

Abstract
Despite remaining uncertainties and ongoing research it is possible to draw up a model for the role of (cancer) stem cells in both the initiation and progression of cancer towards metastasis. The cancer stem cell of origin and the cancer stem cell are, despite phenotypic similarities, genotypically different entities. Given the right circumstances provided by a combination of genomic changes and biochemical and physical interactions with its microenvironment, an epithelial cancer cell may undergo a phenotypic epithelial mesenchymal transition (EMT) towards a cancer stem cell. This transition conveys upon the cell crucial stem cell-like abilities which facilitate migration into the blood circulation as an individual circulating tumor cell, survive there, and subsequently seed into organ tissue where, once more in close interaction with its microenvironment, the process of clonal self renewal may start, leading to a metastatic tumor. Both in the primary tumor as well as in the metastatic tumor, partial differentiation of the cancer stem cell progeny leads to phenotypic heterogeneity. Throughout this complex process of cancer metastasis similarities with the way stem cells function during embryonic development, including the signaling pathways that mediate these functions, are evident. Deeper insight in the EMT process, plasticity of the resulting cancer stem cells, and the role of cancer stem cells in the metastatic process is expected to lead to novel anti-metastatic cancer therapies. Emerging human in vitro cancer models in the form of “organ-on-a-chip” may contribute valuable novel research tools to achieve this aim.
Keywords: Cancer stem cell Wnt signaling pathway circulating tumor evolution metastasis
Introduction
Although ideas on the existence of cancer stem cells as essential contributors to aggressive cancer growth date back far into the last century, the question as to the identity of these cells has proven to be an extremely difficult question to answer. Even with the advanced experimental approaches that we have available today, controversies arise at scientific meetings on the topic, and a consensus on the elusive cancer stem cell is still lacking, although the concept has by now been widely adopted. Confusion often arises when talking about the cancer stem cell of origin versus the cancer stem cell, the latter presumed to be responsible for invasion and metastasis. In recent years, the promise for more effective new cancer therapies has proven a powerful driver of cancer stem cell research, and several theories, supported by sound and sophisticated experimental evidence, now merge into a more consistent picture [1-3] (Figure 1).
Figure 1.
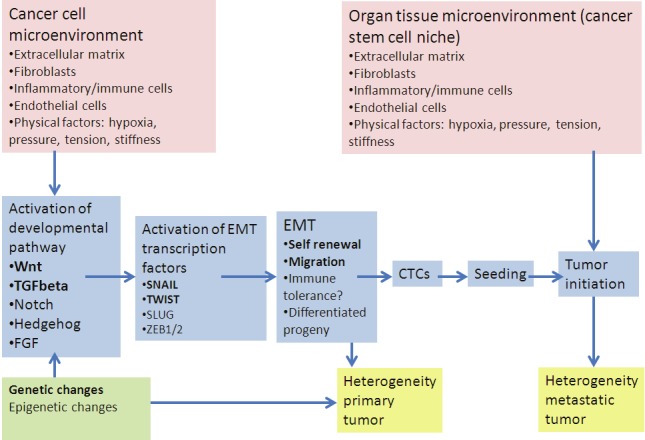
Genetic and epigenetic changes in the cancer cell in interplay with biochemical and physical contextual signals emanating from the microenvironment of the cell may induce aberrant activation of developmental signaling pathways (e.g. TGFbeta, Hedgehog, Wnt), leading to activation of EMT transcription factors like SNAIL/TWIST. Through the EMT transcriptional program mesenchymal stem cell characteristics are acquired, enabling (single) cancer cell migration into the blood stream as part of the circulating tumor cell (CTC) population. After seeding and survival in an appropriate organ niche, growth of a metastatic tumor is initiated, again in interplay with microenvironmental factors, e.g. inflammatory cytokines/growth factors.
Observations and questions on the behavior of cancer
Cancer tissue is morphologically heterogeneous, not only due to the variety of cell types present, endothelial, fibroblast and various immune cells, but cancer cells themselves are not a homogeneous population either. No two tumors look exactly the same. From clinical experience it is evident that a specific type of cancer can behave in very different ways, for example metastasize rapidly in one patient, while not being very aggressive in another patient, or respond very well to a therapy in the one, but not in the other patient. In depth understanding of the cause of this morphological and behavioral tumor heterogeneity, is needed for the development of more effective cancer therapies. In general, patients die from metastasis and not from the primary tumor, and drugs which prevent or block metastatic growth are potentially life-saving. The past decade, interesting insights and theories around stem cells in cancer and their role in cancer metastasis have emerged, explaining some of the intriguing findings on tumor heterogeneity.
A darwinian view: evolution of a tumor leading to genotypic heterogeneity
At least for colon adenoma as an example case, the concept is by now well accepted that an initial genetic defect in a multipotent adult stem cell, which enables the cell to divide more frequently, may give rise to both a larger stem cell population carrying the same mutation, and a faster dividing daughter cell population, or “transit amplifying cells” [4,5]. This population of mutated stem cells among the normal stem cells may function as reservoir of stem cells of origin of cancer. During tumor progression, additional DNA mutations will accumulate in their progeny due to a high rate of cell division, stepwise conferring additional growth advantages to cells by activating relevant signaling pathways. This concept of genetic tumor evolution was introduced first for colon cancer – and is in a sense quite analogous to Darwin’s evolution theory [6-8]. The most rapidly dividing cell clone is supposed to lead in growth of the – sofar still benign- tumor. At least in colon cancer up to several decades may be needed for a tumor to become malignant and metastasize. Arising DNA mutations which interfere with processes like DNA repair and apoptosis result in chromosomal instability, which is invariably associated with accelerated accumulation of genetic defects including aneuploidy. This marks the switch to a genotypically highly heterogeneous tumor and rapidly provides cells with capabilities needed for tissue invasion and metastasis: the hallmark of malignancy. Recently, it was elegantly illustrated using single cancer cell sequencing how accumulation of genetic defects in cancer cells results with time in multiple genetically different clones coexisting in cancer tissue [9]. The evolutionary mechanism underlying tumor heterogeneity and differences in tumor behavior is well accepted and part of cancer heterogeneity has its origin at the genome level, especially in advanced cancer.
Interaction between cancer cells and their environment leading to phenotypic heterogeneity
Aside from genomic abnormalities that may directly influence properties, morphology and behavior of cancer cells, signals emanating from the microenvironment of a cancer cell can also modify morphology and behavior [10,11]. These contextual signals vary depending on the location of a cancer cell in the tumor. At the invasive border between cancer tissue and surrounding normal tissue, cancer cells have maximal interaction with other, non-tumor, cell types, like fibroblasts, and inflammatory and immune cells attracted to the tumor, but also with a variety of extracellular matrix molecules. Specific mutations acquired by a cancer cell may co-determine the functional consequences of interactions with the microenvironment. In this way, contextual factors contribute to cancer heterogeneity and may lead to phenotypic differences between cancer cells with a similar clonal genotype. However, in contrast to genomic heterogeneity, these changes are in principle reversible as they may depend on the continuing presence of specific signals [12].
Stem cells and cancer: the cancer cell of origin versus the cancer stem cell
Where and how do cancer stem cells join the picture? Initial ideas on the potential existence of cancer stem cells emerged in the final decades of the past century, and ran strikingly parallel with upcoming research on embryonic stem cells which yielded a wealth of information on stem cell characteristics and behavior [13]. Crucial insights on the cancer stem cell evolved from research on acute myeloid leukemia (AML) [14]. Leukemic blasts in AML where shown to derive from precursor cells in the bone marrow carrying one specific recognizable DNA mutation in their genome, designated as the leukemia initiating mutation, while during progression of the disease multiple mutations are added. Investigation of the properties of circulating leukemic cells revealed that the majority had lost the capacity to divide, while only a small cell population appeared to be capable of dividing and initiating a novel leukemia in a mouse. These leukemia-initiating cells closely resembled normal multipotent blood stem cells. Normal blood stem cells are rare cells in the bone marrow which can both self renew and generate progenitor cells for the various types of blood cells, supplying these to the blood. Leukemia initiating cells appeared to be highly resistant to chemotherapy and have since been blamed for the nearly unavoidable recurrence of AML after an initial complete remission. Following chemotherapy their stem cell-like nature enables them to self renew and rapidly replenish the leukemic cell population, leading to recurrence of the disease. This process bears similarities to normal regeneration of damaged tissue, for example recruitment of skin stem cells for wound healing [15-17]. The essential insight obtained from this work was that a hierarchy exists among leukemic cells. This hierarchy can be pictured as a pyramid with rare leukemia initiating stem cells at the apex, generating a larger population of rapidly dividing progenitor cells in the middle of the pyramid. Eventually, the base of the pyramid consists of the heterogeneous bulk of more differentiated heterogeneic leukemic blasts, representing immature forms of all blood cell types. The way the leukemic cell population is built up actually resembles normal blood, only completely deregulated and uncontrolled. Having thus defined leukemia initiating cells, or leukemic stem cells, this does not yet address the question as to the origin of these cells. Two explanations are not necessarily mutually exclusive. Either these cells represent a bone marrow-based pool of original blood stem cells carrying (only) the first leukemia-initiating mutation, or they represent leukemic blood cells (unable to divide) that have reverted to a cancer stem cell state, due to specific additional genetic defects and/or interaction with their surrounding environment. In the first case, these abnormal blood stem cells would represent the stem cells of origin of the leukemic disease. In the latter case, they are cancer stem cells which contain multiple gene mutations, have regained the capability to self-renew, and are responsible for leukemic progression and rapid recurrence after treatment. As a third, and maybe not unlikely explanation, the leukemia stem cell population could consist of an as yet indistinguishable mixture of both – only to be resolved by sequencing the genome of of these cells.
Based on the compelling evidence obtained from patients with AML, the search was on for stem cells that could play similar roles in solid cancers. While early research on the topic of cancer stem cells was hampered by lack of appropriate research tools, towards the end of the century fluorescent activated cell sorting (FACS) technologies had sufficiently matured to allow isolation of individual cells from a solid tumor for further study. By that time a unique highly immunodeficient mouse model had been developed, enabling xenotransplantation of human cancer cells. This model was used to determine the minimal number of FACS-sorted cancer cells needed to successfully initiate growth of a new tumor in the mouse. Successful tumor growth and recapture of the heterogeneity of the cancer tissue of origin implicated that stem cell properties of both self renewal and generation of more differentiated progeny had been present. In general only a varying minority of the cancer cells succeeded in initiating a new malignant tumor showing the same morphological heterogeneity as seen in the original cancer. In analogy with the leukemia-initiating cells, these successful cells were tentatively called tumor- initiating cells, which later became synonymous to cancer stem cells, and ever since, questions regarding their origin, identity, and role in the metastatic process in a human patient have been subject to controversy.
How to become a cancer stem cell: epithelial mesenchymal transition (EMT)
According to current thinking, cancer stem cells result from the switch from a polarized epithelial to a non-polarized mesenchymal cell type with stem cell properties, including migratory behavior, self renewal and generation of differentiated progeny, and reduced responsiveness to conventional cancer therapies [3,18,19]. This shows some striking and fascinating similarities to processes taking place during embryonic development when tissues and organ structures are formed and repeated switching between epithelial and mesenchymal cell type is a common phenomenon [20]. Much simplified, switching to a mesenchymal stem cell type, called epithelial-mesenchymal- transition or EMT, may enable cell migration to the right body location in the developing embryo, where the reverse process of mesenchymal-epithelial-transition or MET may take place where needed, allowing differentiation to specialized “secondary” epithelial cell types of the tissue or organ [18,21]. Multiple developmental signaling pathways, especially the TGFbeta and Wnt pathways, but also FGF, Hedgehog and Notch, are cooperatively involved in regulating these developmental processes. During EMT they converge upon activation of specific EMT transcription factors like SNAIL and TWIST which subsequently initiate a stem cell transcriptional program [12]. As a consequence expression of epithelial genes like E-cadherin, is reduced, while expression of characteristic mesenchymal (vimentin, metalloproteases, N-cadherin) and “stemness” genes (e.g. Oct-4, Nanog) increases [18]. Oct-4 and Nanog typically confer stem cell properties like self renewal [22]. Production of metalloproteases, like MMP-2 and MMP-9, enables basal membrane and extracellular matrix degradation, while switching from E-cadherin to N-cadherin results in loss of cell-cell contacts, together enabling cell migration [18,23]. The exact in vivo molecular mechanism behind EMT in cancer is as yet not fully known and likely to be complex. However, available circumstantial evidence points to similarities with developmental EMT, with especially TGFbeta and Wnt pathway activation involved [3,24].
How could developmental signal transduction pathways become active in cancer cells?
As discussed, both genetic changes as well as location-dependent contextual cues may induce changes in behavior of cancer cells. Some DNA mutations may directly lead to abnormal activation of a signaling pathway in the cell; one example is loss of APC in colon cancer leading to constitutive activation of the Wnt pathway [4,24]. Alternatively a genetic or epigenetic change may be associated with enhanced responsiveness of a signaling pathway to an otherwise normal presence of a ligand for its receptor. An example is loss of expression of a Wnt antagonist like the DKK gene, associated with gene promoter methylation [25,26]. The cancer cell microenvironment itself may also be the sole responsible factor for inducing abnormal signaling pathway activation, obviously on the premise that necessary pathway components in the cancer cell constitute a responsive signaling pathway. Both tumor-invading macrophages and other immune cells as well as activated fibroblasts produce a variety of cytokine and growth factor ligands, normally not present in the tumor tissue of origin, and potentially capable of activating EMT-inducing signaling pathways like the Wnt, Hedgehog and TGFbeta pathway [10,27-29]. The most likely location in the tumor for initiation of EMT is probably the invasive front at the border between tumor tissue and normal tissue, where the likelihood of encountering such EMT inducing factors is highest. Indeed, several studies show SNAIL and TWIST expressed in cells at the invasive border of different cancer types, e.g. breast and colon cancer, associated with bad prognosis and increased risk at metastasis [30-33].
Aside from biochemical cues, physical parameters in the tumor may also play a role in the EMT process and associated metastatic cell behavior [34]. Hydrostatic pressure has long been known to be increased in an expanding tumor, while due to increased stiffness of the extracellular matrix network cells in the cancer tissue are exposed to increased shear stress and tension forces [34]. These types of physical forces potentially influence phenotype and function of cells by activation of specific signaling pathways through mechanotransduction, for example by inducing membrane protein conformation changes [35]. Cell membrane-located integrin adhesion molecules, as well as TGFβ and Wnt membrane receptors are among proteins that potentially respond to changes in these forces. Endothelial cells, macrophages and lymphocytes express a variety of integrins, suggesting that it is not only cancer cells which can be modulated by physical cues. As yet, this is to a large extent unexplored territory since controlled introduction of physical cues into cancer model systems is currently not possible.
Cancer stem cells as circulating tumor cells
Circulating tumor cells (CTCs) have been defined as very rare cancer cells in the blood circulation, which can originate from both primary and metastatic lesions, and express the epithelial cancer markers EPCAM (a cell adhesion molecule) on the cell membrane and cytokeratin (a component of the cytoskeleton) in the cell. These markers are in general used for their detection and enrichment or isolation from the multitude of non-epithelial blood cells. However, circulating tumors cells are much more heterogeneous cells than previously thought [36]. Many circulating tumor cells do not express epithelial markers (like EPCAM and cytokeratin) but instead express mesenchymal and stem cell markers (like vimentin, N-cadherin, CD44 and ALD-1) suggesting that they are cancer stem cells which went through EMT in the tumor tissue [37-39]. In general during the EMT process cell-cell connections are lost, while elasticity is gained by getting rid of the relatively rigid epithelial cytoskeleton, facilitating passage as single cells through the endothelial cell layer into the blood – in contrast to EPCAM-expressing epithelial CTCs that are often observed to circulate in cell clusters [36]. Such a single cell status may be a favorable feature to survive (repeated) passage through capillaries and facilitate subsequent extravasation and seeding in an organ tissue. Moreover, this subset of CTCs may have acquired capabilities to suppress and evade immune surveillance, similar to normal mesenchymal stem cells [40,41]. Finally, self-renewal “stemness” properties provide the assets to establish metastatic growth when seeded in a favorable tissue-niche, suitable for supporting survival and capable of providing the right stimuli to induce clonal proliferation.
Initiation of metastatic growth
Initiation of the metastatic tumor growth process is dependent on appropriate growth signals from the niche environment where the cell seeded – in a sense comparable to the normal stem cell niche [42]. Normal stem cells are quiet and rarely divide, however are induced to self renew and start a regenerative “wound-healing” process upon signals indicating loss of differentiated organ cells due to disease or trauma, provided through the associated inflammatory process. Quite analogous, in the cancer stem cell niche inflammatory cells are thought to be able to deliver signals required to initiate tumor growth [43]. Once again key developmental cell signaling pathways are involved in translating these signals towards initiation of cell division. Although direct seeding of CTCs and clonal outgrowth to form a metastatic tumor remains to be demonstrated, at least in mice CTCs have been shown to migrate between different tumors, for example between metastasis and primary tumor or seed a second primary tumor in breast tissue, all depending on favorable local inflammatory conditions: the “reseeding hypothesis” [9,44]. Thus circulating tumor cells may hold many clues to both the identity of cancer stem cells and the process of metastasis, and the field urgently awaits improved methods for isolation of (live) CTCs, especially EPCAM negative CTCs from blood.
The plasticity of a cancer stem cell
Recent developments in stem cell research have convincingly shown that differentiated cells can be forced to revert to a pluripotent state in which they can in principle give rise to all cell types in the body [45]. Would it be possible for a cancer stem cell to give rise to other than epithelial cell types, depending on its microenvironment or niche? Fibroblasts in tumor tissue are quite different from their normal counterparts, and can contain somatic mutations identical to those found in the corresponding cancer cells [46]. More recently the EMT transcription factor SNAIL was reported to be expressed predominantly in stromal cells in the tumor [32]. In melanomas specific cancer markers have been detected in cells with an endothelial phenotype, a phenomenon called “vascular mimicry”, suggesting the possibility that they represent progeny of melanoma cancer stem cells, differentiated to an endothelial cell type under the influence of angiogenetic factors in the tumor microenvironment [47]. Finally, circulating tumor cells in blood may coexpress specific cancer cell markers like HER2 amplification with the leukocyte marker CD45, suggesting partial differentiation along the hematopoietic lineage, for example during temporary residence in the bone marrow niche [36,48]. The tentative conclusion is that at least some cancer stem cells may be much more plastic than originally thought, and give rise to differentiated progeny along quite separate lineages. DNA sequencing of individual cells in cancer tissue may reveal more examples in the near future. If true, this may have major implications both for diagnostics and for drug development, and implies that some of the microenvironment of the cancer cell may be self-generated.
Is EMT reversible? An opportunity for new drugs
A drug which would induce a switch back from mesenchymal cell type to the differentiated epithelial cell type could potentially block the metastatic capabilities of cancer [49-51]. Obviously this is an exciting idea, on the premise that the EMT process is reversible. If we adopt the concept of a phenotypic switch to a mesenchymal cell type caused by interaction with neighboring stroma and non-tumor cells, this condition may be dependent on continuous availability of the proper pathway-activating signals. While signals are initially derived from the environment, with time autocrine Wnt and TGFbeta signaling loops may take over to maintain the cancer stem cell state. Interference with activity of these signaling pathways could conceivably be one therapeutic approach to induce mesenchymal epithelial transition (MET) [3]. To obtain in depth molecular understanding of the complex EMT mechanism, improved EMT model systems are needed which better mimic complex in vivo cancer tissue and enable discovery of novel drug targets. They should preferably allow high throughput screening of drug compound libraries to develop drugs which can prevent or even reverse the EMT process.
Novel cancer model system: filling a gap
In vitro cancer models are currently limited to 2D or 3D cultures in a culture dish on standard culture plastic or in a gel matrix (e.g. tumor spleroids) or scaffold to enable 3D growth with other cell types potentially mixed in [52-54]. While proven to be of high value for cancer research, they lack sophistication to answer all questions regarding the mechanism behind EMT and cancer stem cells in metastatic disease, especially with regard to establishing causal relationships for final proof of the cancer stem cell hypothesis.
The recent advent of novel in vitro model systems approaches to study diseases in the form of “organs-on-chips” might in the near future enable development of cancer models which more closely resemble the in vivo situation, and allow real time monitoring of cellular and biochemical processes [55,56]. This promising new approach, which includes many novel developments in biocompatible and responsive materials, enables 2 or 3D cell culture in a microfluidics-based “micro-incubator” in which in vivo conditions like increased pressure, stroma stiffness, tension forces and tissue hypoxia can be mimicked. [57]. Interactions between different cell types can be controlled, for example separated in the created tissue structure by smart biocompatible membranes or by flowing cells, like immune cells, through microchannels. Membrane substrates can vary in stiffness, may contain pores to enable cell migration and micro- and nanostructures to align cells in specific directions. The chips can be microscope slide-size and compatible with real time monitoring of for example fluorescent signals or cell migration using optical techniques, like a confocal microscope or optical coherence tomography. Development of such “organ-on-chip” type cancer models may facilitate investigation of many aspects of the cancer process, e.g. the intricate interplay between biophysical and biochemical factors in the EMT process, interactions between cancer cells and their microenvironment, including immune cells, and migratory behavior of cells.
Conclusions and research challenges
In the concept described, cancer stem cells do not represent the stem cell of origin of the cancer, but originate from a cancer cell which lost its epithelial properties and instead newly acquired certain stem cell characteristics, enabling it to contribute to tissue invasion and metastasis (Figure 2). Combining experimental evidence from different research areas into a conceptual view on the role of cancer stem cells does not mean that the concept itself has been experimentally proven. Instead, it may provide useful guidance to future design of experiments aiming at elucidating the complex ways in which a cancer metastasizes. Obtaining such in depth knowledge is key to developing more effective drugs to tackle metastatic behavior of cancer – since metastasis causes death.
Figure 2.
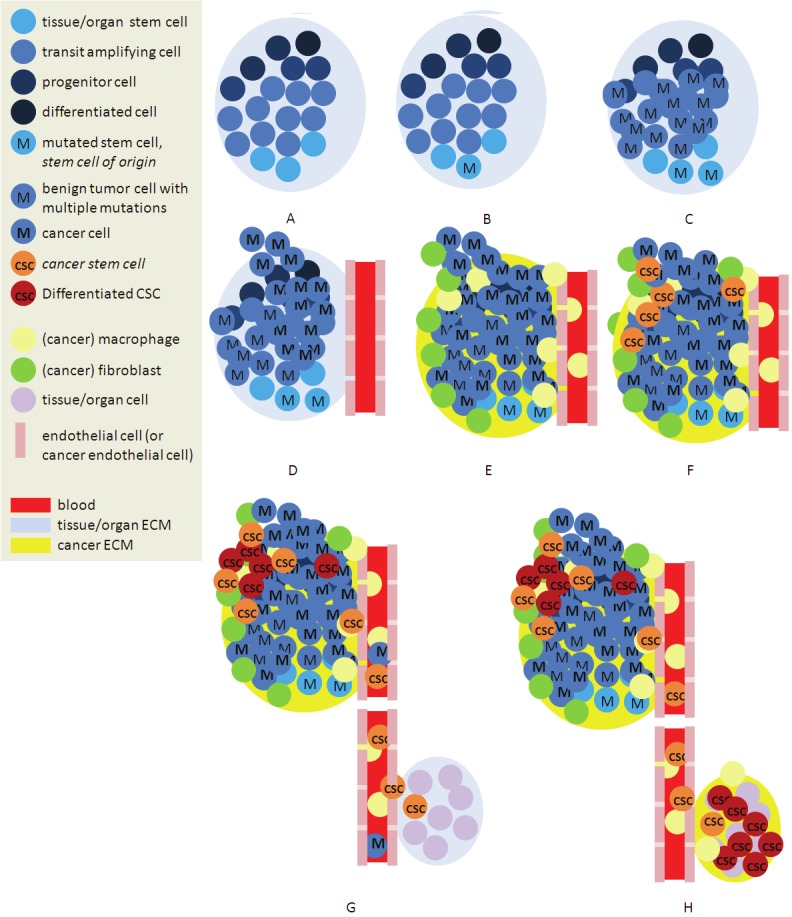
Simplified representation of the described concepts. A. Cells within extracellular matrix (ECM), together form a tissue/organ. Stem cells in a “niche” both self renew and deliver progenitor cells for the differentiated cell types forming the functional tissue/organ. B. A mutation in a signaling pathway gene may occur and C. lead to increased cell division resulting in a benign tumor. D. Mutations accumulate, causing genotypic heterogeneity and initiating transition to invasive cancer. E. Immune cells, among which macrophages infiltrate the tumor. F. Interaction between tumor cells, macrophages, abnormal fibroblasts, in combination with physical factors, like stiffer ECM and increased hydrostatic pressure, induce EMT, resulting in mobile cancer stem cells (CSC). G. CSCs produce phenotypically heterogeneous cancer cells. Among other cancer cells, CSCs migrate into the blood, and –potentially protected from immune attack - seed in other tissue/organ sites. H. Given favorable niche conditions, CSCs start to self renew and produce heterogeneous cancer cells, resulting in a macroscopic metastatic tumor.
Emerging complex human “organ-on-a-chip” cancer models may contribute valuable novel research tools to the field. Many questions need to be answered, here to be named only a few [2]. How does the genome of a cancer stem cell relate to the genome of the cancer cell of origin? Can a metastatic tumor arise from a single seeded cell? Is cell division of cancer stem cells indeed driven by other signaling pathways than their differentiated counterparts? How do microenvironmental factors, both biochemical and physical, induce EMT? Is EMT reversible? Which CTCs establish metastatic tumors, and what are their characteristics? What is the role of the metastatic niche, and of inflammatory factors and the immune system? To which extent can we learn from similar mechanisms in early embryonic development? The ongoing integration between physics, chemistry, mathematics, engineering, and biology opens up a highly challenging and multidisciplinary next level of life sciences research. This development provides many exciting opportunities for creative scientists to contribute to better understanding, and ultimately curing, this complex and multifaceted disease.
References
- 1.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality. Nat Med. 2009;15:1010–1012. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin Cancer Biol. 2012;22:396–403. doi: 10.1016/j.semcancer.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–11. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 5.Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, Clevers H. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337:730–5. doi: 10.1126/science.1224676. [DOI] [PubMed] [Google Scholar]
- 6.Fearon ER, Hamilton SR, Vogelstein B. Clonal analysis of human colorectal tumors. Science. 1987;238:193–7. doi: 10.1126/science.2889267. [DOI] [PubMed] [Google Scholar]
- 7.Vogelstein B, Kinzler KW. “Cancer genes and the pathways they control”. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 8.Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, Traulsen A, Nowak MA, Siegel C, Velculescu VE, Kinzler KW, Vogelstein B, Willis J, Markowitz SD. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008;105:4283–8. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–4. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. 2011;121:3804–9. doi: 10.1172/JCI57099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune microenvironments in solid tumors: new targets for therapy. Genes Dev. 2011;25:2559–72. doi: 10.1101/gad.169029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, Weinberg RA. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145:926–40. doi: 10.1016/j.cell.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mummery C, Wilmut I, van de Stolpe A, Roelen BAJ. Elsevier Books. 2011. Stem Cells, Scientific facts and fiction. ISBN: 978-0-12-3815354. [Google Scholar]
- 14.Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112:4793–4805. doi: 10.1182/blood-2008-08-077941. [DOI] [PubMed] [Google Scholar]
- 15.Battula VL, Evans KW, Hollier BG, Shi Y, Marini FC, Ayyanan A, Wang RY, Brisken C, Guerra R, Andreeff M, Mani SA. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells. 2010;28:1435–1445. doi: 10.1002/stem.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Snippert HJ, Haegebarth A, Kasper M, Jaks V, van Es JH, Barker N, van de Wetering M, van den Born M, Begthel H, Vries RG, Stange DE, Toftgård R, Clevers H. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science. 2010;327:1385–9. doi: 10.1126/science.1184733. [DOI] [PubMed] [Google Scholar]
- 17.Blau HM, Pomerantz JH. Re”evolutionary” regenerative medicine. JAMA. 2011;305:87–8. doi: 10.1001/jama.2010.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 19.Crews LA, Jamieson CH. Selective elimination of leukemia stem cells: Hitting a moving target. Cancer Lett. 2012 Aug 17; doi: 10.1016/j.canlet.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–1449. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, Wong KY, Sung KW, Lee CW, Zhao XD, Chiu KP, Lipovich L, Kuznetsov VA, Robson P, Stanton LW, Wei CL, Ruan Y, Lim B, Ng HH. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–40. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 23.Bae KM, Parker NN, Dai Y, Vieweg J, Siemann DW. E-cadherin plasticity in prostate cancer stem cell invasion. Am J Cancer Res. 2011;1:71–84. [PMC free article] [PubMed] [Google Scholar]
- 24.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 25.Klarmann G, Decker A, Farrar W. Epigenetic gene silencing in the Wnt pathway in breast cancer. Epigenetics. 2008;3:59–63. doi: 10.4161/epi.3.2.5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kocemba KA, Groen RW, van Andel H, Kersten MJ, Mahtouk K, Spaargaren M, Pals ST. Transcriptional silencing of the Wnt-antagonist DKK1 by promoter methylation is associated with enhanced Wnt signaling in advanced multiple myeloma. PLoS One. 2012;7:e30359. doi: 10.1371/journal.pone.0030359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr HA, Delaloye JF, Huelsken J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2011;481:85–9. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- 28.Dawson MR, Chae SS, Jain RK, Duda DG. Direct evidence for lineage-dependent effects of bone marrow stromal cells on tumor progression. Am J Cancer Res. 2011;1:144–54. [PMC free article] [PubMed] [Google Scholar]
- 29.Jinushi M, Baghdadi M, Chiba S, Yoshiyama H. Regulation of cancer stem cell activities by tumor-associated macrophages. Am J Cancer Res. 2012;2:529–39. [PMC free article] [PubMed] [Google Scholar]
- 30.Francí C, Gallén M, Alameda F, Baró T, Iglesias M, Virtanen I, García de Herreros A. Snail1 protein in the stroma as a new putative prognosis marker for colon tumours. PLoS One. 2009;4:e5595. doi: 10.1371/journal.pone.0005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, Oberyszyn TM, Hall BM. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009;28:2940–7. doi: 10.1038/onc.2009.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Nes JG, de Kruijf EM, Putter H, Faratian D, Munro A, Campbell F, Smit VT, Liefers GJ, Kuppen PJ, van de Velde CJ, Bartlett JM. Co-expression of SNAIL and TWIST determines prognosis in estrogen receptor-positive early breast cancer patients. Breast Cancer Res Treat. 2012;133:49–59. doi: 10.1007/s10549-011-1684-y. [DOI] [PubMed] [Google Scholar]
- 33.Wu ZQ, Brabletz T, Fearon E, Willis AL, Hu CY, Li XY, Weiss SJ. Canonical Wnt suppressor, Axin2, promotes colon carcinoma oncogenic activity. Proc Natl Acad Sci U S A. 2012;109:11312–7. doi: 10.1073/pnas.1203015109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu H, Mouw JK, Weaver VM. Forcing form and function: biomechanical regulation of tumor evolution. Trends in Cell Biology. 2011;21:47–56. doi: 10.1016/j.tcb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoffman BD, Grashoff C, Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature. 2011;475:316–323. doi: 10.1038/nature10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van de Stolpe A, Pantel K, Sleijfer S, Terstappen LW, den Toonder JM. Circulating tumor cell isolation and diagnostics: toward routine clinical use. Cancer Res. 2011;71:5955–60. doi: 10.1158/0008-5472.CAN-11-1254. [DOI] [PubMed] [Google Scholar]
- 37.Sieuwerts AM, Kraan J, Bolt J, van der Spoel P, Elstrodt F, Schutte M, Martens JW, Gratama JW, Sleijfer S, Foekens JA. Anti-epithelial cell adhesion molecule antibodies and the detection of circulating normal-like breast tumor cells. J Natl Cancer Inst. 2009;101:61–66. doi: 10.1093/jnci/djn419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hou JM, Krebs M, Ward T, Sloane R, Priest L, Hughes A, Clack G, Ranson M, Blackhall F, Dive C. Circulating tumor cells as a window on metastasis biology in lung cancer. Am J Pathol. 2011;178:989–96. doi: 10.1016/j.ajpath.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balasubramanian P, Lang JC, Jatana KR, Miller B, Ozer E, Old M, Schuller DE, Agrawal A, Teknos TN, Summers TA Jr, Lustberg MB, Zborowski M, Chalmers JJ. Multiparameter analysis, including EMT markers, on negatively enriched blood samples from patients with squamous cell carcinoma of the head and neck. PLoS One. 2012;7:e42048. doi: 10.1371/journal.pone.0042048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dazzi F, Lopes L, Weng L. Mesenchymal stromal cells: a key player in ‘innate tolerance’? Immunology. 2012;137:206–13. doi: 10.1111/j.1365-2567.2012.03621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nombela-Arrieta C, Ritz J, Silberstein LE. The elusive nature and function of mesenchymal stem cells. Nat Rev Mol Cell Biol. 2011;12:126–31. doi: 10.1038/nrm3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Losick VP, Morris LX, Fox DT, Spradling A. Drosophila stem cell niches: a decade of discovery suggests a unified view of stem cell regulation. Dev Cell. 2011;21:159–71. doi: 10.1016/j.devcel.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Comen E, Norton L. Self-seeding in cancer. Recent Results Cancer Res. 2012;195:13–23. doi: 10.1007/978-3-642-28160-0_2. [DOI] [PubMed] [Google Scholar]
- 44.Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, Massagué J. Tumor Self-Seeding by Circulating Cancer Cells. Cell. 2009;139:1315–1326. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takahashi K, Okita K, Nakagawa M, Yamanaka S. Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc. 2007;2:3081–9. doi: 10.1038/nprot.2007.418. [DOI] [PubMed] [Google Scholar]
- 46.Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet. 2002;32:355–7. doi: 10.1038/ng1013. [DOI] [PubMed] [Google Scholar]
- 47.Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, Hendrix MJ. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin Cancer Res. 2012;18:2726–32. doi: 10.1158/1078-0432.CCR-11-3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katz RL, He W, Khanna A, Fernandez RL, Zaidi TM, Krebs M, Caraway NP, Zhang HZ, Jiang F, Spitz MR, Blowers DP, Jimenez CA, Mehran RJ, Swisher SG, Roth JA, Morris JS, Etzel CJ, El-Zein R. Genetically abnormal circulating cells in lung cancer patients: an antigen-independent fluorescence in situ hybridization-based case-control study. Clin Cancer Res. 2010;16:3976–87. doi: 10.1158/1078-0432.CCR-09-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–59. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sachlos E, Risueño RM, Laronde S, Shapovalova Z, Lee JH, Russell J, Malig M, McNicol JD, Fiebig-Comyn A, Graham M, Levadoux-Martin M, Lee JB, Giacomelli AO, Hassell JA, Fischer-Russell D, Trus MR, Foley R, Leber B, Xenocostas A, Brown ED, Collins TJ, Bhatia M. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell. 2012;149:1284–97. doi: 10.1016/j.cell.2012.03.049. [DOI] [PubMed] [Google Scholar]
- 51.Hu Y, Fu L. Targeting cancer stem cells: a new therapy to cure cancer patients. Am J Cancer Res. 2012;2:340–356. [PMC free article] [PubMed] [Google Scholar]
- 52.Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5:675–688. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- 53.Nyga A, Cheema U, Loizidou M. 3D tumour models: novel in vitro approaches to cancer studies. J Cell Commun Signal. 2011;5:239–248. doi: 10.1007/s12079-011-0132-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Staveren WC, Solís DY, Hébrant A, Detours V, Dumont JE, Maenhaut C. Human cancer cell lines: Experimental models for cancer cells in situ? For cancer stem cells? Biochim Biophys Acta. 2009;1795:92–103. doi: 10.1016/j.bbcan.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 55.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting Organ-Level Lung Functions on a Chip. Science. 2010;328:1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baker MA. living system on a chip. Nature. 2011;661:661–665. doi: 10.1038/471661a. [DOI] [PubMed] [Google Scholar]
- 57.Kim J, Hayward RC. Mimicking dynamic in vivo environments with stimuli-responsive materials for cell culture. Trends in Biotechnology. 2012;30:426–438. doi: 10.1016/j.tibtech.2012.04.003. [DOI] [PubMed] [Google Scholar]