

Abstract
Endosulfatases HSulf-1 and -2 (also referred to as Sulf1 and -2) represent a family of enzymes that modulate heparin binding growth factor signaling. Heparan sulfatase 1 (HSulf-1) and heparan sulfatase 2 (HSulf-2) are two important 6-O endosulfatases which remove or edit 6-O sulfate residues of N-glucosamine present on highly sulfated HS. Alteration of heparan sulfatases have been identified in the context of several cancer types. Many cancer types either exhibit increased or decreased HSulfs expression at the transcript levels. Specifically, HSulf-1 was found to be downregulated in early-stage ovarian tumors, hepatocellular carcinoma, and metastatic breast cancer patients. HSulf-2 was found to be upregulated in ductal carcinoma in situ and invasive ductal carcinoma, whereas limited information is present about HSulf-2 expression in different stages of ovarian cancers. Here, we review the important role of these sulfatases play in ovarian and breast cancers in terms of tumorigenesis such as angiogenesis, chemoresistance, apoptosis, growth factor signaling, hypoxia and metastasis. These recent discoveries have added significant understanding about these sulfate editing enzymes.
Keywords: Ovarian and breast cancer, heparin binding growth factor signaling, tumorigenesis, angiogenesis, chemoresponse and metastasis
Introduction
Sulfatases represent a large class of enzymes (17 sulfatase enzymes have been characterized in humans) and majority of them require acidic pH for their activities and are lysosomal resident enzymes [1]. Heparan sulfatases belong to a category of sulfatases which acts at neutral pH conditions and are predominantly localized at ER, golgi apparatus, cell surface and extracellular compartments [1]. These sulfate editing enzymes act on HSPG components present on glycosaminoglycans (GAGs) and glycolipids [2]. Acidic sulfatases are catabolic sulfatases residing in lysosome whereas neutral pH acting sulfatases are regulatory sulfatases [3]. The substrates of these sulfatases, GAGs are classified further into four classes: keratin sulfate (KS), chondroitin sulfate (CS), dermatan sulfate (DS), and heparan sulfate (HS) [4]. These sulfated glycopolymers are attached to a variety of proteins to form proteoglycans (PGs). Heparan sulfates present on these PGs are synthesized by HS biosynthetic enzymes and upon synthesis are subjected to HS editing enzymes heparan sulfatases.
Identification and structure
Heparan sulfatase 1 (HSulf-1) and heparan sulfatase 2 (HSulf-2) are two important 6-O endo-sulfatases which remove or edit 6-O sulfate residues of N-glucosamine present on highly sulfated HS [5]. These are extra cellular sulfatases and hence can act on cell surface as well as extracelullar substrates carrying sulfated HSPGs. HSPGs in turn promote cell signaling by providing binding sites for growth factors and chemokines, which form ternary complex between their cognate receptors and HSPGs acting as co-receptors. N-Glucosamine 6-O sulfate moiety remodeling by heparan sulfatases is achieved by their enzymatic activity which has been shown to regulate the activity of various heparan binding growth factors such as bFGF2, Wnt, VEGF, HGF, amphiregulin and SDF-1 [6]. Among the different sulfate moieties throughout the HS chains, glucosamine 6-O-sulfation has been demonstrated to modify interaction between HSPG and several ligands such as Wnt, BMP, bFGF2, VEGF, SDF-1 alpha, HGF and GDNF [6-11]. It is noteworthy that this is the only sulfate moiety which is regulated post-synthetically. Some of these growth factors form ternary complexes with their cognate receptors as well as surrounding HSPG resulting in ligand-mediated activation. Heparan sulfatases removes 6-O-sulfate moiety and thus modulate bioavailability of these ligands to their receptors- thus either limit or potentiate signaling (Figure 1). Heparan sulfatases acquire enzymatic activities by modification of a critical cysteine residue in its active site and generation of special amino acid C-a-formylglycine via oxidation of cysteine residue [12,13]. This process is highly conserved among the species. Apart from this post-translational modification, heparan sulfatases are also subjected to glycosylation. This glycosylation predominantly occurs at aspargine (N) residues which are spread across the protein. More recently it has been shown that N-glycosylation affects activities and trafficking of quail Sulf1 [14]. For example, non-glycosylated endogenous form of HSulf-2 was detected in renal carcinoma cell lines which was defective in its extracellular secretion [15]. At the molecular level, there is a considerable homology between HSulf-1 and HSulf-2 (64%), and this homology is high in the N-terminal sulfatase domain of HSulfs that harbors C-formylglycine residue, critical for enzymatic activation [5]. HSulfs also exhibit several furin cleavage sites [5,16] .C-terminal domains of HSulfs are less conserved and might play important role in substrate recognition and could account for differential substrate specificities among HSulf-1 and HSulf-2 [5].
Figure 1.

Proposed model of HSulfs in heparin binding growth factor signaling (HB-GF). In the presence of HSulfs the ternary complex formed between the HB-GF, its cognate receptor, and its co-receptor, HSPG is disrupted due to the removal of 6-O sulfation moieties on HSPGs critical for binding HB-GF. This results in attenuated signaling downstream resulting in reduced proliferation and growth. In the absence of HSulfs, the ternary complex formed between the HB-GF, its cognate receptors and its co-receptors HSPG is intact resulting in enhanced downstream signaling.
Function
To understand the physiological roles played by heparan sulfatases during development, HSulf-1 and HSulf-2 knockout mice were generated [17-19]. While no gross changes in either Sulf-1 or Sulf-2 mice were observed, double knockout mice show prenatal lethality [17]. These mice exhibit severe abnormalities in skeletal systems, CNS, kidneys including reduced body size. From these observations, it is clear that both enzymes have overlapping functions. Biochemically, loss of HSulf-1 and HSulf-2, modulated UA(2S)-GlcNS(6S) disaccharide units within the S-domains of HS chains [20]. In addition to 6-O sulfation, changes in 2-O and N-sulfation were also observed which has been ascribed to changes in the levels of sulfotransferases [21].
More recently, HSulf-1 loss has also shown to enhance spontaneous cartilage degeneration and surgically induced osteoarthritis [8]. Sulf-/- mice exhibited increased MMP13 levels which were further induced upon FGF2 treatment. However, in contrast to enhanced FGF2 mediated MMP-13 induction, BMP-7 mediated induction of target genes such as col2a1, aggrecan, and noggin was diminished in Sulf-/- mice.
Role in tumorigenesis
Alteration of heparan sulfatases have been identified in the context of several cancer types. Many cancer types either exhibit increased or decreased HSulfs expression at the transcript levels [22-27]. Specifically, HSulf-1 was found to be downregulated in early-stage ovarian tumors, hepatocellular carcinoma, and metastatic breast cancer patients [28, {Narita, 2007 #26, 29]. HSulf-2 was found to be upregulated in ductal carcinoma in situ and invasive ductal carcinoma [30], whereas limited information is present about HSulf-2 expression in different stages of ovarian cancers. Loss of HSulf-1 is specific for specific histologic subtypes of breast cancer, such as lobular carcinoma [23]. Similarly, increased HSulf-1 expression was associated with increased overall survival [29]. The data obtained from various gene chip arrays, TMA’s and identification of genetic changes associated with cancer types indicates alterations in HSulf-1 or HSulf-2 transcript levels. Subsequently, in vitro examination of role of HSulfs revealed that HSulf-1 and HSulf-2 expression is lacking in majority of the ovarian, breast and renal cancer cells [15,22,29]. Interestingly, highly metastatic ovarian cancer cells such as SKOV3, TOVG21G are devoid of HSulf-1 [22]. Similarly, metastatic breast cancer cells such as MDA231 and MDA468 do not express either HSulf-1 or HSulf-2 [29,31]. Over-expression of HSulf-1 or HSulf-2 in these cells suggest a tumor suppressor activity of HSulf1 in these cells [9,31]. Indeed HSulf-2 was reported as p53 target gene [32]. Significantly, HSulf-1 overexpressing tumor cells exhibit limited or reduced angiogenesis via attenuated VEGF signaling pathway, indicating a key signaling pathway affected by HSulf-1 [9]. Functionally, HSulf-1 overexpression diminishes KDR/VEGF, bFGF2 signaling at the cell surface and promotes sensitivity to various therapeutic agents such as doxorubicin and histone deacetylase inhibitor aphicidin in hepatocellular cancer cells [33]. Therefore, HSulf-1 activity is consistent with the function of a putative tumor suppressor.
HSulfs have also been reported to promote Wnt signaling, which is activated in a variety of tumor types and plays a significant role in tumor growth. Similarly, pro-angiogenic roles of sulfatases have also been reported [24]. At cell surface, sulfatase-2 have shown to upregulate glypican 3 which in turn enhances Wnt pathway in hepatocellular carcinoma [34]. These reports suggest that HSulfs promote tumorigenesis via upregulating autocrine activation of Wnt signaling. In this regard, HSulfs activities are consistent with the function of a putative oncogene. These reports highlight the contrasting roles of HSulfs in specific cancers. At present, it is not clear how and under what conditions heparan sulfatase activities results in distinctive outcomes. Another feature which adds to the complexities, is the distribution of Sulfs in the stromal and epithelial component of the tumors. How HSulfs expression in stroma would influence metastatic breast cancer cells that are devoid of the HSulf? This can be explained by “reservoir hypothesis”. According to this hypothesis, increased affinity of growth factor due to the absence of HSulf-1 would render aggressive growth of the tumors. Another aspect of this theory is stroma usually act as reservoir of the growth factor ligands bound to HSPGs, and that the presence of HSulf-1 in the stroma would result in the release of those growth factors or the increased bioavailability of these ligands and thus enhancing or diminishing adjacent tumor growth depending upon type of growth factor being mobilized. Similarly, it is conceivable that the restoration of HSulf-1 expression inside the tumors by either drugs or chemically active recombinant extracellular protein will have an effect on heparan binding growth factors and consequently on tumor growth. However, tumor type, HSulf isoforms (HSulf-1 and/or HSulf-2) and predominant pathway activation (either Wnt or bFGF2) might result in opposing effect of HSulf’s on tumor growth. Additionally, because of the secretory nature HSulf-1, they could also act on adjacent tumors or proximal tumors cells that do not express HSulf-1. In support of this hypothesis, recent study demonstrates that Wilms tumor (WT1) promotes the expression of Sulf-1 and Sulf-2 in mice podocytes and modulates VEGF-A and bFGF2 signalling in glomerular endothelial cells [35], indicating that Sulfs promote bioavailability of ligands to proximal cells albeit in the context of kidney development. These data indicate that HSulf’s might play important roles in regulating tumor growth by altering the tumor mciroenvironment and heparan binding growth factor signaling depending upon its tissue distribution.
Although both HSulf-1 and HSulf-2 perform catalytically similar activities towards heparan sulfates, whether they have overlapping functions has been addressed in a variety of mouse models as discussed above. Further, differences observed in tissue distribution of HSulf-1 and HSulf-2 might be partly explained by their promoter region. HSulf’s certainly do not share the same promoter and 3’UTR sequences on their mRNA’s. Therefore their expression might be influenced by the bioavailability of transcription factor or mRNA destabilizing or stabilizing factors to account for the differences in expression patterns observed in several cancer types. Apart from the differences in expression patterns of HSulf-1 and HSulf-2 in cancer types, it should also be noted that the presence of HSulfs in stroma might have distinctive roles in tumor development. Indeed one of the studies reported the observation of higher levels of HSulf-2 expression in stroma of metastatic breast cancer than in normal stroma [36]. Molecular mechanisms by which increased levels of HSulf-2 expression in stroma contribute towards breast tumorigenesis remain to be addressed. Paradoxically, HSulfs have been shown to induce Wnt signaling [7]. Various cancer types exhibit increased Wnt signaling, which suggest that HSulf1 expression during carcinogenesis will promote Wnt signaling and hence cancer growth. However, it should be noted that the predominant mechanisms of Wnt pathway activation in various tumor types are the result of mutations in genes involved in Wnt pathway and not due to autocrine Wnt activation. Given that breast cancer lacks any mutation in Wnt pathway but exhibit characteristics of Wnt activation, in these cancers perhaps presence of HSulfs can account for autocrine Wnt activation. Recently, our lab investigated the relationship between HSulf-2 and ductal carcinoma in situ of the breast by utilizing a unique cell line MCF10DCIS.com which expresses HSulf-2 and has the ability to form ductal lesion similar to those found in DCIS pathology of human breast. HSulf-2 knockdown decreased tumor size, promoted apoptosis and retained comedo lesions for longer period of time [37]. Notably, apoptosis was predominantly limited to inner center or luminal area of comedo structures in HSulf-2 depleted xenografts [37]. HSulf-2 knockdown selectively promotes inner luminal cells of the lesions to apoptosis. More importantly, HSulf-2 silencing resulted in the upregulation of the numbers and size of comedo structures with intact basement membrane. A striking feature of HSulf-2 depleted xenografts is the maintenance of integrity of basement membrane even at later stages (week 7) of DCIS to IDC progression. Mechanistically, how might HSulf-2 presence would alter or promote basement membrane disintegration? This is important question that requires further investigation. Interestingly, HSulf-2 depleted xenografts also show reduced expression and activities of MMP9 but not MMP2 and MMP14. Whether HSulf-2 directly regulates MMP9 expression in cell culture model remains to be determined and so is the pathway mediating MMP9 expression that is altered by HSulf-2. Nonetheless, our observation points to the significance of HSulf-2 expression in the basement membrane disintegration in DCIS.com model. While HSulf-2 might have an impact on the invasion and the basement membrane disintegration, it might also play role in luminal filling of comedo structures. It is noteworthy that luminal filling of ductal lesions are generally thought to be driven by activation of oncogene such as Erb2 and EGFR, which propel cell proliferation and concurrently inhibit cell death by resisting anoikis [38]. Similarly, the process of anoikis (matrix de-attachment mediated cell death) results in lumen formation in pre-malignant cells such as MCF10A when cultured in matrigel. Over-expression of BCl-2 have been demonstrated to induce luminal filling in MCF10A cells [39]. Indeed, matrix de-attachment also resulted in the decrease in HSulf-2 expression in multiple breast cancer cell lines. Further HSulf-2 depletion sensitized the cells to matrix deattachment cell death. From these findings, we can envisage that HSulf-2 plays an important role by providing survival signal to the cells in the center of the role of matrix de-attachment. Paradoxically, in vivo analysis of control MCF10DCIS xenograft indicates that HSulf-2 is expressed at center of the ductal lesions in addition to other patterns of its expression as described before. One possibility for this observation is that cells are expressing high levels of HSulf-2 in inner center of the ductal lesions due to nutrient stress and other yet unknown factors participating in in vivo environment. The presence of high levels of HSulf-2 supports the survival of the central cells of the ductal lesions by resisting anoikis mediated cell death. This is further vindicated by our observation that HSulf-2 knockdown increases the number of necrotic comedo ductal lesions with intact basement membrane in the in vivo xenografts.
Endothelial functions of HSulf’s
While much of the focus of the above mentioned studies are on HSulfs expression in cancer types, it should be noted that HSulfs alter heparan binding growth factors, such as VEGF and bFGF2, that promote endothelial functions. HSulf-1 has been shown to attenuate VEGF signaling and hence inhibits the development of tumor vasculature [9]. How normal vasculature and cancer vasculature differ in terms of HSulfs expression is an interesting aspect that can lead to novel differential targeting strategies. Transcriptome-wide analysis of laser captured blood vessels from human skin and chronic wound-edge tissue revealed significant upregulation of HSulf-1 at the wound site along with other candidate genes such as periostin, a highly angiogeneic protein [40]. However, whether HSulf-1 is angiostatic or angiogenic in this particular experimental setting is unknown. However, it is tempting to speculate that wound healing process is tightly regulated both by growth factors as well as cytokines. The inflammation at the wound healing site is implicated in the elaboration of growth factors and cytokines at the wounding site. Thus, it may be possible that the resulting changes are an outcome of net balance of angiostatic and angiogenic switch modulated by inflammation at the wound sites. This is further supported by the fact that various genes identified at the wounding site in fact have shown to be upregulated by inflammation. Therefore, it is likely that HSulf-1 may be upregulated by inflammatory cytokines which then act to restrain growth factor signaling. Another challenging question that remains to be addressed relates to the identification of substrates for HSulfs. Whether there exist overlapping as well as distinct substrates for HSulfs is unknown. However, substrate binding domains are not conserved in HSulf-1 and HSulf-2 whereas heparan binding domain are highly conserved. These unkown aspects of HSulf biology remains an important area of future research.
Regulation of HSulfs expression
Earlier reports have suggested that HSulfs are lost in 70% of the ovarian and 60% of breast cancers [22,23]. Specifically in ovarian cancers, it has been shown that HSulf-1 promoter is methylated in several cancer cell lines as well as primary tumors. Treatment of various demethylating agents such as 5-Aza and LBH589 restores HSulf-1 levels in these cell lines [28] However, more recently, transcriptional factors were identified to regulate HSulf-1 expression. Among them is the variant Hepatocyte Nuclear Factor (vHNF), which acts as a repressor and is highly expressed in clear cell ovarian tumors [41]. Interestingly, among the various histologies of ovarian cancer, clear cell tumors have maximal loss of HSulf-1 and conversely they express vHNF-1 alpha [41]. Subsequently, it has been shown that TGF-b upregulates HSulf-1 expression and alters the heparan sulfate sulfation [42]. HSulf-1 in turn negatively regulates Smad 2/3 phosphorylation and TGF beta target genes such as fibronectin and smooth muscle actin expression [42]. Mechanisms by which TGF beta regulate HSulf-1 transcription or increase the mRNA stability have not been investigated.
Interestingly, it has been shown that hypoxia (a prevalent stress condition of tumor microenvironment resulting from the decreased blood supply and the increased oxygen demand imposed by rapid cell proliferation) downregulates HSulf-1 in breast cancer cells in a hypoxia inducible factor-1 dependent manner [29]. These findings suggest that low oxygen tension can alter heparan sulfate 6-O sulfation by reducing HSulf-1 levels. Similarly, this hypothesis was further tested in clear cell renal carcinoma (ccRCC). ccRCC often exhibit loss of a tumor suppressor von Hippel- Lindau (VHL) [15]. VHL loss results in the development of highly vascular tumors due to the stabilization of the master transcription factor HIF-1 alpha [43]. HIF-1 alpha triggers myriad of cellular responses resulting in changes in metabolism, EMT and cellular resistance to radio- and chemo-therapy [44-46]. Similar to what was observed in breast cancer studies, it was found that both HSulf-2 and HSulf-1 were downregulated under hypoxia in a HIF-1 alpha dependent manner [15,29]. Thus, these recent data point to tumor suppressor role of HSulf-1 in solid cancer. Given hypoxia is widely observed in various cancer types irrespective of VHL status, it can be envisaged that hypoxia might serve as an inhibitory signal for heparan sulfatases in variety of solid cancers. Mechanistically, it has been documented that loss of sulfatases under VHL deficient conditions or due to hypoxia, culminated in the enhanced activation of bFGF2 signaling and the upregulation of vimentin (EMT marker protein) in renal cancer cells [15]. This observation is also supported by finding that under hypoxia, heparan sulfate binding growth factor signaling pathways, such as CXCR4/SDF, FGFR2/bFGF2 and c-Met/HGF cell signaling, are enchanced either due to upregulation of their respective receptors at the cell surface, or defective turnover in case of FGFR2 under hypoxic conditions [47-50]. Furthermore hypoxia triggers invasive and migratory phenotype of the cancer cells [51]. Interestingly, ectopic expression of HSulf-1 under these conditions interfere hypoxia mediated cell migration in response to bFGF2 [29]. This pathway has been implicated in many cancer types including breast and ovarian [52,53]. The proposed model of hypoxia-mediated Sulfs suppression and its effect on growth factor signaling is provided in Figure 2. Further characterization of the effect of HSulf-1 and/or HSulf-2 overexpression on low oxygen-induced changes and its target genes will provide additional insights on the HSulf’s functions in the context of changes occurring in tumor microenvironment. Consistent with its potential role in hypoxia and tumor microenvironment, previous reports suggest that HSulf-1 overexpression in highly aggressive breast cancer cell line MDA468 attenuates angiogenesis [9].
Figure 2.
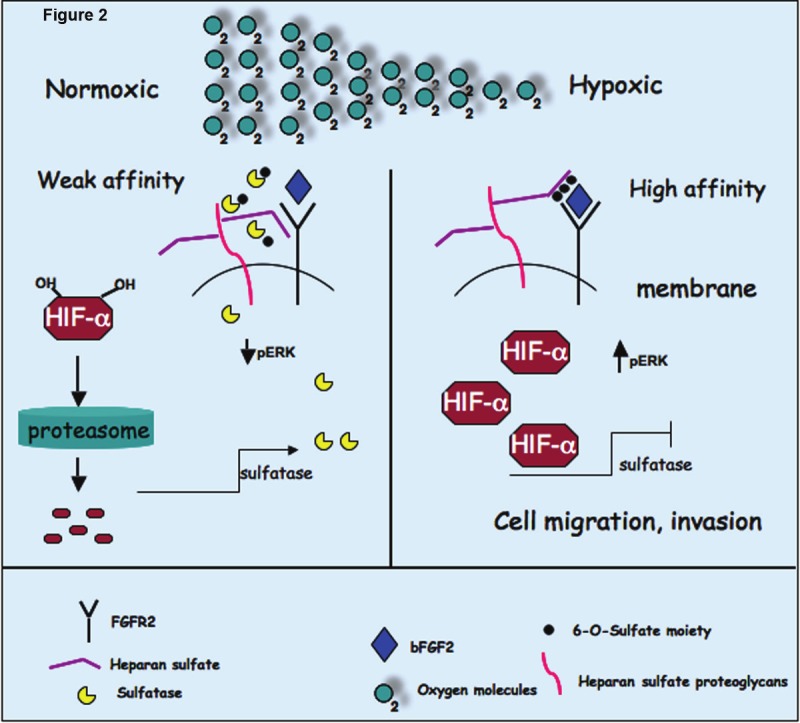
Proposed model of regulation of HSulfs under hypoxic conditions. HSulfs catalytically removes sulfate moiety from 6-O sulfated heparan sulfate on HSPGs. Desulfation of HS results in decreased FGF2 binding sites on co-receptors (HSPGs) and hence decreased signaling. However under low oxygen conditions (a prevalent condition in solid tumors) or when VHL is inactive, HIF-1α is stabilized and shuts down the transcription of HSulfs and decreased its levels resulting in increased sulfation of 6-O-sulfated HS on HSPGs. This increased 6-O sulfation state favors FGF2 signaling, cell migration and invasion.
Unlike HSulf-1, the tumor suppressor function of HSulf-2 in breast cancer is still not clearly defined. Previous report suggests that its expression was found to be increased in breast cancer [24]. However, another study indicated that over expression of HSulf-2 in MDA231 cancer cells markedly attenuated lung metastatic using tail vein model [31]. Both studies have their limitations as well as advantages. Tail vein methodology is well established measure of metastatic potential of candidate genes. However, it does not reflect the full spectrum of dissemination of cancer cells. Moreover, mechanistically how HSulf-2 modulate metastasis is not clear. Similarly, microarray analysis of candidate genes does not faithfully reflect corresponding changes in HSulf-2 at protein levels. Therefore, further studies are needed to define the exact role of HSulf-2 in breast cancer tumorigenesis.
Mechanistically, HSulfs have been shown to attenuate bFGF2 signaling. bFGF2-FGFR2 pathway is critical in cell proliferation and hence tumor growth. Presence of HSulf-1 in tumors might inhibit FGFR2 signalling and hence inhibit tumor growth. Alternatively, HSulfs are activators of Wnt signaling, therefore their overexpression in tumors might explain autocrine activation of Wnt signaling. Another important aspect of recent findings is the demonstration that HSulf2 knockdown upregulates vimentin expression. Vimentin is a mesenchymal marker and is induced in epithelial to mesenchymal transition (EMT) that is also upregulated under hypoxic conditions [54]. Therefore, hypoxiamediated downregulation of HSulf2 may represent a mechanism by which vimentin is upregulated by hypoxia, and HSulfs may play an important role in EMT process. However, caution should be exercised while interpreting these data generated through in vitro use of long passaged cell lines. Therefore, further studies utilizing more appropriate mouse models and human derived cancer tissues are warranted to strengthen above findings and to validate whether similar phenomenon operate in cancer progression.
HSulf-1 promoter and genomic structure
The HSulf-1 gene spans 211-kb genomic fragment on chromosome 8q13.3. We previously reported that our analysis of the cDNA and gene agreed with that reported by Morimoto-Tomita et al. [5] with one notable exception. Morimoto-Tomita et al. reported the presence of a 280-bp noncoding exon in the 5’-UTR that they called exon 1 [5], and we denote here as exon 1A. In addition to this sequence, we have identified two other noncoding exons, one 5’ to this exon (314 bases, which we denote as exon 1) and another 3’ to this sequence (165 bases, which we noted as exon 2). To determine which of these exons, 1, 1A, or 2, are present in transcribed message, primers in exons 1 and 5 were utilized to amplify a 950-bp 5’-UTR from normal ovarian epithelial cells, HMEC, and normal kidney. All of the cDNAs amplified lack exon 1A. Instead, two different splice variants were amplified, a less abundant one lacking exon 1A but containing exons 1 and 2 (corresponding to RefSeq Accession: NM_015170), and a more abundant one lacking both exon 1A and exon 2 (corresponding to RefSeq Accession: NM_001128204) (data not shown). Exon 1A contains the CpG island and acts as a promoter which we will designate as promoter P2. Methylation of this region in ovarian cancer cell lines and primary tumors is associated with the loss of HSulf-1 transcript [55]. An alternate promoter P1, upstream of exon 1 [22] contains specific transcription factor binding sites that play a critical role in regulating HSulf-1 transcript (Figure 3B). UCSC Genome Browser View of 5’ Region of HSulf-1 is shown in Figure 3A. As indicated, we have shown that hypoxia-responsive elements within the P1 promoter play a critical role in hypoxia-mediated downregulation of HSulf-1 in breast cancer [29]. Our additional studies in ovarian cancer have shown the presence of vHNF binding elements negatively regulating the expression of HSulf-1 in clear cell cancers of the ovary [41]. Our unpublished data also indicate another transcription factor, CEBP beta that is negatively modulated by flavopiridol, represses HSulf-1 transcription. Very little is known about the differential usage of these promoters in cancer. In general, when a common downstream exon contains the translation initiation site resulting in the same open reading frame, no variation in the resulting proteins is generated, as is the case with HSulf-1 protein. It is well established that alternate promoter usage may be tissue specific or developmentally regulated. Alternate promoter usage may also play a role in stress response (chemotherapeutic and /or metabolic stress) and may result in differences in their translational efficiencies due to secondary structure of the mRNAs affecting their stability and translation. In the quail model, Sahota and Dhoot reported the presence of a shorter alternatively sliced isoform of Sulf1B that inhibits wnt signaling as opposed to the previously described Sulf1A isoform that promotes wnt signaling. No such isoform has been described in humans [56].
Figure 3.

A: UCSC Genome Browser View of 5’ Region of HSulf-1. Coordinates are based on human reference genome GRCh37/hg19 Assembly. RefSeq annotations of exons are presented in the top track. Encode data, consisting of RNA sequencing, enhancer/promoter-associated histone mark (H3K4Me1), Promoter-associated histone mark (H3K4Me3), DNaseI hypersensitive clusters, and select transcription factor ChIP sequencing tracks are also shown. Collectively, these data indicate two alternative promoters for HSulf-1. B: Schematic representation of Sulf-1 gene. P1- is promoter1 5’ of Exon 1 and P2 is promoter 2 in Exon 1A that contains the CpG island. Both P1 and P2 are underlined.
Therapeutic targeting
HSulfs have also been reported to promote chemosensitivity towards paclitaxel and cisplatin [55]. Indeed HSulf-1 was identified to be lost in recurrent tumors which were resistant to these drugs. In vitro and in vivo analysis suggests that tumor cells expressing HSulf-1 rapidly undergo cell death upon treatment with paclitaxel and cisplatin. Consensus for HSulf-2 as a target gene has been developed in breast cancer, where it has been found to be overexpressed. In this context, compound OKN007 has been shown to inhibit HSulf-2 activities and a potent inhibitor of tumor growth in MCF-7 derived mouse xenografts [57]. Further validation of this agent as a single and/or adjauvant therapy may reveal novel role of this enzyme in breast tumorigenesis. Recently, in an attempt to identify inhibitors of HSulf-2, by serendipity we found that proteasomal inhibitors such as MG132, lactacystine and FDA approved drug Velcade (Bortezomib) abolished HSulf-2 expression in multiple cancer cells lines including cell lines of breast cancer. Further, bortezomib treatment effectively attenuated tumor size and decreased HSulf-2 expression with concurrent accumulation of caspase 3, cleaved PARP and Bim. Brtezomib mediated repression of HSulf-2 could not be reversed upon addition of pan-caspase inhibitor or reactive oxygen scavenger (NAC), suggesting that suppression was not the consequence of cell death program. How bortezomib treatment leads to HSulf-2 downregulation remains to be determined. Also, these effects of bortezomib can result in alteration of many proteins or transcription factors that are actively degraded or have high protein turnover rate.
Conclusion
Although contrasting roles of both HSulf’s have been documented in the literature, it has been largely accepted that HSulf’s represent a unique extracellular sulfate editing enzymes which are differentially expressed depending upon cancer type. It has been well established that HSulfs promote Wnt signaling and attenuate heparan binding growth factor signaling such as bFGF2. More recently HSulfs has been shown to be negatively regulated by master transcription factor hypoxia inducible factor-1 alpha and positively by von Hippel-Lindau tumor suppressor gene. This further lends support to our notion that these enzymes might play unique role under altered tumor microenvironment and hence tumor progression. HSulfs have been linked to important aspects of tumorigenesis such as angiogenesis, chemoresistance, apoptosis, growth factor signaling, hypoxia and metastasis. These recent discoveries have added significant understanding about these sulfate editing enzymes. Strategies either to restore HSulfs in aggressive tumors deficient of HSulfs or development of small molecular inhibitors of HSulf for tumors that overexpress HSulfs which further potentiate Wnt signalling might pave way for novel ways of treatment of cancer.
Acknowledgement
This work was supported, in whole or in part, by CA106954-04 (VS) and P50 CA13639 National Institutes of Health Grants. XH is the recipient of the T32 training grant, 5T32 CA148073.
References
- 1.Diez-Roux G, Ballabio A. Sulfatases and human disease. Annu Rev Genomics Hum Genet. 2005;6:355–379. doi: 10.1146/annurev.genom.6.080604.162334. [DOI] [PubMed] [Google Scholar]
- 2.Buono M, Cosma MP. Sulfatase activities towards the regulation of cell metabolism and signaling in mammals. Cell Mol Life Sci. 2010;67:769–780. doi: 10.1007/s00018-009-0203-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanson SR, Best MD, Wong CH. Sulfatases: structure, mechanism, biological activity, inhibition, and synthetic utility. Angew Chem Int Ed Engl. 2004;43:5736–5763. doi: 10.1002/anie.200300632. [DOI] [PubMed] [Google Scholar]
- 4.Bulow HE, Hobert O. The molecular diversity of glycosaminoglycans shapes animal development. Annu Rev Cell Dev Biol. 2006;22:375–407. doi: 10.1146/annurev.cellbio.22.010605.093433. [DOI] [PubMed] [Google Scholar]
- 5.Morimoto-Tomita M, Uchimura K, Werb Z, Hemmerich S, Rosen SD. Cloning and characterization of two extracellular heparin-degrading endosulfatases in mice and humans. J Biol Chem. 2002;277:49175–49185. doi: 10.1074/jbc.M205131200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uchimura K, Morimoto-Tomita M, Bistrup A, Li J, Lyon M, Gallagher J, Werb Z, Rosen SD. HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparinbound growth factors and chemokines: effects on VEGF, FGF-1, and SDF-1. BMC Biochem. 2006;7:2. doi: 10.1186/1471-2091-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nawroth R, van Zante A, Cervantes S, McManus M, Hebrok M, Rosen SD. Extracellular sulfatases, elements of the Wnt signaling pathway, positively regulate growth and tumorigenicity of human pancreatic cancer cells. PLoS One. 2007;2:e392. doi: 10.1371/journal.pone.0000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Otsuki S, Hanson SR, Miyaki S, Grogan SP, Kinoshita M, Asahara H, Wong CH, Lotz MK. Extracellular sulfatases support cartilage homeostasis by regulating BMP and FGF signaling pathways. Proc Natl Acad Sci U S A. 2010;107:10202–10207. doi: 10.1073/pnas.0913897107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narita K, Staub J, Chien J, Meyer K, Bauer M, Friedl A, Ramakrishnan S, Shridhar V. HSulf-1 inhibits angiogenesis and tumorigenesis in vivo. Cancer Res. 2006;66:6025–6032. doi: 10.1158/0008-5472.CAN-05-3582. [DOI] [PubMed] [Google Scholar]
- 10.Lai JP, Chien J, Strome SE, Staub J, Montoya DP, Greene EL, Smith DI, Roberts LR, Shridhar V. HSulf-1 modulates HGF-mediated tumor cell invasion and signaling in head and neck squamous carcinoma. Oncogene. 2004;23:1439–1447. doi: 10.1038/sj.onc.1207258. [DOI] [PubMed] [Google Scholar]
- 11.Ai X, Kitazawa T, Do AT, Kusche-Gullberg M, Labosky PA, Emerson CP Jr. SULF1 and SULF2 regulate heparan sulfate-mediated GDNF signaling for esophageal innervation. Development. 2007;134:3327–3338. doi: 10.1242/dev.007674. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt B, Selmer T, Ingendoh A, von Figura K. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell. 1995;82:271–278. doi: 10.1016/0092-8674(95)90314-3. [DOI] [PubMed] [Google Scholar]
- 13.von Bulow R, Schmidt B, Dierks T, von Figura K, Uson I. Crystal structure of an enzymesubstrate complex provides insight into the interaction between human arylsulfatase A and its substrates during catalysis. J Mol Biol. 2001;305:269–277. doi: 10.1006/jmbi.2000.4297. [DOI] [PubMed] [Google Scholar]
- 14.Ambasta RK, Ai X, Emerson CP Jr. Quail Sulf1 function requires asparagine-linked glycosylation. J Biol Chem. 2007;282:34492–34499. doi: 10.1074/jbc.M706744200. [DOI] [PubMed] [Google Scholar]
- 15.Khurana A, Tun HW, Marlow L, Copland JA, Dredge K, Shridhar V. Hypoxia negatively regulates heparan sulfatase 2 expression in renal cancer cell lines. Mol Carcinog. 2012 Jul;51:565–75. doi: 10.1002/mc.20824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagamine S, Keino-Masu K, Shiomi K, Masu M. Proteolytic cleavage of the rat heparan sulfate 6-O-endosulfatase SulfFP2 by furintype proprotein convertases. Biochem Biophys Res Commun. 2010 Jan 1;391:107–12. doi: 10.1016/j.bbrc.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 17.Lum DH, Tan J, Rosen SD, Werb Z. Gene trap disruption of the mouse heparan sulfate 6-O-endosulfatase gene, Sulf2. Mol Cell Biol. 2007;27:678–688. doi: 10.1128/MCB.01279-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holst CR, Bou-Reslan H, Gore BB, Wong K, Grant D, Chalasani S, Carano RA, Frantz GD, Tessier-Lavigne M, Bolon B, French DM, Ashkenazi A. Secreted sulfatases Sulf1 and Sulf2 have overlapping yet essential roles in mouse neonatal survival. PLoS One. 2007;2:e575. doi: 10.1371/journal.pone.0000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ratzka A, Kalus I, Moser M, Dierks T, Mundlos S, Vortkamp A. Redundant function of the heparan sulfate 6-O-endosulfatases Sulf1 and Sulf2 during skeletal development. Dev Dyn. 2008;237:339–353. doi: 10.1002/dvdy.21423. [DOI] [PubMed] [Google Scholar]
- 20.Lamanna WC, Kalus I, Padva M, Baldwin RJ, Merry CL, Dierks T. The heparanome--the enigma of encoding and decoding heparan sulfate sulfation. J Biotechnol. 2007;129:290–307. doi: 10.1016/j.jbiotec.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 21.Lamanna WC, Frese MA, Balleininger M, Dierks T. Sulf loss influences N-, 2-O-, and 6-Osulfation of multiple heparan sulfate proteoglycans and modulates fibroblast growth factor signaling. J Biol Chem. 2008;283:27724–27735. doi: 10.1074/jbc.M802130200. [DOI] [PubMed] [Google Scholar]
- 22.Lai J, Chien J, Staub J, Avula R, Greene EL, Matthews TA, Smith DI, Kaufmann SH, Roberts LR, Shridhar V. Loss of HSulf-1 up-regulates heparin-binding growth factor signaling in cancer. J Biol Chem. 2003;278:23107–23117. doi: 10.1074/jbc.M302203200. [DOI] [PubMed] [Google Scholar]
- 23.Narita K, Chien J, Mullany SA, Staub J, Qian X, Lingle WL, Shridhar V. Loss of HSulf-1 expression enhances autocrine signaling mediated by amphiregulin in breast cancer. J Biol Chem. 2007;282:14413–14420. doi: 10.1074/jbc.M611395200. [DOI] [PubMed] [Google Scholar]
- 24.Morimoto-Tomita M, Uchimura K, Bistrup A, Lum DH, Egeblad M, Boudreau N, Werb Z, Rosen SD. Sulf-2, a proangiogenic heparan sulfate endosulfatase, is upregulated in breast cancer. Neoplasia. 2005;7:1001–1010. doi: 10.1593/neo.05496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bret C, Moreaux J, Schved JF, Hose D, Klein B. SULFs in human neoplasia: implication as progression and prognosis factors. J Transl Med. 2011 May 21;9:72. doi: 10.1186/1479-5876-9-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemjabbar-Alaoui H, van Zante A, Singer MS, Xue Q, Wang YQ, Tsay D, He B, Jablons DM, Rosen SD. Sulf-2, a heparan sulfate endosulfatase, promotes human lung carcinogenesis. Oncogene. 2010 Feb 4;29:635–46. doi: 10.1038/onc.2009.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai Y, Yang Y, MacLeod V, Yue X, Rapraeger AC, Shriver Z, Venkataraman G, Sasisekharan R, Sanderson RD. HSulf-1 and HSulf-2 are potent inhibitors of myeloma tumor growth in vivo. J Biol Chem. 2005;280:40066–40073. doi: 10.1074/jbc.M508136200. [DOI] [PubMed] [Google Scholar]
- 28.Lai JP, Chien JR, Moser DR, Staub JK, Aderca I, Montoya DP, Matthews TA, Nagorney DM, Cunningham JM, Smith DI, Greene EL, Shridhar V, Roberts LR. hSulf1 Sulfatase promotes apoptosis of hepatocellular cancer cells by decreasing heparin-binding growth factor signaling. Gastroenterology. 2004;126:231–248. doi: 10.1053/j.gastro.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 29.Khurana A, Liu P, Mellone P, Lorenzon L, Vincenzi B, Datta K, Yang B, Linhardt RJ, Lingle W, Chien J, Baldi A, Shridhar V. HSulf-1 modulates FGF2- and hypoxia-mediated migration and invasion of breast cancer cells. Cancer Res. 2011 Mar 15;71:2152–61. doi: 10.1158/0008-5472.CAN-10-3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abba MC, Drake JA, Hawkins KA, Hu Y, Sun H, Notcovich C, Gaddis S, Sahin A, Baggerly K, Aldaz CM. Transcriptomic changes in human breast cancer progression as determined by serial analysis of gene expression. Breast Cancer Res. 2004;6:R499–513. doi: 10.1186/bcr899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peterson SM, Iskenderian A, Cook L, Romashko A, Tobin K, Jones M, Norton A, Gomez-Yafal A, Heartlein MW, Concino MF, Liaw L, Martini PG. Human Sulfatase 2 inhibits in vivo tumor growth of MDA-MB-231 human breast cancer xenografts. BMC Cancer. 2010 Aug 13;10:427. doi: 10.1186/1471-2407-10-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chau BN, Diaz RL, Saunders MA, Cheng C, Chang AN, Warrener P, Bradshaw J, Linsley PS, Cleary MA. Identification of SULF2 as a novel transcriptional target of p53 by use of integrated genomic analyses. Cancer Res. 2009;69:1368–1374. doi: 10.1158/0008-5472.CAN-08-2742. [DOI] [PubMed] [Google Scholar]
- 33.Lai JP, Yu C, Moser CD, Aderca I, Han T, Garvey TD, Murphy LM, Garrity-Park MM, Shridhar V, Adjei AA, Roberts LR. SULF1 inhibits tumor growth and potentiates the effects of histone deacetylase inhibitors in hepatocellular carcinoma. Gastroenterology. 2006;130:2130–2144. doi: 10.1053/j.gastro.2006.02.056. [DOI] [PubMed] [Google Scholar]
- 34.Lai JP, Oseini AM, Moser CD, Yu C, Elsawa SF, Hu C, Nakamura I, Han T, Aderca I, Isomoto H, Garrity-Park MM, Shire AM, Li J, Sanderson SO, Adjei AA, Fernandez-Zapico ME, Roberts LR. The oncogenic effect of sulfatase 2 in human hepatocellular carcinoma is mediated in part by glypican 3-dependent Wnt activation. Hepatology. 2010 Nov;52:1680–9. doi: 10.1002/hep.23848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schumacher VA, Schlotzer-Schrehardt U, Karumanchi SA, Shi X, Zaia J, Jeruschke S, Zhang D, Pavenstaedt H, Drenckhan A, Amann K, Ng C, Hartwig S, Ng KH, Ho J, Kreidberg JA, Taglienti M, Royer-Pokora B, Ai X. WT1-Dependent Sulfatase Expression Maintains the Normal Glomerular Filtration Barrier. J Am Soc Nephrol. 2011 Jul;22:1286–96. doi: 10.1681/ASN.2010080860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, Hallett M, Park M. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 37.Khurana A, McKean H, Kim H, Kim SH, mcguire J, Roberts LR, Goetz MP, Shridhar V. Silencing of HSulf-2 expression in MCF10DCIS. com cells attenuate ductal carcinoma in situ progression to invasive ductal carcinoma in vivo. Breast Cancer Res. 2012 Mar 12;14:R43. doi: 10.1186/bcr3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 2002;111:29–40. doi: 10.1016/s0092-8674(02)01001-2. [DOI] [PubMed] [Google Scholar]
- 39.Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5:675–688. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- 40.Roy S, Patel D, Khanna S, Gordillo GM, Biswas S, Friedman A, Sen CK. Transcriptome-wide analysis of blood vessels laser captured from human skin and chronic wound-edge tissue. Proc Natl Acad Sci U S A. 2007;104:14472–14477. doi: 10.1073/pnas.0706793104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu P, Khurana A, Rattan R, He X, Kalloger S, Dowdy S, Gilks B, Shridhar V. Regulation of HSulf-1 expression by variant hepatic nuclear factor 1 in ovarian cancer. Cancer Res. 2009;69:4843–4850. doi: 10.1158/0008-5472.CAN-08-3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yue X, Li X, Nguyen HT, Chin DR, Sullivan DE, Lasky JA. Transforming growth factor-beta1 induces heparan sulfate 6-O-endosulfatase 1 expression in vitro and in vivo. J Biol Chem. 2008;283:20397–20407. doi: 10.1074/jbc.M802850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 44.Zhou J, Schmid T, Schnitzer S, Brune B. Tumor hypoxia and cancer progression. Cancer Lett. 2006;237:10–21. doi: 10.1016/j.canlet.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 45.Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10:295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- 46.Cosse JP, Michiels C. Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anticancer Agents Med Chem. 2008;8:790–797. doi: 10.2174/187152008785914798. [DOI] [PubMed] [Google Scholar]
- 47.Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–311. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- 48.Nakaigawa N, Yao M, Baba M, Kato S, Kishida T, Hattori K, Nagashima Y, Kubota Y. Inactivation of von Hippel-Lindau gene induces constitutive phosphorylation of MET protein in clear cell renal carcinoma. Cancer Res. 2006;66:3699–3705. doi: 10.1158/0008-5472.CAN-05-0617. [DOI] [PubMed] [Google Scholar]
- 49.Koochekpour S, Jeffers M, Wang PH, Gong C, Taylor GA, Roessler LM, Stearman R, Vasselli JR, Stetler-Stevenson WG, Kaelin WG Jr, Linehan WM, Klausner RD, Gnarra JR, Vande Woude GF. The von Hippel-Lindau tumor suppressor gene inhibits hepatocyte growth factor/scatter factor-induced invasion and branching morphogenesis in renal carcinoma cells. Mol Cell Biol. 1999;19:5902–5912. doi: 10.1128/mcb.19.9.5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hsu T, Adereth Y, Kose N, Dammai V. Endocytic function of von Hippel-Lindau tumor suppressor protein regulates surface localization of fibroblast growth factor receptor 1 and cell motility. J Biol Chem. 2006;281:12069–12080. doi: 10.1074/jbc.M511621200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peinado H, Cano A. A hypoxic twist in metastasis. Nat Cell Biol. 2008;10:253–254. doi: 10.1038/ncb0308-253. [DOI] [PubMed] [Google Scholar]
- 52.Meyer KB, Maia AT, O’Reilly M, Teschendorff AE, Chin SF, Caldas C, Ponder BA. Allelespecific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 2008;6:e108. doi: 10.1371/journal.pbio.0060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Katoh M. Cancer genomics and genetics of FGFR2 (Review) Int J Oncol. 2008;33:233–237. [PubMed] [Google Scholar]
- 54.Vuoriluoto K, Haugen H, Kiviluoto S, Mpindi JP, Nevo J, Gjerdrum C, Tiron C, Lorens JB, Ivaska J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene. 2011 Mar 24;30:1436–48. doi: 10.1038/onc.2010.509. [DOI] [PubMed] [Google Scholar]
- 55.Staub J, Chien J, Pan Y, Qian X, Narita K, Aletti G, Scheerer M, Roberts LR, Molina J, Shridhar V. Epigenetic silencing of HSulf-1 in ovarian cancer: implications in chemoresistance. Oncogene. 2007;26:4969–4978. doi: 10.1038/sj.onc.1210300. [DOI] [PubMed] [Google Scholar]
- 56.Sahota AP, Dhoot GK. A novel SULF1 splice variant inhibits Wnt signalling but enhances angiogenesis by opposing SULF1 activity. Exp Cell Res. 2009;315:2752–2764. doi: 10.1016/j.yexcr.2009.06.029. [DOI] [PubMed] [Google Scholar]
- 57.Zheng X, Gai X, Han S, Moser CD, Hu C, Shire AM, Floyd RA, Roberts LR. The human sulfatase 2 inhibitor 2,4-disulfonylphenyl-tert-butylnitrone (OKN-007) has an antitumor effect in hepatocellular carcinoma mediated via suppression of TGFB1/SMAD2 and Hedgehog/GLI1 signaling. Genes Chromosomes Cancer. 2013;52:225–36. doi: 10.1002/gcc.22022. [DOI] [PMC free article] [PubMed] [Google Scholar]