

Abstract
The Ikaros transcription factor is crucial for many aspects of hematopoiesis. Loss of function mutations in IKZF1, the gene encoding Ikaros, have been implicated in adult and pediatric B cell acute lymphoblastic leukemia (B-ALL). These mutations result in haploinsufficiency of the Ikaros gene in approximately half of the cases. The remaining cases contain more severe or compound mutations that lead to the generation of dominant-negative proteins or complete loss of function. All IKZF1 mutations are associated with a poor prognosis. Here we review the current genetic, clinical and mechanistic evidence for the role of Ikaros as a tumor suppressor in B-ALL.
Keywords: B cell leukemia, Ikaros, tumor suppressor
From T-ALL to center stage in B-ALL
Ikaros is a zinc finger transcription factor encoded by the IKZF1 gene. First characterized as a protein that binds to regulatory elements in the genes encoding CD3Δ and deoxynucleotidyl terminal transferase in T lymphocytes [1,2], knock-out studies in animals have revealed a wide-ranging role for Ikaros in many aspects of hematopoiesis. Ikaros null mice show a profound reduction in hematopoietic stem cell activity, lack numerous hematopoietic cell types such as B cells, NK cells and dendritic cells, are defective in erythropoiesis and show a differentiation bias towards the CD4+ T cell lineage in the thymus [3-8]. At the molecular level, Ikaros both represses and activates gene transcription by binding to target sequences containing the core TGGGAA sequence, and it interacts with chromatin remodeling complexes to modulate gene expression [9-11].
Mouse studies also revealed Ikaros to be a tumor suppressor in T cells. Indeed, mice heterozygous for a dominant-negative (dn) mutation of Ikzf1, or homozygous for a knock-down Ikzf1 mutation, develop T cell lymphomas/leukemias with 100% penetrance [12-15]. Other studies have confirmed the prominent status of Ikaros as a major tumor suppressor in T cells in the mouse (see [16] for review). These results prompted several teams to investigate if Ikaros mutations were implicated in human T cell malignancies. However, Ikaros defects are rare in T cell acute lymphoblastic leukemias (T-ALL) and are observed in approximately 5% of cases [17-19].
Interestingly, Ikaros mutations are frequent in B-ALL. These studies can be grossly divided into those done before, versus those done after, the age of pan-genomic technology. Early results, based primarily on RT-PCR analysis of the IKZF1 coding sequence, revealed the presence of Ik6, a dn Ikaros isoform, in a subset of pediatric and adult B-ALL [20-24]. Nakase et al and Tonnelle et al reported that Ik6 is detected in ~30% of adult B-ALL, and is prevalent in those presenting the t(9; 22) translocation that generates the BCR-ABL fusion oncoprotein [20,22]. Ik6 is also associated with BCR-ABL positive chronic myeloid leukemia (CML) cells in blast crisis [25]. As Ik6 expression is accompanied by the appearance of novel restriction fragments in Southern blot analyses of the IKZF1 locus, it was suggested that genomic rearrangements occur in this region [20,25]. Other mechanisms such as alternative splicing of IKZF1 transcripts in response to BCR-ABL signaling were also proposed [26]. With the advent of large-scale comparative genomic hybridization (CGH) technology using high-density microarrays, it finally became clear that IKZF1 is subject to frequent somatic mutations/deletions in B-ALL [27-30]. These studies showed that 15-20% of pediatric B-ALL and >75% of BCR-ABL positive B-ALL are affected by genomic deletions in the IKZF1 locus (Table 1). Fewer studies have investigated adult B-ALL, but available data suggest that IKZF1 mutations might occur at an even higher frequency in adults (close to 50%, Table 1) [31,32]. The deletions fall into several categories (see below), with one being an intragenic deletion of IKZF1 exons 4-7. Transcription from these alleles produces Ikaros transcripts lacking exons 4-7 and corresponding to Ik6. In addition, Mullighan et al showed an absolute correlation between exon 4-7 deletions, and Ik6 transcripts and proteins [29]. Thus genomic deletion is the dominant mechanism behind the synthesis of dn Ik6 isoforms in B-ALL. Collectively, these studies identify Ikaros loss of function as a recurrent anomaly in human B-ALL.
Table 1.
Summary of recent studies that addressed Ikaros loss of function in B-ALL
| Study | Patient cohort | Main results |
|---|---|---|
| Mullighan 2008 [29] | 43 Pediatric BCR-ABL positive B-ALL | • IKZF1 deletions in 84% of cases |
| • IKZF1 mutations frequent in lymphoid blast crisis of CML patients | ||
| Mullighan 2009 [35] | • 221 pediatric high risk BCR-ABL negative B-ALL | • IKZF1 mutations in 28.6% of high risk cases and 24.8% of validation cohort |
| • 258 pediatric B-ALL | • IKZF1 mutations are predictive of adverse prognosis | |
| Iacobucci 2009 [30] | 106 Adult BCR-ABL positive B-ALL | • IKZF1 mutations in 75% of patients |
| • IKZF1 mutations in 66% of lymphoid blast crisis of CML patients | ||
| • 34 selected diagnosis/ relapse pairs of pediatric B-ALL | • 18/131 mutations in the unselected cohort (13.7%) | |
| Kuiper 2010 [36] | • 13/34 IKZF1 mutations in the relapse prone cohort | |
| • 131 unselected pediatric B-ALL | • 2 patients with IKZF1 mutations that appeared at relapse | |
| Martinelli 2009 [56] | 83 BCR-ABL positive adult patients | • 63% with IKZF1 deletions |
| • shorter disease-free survival of patients with IKZF1 mutations | ||
| Dupuis 2012 [31 | 139 unselected adult (46) and pediatric (93) B-ALL | • IKZF1 deletions in 52% of adult and 27% of pediatric patients |
| • biallelic deletions in 15% of patients with IKZF1 deletions | ||
| • several cases of biclonal deletions | ||
| • IKZF1 deletions are associated with adverse prognosis in adult BCR-ABL negative patients | ||
| Cayé 2012 [34] | • 60 BCR-ABL positive pediatric B-ALL | • 75% of IKZF1 deletions in BCR-ABL positive B-ALL |
| • 512 BCR-ABL negative pediatric B-ALL | • 16% of IKZF1 deletions in BCR-ABL negative B-ALL | |
| • multiple cases of subclonal IKZF1 deletions | ||
| Harvey 2010 [54] | 207 high risk pediatric B-ALL | • 30% of IKZF1 mutations |
| • Strong association with JAK1 or JAK2 mutations and CRLF2 rearrangements | ||
| Mi 2012 [32] | • 203 unselected adult B-ALL | • 56 pediatric cases with the Δ4-7 deletion (14.7%) |
| • 379 unselected pediatric B-AL | • 64 adult cases with the Δ4-7 deletion (31.3%) | |
| • Other types of IKZF1 mutations not studied |
The genetics of Ikaros loss of function in B-ALL
Nature of somatic IKZF1 mutations in B-ALL
The human IKZF1 locus spans over 120 kilobases (kb) on chromosome 7p12.2, and comprises 8 exons. Mutations in this locus in B-ALL can be grouped into 3 main categories (Figure 1A). The first corresponds to monosomy 7 or large deletions of the 7p arm, features found in ~4% of all B-ALL [33]. This defect is always associated with the complete loss of one IKZF1 allele. As none of the other known tumor suppressors in B-ALL is located on chromosome 7, it is reasonable to assume that loss of Ikaros might be a driving force in selecting for this anomaly. The second category consists of deletions of 15-200 kb which are restricted to the IKZF1 gene. The most common deletion is one of 50.7 kb that comprises exons 4 to 7 (Δ4-7). The 5’ and 3’ breakpoints are highly conserved and are flanked by recombination signal sequences (RSS), suggesting that this deletion is mediated by off-target effects of the recombination activating gene (RAG) proteins during VDJ recombination in B cell progenitors [29,30]. Δ4-7 deletions lead to in-frame splicing of exon 3 to exon 8, and the synthesis of Ik6. A second common deletion comprises exons 2-7 (Δ2-7). These deletions have the same 3’ breakpoint as the Δ4-7 deletion but more variable 5’ breakpoints [30,34]. They are also likely to be mediated by the RAG machinery, as the breakpoints are flanked by RSS motifs. Δ2-7 deletions probably lead to null alleles, since exon 2 encodes the initiation ATG. In addition, other types of deletions have been reported. These delete the non-coding exon 1 and various lengths of 5’ sequences, and other combinations of coding exons, like exons 2-8 [30,31,34]. All of the deletions are likely to result in null mutations. Finally the last category corresponds to point mutations [35-37], usually frameshift mutations leading to premature stop codons; they account for 11 of the combined 97 IKZF1 mutations (~10%) reported in these studies. One case is a point mutation that results in a G to S amino acid change in the second zinc finger of the DNA binding domain (DBD) of Ikaros [35]. Point mutations affecting the DBD are frequent spontaneous mutations in murine T-ALL where they are predicted to impair Ikaros function (see [16] for review). Other studies, however, have failed to detect point mutations in large B-ALL cohorts [29,30,38]. These discrepancies may reflect technical issues, or an overall low incidence of IKZF1 point mutations if they are less frequent in BCR-ABL positive cases (the focus of 2 of the negative studies) [29,30]. It is noteworthy that the highest frequency of point mutations (3 out of 12 IKZF1 mutations) was found using deep sequencing of genomic DNA and mRNA, which seems to provide the most accurate data [37]. Thus, the true frequency of IKZF1 point mutations in B-ALL remains uncertain and must be reassessed with deep sequencing techniques on large cohorts of patients.
Figure 1.
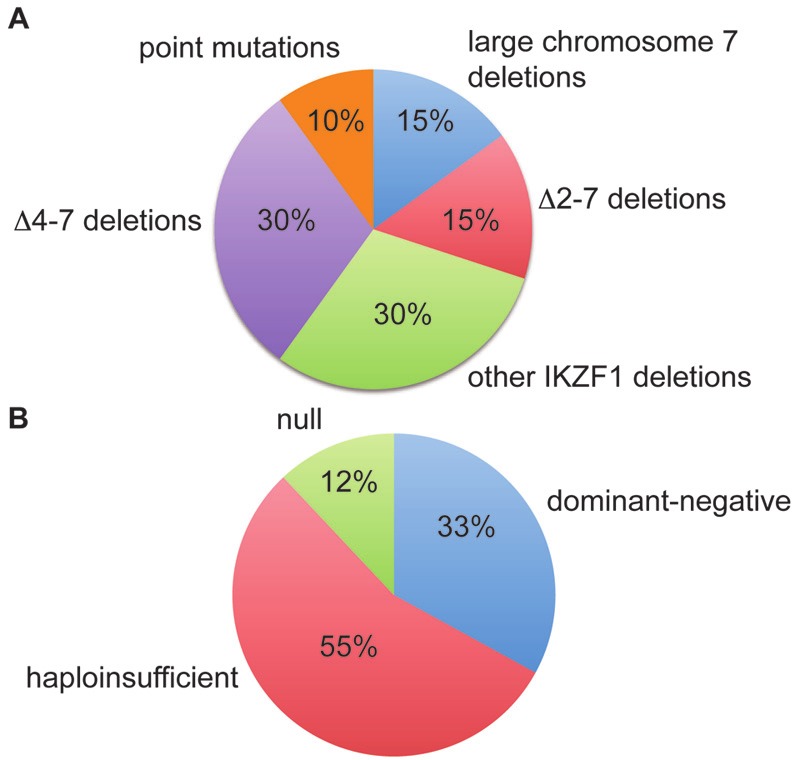
Distribution of the various types of mutations in B-ALL cases with IKZF1 defects. A. Distribution of the genetic defects in B-ALL cases with IKZF1 abnormalities. Numbers are estimates and derived from compiled data from the studies of Mullighan et al. and Dupuis et al [31,35]. B. Distribution of functional classes of mutations. Data are derived from the study by Dupuis et al [31].
Functional classes of IKZF1 mutations
Approximately 55% of B-ALL with IKZF1 mutations exhibit a monoallelic null mutation. These mutations lead to haploinsufficiency, with reduced protein levels of Ikaros as detected by Western blot and immunofluorescence (Figure 2) [31]. Δ4-7 deletions account for ~33% of IKZF1 mutations. These deletions lead to the production of the Ik6 isoform, which is synthesized abundantly and localized in the cytoplasm due to the loss of the DBD and nuclear localization sequences (Figure 2) [21,24,31]. Because Ik6 can dimerize with wild type (WT) Ikaros proteins synthesized from the intact allele, it is thought to exert a dominant-negative effect by sequestering normal proteins in the cytoplasm, as well as the related Aiolos, Helios or Eos proteins. Indeed, a similar deletion in the mouse leads to a phenotype that is more severe than a null mutation [39]. About 12% of B-ALL with IKZF1 mutations have biallelic deletions [29-31,37]. Most of these correspond to 2 null alleles which lead to the complete absence of Ikaros proteins (Figure 2) [29,31]. Thus B-ALL with IKZF1 mutations can be subdivided into 3 functional categories with haploinsufficient, dominantnegative and null Ikaros phenotypes (Figure 2). The occurrence of biallelic null mutations indicates that some leukemic cells that have lost one IKZF1 allele will gain a selective advantage by losing the second allele. Indeed, some B-ALL exhibit both a null and a Δ4-7 deletion [29-31], suggesting that the Δ4-7 deletion is acquired to further reduce Ikaros activity from a haploinsufficient state.
Figure 2.
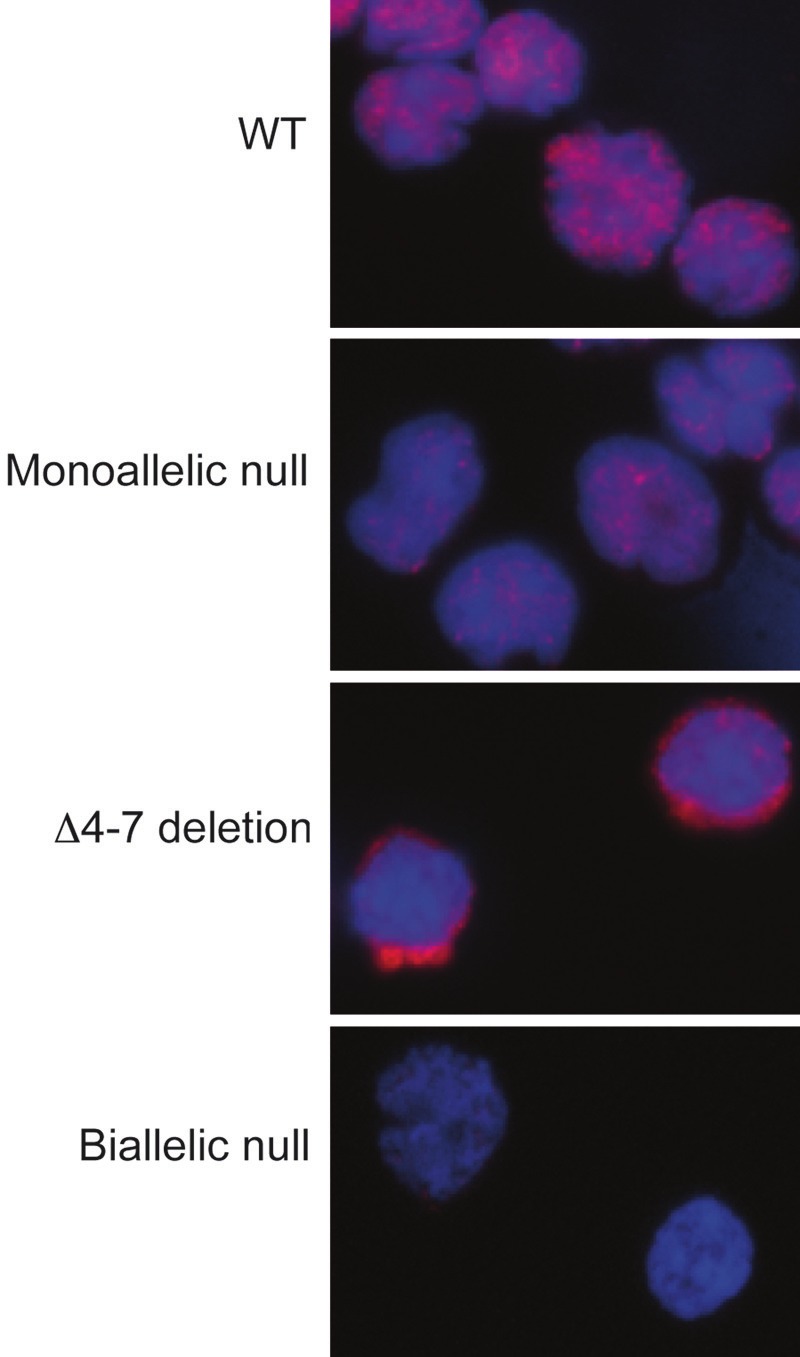
Three functional classes of IKZF1 mutations in B-ALL are associated with distinct quantities and localizations of Ikaros proteins. Immunofluorescence detection of Ikaros proteins in B-ALL cells from WT, haploinsufficient (Δ2-7 deletion), dominant-negative (Δ4-7 deletion) or biallelic null mutations, as indicated.
It is currently unclear if different classes of mutations are associated with different molecular signatures or clinical states. As the loss of one or two IKZF1 alleles results in dramatically different consequences in the mouse, it is likely that haploinsufficient and severe (Δ4-7 deletions or biallelic mutations) mutations will also be associated with distinct phenotypes. Various studies have indicated that BCR-ABL positive B-ALL predominantly exhibit severe IKZF1 mutations. These mutations account for 70%, 65% and 59% of the IKZF1 mutations examined by Dupuis et al, Mullighan et al, and Iacobucci et al, respectively [29-31]. They are in agreement with earlier studies that reported high frequencies of Ik6 mRNA expression in BCR-ABL positive B-ALL [22,24]. In contrast, haploinsufficiency is predominant in BCR-ABL negative B-ALL (72% of the IKZF1 mutations in the adult and pediatric cases studied by Dupuis et al [31], and 57% of the IKZF1 mutations in the high risk pediatric cases studied by Mullighan et al [35]). These observations suggest that the selective pressure to acquire severe Ikaros mutations is higher in BCR-ABL positive B-ALL than other subtypes. More studies are required to determine if there is a functional basis for this difference, and if distinct classes of Ikaros mutations are correlated with other clinical features.
IKZF1 mutations as secondary hits during BALL progression?
Recent studies have suggested that IKZF1 mutations may occur as secondary mutations in B-ALL. Cazzaniga et al reported cases of B-ALL that developed in two pairs of monozygotic twins [40]. In the first pair, both twins developed B-ALL that exhibited the same BCRABL rearrangement, but only one had a IKZF1 deletion. In the second pair, one twin developed a leukemia that exhibited a BCR-ABL fusion gene and a loss of IKZF1. The other twin also had cells with the same BCR-ABL fusion but no IKZF1 deletion, and was healthy. In these cases, the t(9; 22) translocation was the initiating event, while the IKZF1 deletion was acquired secondarily. Dupuis et al identified a BCR-ABL positive B-ALL with both a complete deletion of the IKZF1 gene and a Δ4-7 deletion at diagnosis, but only the complete deletion at relapse [31], suggesting that 2 leukemic clones existed at diagnosis, and only one remained at relapse. Cayé et al employed multiplex qPCR to detect common IKZF1 intragenic rearrangements in a cohort of B-ALL [34], and identified 4 cases with at least two IKZF1 rearrangements at the subclonal level. 15 other cases had a single IKZF1 rearrangement that was present in only a fraction of the leukemic cells (1-20%). These data indicate that IKZF1 mutations can arise as secondary mutations during disease progression, and that independent mutations can be selected for in different clones. IKZF1 mutations can also be acquired late, as some cases have been reported where the IKZF1 mutation was not detected at diagnosis but only at relapse [36,41,42]. An important question is whether IKZF1 mutations are oncogenic alone or only in synergy with other mutations. In this regard, mice haploinsufficient for Ikzf1 in their germline never develop B-ALL on their own, but those heterozygous for Ikzf1 display accelerated B cell leukemogenesis in the presence of a Bcr-Abl transgene [43]. Reconstitution of the phylogenic mutational tree will be required to define how IKZF1 mutations fit into the genetic hierarchy of B-ALL [44].
Inherited polymorphisms of IKZF1 predispose to B-ALL
In addition to somatic mutations, polymorphisms of the IKZF1 locus significantly predispose to childhood B-ALL according to 2 genome-wide association studies (GWAS), suggesting that inherited variations in the IKZF1 gene may promote early B-ALL. Papaemmanuil et al found that the “C” variant of SNP rs4132601 is a risk factor for B-ALL [45]. This SNP is located in exon 8, in the sequence encoding the 3’ untranslated region of Ikaros, and may interfere with mRNA stability. Indeed, the C variant is associated with reduced Ikaros mRNA levels in blood leucocytes. The correlation between this variant and pediatric B-ALL was independently confirmed in cohorts of patients from Poland [46], and from Germany and the United Kingdom [47]. In all 3 studies, heterozygosity for the risk allele is associated with increased ALL frequency, and the risk increases with homozygosity. Another GWAS study by Trevino et al identified a second SNP (rs11978267) located in intron 7 of IKZF1 [48]. The “C” variant of this SNP is linked to an increased predisposition to ALL, with an increased risk from the heterozygous to the homozygous condition. The relevance of this SNP was also investigated by Ross et al in infant ALL and acute myeloid leukemia (AML) [49]. These authors found that the G variant significantly increases the risk for ALL lacking MLL rearrangements, but not for ALL with MLL rearrangements. Interestingly, the G variant also increases the risk for AML. Why a single nucleotide change in an intronic region should increase the risk of leukemia development remains obscure. It is noteworthy, however, that the location of the rs11978267 SNP borders a predicted enhancer in the human GM12878 B lymphoblastoid cell line, but not in non-B cell lines, as delineated by the presence of mono- or tri-methylated lysine 4 of histone H3 in this region (data from ENCODE; Figure 3). It is therefore possible that this sequence variation modulates the activity of a regulatory element important for Ikaros expression in B cells.
Figure 3.
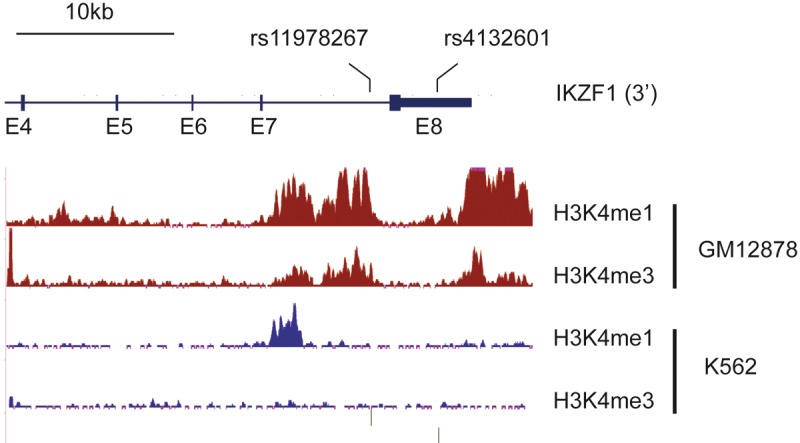
Localization of SNPs associated with increased susceptibility for childhood B-ALL and chromatin features of active regulatory elements in the B lymphoblastoid GM12878 and erythro-myeloid K562 cell lines. ChIP-seq data for mono- and tri-methylated lysine 4 of histone H3 (H3K4me1 and H3K4me3, respectively) are from publicly available tracks from the ENCODE consortium. The rs11978267 SNP borders a region specifically associated with active chromatin marks in the GM12878 cells.
Clinical significance of IKZF1 mutations
Association with genetic defects that activate the JAK-STAT pathway
As noted, IKZF1 mutations strongly associate with the BCR-ABL fusion oncogene. This is true both for B-ALL and for lymphoid blast crisis CML where IKZF1 mutations are frequently detected [25,29,30,50,51], suggesting that Ikaros deficiency plays a role in modulating the oncogenic activity of the BCR-ABL kinase.
The potent synergy between IKZF1 loss of function and BCR-ABL was recently demonstrated in a mouse model of B-ALL. Deletion of a single allele of Ikzf1 in mice expressing a BCR-ABL transgene leads to a dramatic acceleration of B-ALL development, with a decrease in median survival time from 14.7 to 5.7 weeks [43]. Further, Ikzf1 haploinsufficiency is associated with a polyclonal disease, while leukemias that develop in mice with WT Ikzf1 alleles are monoor oligoclonal. Importantly, leukemic cells from the double mutant mice retain the intact Ikzf1 allele and express Ikaros proteins at about 50% of WT levels. Thus a reduction in Ikaros activity is sufficient to accelerate B-ALL development.
Interestingly, IKZF1 mutations are also frequent in BCR-ABL negative leukemias that share a similar gene expression signature with BCRABL positive B-ALL [52]. A point in common may be the JAK-STAT pathway. BCR-ABL signaling activates the JAK-STAT pathway [53], as do amplifications of CRFL2 (which encodes the receptor for the cytokine TSLP), activating mutations of JAK2 or other types of rearrangements/mutations involving genes encoding receptors that signal through this pathway, like IL7R, FLT3 and PDGFR [37,54,55]. These data suggest that Ikaros deficiency may synergize with JAK-STAT activation in the presence or absence of BCR-ABL.
IKZF1 mutations are predictive of a poor prognosis
IKZF1 mutations are associated with a poor prognosis in terms of overall survival and frequency of relapse. This has been studied mostly in pediatric B-ALL, but also appears to be true for adult B-ALL (Table 1) [31,32,35,36,56-58]. In children, IKZF1 mutations are a better predictor of relapse than other genetic defects (ie. EBF1 or PAX5 mutations), or the conventional classification based on clinical and cytogenetic criteria [35]. Interestingly, the combination of IKZF1 mutations and minimal residual disease (MRD) status is a better predictor of relapse than either marker alone [59], suggesting that risk stratification may be improved by comparing IKZF1 status with other criteria of adverse prognosis. In this regard, we have observed that IKZF1 deficiency appears to synergize with a lack of IgH rearrangements, as these parameters point to a very pejorative prognosis (Figure 4). These results were obtained using multivariate analysis to identify clinical parameters in a cohort of 139 adult and pediatric B-ALL, and must be confirmed on more patients. Collectively, these studies suggest that loss of IKZF1 may synergize with other biological or clinical features to define subsets of particularly aggressive leukemias.
Figure 4.
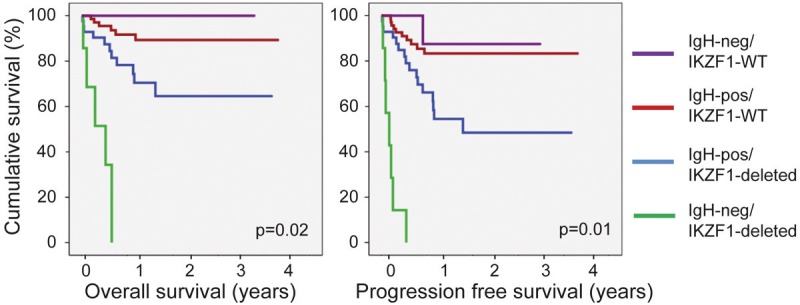
Overall survival curves of patients from the cohort of 136 adult and pediatric patients described by Dupuis et al [31], according to their IKZF1 status and IgH rearrangement (as determined by the Biomed2 protocol). Statistical significance of the differences among the 4 groups of patients was estimated using a Cox Model. The number of patients in the IgH-neg/IKZF1-deleted, IgH-pos/IKZF1-deleted, IgH-neg/IKZF1-WT and IgH-pos/IKZF1-WT were respectively 7, 42, 9 and 78. The group of patients (3 pediatric and 4 adults) who had IKZF1 deletions and lacked IgH rearrangements had a particularly poor survival, suggesting a synergistic predictive value of these two features (note that only 6 IgH-neg/IKZF1-deleted patients could be included in the overall survival analysis as the fate of one patient that had relapsed after 178 days is unknown).
Ikaros mutations to assess MRD
Two groups have proposed that IKZF1 mutations, when detected at diagnosis, may be reliable markers to follow MRD [34,60]. The strong association of IKZF1 deletions with aggressive leukemias and relapse, as well as the relatively well conserved breakpoints of the deletions, provide a strong case using IKZF1 rearrangements as MRD markers. Venn et al used qPCR to quantify the Δ4-6 deletion, while Cayé et al employed multiplex qPCR to simultaneously detect the various IKZF1 breakpoints that may occur in distinct leukemic subclones. Both studies showed that IKZF1 deletions can be sensitive markers to monitor leukemias in remission. It should be noted, however, that some leukemias may escape detection using these methods, as was true for a leukemia with biclonal IKZF1 mutations (chromosome 7 rearrangement, Δ4-7 deletion) at diagnosis and which lost the Δ4-7 deletion at relapse (see above) [31].
The challenge of identifying IKZF1 mutations at diagnosis
The strong association of IKZF1 mutations with a poor prognosis has prompted calls for IKZF1 screening at diagnosis [61]. This raises the question of which method to use. Multiplex Ligation Probe-dependent Amplification (MLPA) is powerful for detecting genomic deletions [62], but it cannot distinguish between monoand biallelic deletions, or deletions that affect leukemic subclones [31]. This conclusion was also reached by Cayé et al who found that MLPA did not detect IKZF1 rearrangements in 13 of 82 leukemias (15%) with IKZF1 deletions [34]. We have developed PCR-based methods to analyze Δ4-7 and larger deletions [31], but these miss unconventional deletions affecting the 5’ IKZF1 region. The direct multiplex PCR amplification developed by Cayé et al to detect various IKZF1 breakpoints is very sensitive and can detect IKZF1 mutations in even minor leukemic subclones [34], but this method will not detect 5’ deletions. Further, no method has been developed that can identify point mutations. Thus the development of a robust and rapid method for the identification of IKZF1 mutations remains a challenge.
Possible functions of IKZF1 mutations in B cell leukemogenesis
Maintenance of a stem cell phenotype
The molecular events affected by the loss of Ikaros remain poorly understood. Gene expression signatures associated with loss of IKZF1 in pediatric and adult B-ALL have revealed an overexpression of genes indicative of a stem cell phenotype [35,63]. This possibility was first proposed by Tonnelle et al who found that Ik6 expressing adult B-ALL show higher levels of CD34 [22]. In animal models, Ng et al have reported that certain stem cell genes are less efficiently silenced in lymphoid progenitors from Ikaros null mice [8]. It remains to be seen if Ikaros is directly involved in the suppression of a stem cell signature. Thus Ikaros deficiency might lead to the persistence or activation of a stem cell gene expression program which may favor the self-renewal or formation of leukemic stem cells.
Blocking B cell differentiation
Another role for Ikaros deficiency may be to block B cell differentiation. In the mouse, Ikaros is required for B cell commitment, as Ikaros null mice lack B cells [3]. In addition, Ikaros is needed during B cell differentiation, as animals expressing low levels of Ikaros show a partial block in the transition between the pro- and pre-B cell stage [64]. Ikaros binds to regulatory elements in the IgH locus and is required for the rearrangement of the heavy chain of the B cell receptor (BCR) and pre-BCR [65-67]. Ikaros is also important for silencing the Igll1 gene, which encodes the Lambda 5 surrogate light chain of the pre-BCR [68-70]. These results suggest that Ikaros plays a role in regulating both the up- and downregulation of the pre- BCR. Other studies have suggested that Ikaros functions downstream of the pre-BCR, by contributing to the cell cycle arrest induced by pre- BCR signaling in pro-B cells [71], through downregulation of c-Myc [72], and induction of kappa light chain expression [67]. Thus, Ikaros appears to be a key regulator of the pre-BCR checkpoint during B cell differentiation.
Regulation of JAK-STAT signaling
As discussed, IKZF1 mutations are prevalent in leukemias with activated JAK-STAT signaling, suggesting that Ikaros may repress the activity of this pathway. How this is achieved remains to be determined. Ikaros has been shown to antagonize the Notch pathway in T cells as well as Ebf1 in B cells [69,73]. Since downregulation of Stat5-mediated gene expression is crucial for B cell differentiation after the pro-B cell stage [74], Ikaros repression at this juncture might explain why Ikaros is important for the pro- to pre-B cell transition. Thus, it will be important to study if and how Ikaros modulates STAT5-mediated target gene regulation.
Conclusion and future challenges
It is now clear that IKZF1 loss of function mutations are important genetic events associated with aggressive B cell leukemias. Several outstanding issues need to be clarified in the future. It will be important to understand the sequence of events leading to Ikaros loss of function. In particular, the frequency of point mutations as well as the possibility of epigenetic, transcriptional or post-transcriptional silencing need to be addressed. It will also be important to determine if B-ALL with severe Ikaros deficiencies (eg. loss of both alleles or expression of dn proteins) exhibit distinct clinical and molecular features compared with those haploinsufficient for IKZF1. Standardized diagnostic tools to genotype IKZF1 mutations need to be defined. Finally, the mechanism of tumor suppression by Ikaros needs to be better understood. The integration of fundamental and clinical studies will be needed for these aims to be achieved.
Acknowledgements
This work was supported by a grant from the Institut National du Cancer (#2011-144), the Ligue Nationale contre le Cancer (équipe labellisée) and institute funding from INSERM, CNRS and the Université de Strasbourg. AD was supported by a fellowship from the Fondation ARC.
References
- 1.Lo K, Landau NR, Smale ST. LyF-1, a transcriptional regulator that interacts with a novel class of promoters for lymphocyte-specific genes. Mole Cell Biol. 1991;11:5229–5243. doi: 10.1128/mcb.11.10.5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Georgopoulos K, Moore DD, Derfler B. Ikaros, an early lymphoid-specific transcription factor and a putative mediator for T cell commitment. Science. 1992;258:808–812. doi: 10.1126/science.1439790. [DOI] [PubMed] [Google Scholar]
- 3.Wang JH, Nichogiannopoulou A, Wu L, Sun L, Sharpe AH, Bigby M, Georgopoulos K. Selective defects in the development of the fetal and adult lymphoid system in mice with an Ikaros null mutation. Immunity. 1996;5:537–549. doi: 10.1016/s1074-7613(00)80269-1. [DOI] [PubMed] [Google Scholar]
- 4.Wu L, Nichogiannopoulou A, Shortman K, Georgopoulos K. Cell-autonomous defects in dendritic cell populations of Ikaros mutant mice point to a developmental relationship with the lymphoid lineage. Immunity. 1997;7:483–492. doi: 10.1016/s1074-7613(00)80370-2. [DOI] [PubMed] [Google Scholar]
- 5.Nichogiannopoulou A, Trevisan M, Neben S, Friedrich C, Georgopoulos K. Defects in hemopoietic stem cell activity in Ikaros mutant mice. J Exp Med. 1999;190:1201–1214. doi: 10.1084/jem.190.9.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lopez RA, Schoetz S, DeAngelis K, O’Neill D, Bank A. Multiple hematopoietic defects and delayed globin switching in Ikaros null mice. Proc Natl Acad Sci U S A. 2002;99:602–607. doi: 10.1073/pnas.022412699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allman D, Dalod M, Asselin-Paturel C, Delale T, Robbins SH, Trinchieri G, Biron CA, Kastner P, Chan S. Ikaros is required for plasmacytoid dendritic cell differentiation. Blood. 2006;108:4025–4034. doi: 10.1182/blood-2006-03-007757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng SY, Yoshida T, Zhang J, Georgopoulos K. Genome-wide lineage-specific transcriptional networks underscore Ikaros-dependent lymphoid priming in hematopoietic stem cells. Immunity. 2009;30:493–507. doi: 10.1016/j.immuni.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molnar A, Georgopoulos K. The Ikaros gene encodes a family of functionally diverse zinc finger DNA-binding proteins. Mol Cell Biol. 1994;14:8292–8303. doi: 10.1128/mcb.14.12.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J, Sif S, Jones B, Jackson A, Koipally J, Heller E, Winandy S, Viel A, Sawyer A, Ikeda T, Kingston R, Georgopoulos K. Ikaros DNAbinding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity. 1999;10:345–355. doi: 10.1016/s1074-7613(00)80034-5. [DOI] [PubMed] [Google Scholar]
- 11.Sridharan R, Smale ST. Predominant interaction of both Ikaros and Helios with the NuRD complex in immature thymocytes. J Biol Chem. 2007;282:30227–30238. doi: 10.1074/jbc.M702541200. [DOI] [PubMed] [Google Scholar]
- 12.Winandy S, Wu P, Georgopoulos K. A dominant mutation in the Ikaros gene leads to rapid development of leukemia and lymphoma. Cell. 1995;83:289–299. doi: 10.1016/0092-8674(95)90170-1. [DOI] [PubMed] [Google Scholar]
- 13.Winandy S, Wu L, Wang JH, Georgopoulos K. Pre-T cell receptor (TCR) and TCR-controlled checkpoints in T cell differentiation are set by Ikaros. J Exp Med. 1999;190:1039–1048. doi: 10.1084/jem.190.8.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papathanasiou P, Perkins AC, Cobb BS, Ferrini R, Sridharan R, Hoyne GF, Nelms KA, Smale ST, Goodnow CC. Widespread failure of hematolymphoid differentiation caused by a recessive niche-filling allele of the Ikaros transcription factor. Immunity. 2003;19:131–144. doi: 10.1016/s1074-7613(03)00168-7. [DOI] [PubMed] [Google Scholar]
- 15.Dumortier A, Jeannet R, Kirstetter P, Kleinmann E, Sellars M, dos Santos NR, Thibault C, Barths J, Ghysdael J, Punt JA, Kastner P, Chan S. Notch activation is an early and critical event during T-Cell leukemogenesis in Ikarosdeficient mice. Mol Cell Biol. 2006;26:209–220. doi: 10.1128/MCB.26.1.209-220.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kastner P, Chan S. Role of Ikaros in T-cell acute lymphoblastic leukemia. World J Biol Chem. 2011;2:108–114. doi: 10.4331/wjbc.v2.i6.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marcais A, Jeannet R, Hernandez L, Soulier J, Sigaux F, Chan S, Kastner P. Genetic inactivation of Ikaros is a rare event in human T-ALL. Leuk Res. 2010;34:426–429. doi: 10.1016/j.leukres.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 18.Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A, O’Neil J, Gutierrez A, Ivanova E, Perna I, Lin E, Mani V, Jiang S, Mc-Namara K, Zaghlul S, Edkins S, Stevens C, Brennan C, Martin ES, Wiedemeyer R, Kabbarah O, Nogueira C, Histen G, Aster J, Mansour M, Duke V, Foroni L, Fielding AK, Goldstone AH, Rowe JM, Wang YA, Look AT, Stratton MR, Chin L, Futreal PA, DePinho RA. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007;447:966–971. doi: 10.1038/nature05886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen SC, Wei L, Collins-Underwood JR, Ma J, Roberts KG, Pounds SB, Ulyanov A, Becksfort J, Gupta P, Huether R, Kriwacki RW, Parker M, McGoldrick DJ, Zhao D, Alford D, Espy S, Bobba KC, Song G, Pei D, Cheng C, Roberts S, Barbato MI, Campana D, Coustan-Smith E, Shurtleff SA, Raimondi SC, Kleppe M, Cools J, Shimano KA, Hermiston ML, Doulatov S, Eppert K, Laurenti E, Notta F, Dick JE, Basso G, Hunger SP, Loh ML, Devidas M, Wood B, Winter S, Dunsmore KP, Fulton RS, Fulton LL, Hong X, Harris CC, Dooling DJ, Ochoa K, Johnson KJ, Obenauer JC, Evans WE, Pui CH, Naeve CW, Ley TJ, Mardis ER, Wilson RK, Downing JR, Mullighan CG. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakase K, Ishimaru F, Avitahl N, Dansako H, Matsuo K, Fujii K, Sezaki N, Nakayama H, Yano T, Fukuda S, Imajoh K, Takeuchi M, Miyata A, Hara M, Yasukawa M, Takahashi I, Taguchi H, Matsue K, Nakao S, Niho Y, Takenaka K, Shinagawa K, Ikeda K, Niiya K, Harada M. Dominant negative isoform of the Ikaros gene in patients with adult B-cell acute lymphoblastic leukemia. Cancer Res. 2000;60:4062–4065. [PubMed] [Google Scholar]
- 21.Nishii K, Katayama N, Miwa H, Shikami M, Usui E, Masuya M, Araki H, Lorenzo F, Ogawa T, Kyo T, Nasu K, Shiku H, Kita K. Non-DNA-binding Ikaros isoform gene expressed in adult Bprecursor acute lymphoblastic leukemia. Leukemia. 2002;16:1285–1292. doi: 10.1038/sj.leu.2402533. [DOI] [PubMed] [Google Scholar]
- 22.Tonnelle C, Imbert MC, Sainty D, Granjeaud S, N’Guyen C, Chabannon C. Overexpression of dominant-negative Ikaros 6 protein is restricted to a subset of B common adult acute lymphoblastic leukemias that express high levels of the CD34 antigen. Hematol J. 2003;4:104–109. doi: 10.1038/sj.thj.6200235. [DOI] [PubMed] [Google Scholar]
- 23.Ruiz A, Jiang J, Kempski H, Brady HJ. Overexpression of the Ikaros 6 isoform is restricted to t(4; 11) acute lymphoblastic leukaemia in children and infants and has a role in B-cell survival. Br J Haematol. 2004;125:31–37. doi: 10.1111/j.1365-2141.2004.04854.x. [DOI] [PubMed] [Google Scholar]
- 24.Iacobucci I, Lonetti A, Messa F, Cilloni D, Arruga F, Ottaviani E, Paolini S, Papayannidis C, Piccaluga PP, Giannoulia P, Soverini S, Amabile M, Poerio A, Saglio G, Pane F, Berton G, Baruzzi A, Vitale A, Chiaretti S, Perini G, Foa R, Baccarani M, Martinelli G. Expression of spliced oncogenic Ikaros isoforms in Philadelphia-positive acute lymphoblastic leukemia patients treated with tyrosine kinase inhibitors: implications for a new mechanism of resistance. Blood. 2008;112:3847–3855. doi: 10.1182/blood-2007-09-112631. [DOI] [PubMed] [Google Scholar]
- 25.Nakayama H, Ishimaru F, Avitahl N, Sezaki N, Fujii N, Nakase K, Ninomiya Y, Harashima A, Minowada J, Tsuchiyama J, Imajoh K, Tsubota T, Fukuda S, Sezaki T, Kojima K, Hara M, Takimoto H, Yorimitsu S, Takahashi I, Miyata A, Taniguchi S, Tokunaga Y, Gondo H, Niho Y, Harada M, et al. Decreases in Ikaros activity correlate with blast crisis in patients with chronic myelogenous leukemia. Cancer Res. 1999;59:3931–3934. [PubMed] [Google Scholar]
- 26.Klein F, Feldhahn N, Herzog S, Sprangers M, Mooster JL, Jumaa H, Muschen M. BCRABL1 induces aberrant splicing of IKAROS and lineage infidelity in pre-B lymphoblastic leukemia cells. Oncogene. 2006;25:1118–1124. doi: 10.1038/sj.onc.1209133. [DOI] [PubMed] [Google Scholar]
- 27.Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, Pui CH, Relling MV, Evans WE, Shurtleff SA, Downing JR. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 28.Kuiper RP, Schoenmakers EF, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, Hoogerbrugge PM. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007;21:1258–1266. doi: 10.1038/sj.leu.2404691. [DOI] [PubMed] [Google Scholar]
- 29.Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, White D, Hughes TP, Le Beau MM, Pui CH, Relling MV, Shurtleff SA, Downing JR. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110–114. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 30.Iacobucci I, Storlazzi CT, Cilloni D, Lonetti A, Ottaviani E, Soverini S, Astolfi A, Chiaretti S, Vitale A, Messa F, Impera L, Baldazzi C, D’Addabbo P, Papayannidis C, Lonoce A, Colarossi S, Vignetti M, Piccaluga PP, Paolini S, Russo D, Pane F, Saglio G, Baccarani M, Foa R, Martinelli G. Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1-positive acute lymphoblastic leukemia patients: on behalf of Gruppo Italiano Malattie Ematologiche dell’Adulto Acute Leukemia Working Party (GIMEMA AL WP) Blood. 2009;114:2159–2167. doi: 10.1182/blood-2008-08-173963. [DOI] [PubMed] [Google Scholar]
- 31.Dupuis A, Gaub MP, Legrain M, Drenou B, Mauvieux L, Lutz P, Herbrecht R, Chan S, Kastner P. Biclonal and biallelic deletions occur in 20% of B-ALL cases with IKZF1 mutations. Leukemia. 2012 doi: 10.1038/leu.2012.204. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 32.Mi JQ, Wang X, Yao Y, Lu HJ, Jiang XX, Zhou JF, Wang JH, Jiao B, Shen SH, Tang JY, Gu LJ, Jiang H, Ma LY, Hao SG, Chen FY, Xiong SM, Shen ZX, Chen Z, Chen B, Chen SJ. Newly diagnosed acute lymphoblastic leukemia in China (II): prognosis related to genetic abnormalities in a series of 1091 cases. Leukemia. 2012;26:1507–1516. doi: 10.1038/leu.2012.23. [DOI] [PubMed] [Google Scholar]
- 33.Heerema NA, Nachman JB, Sather HN, La MK, Hutchinson R, Lange BJ, Bostrom B, Steinherz PG, Gaynon PS, Uckun FM. Deletion of 7p or monosomy 7 in pediatric acute lymphoblastic leukemia is an adverse prognostic factor: a report from the Children’s Cancer Group. Leukemia. 2004;18:939–947. doi: 10.1038/sj.leu.2403327. [DOI] [PubMed] [Google Scholar]
- 34.Caye A, Beldjord K, Mass Malo K, Drunat S, Soulier J, Gandemer V, Baruchel A, Bertrand Y, Cave H, Clappier E. Breakpoint-specific multiplex PCR allows the detection of IKZF1 intragenic deletions and minimal residual disease monitoring in B-cell precursor acute lymphoblastic leukemia. Haematologica. 2012 doi: 10.3324/haematol.2012.073965. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, Ma J, Liu W, Cheng C, Schulman BA, Harvey RC, Chen IM, Clifford RJ, Carroll WL, Reaman G, Bowman WP, Devidas M, Gerhard DS, Yang W, Relling MV, Shurtleff SA, Campana D, Borowitz MJ, Pui CH, Smith M, Hunger SP, Willman CL, Downing JR. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–480. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuiper RP, Waanders E, van der Velden VH, van Reijmersdal SV, Venkatachalam R, Scheijen B, Sonneveld E, van Dongen JJ, Veerman AJ, van Leeuwen FN, van Kessel AG, Hoogerbrugge PM. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia. 2010;24:1258–1264. doi: 10.1038/leu.2010.87. [DOI] [PubMed] [Google Scholar]
- 37.Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, Chen SC, Payne-Turner D, Churchman ML, Harvey RC, Chen X, Kasap C, Yan C, Becksfort J, Finney RP, Teachey DT, Maude SL, Tse K, Moore R, Jones S, Mungall K, Birol I, Edmonson MN, Hu Y, Buetow KE, Chen IM, Carroll WL, Wei L, Ma J, Kleppe M, Levine RL, Garcia-Manero G, Larsen E, Shah NP, Devidas M, Reaman G, Smith M, Paugh SW, Evans WE, Grupp SA, Jeha S, Pui CH, Gerhard DS, Downing JR, Willman CL, Loh M, Hunger SP, Marra MA, Mullighan CG. Genetic alterations activating kinase and cytokine receptor signaling in highrisk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153–166. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang YL, Hung CC, Chen JS, Lin KH, Jou ST, Hsiao CC, Sheen JM, Cheng CN, Wu KH, Lin SR, Yu SL, Chen HY, Lu MY, Wang SC, Chang HH, Lin SW, Su YN, Lin DT. IKZF1 deletions predict a poor prognosis in children with B-cell progenitor acute lymphoblastic leukemia: a multicenter analysis in Taiwan. Cancer Sci. 2011;102:1874–1881. doi: 10.1111/j.1349-7006.2011.02031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Georgopoulos K, Bigby M, Wang JH, Molnar A, Wu P, Winandy S, Sharpe A. The Ikaros gene is required for the development of all lymphoid lineages. Cell. 1994;79:143–156. doi: 10.1016/0092-8674(94)90407-3. [DOI] [PubMed] [Google Scholar]
- 40.Cazzaniga G, van Delft FW, Lo Nigro L, Ford AM, Score J, Iacobucci I, Mirabile E, Taj M, Colman SM, Biondi A, Greaves M. Developmental origins and impact of BCR-ABL1 fusion and IKZF1 deletions in monozygotic twins with Ph+ acute lymphoblastic leukemia. Blood. 2011;118:5559–5564. doi: 10.1182/blood-2011-07-366542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, Downing JR. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–1380. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davidsson J, Paulsson K, Lindgren D, Lilljebjorn H, Chaplin T, Forestier E, Andersen MK, Nordgren A, Rosenquist R, Fioretos T, Young BD, Johansson B. Relapsed childhood high hyperdiploid acute lymphoblastic leukemia: presence of preleukemic ancestral clones and the secondary nature of microdeletions and RTK-RAS mutations. Leukemia. 2010;24:924–931. doi: 10.1038/leu.2010.39. [DOI] [PubMed] [Google Scholar]
- 43.Virely C, Moulin S, Cobaleda C, Lasgi C, Alberdi A, Soulier J, Sigaux F, Chan S, Kastner P, Ghysdael J. Haploinsufficiency of the IKZF1 (IKAROS) tumor suppressor gene cooperates with BCR-ABL in a transgenic model of acute lymphoblastic leukemia. Leukemia. 2010;24:1200–1204. doi: 10.1038/leu.2010.63. [DOI] [PubMed] [Google Scholar]
- 44.Clappier E, Gerby B, Sigaux F, Delord M, Touzri F, Hernandez L, Ballerini P, Baruchel A, Pflumio F, Soulier J. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J Exp Med. 2011;208:653–661. doi: 10.1084/jem.20110105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, Kinsey SE, Lightfoot T, Roman E, Irving JA, Allan JM, Tomlinson IP, Taylor M, Greaves M, Houlston RS. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009;41:1006–1010. doi: 10.1038/ng.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pastorczak A, Gorniak P, Sherborne A, Hosking F, Trelinska J, Lejman M, Szczepanski T, Borowiec M, Fendler W, Kowalczyk J, Houlston RS, Mlynarski W. Role of 657del5 NBN mutation and 7p12.2 (IKZF1), 9p21 (CDKN2A), 10q21.2 (ARID5B) and 14q11.2 (CEBPE) variation and risk of childhood ALL in the Polish population. Leuk Res. 2011;35:1534–1536. doi: 10.1016/j.leukres.2011.07.034. [DOI] [PubMed] [Google Scholar]
- 47.Prasad RB, Hosking FJ, Vijayakrishnan J, Papaemmanuil E, Koehler R, Greaves M, Sheridan E, Gast A, Kinsey SE, Lightfoot T, Roman E, Taylor M, Pritchard-Jones K, Stanulla M, Schrappe M, Bartram CR, Houlston RS, Kumar R, Hemminki K. Verification of the susceptibility loci on 7p12.2, 10q21.2, and 14q11.2 in precursor B-cell acute lymphoblastic leukemia of childhood. Blood. 2010;115:1765–1767. doi: 10.1182/blood-2009-09-241513. [DOI] [PubMed] [Google Scholar]
- 48.Trevino LR, Yang W, French D, Hunger SP, Carroll WL, Devidas M, Willman C, Neale G, Downing J, Raimondi SC, Pui CH, Evans WE, Relling MV. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009;41:1001–1005. doi: 10.1038/ng.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ross JA, Linabery AM, Blommer CN, Langer EK, Spector LG, Hilden JM, Heerema NA, Radloff GA, Tower RL, Davies SM. Genetic variants modify susceptibility to leukemia in infants: A Children’s Oncology Group report. Pediatric Blood Cancer. 2013;60:31–4. doi: 10.1002/pbc.24131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nacheva EP, Brazma D, Virgili A, Howard-Reeves J, Chanalaris A, Gancheva K, Apostolova M, Valganon M, Mazzullo H, Grace C. Deletions of immunoglobulin heavy chain and T cell receptor gene regions are uniquely associated with lymphoid blast transformation of chronic myeloid leukemia. BMC Genomics. 2010;11:41. doi: 10.1186/1471-2164-11-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grossmann V, Kohlmann A, Zenger M, Schindela S, Eder C, Weissmann S, Schnittger S, Kern W, Muller MC, Hochhaus A, Haferlach T, Haferlach C. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9% of cases. Leukemia. 2011;25:557–560. doi: 10.1038/leu.2010.298. [DOI] [PubMed] [Google Scholar]
- 52.Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, Van Zutven LJ, Beverloo HB, Van der Spek PJ, Escherich G, Horstmann MA, Janka-Schaub GE, Kamps WA, Evans WE, Pieters R. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genomewide classification study. Lancet Oncol. 2009;10:125–134. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoelbl A, Kovacic B, Kerenyi MA, Simma O, Warsch W, Cui Y, Beug H, Hennighausen L, Moriggl R, Sexl V. Clarifying the role of Stat5 in lymphoid development and Abelsoninduced transformation. Blood. 2006;107:4898–4906. doi: 10.1182/blood-2005-09-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harvey RC, Mullighan CG, Chen IM, Wharton W, Mikhail FM, Carroll AJ, Kang H, Liu W, Dobbin KK, Smith MA, Carroll WL, Devidas M, Bowman WP, Camitta BM, Reaman GH, Hunger SP, Downing JR, Willman CL. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric Bprogenitor acute lymphoblastic leukemia. Blood. 2010;115:5312–5321. doi: 10.1182/blood-2009-09-245944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen IM, Harvey RC, Mullighan CG, Gastier-Foster J, Wharton W, Kang H, Borowitz MJ, Camitta BM, Carroll AJ, Devidas M, Pullen DJ, Payne-Turner D, Tasian SK, Reshmi S, Cottrell CE, Reaman GH, Bowman WP, Carroll WL, Loh ML, Winick NJ, Hunger SP, Willman CL. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children’s Oncology Group study. Blood. 2012;119:3512–3522. doi: 10.1182/blood-2011-11-394221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martinelli G, Iacobucci I, Storlazzi CT, Vignetti M, Paoloni F, Cilloni D, Soverini S, Vitale A, Chiaretti S, Cimino G, Papayannidis C, Paolini S, Elia L, Fazi P, Meloni G, Amadori S, Saglio G, Pane F, Baccarani M, Foa R. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report. J. Clin. Oncol. 2009;27:5202–5207. doi: 10.1200/JCO.2008.21.6408. [DOI] [PubMed] [Google Scholar]
- 57.Krentz S, Hof J, Mendioroz A, Vaggopoulou R, Dorge P, Lottaz C, Engelmann JC, Groeneveld TW, Korner G, Seeger K, Hagemeier C, Henze G, Eckert C, von Stackelberg A, Kirschner-Schwabe R. Prognostic value of genetic alterations in children with first bone marrow relapse of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia. 2012 doi: 10.1038/leu.2012.155. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 58.Dorge P, Meissner B, Zimmermann M, Moericke A, Schrauder A, Bourquin JP, Schewe D, Harbott J, Teigler-Schlegel A, Ratei R, Ludwig WD, Kohler R, Bartram CR, Schrappe M, Stanulla M, Cario G. IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica. 2012 doi: 10.3324/haematol.2011.056135. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Waanders E, van der Velden VH, van der Schoot CE, van Leeuwen FN, van Reijmersdal SV, de Haas V, Veerman AJ, van Kessel AG, Hoogerbrugge PM, Kuiper RP, van Dongen JJ. Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia. 2011;25:254–258. doi: 10.1038/leu.2010.275. [DOI] [PubMed] [Google Scholar]
- 60.Venn NC, van der Velden VH, de Bie M, Waanders E, Giles JE, Law T, Kuiper RP, de Haas V, Mullighan CG, Haber M, Marshall GM, Md N, van Dongen JJ, Sutton R. Highly sensitive MRD tests for ALL based on the IKZF1 Delta3-6 microdeletion. Leukemia. 2012;26:1414–1416. doi: 10.1038/leu.2011.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mullighan CG. Genomic profiling of B-progenitor acute lymphoblastic leukemia. Best Pract Res Clin Haematol. 2011;24:489–503. doi: 10.1016/j.beha.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schwab CJ, Jones LR, Morrison H, Ryan SL, Yigittop H, Schouten JP, Harrison CJ. Evaluation of multiplex ligation-dependent probe amplification as a method for the detection of copy number abnormalities in B-cell precursor acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2010;49:1104–1113. doi: 10.1002/gcc.20818. [DOI] [PubMed] [Google Scholar]
- 63.Iacobucci I, Iraci N, Messina M, Lonetti A, Chiaretti S, Valli E, Ferrari A, Papayannidis C, Paoloni F, Vitale A, Storlazzi CT, Ottaviani E, Guadagnuolo V, Durante S, Vignetti M, Soverini S, Pane F, Foa R, Baccarani M, Muschen M, Perini G, Martinelli G. IKAROS deletions dictate a unique gene expression signature in patients with adult B-cell acute lymphoblastic leukemia. PloS One. 2012;7:e40934. doi: 10.1371/journal.pone.0040934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kirstetter P, Thomas M, Dierich A, Kastner P, Chan S. Ikaros is critical for B cell differentiation and function. Eur J Immunol. 2002;32:720–730. doi: 10.1002/1521-4141(200203)32:3<720::AID-IMMU720>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 65.Reynaud D, Demarco IA, Reddy KL, Schjerven H, Bertolino E, Chen Z, Smale ST, Winandy S, Singh H. Regulation of B cell fate commitment and immunoglobulin heavy-chain gene rearrangements by Ikaros. Nature Immunol. 2008;9:927–936. doi: 10.1038/ni.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sellars M, Reina-San-Martin B, Kastner P, Chan S. Ikaros controls isotype selection during immunoglobulin class switch recombination. J Exp Med. 2009;206:1073–1087. doi: 10.1084/jem.20082311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alkhatib A, Werner M, Hug E, Herzog S, Eschbach C, Faraidun H, Kohler F, Wossning T, Jumaa H. FoxO1 induces Ikaros splicing to promote immunoglobulin gene recombination. J Exp Med. 2012;209:395–406. doi: 10.1084/jem.20110216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sabbattini P, Lundgren M, Georgiou A, Chow C, Warnes G, Dillon N. Binding of Ikaros to the lambda5 promoter silences transcription through a mechanism that does not require heterochromatin formation. EMBO J. 2001;20:2812–2822. doi: 10.1093/emboj/20.11.2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thompson EC, Cobb BS, Sabbattini P, Meixlsperger S, Parelho V, Liberg D, Taylor B, Dillon N, Georgopoulos K, Jumaa H, Smale ST, Fisher AG, Merkenschlager M. Ikaros DNA-binding proteins as integral components of B cell developmental-stage-specific regulatory circuits. Immunity. 2007;26:335–344. doi: 10.1016/j.immuni.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 70.Ma S, Pathak S, Trinh L, Lu R. Interferon regulatory factors 4 and 8 induce the expression of Ikaros and Aiolos to down-regulate pre-B-cell receptor and promote cell-cycle withdrawal in pre-B-cell development. Blood. 2008;111:1396–1403. doi: 10.1182/blood-2007-08-110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Trageser D, Iacobucci I, Nahar R, Duy C, von Levetzow G, Klemm L, Park E, Schuh W, Gruber T, Herzog S, Kim YM, Hofmann WK, Li A, Storlazzi CT, Jack HM, Groffen J, Martinelli G, Heisterkamp N, Jumaa H, Muschen M. Pre-B cell receptor-mediated cell cycle arrest in Philadelphia chromosome-positive acute lymphoblastic leukemia requires IKAROS function. J Exp Med. 2009;206:1739–1753. doi: 10.1084/jem.20090004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ma S, Pathak S, Mandal M, Trinh L, Clark MR, Lu R. Ikaros and Aiolos inhibit pre-B-cell proliferation by directly suppressing c-Myc expression. Mol Cell Biol. 2010;30:4149–4158. doi: 10.1128/MCB.00224-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kleinmann E, Geimer Le Lay AS, Sellars M, Kastner P, Chan S. Ikaros represses the transcriptional response to Notch signaling in T-cell development. Mol Cell Biol. 2008;28:7465–7475. doi: 10.1128/MCB.00715-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Malin S, McManus S, Busslinger M. STAT5 in B cell development and leukemia. Curr Opin Immunol. 2010;22:168–176. doi: 10.1016/j.coi.2010.02.004. [DOI] [PubMed] [Google Scholar]