual innovations. They provide a new insight into formaldehyde detoxification in bacteria that do not generate formaldehyde during the catabolism of methanol. Our results also indicate that the conserved cysteine, found in all EstD enzymes from humans to microbes, is a site of enzyme regulation, probably via S-glutathionylation. Conclusion: The adhc-estD system protects against formaldehyde produced during endogenous metabolism. Antioxid. Redox Signal. 18, 743–755.
Introduction
The highly cytotoxic compound formaldehyde is a by-product of numerous environmental processes (16). This strong electrophile undergoes indiscriminate, nonenzymatic, and rapid reactions with biological macromolecules, leading to irreversible alkylations and cross-linkages, with resulting damage to proteins and DNA (7). Thus, it comes as no surprise that formaldehyde detoxification systems are diverse and widespread across the biological world. These systems often depend on the glutathione (GSH) pool, as it reacts spontaneously with formaldehyde to form S-hydroxymethylglutathione (HMGSH, Fig. 1) (7). This adduct is in turn oxidized by a class III, zinc-containing, NAD+-dependent alcohol dehydrogenase (AdhC) to generate S-formylglutathione (SFG) (48). GSH is regenerated via subsequent hydrolysis of SFG by a thioesterase, which also generates formate as a by-product (Fig. 1) (49).
FIG. 1.
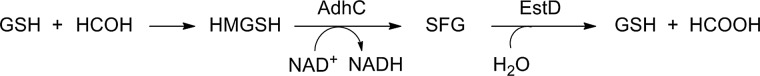
Proposed reaction mechanism of formaldehyde detoxification by AdhC and EstD.
Innovation.
The adhc-estD detoxification system for formaldehyde is commonly understood to remove formaldehyde generated from exogenous sources or as a by-product of methanol metabolism. Our results suggest that the adhc-estD system is required for protection against formaldehyde that is generated during endogenous metabolism. They provide a new insight into formaldehyde detoxification in bacteria that do not generate formaldehyde during the catabolism of methanol.
It has been known for decades that formaldehyde is also produced endogenously during cellular metabolism of ethanol and methanol. As a result, there is a wealth of literature on this GSH-dependent detoxification system from the mammalian liver and plants (12, 48). More recently, it was recognized that Gram-negative methylotrophic bacteria also employ a class-III alcohol dehydrogenase (AdhC) and a thioesterase (EstD) to detoxify formaldehyde during methanol metabolism (5, 6, 14, 50). Intriguingly, analysis of bacterial genomes has revealed that adhC and estD genes are well distributed in bacteria that do not oxidize methanol. An example is Escherichia coli, where formaldehyde induces the expression of the operon consisting of frmR, encoding a formaldehyde-responsive repressor, frmA (adhC), and frmB (estD) (13).
The bacterial pathogen Neisseria meningitidis is the etiological agent of meningococcal meningitis. It is closely related to Neisseria gonorrhoeae, the causative agent of gonorrhoea. In both organisms, adhC and estD are found as an operon that is controlled by NmlR, a MerR-like regulator that was recently characterized in the gonococcus (24). While adhC is not functional in all strains of N. gonorrhoeae due to a frameshift mutation in the gene, the adhC gene in N. meningitidis has remained intact (36). Like E. coli, these pathogenic Neisseria species are not able to use methanol as a carbon source, and thus the presence and function of these formaldehyde detoxification genes in their genome remain unclear. In this study, we report the effect of mutation of adhC and estD on the ability of N. meningitidis to defend against reactive aldehyde stress. To understand the molecular basis of formaldehyde defense in N. meningitidis, we carried out a biochemical characterization of the esterase encoded by the adhC-estD operon. We also report the crystal structure of EstD and describe its role in formaldehyde detoxification. Our studies provide new insights into the covalent modification of a conserved cysteine present at the enzyme active site of EstD. Our results also provide an understanding of the possible biological role of adhC and estD in defense against endogenously produced formaldehyde.
Results
NMB1304 (adhC) and NMB1305 (estD) are required for resistance to formaldehyde in N. meningitidis
The operon consisting of NMB1304 and NMB1305 within the N. meningitidis MC58 genome (44) contains genes that were annotated as a adhC and an S-formylglutathione hydrolase (SFGH, EstD), respectively (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars). This operon is also present in the meningococcal C311#3 strain. We have sequenced these genes in both strains and found them to be identical (data not shown). To assess the functional role of this operon in the meningococcus, we constructed mutant strains in which the entire bicistron or each of the individual genes was replaced with a kanamycin-resistance cassette (Supplementary Fig. S1), creating C311#3adhC, C311#3estD, and C311#3adhC-estD. These engineered strains were subsequently challenged with a range of reactive aldehydes: formaldehyde as well as glutaraldehyde, glycolaldehyde, glyoxal, and methylglyoxal. Figure 2A shows the results from a disc-diffusion killing assay, where the wild-type and mutant strains displayed similar zones of growth inhibition for all chemical challenges, with the exception of formaldehyde. In this case, a significant increase in the zone of inhibition was observed for the single and double NMB1304 and NMB1305 mutant strains when compared to wild-type meningococci. Additional short-chain aldehydes, such as acetaldehyde and propionaldehyde, were also tested, but no difference between the mutant strains was observed (Supplementary Fig. S2).
FIG. 2.

The adhC-estD operon in Neisseria meningitidis confers resistance to formaldehyde. (A) Sensitivity of meningococci to reactive aldehydes, as determined by disc diffusion assay. Black bars, wild type; white bars, adhC mutant; dark gray bars, estD mutant; light gray bars, adhC-estD mutant. MG, methylglyoxal; GTA, glutaraldehyde; GCA, glycoaldehyde; FA, formaldehyde. Six independent experiments were performed. Y-error bars indicate±standard error from the mean. *p<0.0002; **p<0.0005; ***p<0.0007. (B) Growth of meningococci on a solid medium containing increasing concentrations of formaldehyde as indicated. The experiment was repeated at least three times, and representative results are shown. Number of cells inoculated is shown on the Y-axis. (C) Bacterial count after overnight growth on a formaldehyde-containing medium as in panel B. Filled circles (●), wild type; empty circles (○), adhC mutant; filled triangles (▼), estD mutant; empty triangles (△), adhC-estD mutant. Three independent experiments were performed. Y-error bars indicate±standard error from the mean. (D) Recovery of resistance to formaldehyde by chromosomal complementation with an intact adhC-estD operon. Filled circles (●), wild type; empty circles (○), estD mutant; filled triangles (▼), estD adhC-estD+ complemented mutant; empty triangles (△) adhC-estD mutant; filled squares (■), adhC-estD adhC-estD+ complemented mutant.
To confirm this formaldehyde-sensitive phenotype, we developed a killing assay where serial dilutions of meningococci were plated on a solid medium containing increasing concentrations of formaldehyde (0–1.6 mM). Figure 2B confirmed that mutants of the NMB1304-1305 operon were more sensitive to killing by formaldehyde than wild-type cells. The data also indicated that this sensitivity was most pronounced for the NMB1305 single mutant. This observation was confirmed when the colony counts of cells grown in the presence of formaldehyde were determined. Figure 2C shows the recovery of colony-forming units (CFUs) after incubation with decreasing concentrations of formaldehyde for the wild-type cells. Compared to the wild type, there was a decline in CFU recovery as a function of formaldehyde concentration for the NMB1304 single and NMB1304-1305 double mutants that followed an almost identical pattern. However, the CFU recovery for the NMB1305 single mutant was notably lower than for the other two mutants, and this loss of CFU also began at lower formaldehyde concentrations (Fig. 2C). Complementation of each mutant by insertion of a single copy of the entire NMB1304-1305 operon into the meningococcal chromosome led to a complete recovery of the wild-type formaldehyde-resistant phenotype (Fig. 2D). Taken together, these results confirmed the annotation of NMB1304 and NMB1305 as adhC and estD, respectively, and confirmed the role of these gene products in the protection against formaldehyde toxicity in N. meningitidis.
adhC and estD are required for survival in biofilms
N. meningitidis is an obligate human pathogen that frequently colonizes the nasopharynx, and formation of meningococcal biofilms on the nasopharyngeal epithelial cells is a key virulence factor (33). We examined biofilm formation by wild-type meningococci and adhC, estD, and adhC-estD mutant strains on glass coverslips over a period of 24 h. No exogenous formaldehyde was added in these experiments. COMSTAT analyses found no significant difference in the abilities of these strains to form biofilms, as indicated by biofilm thickness and average biomass (Fig. 3A). However, when we evaluated the survival of meningococci within the biofilm matrix using a viability stain, we observed that while wild-type cells survived, and the adhC, estD, and adhC-estD mutant strains were nonviable (Fig. 3B and Supplementary Fig. S3). This apparent aging of biofilms points to a possible role of the adhC-estD operon in the survival of N. meningitidis.
FIG. 3.

The adhC-estD operon is required for biofilm viability. (A) COMSTAT analysis of biofilm biomass (black bars) and average thickness (white bars). The error bars represent±standard error of the mean. No statistical difference (p≤0.05) was observed in either the biomass or thickness of the biofilms formed by the adhC, estD, and adhC-estD mutants relative to the wild-type strain. These results are the combination of three independent experiments. (B) Three-dimensional reconstructions of stacked z-series of meningococcal wild-type, adhC, estD, and adhC-estD mutants grown over glass taken at 200×magnification and rendered using EZ-C1 viewer (Nikon). Cells staining green represent viable cells, while cells staining red represent dead cells. These experiments were performed three times, and representative results are shown. (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
NmEstD is an SFGH
The gene product of estD (herein referred to as NmEstD) exhibits high sequence identity (40%–60%) with several SFGHs from human (ESD), Saccharomyces cerevisiae (YJG8), and E. coli (FrmB and YeiG). In particular, the serine catalytic triad (Ser145-Asp221-His254) in NmEstD is fully conserved (Supplementary Fig. S4). To confirm the expectation that NmEstD functions as a typical SFGH, it was expressed as a recombinant protein in E. coli with a hexa-His epitope at the N-terminus. The enzyme was obtained routinely in high yield (∼300 mg/L culture) and purity (>95% pure) in a dimeric and fully reduced form (Supplementary Fig. S5 and Supplementary Table S1, respectively). The purified enzyme was subsequently assayed for activity against SFG. We also tested S-lactoylglutathione (SLG), a naturally occurring thioester that is produced during the removal of methylglyoxal using the glyoxalase system. The synthetic carboxyl ester p-nitrophenyl acetate (pNPAC) was used as a positive control for the reaction. NmEstD was active toward all tested substrates, and Table 1 lists the kinetic parameters obtained for the NmEstD-catalyzed hydrolysis of these substrates under the same conditions. The catalytic efficiencies toward pNPAC (kcat/Km 3.2×103 M−1 s−1) and SLG (kcat/Km 2.90×103 M−1 s−1) were comparable to those reported previously for other SFGHs (Table 2). However, the highest activity of NmEstD was obtained with SFG (kcat/Km 1.95×106 M−1 s−1) (Fig. 4), with a catalytic efficiency of at least three orders of magnitude higher than those obtained for pNPAC and SLG. It was also noted that the specificity rate constant toward SFG was at least 25-fold higher than those reported previously for other SFGHs (Table 2). These results are consistent with the conclusion that SFG is the likely physiological substrate of NmEstD.
Table 1.
Kinetic Parameters of NmEstD and Its Variants
| Protein | Substrate | Km (mM) | Vmax (μmol.min−1.μmol protein−1) | kcat (s−1) | kcat/Km (M−1s−1) |
|---|---|---|---|---|---|
| WT | SFG | 2.21±0.35 | 2.6×105±1.8×104 | 4300 | 1.95×106 |
| SLG | 2.69±0.47 | 4.8×102±0.3×102 | 7.90 | 2.90×103 | |
| pNPAC | 0.62±0.06 | 1.2×102±0.3×102 | 2.00 | 3.20×103 | |
| C54A | SFG | 2.77±0.26 | 2.1×105±9.4×103 | 3500 | 1.26×106 |
| SLG | 2.08±0.52 | 1.6×102±0.1×102 | 2.70 | 1.30×103 | |
| pNPAC | 0.79±0.06 | 7.8×101±2.7×101 | 1.30 | 1.65×103 | |
| C26A | SFG | 4.23±0.66 | 1.6×105±1.4×104 | 2600 | 6.15×105 |
| SLG | 3.00±0.90 | 5.2×102±0.6×102 | 8.70 | 2.90×103 | |
| pNPAC | 0.48±0.05 | 9.5×101±2.9×101 | 1.60 | 3.30×103 | |
| S145A | SFG | ND | ND | ND | ND |
| SLG | ND | ND | ND | ND | |
| pNPAC | ND | ND | ND | ND |
ND, not detectable; pNPAC, p-nitrophenyl acetate; SFG, S-formylglutathione; SLG, S-lactoylglutathione.
Table 2.
Kinetic Parameters of EstD Homologs
| |
SFG |
SLG |
pNPAC |
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Organism | Km (mM) | kcat (s−1) | kcat/Km (M−1s−1) | Km (mM) | kcat (s−1) | kcat/Km (M−1s−1) | Km (mM) | kcat (s−1) | kcat/Km (M−1s−1) | Ref |
| Neisseria meningitidis | 2.21 | 4300 | 2.0×106 | 2.69 | 7.9 | 2900 | 0.62 | 2 | 3200 | This study |
| Saccharomyces cerevisiae | ND | ND | ND | >1.25 | ND | ND | 0.4 | 1.8a | 4500a | (29) |
| Escherichia coli FrmB | 0.41 | 28.5 | 6.9×104 | 0.6 | 0.5 | 800 | 0.29 | 0.3 | 1030 | (13) |
| E. coli YeiG | 0.43 | 6.51 | 1.5×104b | 0.58 | 1.02 | 1750 | 0.45 | 0.26 | 570 | (13) |
| Arabidopsis thaliana | 0.13 | 10.4c | 4.6×104c | ND | ND | ND | 1.02 | 2.9c | 2900c | (27) |
| Agrobacterium tumefaciens | ND | ND | ND | ND | ND | ND | 0.48 | 1.95 | 4070 | (51) |
Converted from min−1.
Corrected value obtained from the data published.
Converted from mM.s−1 and kcat value as published.
FIG. 4.

NmEstD is a SFGH. Activity of NmEstD was assayed against SFG (filled circles, ●), SLG (empty circles, ○), and pNPAC (filled triangles, ▼). The results were plotted as a function of substrate concentration and fitted to a rectangular hyperbola. All assays were performed in HEPES buffer (50 mM, pH 7.0) at 37°C using 0.4 μM EstD for SLG and pNPAC or 4 nM EstD for SFG. Units=μmol.min−1. pNPAC, p-nitrophenyl acetate; SFGH, S-formylglutathione hydrolase; SLG, S-lactoylglutathione.
Despite the presence of a serine catalytic triad, the absolute conservation of a cysteine residue (Cys54 in NmEstD; Supplementary Fig. S4) among various SFGH homologs and the sensitivity of these enzymes to thiol-modifying reagents led to their initial misclassification as cysteine hydrolases (28). In this study, we confirmed that NmEstD functions using a typical serine hydrolase active site. Substitution of the active-site serine (Ser145 in NmEstD) with alanine completely abolished enzyme activity toward all tested substrates (Table 1). We also tested the effect of replacing Cys54 with alanine on enzyme activity. The C54A mutant had Km values comparable to those of the wild-type enzyme toward all tested substrates (Table 1). However, we noted that this mutant exhibited a small (19%) decrease in its kcat value toward SFG compared to wild-type NmEstD. Similarly, alanine substitution of a second, solvent-exposed cysteine (Cys26) did not affect enzyme activity toward SLG or pNPAC, and only a minimal decrease in kcat (1.7-fold) was detected for SFG (Table 1). These results confirmed that Cys54 is not involved in a direct catalytic role.
Structural characterization of NmEstD
To characterize the molecular basis of the apparent specificity for SFG, we determined the crystal structure of NmEstD. Crystals of NmEstD had the symmetry of the hexagonal space group (P6522), two molecules per asymmetric unit, and they diffracted to 1.3 Å resolution. Statistics for data processing and structure refinement are listed in Table 3. The NmEstD structure reveals a typical α/β hydrolase fold displayed by other members of the SFGH family (35). Nine central mixed β-strands (β1–β9) form a twisted β-sheet that is surrounded by nine α-helices [αA-αI; secondary structure notation as in (35)] and three 310 helices (Fig. 5A). There is one large dimer interface in the NmEstD crystals (occluding ∼900 Å2 of solvent accessible area per monomer), and it involves polar and nonpolar interactions (Fig. 5D) between residues in β1–β2, αA–αB, and αI. Electrostatic interactions involve the side chains of Asp253 and His7 in one monomer and the hydroxyl group of Tyr252 and the main-chain N-atom of Gly11 in the other monomer. Further contacts involve cation-π interaction between Arg67 in one monomer and Phe10 and Tyr68 in the other (Fig. 5C). These residues are conserved among other bacterial SFGHs, but have been substituted in eukaryotic proteins (Supplementary Fig. S4).
Table 3.
Data Collection and Refinement Statistics for NmEstD
| Data collection and processing | |
|---|---|
| X-ray source | MX-2 Australian Synchrotron |
| Wavelength (Å) | 0.954 |
| Space group | P6122 |
| Cell dimensions [a, b, c (Å)] | 111.3, 111.3, 169.9 |
| Resolution range (Å)a | 19.84–1.30 (1.30–1.37) |
| Rmerge (%)a | 6.7 (80.8) |
| <I/σI>a | 10.0 (1.0) |
| Completeness (%)a | 99.3 (97.0) |
| Multiplicitya | 5.2 (5.0) |
| Number of total reflectionsa | 783,482 (106,886) |
| Number of unique reflectionsa | 150,461 (21,149) |
| Refinement | |
|---|---|
| Space group | P6522 |
| Resolution range (Å2)a | 19.84–1.40 (1.42–1.40) |
| Rwork/Rfree (%)b | 14.34/15.39 |
| Average B-factor protein/solvent (Å2) | 20.16/31.46 |
| r.m.s. deviation bonds (lengths/angles) | 0.011 Å/1.22° |
| Ramachandran (%) favored/allowed | 96.8/100.0 |
Numbers in parentheses refer to the statistics for the highest resolution shell.
R=Σhkl(||Fobshkl|-|Fcalchkl||)/|Fobshkl|, where |Fobshkl| and |Fcalchkl| represent the observed and calculated structure factor amplitudes, respectively.
FIG. 5.

X-ray crystal structure of NmEstD. (A) Cartoon representation of the NmEstD dimer. Active site residues and those involved in the dimer interface are shown as ball-and-stick models. (B) Organization of the active site. Zoom-in shows residues making up the catalytic triad (S145, H254, and D221) and acyl pocket as highlighted in panel A. Hydrogen bonds between residues in the catalytic triad are shown as dashed lines. (C) Surface representation of the active site as colored by electrostatic potential (negative [red], to positive [blue]). Residues contributing to the overall positive charge are shown as ball-and-stick models together with those in the catalytic triad. Primed numbers indicate residues from the second monomer. (D) Details of the dimer interface. The protein backbone is shown as a ribbon, and residues taking part in cation-π interactions are shown as ball-and-stick models. Individual NmEstD monomers are shown in different colors (green and blue). Primed numbers indicate residues from the second monomer. (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
The structure further revealed that the substrate-binding pocket of NmEstD displayed an overall positive charge due to residues Asn58, Lys62, and His144 (Fig. 5C). These residues are conserved among NmEstD homologs and may be important for orienting the GSH moiety within the active site. Indeed, the dimensions of this site (roughly 14×8×6 Å) can easily accommodate the GSH moiety in both SFG and SLG substrates. However, despite various attempts, we were unable to cocrystallize NmEstD with its substrates. Thus, we employed a simple docking protocol to model binding of SFG in this substrate pocket. These in silico analyses confirmed that atoms from residues Asn58 and Lys62 are well positioned to form hydrogen bonds to charged groups from the GSH moiety (Fig. 6A, B). These residues are likely to be important for the proper orientation of the substrate into the active site. However, the size of the side chain attached to GSH is central in defining substrate specificity. Previous studies have shown that the size of the substrate that can be accommodated in the active site is defined by the acyl pocket (9, 10, 29). The acyl pocket of NmEstD is highly similar to those previously seen for other SFGHs and is formed by residues Leu52, Tyr94, Ile171, Trp179, Phe223, and Leu228 (Fig. 5B). It is likely that this shallow acyl pocket of NmEstD is responsible for its preference for SFG over bulkier substrates such as SLG, or those with aromatic moieties like pNPAC. The presence of an ordered water molecule (B factor 12.4 Å2) in close proximity to the enzyme active site further allowed us to identify the oxyanion hole in NmEstD. Based on the proposed reaction mechanism for SFGHs (9, 29), the O atom from the formate moiety in SFG interacts with backbone amide groups from the oxyanion hole. In the NmEstD structure, the O atom from the water molecule is within 3.1 Å of the backbone amide groups of Leu52 and Met146, and therefore these residues are the most likely candidates to for the oxyanion hole in NmEstD.
FIG. 6.

In silico models of NmEstD with its SFG substrate and S-glutathionylated Cys54. (A) Surface as colored by electrostatic potential (negative [red], to positive [blue]) showing a cross-section of the substrate-binding pocket with the docked substrate SFG. Residues making up the oxyanion hole are shown as ball-and-stick representation. Atoms from the substrate are also shown as ball-and-stick representation. Potential hydrogen bonds are indicated by dashed lines. (B) Top view of the substrate-binding pocket (as shown in A) with docked SFG. Potential hydrogen bonds between substrate and enzyme atoms as suggested by the docking analysis are shown as dashed lines. (C) Modification of Cys54 by GSH blocks the substrate-binding pocket as suggested by the docking analysis. GSH is shown as a stick model; SFG is also shown in the background as a semitransparent ball-and-stick model. Docking of GSH was restrained by enforcing a disulfide bond between the thiol groups from Cys54 and GSH (dark-yellow dashed line). GSH, glutathione. (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
Cys54 is S-glutathionylated by reaction with S-nitrosoglutathione
There is increasing evidence that cysteine residues can function as redox switches and control the catalytic activity of enzymes (26). Thiol-modifying compounds have been shown previously to target the conserved cysteine residue in SFGH homologs from S. cerevisiae, Arabidopsis thaliana, and E. coli, which has led to the suggestion that this residue (Cys54 in NmEstD) may play a role in the secondary regulation of enzyme activity (9, 13, 29). In this work, we tested the reactivity of the cysteines in NmEstD toward synthetic alkylating agents and analyzed the reaction products by electrospray ionization mass spectrometry (ESI MS). Incubation with a large excess of iodoacetamide (IA; 50 molar equivalents) led to a mass increase of +57 Da compared to the untreated reduced protein (Fig. 7A). These results are consistent with the alkylation of only one of the two solvent-exposed Cys residues in NmEstD. We extended this work to determine if NmEstD could be covalently modified via S-glutathionylation. Incubation with various molar ratios of the naturally occurring reagent S-nitrosoglutathione (GSNO) increased the mass of NmEstD by +305 Da, corresponding to the attachment of a single GSH moiety to one Cys residue (Fig. 7B). A second biologically relevant S-glutathionylation agent, oxidized glutathione (GSSG), was also used in these experiments, but it did not modify the enzyme with the same efficiency as GSNO (data not shown). To identify the reactive cysteine in NmEstD, two protein variants were produced in which each of the surface Cys residues was replaced with an alanine. In the C26A variant, only Cys54 is accessible to the solvent. Likewise, in the C54A variant, only Cys26 is present at the surface. GSNO treatment followed by ESI MS analyses revealed that the C26A variant was modified almost to completion, while only a third of the C54A mutant was modified under the same conditions (Fig. 7B). In addition, the C54A mutant was also resistant to S-alkylation by IA (Fig. 7A). Taken together, the above results suggest that Cys54 is in fact the reactive Cys residue in NmEstD.
FIG. 7.

S-Glutathionylation of Cys54 by GSNO. (A) Mass spectra of NmEstD wild-type and C54A variant before and after treatment with 50 equivalents of IA. (B) Mass spectra of NmEstD wild-type, C26A, and C54A variants after treatment with 0 or 15 molar equivalents of GSNO. S-glutathionylated forms (-SSG) are indicated on top of each spectrum. The mass spectrum of the C26A mutant after additional treatment with DTT is shown. The peak indicated by an asterisk (+178 Da) represents a modified form of NmEstD due to the spontaneous alpha-N-6-phosphogluconoylation of the hexa-His epitope. (C) Effect of GSNO treatment on the SFGH activity of NmEstD. The C26A and C54A variants from panel (A) were assayed for activity using 1 mM SFG as substrate. Black bars, C26A mutant protein; white bars, C54A mutant protein. (D) Recovery of activity by DTT. Black bars, wild-type protein; white bars, C26A mutant. DTT, dithiothreitol; GSNO, S-nitrosoglutathione; IA, iodoacetamide. The error bars represent±standard error of the mean. These experiments were performed three times and representative results are shown.
We further examined the effect of Cys54 modification on the thioesterase activity of NmEstD. For simplicity, we used the C26A and C54A mutants of NmEstD in these studies, as each mutant only contained one solvent-accessible Cys residue. Figure 7C shows that activity of the C54A mutant remained relatively unaffected by GSNO. In contrast, the C26A mutant was inhibited under the same conditions. The loss of activity was proportional to the amount of GSNO added, and the activity was recovered by incubation with dithiothreitol (DTT) (Fig. 7C). These results supported the proposal that S-glutathionylation of Cys54 in NmEstD could modulate the activity of this enzyme. However, this covalent modification of Cys54 appeared to have no effect on its secondary structure (Supplementary Fig. S6) or dimeric state (Supplementary Fig. S5). To this end, we have not been able to crystallize the GSNO-modified form of NmEstD. Nevertheless, examination of the crystal structure showed that Cys54 is located in a loop between β4 and αA and sits at the entrance to the catalytic site of NmEstD (Fig. 5C). We were able to model the impact of S-glutathionylation using a docking protocol as described in the Materials and Methods section. These in silico results suggest that this modification would in fact block access to the substrate-binding pocket of NmEstD, thus providing a rationale behind the inhibition observed above (Fig. 6C).
To further characterize the role of Cys54 in the NmEstD-mediated formaldehyde detoxification in vivo, we constructed a C54AestD mutant strain by complementing the estD deletion strain with a single copy of the same operon expressing a C54A mutant of NmEstD. Our killing assay showed that the C54AestD mutant was resistant to formaldehyde to the level seen in wild-type meningococci (Fig. 8). The same was observed when a C26A mutation was introduced. By contrast, the S145AestD mutant was sensitive to killing by formaldehyde, consistent with our observation that the same mutation in the NmEstD enzyme led to a complete loss of SFGH activity (cf. Table 1).
FIG. 8.

A C54AestD mutant, but not S145AestD, is resistant to formaldehyde. Bacterial counts after overnight growth on a solid medium containing formaldehyde (0–1.6 mM). Filled circles (●), wild type; empty circles (○), estD mutant; filled triangles (▼), estD adhC-estD C26A+ complemented mutant; empty triangles (△), estD adhC-estD C54A+ complemented mutant; filled squares (■), estD adhC-estD S145A+ complemented mutant.
Discussion
The combination of an alcohol dehydrogenase and a serine thioesterase represents an established system for the GSH-dependent detoxification of formaldehyde across the biological world. Although the first bacterial systems to be described were those in gram-negative methylotrophs (5, 6, 14, 50), it has become clear that adhC and estD are present in a wide variety of bacteria, the majority of which do not generate formaldehyde via methanol oxidation. N. meningitidis is one such bacterium, and our in vitro killing assays demonstrate that the primary function of the meningococcal adhC-estD system is in fact to protect this bacterium against the toxic effects of formaldehyde. This observation is similar to those made for E. coli (13), a bacterium that can survive in a variety of environments where it might be expected to encounter formaldehyde. By contrast, N. meningitidis is an obligate human host-adapted bacterium, and this raises the question about the circumstances under which this bacterium would encounter this toxic aldehyde.
It is known that formaldehyde is produced endogenously in host tissues from methylamine (8, 55). In addition, it also appears to be formed during lipid peroxidation and hydroxyl radical formation (40, 45), two processes that occur during inflammation (32). Inflammation of the meninges of the brain or spinal cord (meningitis) is associated with meningococcal infection, and so it may be the case that the adhC-estD system has a protective role during the systemic dissemination of N. meningitidis. It is notable that a highly similar NmlR-regulated adhC-estD system is found in Haemophilus influenzae, a bacterium that can also spread systemically and also causes inflammation and meningitis (23). A number of reactive aldehydes are produced as part of the innate immune response to infection. Methylglyoxal and glycoaldehyde are produced by neutrophils as a consequence of the myeloperoxidase-driven oxidation of threonine and serine, respectively (4). However, as observed for E. coli, mutation of estD (and adhC) did not alter the susceptibility of N. meningitidis to methylglyoxal. Furthermore, in the case of the meningococcal mutant, we observed no change in sensitivity to these other reactive aldehydes. This observation may also be explained by the low specificity displayed by NmEstD toward SLG, a product of methylglyoxal detoxification. It is likely that the GloA/GloB system that is widespread among bacteria (46), including N. meningitidis, is responsible for defense against methylglyoxal toxicity.
Another possibility is that formaldehyde is generated endogenously within N. meningitidis during carbon metabolism. In H. influenzae, it is known that AdhC is most highly expressed under highly aerobic conditions (11), and we have previously observed that an adhC mutant of this bacterium grows more slowly under conditions of high oxygen tension than wild-type cells (23). Under these conditions, intermediary carbon metabolism may generate higher internal levels of sugar phosphates as a consequence of a switch to the pentose phosphate cycle. The short-chain sugars of the pentose phosphate cycle (erythrose and glyceraldehyde) have the potential to form toxic dicarbonyl compounds (34), which can drive formation of aldehydes from α-amino acids via a process known as Strecker degradation (53). In the case of glycine, the product formed would be formaldehyde. If this is the case, then the adhC-estD system may be considered a detoxification system that is linked more closely to intermediary metabolism rather than a defense system associated with protection against formaldehyde generated from exogenous sources. That the former might be true comes from our observation that the viability of N. meningitidis cells within biofilm communities was reduced in the adhC and estD mutants compared to wild-type cells. This reduced viability or premature aging of biofilms may be a consequence of the accumulation of the toxic metabolites due to the inactivation of the adhC-estD formaldehyde detoxification system. This would be consistent with endogenous formaldehyde stress in this bacterium.
Although EstD is recognized as a part of the GSH-dependent formaldehyde detoxification system, most studies have focused on AdhC, based perhaps on the assumption that this enzyme would be the more critical component of the defense system. However, our observation that the estD mutant was more sensitive to formaldehyde than the adhC single mutant or even the adhC-estD double mutant may suggest the following: (i) SFG itself is toxic, and/or (ii) the failure to regenerate reduced GSH in the estD mutant leads to GSH depletion and cell death as a consequence of loss of intracellular thiol-redox poise. That the latter might be the case is supported by observations that mutations in systems that fail to generate NADPH and thereby were unable to maintain the GSH pool in a reduced state lead to increased sensitivity to formaldehyde (19). Further work is needed to test if this is the case in N. meningitidis.
The biochemistry of NmEstD described in this study has revealed features that were previously unrecognized. NmEstD displayed a dramatic preference (50-fold) for SFG compared to other possible substrates, including SLG and pNPAC, which is presumably dictated by its small catalytic site. Yet, despite the high conservation of residues that make up the active site, such a high specificity for SFG by a prokaryotic or eukaryotic thioesterase has not been previously reported. It is tempting to suggest that this feature is a part of an adaptation mechanism by N. meningitidis, which thrives under only a limited range of environment when compared to that of other less-fastidious bacteria such as E. coli. The same may be true for other host-adapted pathogens that carry this thioesterase, such as N. gonorrhoeae and H. influenzae.
Previous reports identifying the sensitivity of EstD toward cysteine-modifying agents lead to the incorrect suggestion that the conserved Cys54 in NmEstD and its homologs played an important a role in catalysis (28, 49). This idea has been contradicted by a substantial body of evidence that SFGHs are serine hydrolases (3, 9, 13, 51, 54), and our observation that mutation of Ser154 inactivates NmEstD is entirely consistent with the present models for SFGH mechanism. Nevertheless, the possibility that Cys54 might have a biological function is intriguing. The previous observation that modification of Cys54 with thiol-specific reagents resulted in enzyme inactivation led to the suggestion that Cys54 may act as a gatekeeper (9). Such a model is consistent with the structure of the active site and the position of Cys54. We have extended this line of thinking and demonstrated that NmEstD was inactivated via S-glutathionylation by a naturally occurring reagent GSNO. This covalent modification is becoming increasingly recognized to play a key regulatory role in biology (25). Unfortunately, we were not able to generate diffracting crystals of the GSH-modified form of NmEstD. However, our modeling of the NmEstD structure with a GSH-modified Cys54 indicates that the active site would indeed be blocked by such a modification.
It is possible that this modulation of activity via S-glutathionylation may be linked to complex metabolic signaling associated with GSNO. It has been suggested that the interplay between the metabolism of GSH and its derivatives, including GSNO and HMGSH, could have a role in regulation of NO signaling via S-nitrosylation of proteins (41). In fact, in mammals, yeast, and E. coli, AdhC is known to catalyze the NADH-dependent reduction of GSNO (30), and it has been proposed that this enzyme protects against nitrosative stress (17, 30, 42). Interestingly, EstD is not generally considered to play a role in the removal of the products of GSNO reduction (41). In the present study, we found that compared to the wild-type strain, neither the adhC nor the estD mutants of N. meningitidis were any more sensitive to killing by generators of nitrosative stress such as nitrite and GSNO (Chen, Djoko, and McEwan, unpublished observations). In addition, purified NmEstD was unable to remove the products of AdhC-catalyzed detoxification of GSNO (Chen, Djoko, and McEwan, unpublished observations). Our observation that NmEstD is highly specific for SFG supports the idea that the AdhC-EstD system in the meningococcus is dedicated solely to the removal of formaldehyde and not any other toxic species. Our biofilm results further suggest that this pathway represents an innate mechanism for survival in the human host.
Materials and Methods
Chemicals and reagents
Unless otherwise specified, chemicals were obtained from commercial suppliers. GSNO (15) and SFG (47) were synthesized as described previously.
Bacterial strains and culture conditions
Uncapsulated N. meningitidis strain C311#3 (52) was propagated on a solid brain–heart infusion (BHI) medium (Oxoid, Adelaide, South Australia) supplemented with 10% (v/v) Levinthal's base (2), and grown by incubation at 37 °C and 5% CO2. E. coli strains DH5α and BL21(DE3) were cultured on a Luria-Bertani (LB) medium. Where appropriate, kanamycin (100 μg/ml), spectinomycin (50 μg/ml), or ampicillin (100 μg/ml) was used.
Construction of mutant meningococcal strains
Meningococcal adhC (NMB1304) gene, estD (NMB1305) gene, and the adhC-estD operon were inactivated by deletion and subsequent insertion of a kanamycin-resistance cassette as illustrated in Supplementary Figure S1. Briefly, the upstream and downstream flanking regions of the target gene (H1 and H2, respectively) were amplified separately by polymerase chain reaction (PCR) using primers listed in Supplementary Table S2 and combined with the kanamycin cassette via splice-overlap PCR. The resulting constructs were cloned into the SmaI site of pUC19 to generate pUC19::adhC::kan, pUC19::estD::kan, and pUC19::adhC-estD::kan. Plasmids were linearized with EcoRI and transformed into N. meningitidis C311#3 wild-type strain. Kanamycin-resistant transformants were selected, and correct insertion of the kanamycin cassette was confirmed by PCR using primers that annealed to regions flanking each insertion (Supplementary Table S2).
Mutant meningococci were complemented by inserting a wild-type copy of the entire adhC-estD operon and a spectinomycin-resistance cassette into the proB gene in the N. meningitidis chromosome (43). Briefly, a region containing the adhC-estD operon was amplified by PCR using the primers AEnm_comp_XmaI_F and AEnm_comp_AflII_R (Supplementary Table S2), and the product was inserted between the XmaI and AflII sites of pCTS32. The resulting plasmid pCTS32::adhC-estD was linearized with NdeI before transformation. Positive transformants were checked with PCR using the primer pair proBnm-F/R (Supplementary Table S2).
To generate site-directed variants of the estD gene, the estD single-mutant strain was complemented as described above with the C26AestD, C54AestD, or S145AestD gene. The desired mutation was introduced by splice-overlap PCR using primers as listed in Supplementary Table S2. All mutant strains constructed in this study were confirmed through DNA sequencing.
Bacterial sensitivity assays
Sensitivity of N. meningitidis C311#3 to reactive aldehydes was tested using methylglyoxal (40% w/v), glyoxal (40% w/v), glutaraldehyde (25% w/v), glycoaldehyde (1 M), and formaldehyde (35%–40% w/v). Disc diffusion susceptibility assays were performed by placing paper discs containing 10 μl of each stress reagent onto a lawn of meningococci on a BHI medium. After overnight incubation, the zone of clearing around the disc was measured. Sensitivity to formaldehyde was also examined by a modified killing assay. Briefly, fresh lawns of meningococci from an overnight agar plate were resuspended in phosphate-buffered saline (PBS) to an OD600 of 0.4 (∼8×107 CFU/ml). Serial dilutions (5 μl each) were plated on a BHI solid medium containing increasing concentrations of formaldehyde (0–1.6 mM). Survival was assessed by counting the number of visible colonies after overnight growth.
Growth of biofilms in continuous-flow chambers over glass
N. meningitidis C311#3 wild-type, adhC, estD, and adhC-estD mutants were assayed for their abilities to form biofilms over glass coverslips as described previously (39). The strains were grown to an OD600 of 0.25 in a GC medium supplemented with 1% IsoVitaleX (Becton Dickinson, North Ryde, NSW, Australia), Kellogg's supplements (22), and 0.042% w/v sodium bicarbonate. This cell suspension was used to inoculate a continuous-flow chamber. Cells were allowed to adhere to the glass surface for 1 h at 37°C before the chambers were incubated for 24 h under a flow rate of 180 μl biofilm medium (a GC medium diluted 1:4 in PBS with 1% IsoVitaleX and Kellogg's supplements added) per minute. At the end of the experiment, the effluent was cultured to check for contamination, and the biofilm was stained using LIVE/DEAD BacLight bacterial viability stain (Invitrogen, Mulgrave, VIC, Australia). Z-series photomicrographs were scanned with a Nikon C1 confocal microscope, and three-dimensional images were rendered using EZ-C1 software (Nikon, Lidcombe, NSW, Australia). COMSTAT analyses were performed on each z-series to quantify the average biomass and thickness (18).
Expression and purification of NmEstD
The estD gene was amplified from the chromosomal DNA of N. meningitidis C311#3 using the cloning primer pair estDnm_NdeI_F/R (Supplementary Table S2). Site-directed Cys variants were constructed by splice-overlap PCR using primers carrying the desired mutation (Supplementary Table S2). The resulting PCR products were inserted between NdeI and BamHI sites of plasmid pET-15b (Novagen, Kilsyth, VIC, Australia) and transformed into E. coli strain BL21(DE3) for protein overexpression. Bacteria were grown in an LB medium (1 L) containing ampicillin at 37°C with vigorous shaking at 200 rpm. When the OD600 reached 0.4, isopropyl 1-thio-β-D-galactopyranoside (0.4 mM) was added. Cells were harvested after 5 h of further growth. Cell pellets were resuspended in 50 ml of Buffer A (25 mM Tris–HCl, 15 mM imidazole, 300 mM NaCl, and 5 mM β-mercaptoethanol, pH 8.0) and lysed by three passages through a French pressure cell (16,000 psi) (Thermo Fisher, Scorseby, VIC, Australia).
NmEstD proteins were purified by metal ion affinity chromatography using a HiTrap Chelating Sepharose column (5 ml; GE Healthcare, Rydalmere, NSW, Australia) in Buffer A. Briefly, crude lysates were cleared by centrifugation and loaded on the column. Unbound proteins were removed by washing with Buffer A, and bound proteins were eluted using a linear gradient of 0–500 mM imidazole in the same buffer. Fractions containing NmEstD were pooled, and dialyzed into the Tris–HCl buffer (20 mM, 1 mM DTT, pH 8.0). The purity of each protein preparation was checked routinely by SDS-PAGE. The identity of each protein was confirmed by ESI MS as described below (Supplementary Table S3). Protein concentrations were quantified using solution absorbances at 280 nm (ɛ280, 51,300 M−1cm−1).
Generation of S-glutathionylated forms of NmEstD
Reduced forms of NmEstD (50 μM) were prepared by incubation with excess DTT (100 molar equivalents) for 30 min, followed by desalting into the HEPES buffer (50 mM, pH 7.0). To generate S-glutathionylated forms, GSNO was added, and the mixture was incubated for 1 h at 37°C in the dark. Unreacted GSNO was removed by elution on a PD-10 (GE Healthcare) column. Each modified form of NmEstD was identified using ESI MS. The esterase activity was also assayed immediately using 2 mM pNPAC and 1 mM SFG as described below.
Measurement of esterase activity
Carboxylesterase activities of wild-type Cys variants and S-glutathionylated forms of NmEstD were assayed using pNPAC as a substrate. Briefly, to a solution of pNPAC (0–4 mM) in the HEPES buffer (50 mM, pH 7), NmEstD (0.4 μM) was added, and generation of p-nitrophenol was monitored at 37°C at 400 nm (ɛ, 16,400 M−1 cm−1). Initial rates of reactions were calculated from the rate obtained 30 s after the addition of NmEstD, and the results were plotted as a function of substrate concentration. Michaelis–Menten parameters were obtained by nonlinear regression to a rectangular hyperbola.
Thioesterase activity was measured as above, but using SLG (0–10 mM) or SFG (0–4 mM) as substrates. Hydrolysis of the thioester bond was monitored at 240 nm (ɛ, 3100 and 3000 M−1 cm−1 for SLG and SFG, respectively) (47), except when higher concentrations of SLG were used (5–10 mM), in which case the solution absorbance at 247 nm was monitored (ɛ, 1675 M−1cm−1). Due to the high activity of EstD toward SFG, only 4 nM of NmEstD was used in these assays.
Electrospray ionization mass spectrometry
NmEstD samples (10–20 μM) were prepared in 50% acetonitrile containing 0.01% formic acid. Spectral data were collected on QSTAR Pulsar ESI-QqTOF (Applied Biosystems, Mulgrave, VIC, Australia) using the positive ion (ESI+) mode with a cone voltage 25–35 V and a flow rate of 5 μl acetonitrile per min. The average molar mass of each sample was obtained by applying a deconvolution algorithm to the recorded spectrum, which was calibrated with horse heart myoglobin (16,951 Da).
Crystal structure determination and structural analysis
NmEstD was crystallized by hanging-drop vapor diffusion in 25 mM bis-Tris (pH 6.5), 200 mM lithium sulfate, and 25% PEG 3350. Crystallization drops consisted of 2 μl of purified EstD at 20 mg/ml in 10 mM HEPES, pH 7.0, 1 mM DTT, and 2.0 μl reservoir solution. Large (0.3×0.1×0.1 mm), hexagonal crystals were grown at 293 K. Before X-ray diffraction analysis, crystals were mounted on nylon loops (Hampton Research, Aliso Viejo, CA) and flash-cooled by plunging into a liquid nitrogen bath. To preserve the diffraction quality of the crystals after flash cooling, 1 μl of a solution containing the reservoir solution supplemented with 20% glycerol was added directly to the crystallization drop. A complete diffraction dataset from the EstD crystals was collected using a frame width of 0.5 ° at the Australian Synchrotron (beamline MX2) at 0.954 Å wavelength, using Blu-Ice software (31). Images were processed using XDS (21), and scaled and merged using software within the CCP4 suite (37). The crystallographic phase problem was solved by molecular replacement using the human EstD as the search model (PDB ID 3FCX). Molecular replacement and structure refinement were performed using the programs within PHENIX (1). Data processing and structure refinement statistics can be found in Table 3. The structure has been deposited in the Protein Data Bank (PDB ID 4B6G).
In silico modeling of SFG binding to NmEstD and S-glutathionylation of Cys54
Models for SFG and GSH were generated using the Dundee PRODRUG server (38). All water molecules and ligands were removed from the coordinate file of NmEstD before docking. The program GOLD (20) was used for all docking calculations. All hydrogen atoms were added to the protein within GOLD. For docking of SFG, the coordinates of the oxygen atom from Ser145 defined a search radius of 15 Å. Docking of SFG to NmEstD was restrained based on ordered solvent molecules found within hydrogen bond distances of the main-chain nitrogen atoms from Met144 and Leu52 (oxyanion hole) and the oxygen atom from Ser145. A similar protocol was employed for the in silico modeling of S-glutathionylated Cys54, but in this case, a disulfide bond was introduced between the sulfur atoms from the protein residue and that from GSH. All other bonds in the GSH ligand were kept unrestrained. For each ligand, 20 independent docking searches were undertaken and scored with the default setting, ChemPLP.
Supplementary Material
Abbreviations Used
- ADH
alcohol dehydrogenase
- CFU
colony-forming unit
- DTT
dithiothreitol
- ESI MS
electrospray ionization mass spectrometry
- FA
formaldehyde
- GCA
glycoaldehyde
- GSH
glutathione
- GSNO
S-nitrosoglutathione
- GSSG
oxidized glutathione
- GTA
glutaraldehyde
- HMGSH
S-hydroxymethylglutathione
- IA
iodoacetamide
- LB
Luria-Bertani
- MG
methylglyoxal
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- pNPAC
p-nitrophenyl acetate
- SFG
S-formylglutathione
- SFGH
S-formylglutathione hydrolase
- SLG
S-lactoylglutathione
Acknowledgments
Our work was supported by the Australian Research Council (ARC) grant DP0986578 to A.G.M. and the National Health & Medical Research Council (NHMRC) Program grant 565526 to A.G.M., M.P.J., and B.K. N.H.C. is a recipient of the Australian Postgraduate Award. The biofilm experiments were performed during a visit to the University of Iowa supported by the University of Queensland Graduate School International Travel Award to N.H.C. B.K. is an NHMRC Senior Research Fellow. We thank Prof. James DeVoss (University of Queensland) for his assistance with SFG synthesis and Dr. Alun Jones (Institute for Molecular Bioscience) for his assistance with ESI MS.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Adams PD. Afonine PV. Bunkoczi G. Chen VB. Davis IW. Echols N. Headd JJ. Hung LW. Kapral GJ. Grosse-Kunstleve RW. McCoy AJ. Moriarty NW. Oeffner R. Read RJ. Richardson DC. Richardson JS. Terwilliger TC. Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander H The Haemophilus group. In: Bacterial and Mycotic Infection in Man. Dubos R, editor; Hirsch J, editor. London, United Kingdom: Pitman Medical Publishing; 1965. pp. 724–741. [Google Scholar]
- 3.Alterio V. Aurilia V. Romanelli A. Parracino A. Saviano M. D'Auria S. De Simone G. Crystal Structure of an S-formylglutathione Hydrolase from Pseudoalteromonas haloplanktis TAC125. Biopolymers. 2010;93:669–677. doi: 10.1002/bip.21420. [DOI] [PubMed] [Google Scholar]
- 4.Anderson MM. Hazen SL. Hsu FF. Heinecke JW. Human neutrophils employ the myeloperoxidase-hydrogen peroxide-chloride system to convert hydroxy-amino acids into glycolaldehyde, 2-hydroxypropanal, and acrolein—a mechanism for the generation of highly reactive alpha-hydroxy and alpha,beta-unsaturated aldehydes by phagocytes at sites of inflammation. J Clin Invest. 1997;99:424–432. doi: 10.1172/JCI119176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barber RD. Donohue TJ. Pathways for transcriptional activation of a glutathione-dependent formaldehyde dehydrogenase gene. J Mol Biol. 1998;280:775–784. doi: 10.1006/jmbi.1998.1900. [DOI] [PubMed] [Google Scholar]
- 6.Barber RD. Rott MA. Donohue TJ. Characterization of a glutathione-dependent formaldehyde dehydrogenase from Rhodobacter sphaeroides. J Bacteriol. 1996;178:1386–1393. doi: 10.1128/jb.178.5.1386-1393.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolt HM. Experimental toxicology of formaldehyde. J Cancer Res Clin Oncol. 1987;113:305–309. doi: 10.1007/BF00397713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boor PJ. Trent MB. Lyles GA. Tao M. Ansari GAS. Methylamine metabolism to formaldehyde by vascular semicarbazide-sensitive amine oxidase. Toxicology. 1992;73:251–258. doi: 10.1016/0300-483x(92)90067-o. [DOI] [PubMed] [Google Scholar]
- 9.Cummins I. McAuley K. Fordham-Skelton A. Schwoerer R. Steel PG. Davis BG. Edwards R. Unique regulation of the active site of the serine esterase S-formylglutathione hydrolase. J Mol Biol. 2006;359:422–432. doi: 10.1016/j.jmb.2006.03.048. [DOI] [PubMed] [Google Scholar]
- 10.Degrassi G. Uotila L. Klima R. Venturi V. Purification and properties of an esterase from the yeast Saccharomyces cerevisiae and identification of the encoding gene. Appl Environ Microbiol. 1999;65:3470–3472. doi: 10.1128/aem.65.8.3470-3472.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edwards JS. Palsson BO. Systems properties of the Haemophilus influenzae Rd metabolic genotype. J Biol Chem. 1999;274:17410–17416. doi: 10.1074/jbc.274.25.17410. [DOI] [PubMed] [Google Scholar]
- 12.Fall R. Benson AA. Leaf methanol—the simplest natural product from plants. Trends Plant Sci. 1996;1:296–301. [Google Scholar]
- 13.Gonzalez C. Proudfoot M. Brown G. Korniyenko Y. Mori H. Savchenko A. Yakuin A. Molecular basis of formaldehyde detoxification: characterization of two S-formylglutathione hydrolases from Escherichia coli, FrmB and YeiG. J Biol Chem. 2006;281:14514–14522. doi: 10.1074/jbc.M600996200. [DOI] [PubMed] [Google Scholar]
- 14.Harms N. Ras J. Reijnders WNM. van Spanning RJM. Stouthamer AH. S-formylglutathione hydrolase of Paracoccus denitrificans is homologous to human esterase D: a universal pathway for formaldehyde detoxification? J Bacteriol. 1996;178:6296–6299. doi: 10.1128/jb.178.21.6296-6299.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hart TW. Some observations concerning the S-nitroso and S-phenylsulphonyl derivatives of L-cysteine and glutathione. Tetrahedron Lett. 1985;26:2013–2016. [Google Scholar]
- 16.Heck HD. Casanova M. Starr TB. Formaldehyde toxicity—new understanding. Criti Rev Toxicol. 1990;20:397–426. doi: 10.3109/10408449009029329. [DOI] [PubMed] [Google Scholar]
- 17.Hedberg JJ. Griffiths WJ. Nilsson SJF. Hoog JO. Reduction of S-nitrosoglutathione by human alcohol dehydrogenase 3 is an irreversible reaction as analysed by electrospray mass spectrometry. Eur J Biochem. 2003;270:1249–1256. doi: 10.1046/j.1432-1033.2003.03486.x. [DOI] [PubMed] [Google Scholar]
- 18.Heydorn A. Nielsen AT. Hentzer M. Sternberg C. Givskov M. Ersboll BK. Molin S. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology UK. 2000;146:2395–2407. doi: 10.1099/00221287-146-10-2395. [DOI] [PubMed] [Google Scholar]
- 19.Hickman JW. Barber RD. Skaar EP. Donohue TJ. Link between the membrane-bound pyridine nucleotide transhydrogenase and glutathione-dependent processes in Rhodobacter sphaeroides. J Bacteriol. 2002;184:400–409. doi: 10.1128/JB.184.2.400-409.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones G. Willett P. Glen RC. Leach AR. Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 21.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- 22.Kellogg DS. Peacock WL. Pirkle CI. Deacon WE. Brown L. Neisseria gonorrhoeae. 1. Virulence genetically linked to clonal variation. J Bacteriol. 1963;85:1274–1279. doi: 10.1128/jb.85.6.1274-1279.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kidd SP. Jiang D. Jennings MP. McEwan AG. Glutathione-dependent alcohol dehydrogenase AdhC is required for defense against nitrosative stress in Haemophilus influenzae. Infect Immun. 2007;75:4506–4513. doi: 10.1128/IAI.00487-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kidd SP. Potter AJ. Apicella MA. Jennings MP. McEwan AG. NmlR of Neisseria gonorrhoeae: a novel redox responsive transcription factor from the MerR family. Mol Microbiol. 2005;57:1676–1689. doi: 10.1111/j.1365-2958.2005.04773.x. [DOI] [PubMed] [Google Scholar]
- 25.Klatt P. Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 26.Klomsiri C. Karplus PA. Poole LB. Cysteine-based redox switches in enzymes. Antioxid Redox Signal. 2011;14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kordic S. Cummins I. Edwards R. Cloning and characterization of an S-formylglutathione hydrolase from Arabidopsis thaliana. Arch Biochem Biophys. 2002;399:232–238. doi: 10.1006/abbi.2002.2772. [DOI] [PubMed] [Google Scholar]
- 28.Lee WH. Wheatley W. Benedict WF. Huang CM. Lee E. Purification, biochmical-characterization, and biological function of human Esterase-D. Proc Natl Acad Sci U S A. 1986;83:6790–6794. doi: 10.1073/pnas.83.18.6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Legler PN. Kumaran D. Swaminathan S. Studier FW. Millard CB. Structural characterization and reversal of the natural organophosphate resistance of a D-type esterase, Saccharomyces cerevisiae S-formylglutathione hydrolase. Biochemistry. 2008;47:9592–9601. doi: 10.1021/bi8010016. [DOI] [PubMed] [Google Scholar]
- 30.Liu LM. Hausladen A. Zeng M. Que L. Heitman J. Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 31.McPhillips TM. McPhillips SE. Chiu HJ. Cohen AE. Deacon AM. Ellis PJ. Garman E. Gonzalez A. Sauter NK. Phizackerley RP. Soltis SM. Kuhn P. Blu-Ice and the distributed control system: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 32.Morris CJ. Earl JR. Trenam CW. Blake DR. Reactive oxygen species and iron—a dangerous partnership in inflammation. Int J Biochem Cell Biol. 1995;27:109–122. doi: 10.1016/1357-2725(94)00084-o. [DOI] [PubMed] [Google Scholar]
- 33.Neil RB. Apicella MA. Clinical and laboratory evidence for Neisseria meningitidis biofilms. Future Microbiol. 2009;4:555–563. doi: 10.2217/fmb.09.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okado-Matsumoto A. Fridovich I. The role of alpha,beta-dicarbonyl compounds in the toxicity of short chain sugars. J Biol Chem. 2000;275:34853–34857. doi: 10.1074/jbc.M005536200. [DOI] [PubMed] [Google Scholar]
- 35.Ollis DL. Cheah E. Cygler M. Dijkstra B. Frolow F. Franken SM. Harel M. Remington SJ. Silman I. Schrag J. Sussman JL. Verschueren KHG. Goldman A. The alpha/beta-hydrolase fold. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 36.Potter AJ. Kidd SP. Jennings MP. McEwan AG. Evidence for distinctive mechanisms of S-nitrosoglutathione metabolism by AdhC in two closely related species, Neisseria gonorrhoeae and Neisseria meningitidis. Infect Immun. 2007;75:1534–1536. doi: 10.1128/IAI.01634-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Potterton E. Briggs P. Turkenburg M. Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr D Biol Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 38.Schuttelkopf AW. van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 39.Seib KL. Wu HJ. Srikhanta YN. Edwards JL. Falsetta ML. Hamilton AJ. Maguire TL. Grimmond SM. Apicella MA. McEwan AG. Jennings MP. Characterization of the OxyR regulon of Neisseria gonorrhoeae. Mol Microbiol. 2007;63:54–68. doi: 10.1111/j.1365-2958.2006.05478.x. [DOI] [PubMed] [Google Scholar]
- 40.Spiteller G. Peroxyl radicals are essential reagents in the oxidation steps of the Maillard reaction leading to generation of advanced glycation end products. Ann N Y Acad Sci. 2008;1126:128–133. doi: 10.1196/annals.1433.031. [DOI] [PubMed] [Google Scholar]
- 41.Staab CA. Alander J. Brandt M. Lengqvist J. Morgenstern R. Grafstrom RC. Hoog JO. Reduction of S-nitrosoglutathione by alcohol dehydrogenase 3 is facilitated by substrate alcohols via direct cofactor recycling and leads to GSH-controlled formation of glutathione transferase inhibitors. Biochem J. 2008;413:493–504. doi: 10.1042/BJ20071666. [DOI] [PubMed] [Google Scholar]
- 42.Staab CA. Hellgren M. Hoog JO. Dual functions of alcohol dehydrogenase 3: implications with focus on formaldehyde dehydrogenase and S-nitrosoglutathione reductase activities. Cell Mol Life Sci. 2008;65:3950–3960. doi: 10.1007/s00018-008-8592-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steichen CT. Shao JQ. Ketterer MR. Apicella MA. Gonococcal cervicitis: a role for biofilm in pathogenesis. J Infect Dis. 2008;198:1856–1861. doi: 10.1086/593336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tettelin H. Saunders NJ. Heidelberg J. Jeffries AC. Nelson KE. Eisen JA. Ketchum KA. Hood DW. Peden JF. Dodson RJ. Nelson WC. Gwinn ML. DeBoy R. Peterson JD. Hickey EK. Haft DH. Salzberg SL. White O. Fleischmann RD. Dougherty BA. Mason T. Ciecko A. Parksey DS. Blair E. Cittone H. Clark EB. Cotton MD. Utterback TR. Khouri H. Qin HY. Vamathevan J. Gill J. Scarlato V. Masignani V. Pizza M. Grandi G. Sun L. Smith HO. Fraser CM. Moxon ER. Rappuoli R. Venter JC. Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science. 2000;287:1809–1815. doi: 10.1126/science.287.5459.1809. [DOI] [PubMed] [Google Scholar]
- 45.Thornalley P. Wolff S. Crabbe J. Stern A. The autoxidation of glyeraldehyde and other simple monosaccharides under physiological conditions catalyzed by buffer ions. Biochim Biophys Acta. 1984;797:276–287. doi: 10.1016/0304-4165(84)90131-4. [DOI] [PubMed] [Google Scholar]
- 46.Thornalley PJ. The glyoxalase system in health and disease. Mol Aspects Med. 1993;14:287–371. doi: 10.1016/0098-2997(93)90002-u. [DOI] [PubMed] [Google Scholar]
- 47.Uotila L. Thioesters of glutathione. Methods Enzymol. 1981;77:424–430. [Google Scholar]
- 48.Uotila L. Koivusal M. Formaldehyde dehydrogenase from human liver—purification, properties, and evidence for for formation of glutathione thiol esters by enzyme. J Biol Chem. 1974;249:7653–7663. [PubMed] [Google Scholar]
- 49.Uotila L. Koivusal M. Purification and properties of S-formylglutathione hydrolase from human liver. J Biol Chem. 1974;249:7664–7672. [PubMed] [Google Scholar]
- 50.van Ophem PW. Duine JA. NAD-Substrate and cosubstrate (GSH or Factor)-dependent formaldehyde dehydrogenases from methylotrophic microorganisms act as a class-III alcohol dehydrogenase. FEMS Microbiol Lett. 1994;116:87–93. [Google Scholar]
- 51.van Straaten KE. Gonzalez CF. Valladares RB. Xu XH. Savchenko AV. Sanders DAR. The structure of a putative S-formylglutathione hydrolase from Agrobacterium tumefaciens. Protein Sci. 2009;18:2196–2202. doi: 10.1002/pro.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Virji M. Kayhty H. Ferguson DJP. Alexandrescu C. Heckels JE. Moxon ER. The role of pili in the interactions of pathogenic Neisseria with cultured human endothelial cells. Mol Microbiol. 1991;5:1831–1841. doi: 10.1111/j.1365-2958.1991.tb00807.x. [DOI] [PubMed] [Google Scholar]
- 53.Wang Y. Ho C-T. Flavour chemistry of methylglyoxal and glyoxal. Chem Soc Rev. 2012;41:4140–4149. doi: 10.1039/c2cs35025d. [DOI] [PubMed] [Google Scholar]
- 54.Wu D. Li Y. Song GJ. Zhang D. Shaw N. Liu ZJ. Crystal structure of human esterase D: a potential genetic marker of retinoblastoma. FASEB J. 2009;23:1441–1446. doi: 10.1096/fj.08-125286. [DOI] [PubMed] [Google Scholar]
- 55.Yu PH. Zuo DM. Oxidative deamination of methylamine by semicarbazide-sensitive amine oxidase leads to cytotoxic damage in endothelial cells - possible consequences for diabetes. Diabetes. 1993;42:594–603. doi: 10.2337/diab.42.4.594. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.