

Abstract
Acute lung injury (ALI) is a devastating clinical condition associated with pulmonary and systemic inflammation and characterized by incompetence of the pulmonary microvascular barrier culminating in noncardiogenic pulmonary edema. An understanding of the mechanisms underlying endothelial barrier dysfunction in ALI has been facilitated by study of the effects of statins in relevant cellular and animals models. Many of the pleotropic properties of these drugs, including direct effects on endothelial cell (EC) cytoskeletal rearrangement, NADPH oxidase, and nitric oxide activity, as well as effects on differential EC gene expression, are relevant to the pathobiology of ALI and suggest a potential therapeutic role for statins in this context. Moreover, results from preclinical studies and observations in relevant patient populations support the protective potential of statins in ALI, paving the way now for definitive clinical trials.
Keywords: acute lung injury, pulmonary endothelium, statins
In 1971, Dr. Akira Endo and colleagues correctly surmised that inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme of the cholesterol biosynthetic pathway, were likely present in the arsenal of select micro-organisms. There they might be useful for inhibiting the growth of competing microbes which require sterols or other isoprenoids for cytoskeletal and cell wall maintenance.[1] In the following decade, the HMG-CoA reductase inhibitors mevastatin and lovastatin were successfully isolated from the fungal species Penicillium citrinum and Aspergillus terreus, respectively.[2,3] Subsequent refinements to the molecular structure of these compounds led to the establishment of a class of medications known as statins that is now widely used to reduce serum cholesterol levels in patients either at risk for or with coronary artery disease (CAD).[4] In recent years, it has become increasingly apparent that reductions in ischemic cardiac events and mortality associated with the use of statins in CAD patients is markedly out of proportion to their influence on the availability of serum cholesterol as a substrate for atheroma formation.[5,6] Research into the mechanisms underlying these effects of statins has revealed a broad impact of the cholesterol biosynthetic pathway on mammalian cell biology ranging from modulation of cell signaling, to post-translational modification of proteins, and regulation of gene expression.[7] Furthermore, the study of statins has contributed to the emerging knowledge of the role of inflammation and its associated effects on endothelial cell (EC) function in the pathogenesis of vascular disease.[8,9] Indeed, Dr. Endo's original assertion concerning the utility of HMG-CoA reductase inhibition for microbial survival can now be expanded to include attenuation of various facets of host inflammatory defenses.[10]
Statins have been utilized in the study of other inflammatory settings associated with vascular diseases including the disorders of the pulmonary circulation. Of these, significant mechanistic insight and therapeutic promise has come from the study of statins as a potential therapy for patients with acute lung injury (ALI). ALI develops during critical illnesses characterized by pulmonary and/or systemic inflammation, and contributes to an estimated 75,000 yearly deaths in the United States.[11] It is defined by noncardiogenic pulmonary edema resulting from incompetence of the microvascular endothelial barrier.[12,13] Analysis of the complex relationship between inflammation and endothelial barrier dysfunction during ALI has been facilitated by the introduction of statins into cellular and animal models of the disease. The findings of these analyses as well as observations stemming from human studies will be reviewed here.
Cholesterol biosynthesis, statins, and endothelial cell biology
Statins competitively inhibit HMG-CoA reductase which mediates the rate-limiting step of mevalonate production. In addition to cholesterol, mevalonate serves as a substrate for the generation of the isoprenoid intermediates, farnesyl pyrophosphate (FPP), and geranylgeranyl pyrophosphate (GGPP; Fig. 1). These isoprenoid chains, consisting of 15- and 20-carbons respectively, serve as the ingredients for post-translational protein prenylation, the addition of hydrophobic molecules (isoprenoid chains) to cysteine residues.[14] Prenylation is thought to facilitate attachment to cell membranes but has also been shown to be pertinent to protein-protein binding interactions.[15]
Figure 1.
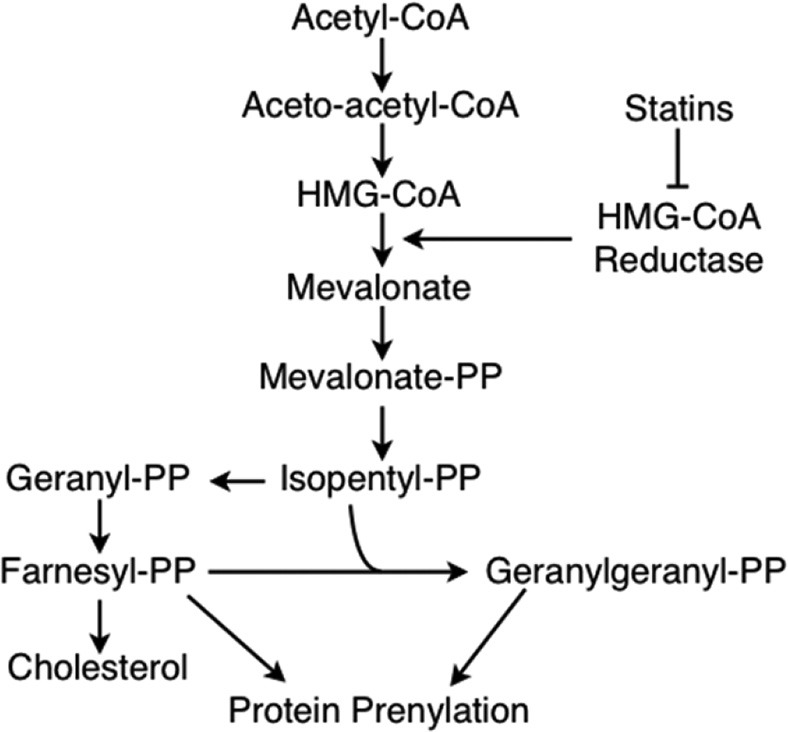
Statin mechanism of action. Statins are competitive inhibitors of HMG-CoA reductase, an enzyme which mediates the rate-limiting step of mevalonate production during cholesterol biosynthesis. In addition to cholesterol, this pathway also generates the isoprenoid intermediates, farnesyl pyrophosphate (PP) and geranylgeranyl pyrophosphate. Both of these products are used in post-translational modification of proteins via prenylation.
Among the known prenylated proteins and particularly relevant to EC function are the Rho family of small GTPases, including Rho and Rac. These small GTPases function as “molecular switches” whose downstream effectors impact upon fundamental cellular processes such as signal transduction and actin cytoskeletal regulation. Their “switch” property is characterized by a cycling between an active, GTP-bound state, and an inactive, GDP-bound state (Fig. 2). This cycling may be further regulated by three types of mediators: GTPase activating proteins (GAPs), guanine nucleotide exchange factors (GEFs), and guanine nucleotide dissociation inhibitors (GDIs). GAPs control GTP hydrolysis to GDP thus directing the movement between the active and inactive conformations, while GEFs direct the opposite movement by accelerating the exchange of GDP for GTP.[16] GDIs oppose the action of GEFs and also, interestingly, provide a level of spatial control by preventing small GTPases from localizing to membranes where both GEFs and effector targets of Rho GTPases commonly reside. GDI-mediated inhibition of membrane localization arises from the insertion of the isoprenoid moeity of the prenylated GTPase into a hydrophobic pocket of the GDI thus maintaining the Rho GTPase as a soluble cytosolic protein. The inhibition of prenylation by statins, therefore, would be expected to inhibit GTPase access to membrane-associated GEFs or downstream effector targets, and lead to an increase in inactivated GDP-bound cytosolic proteins. However, the effects of statins on GTPase regulation are in fact complex, dependent on both spatial and temporal factors, and not entirely understood.[17]
Figure 2.
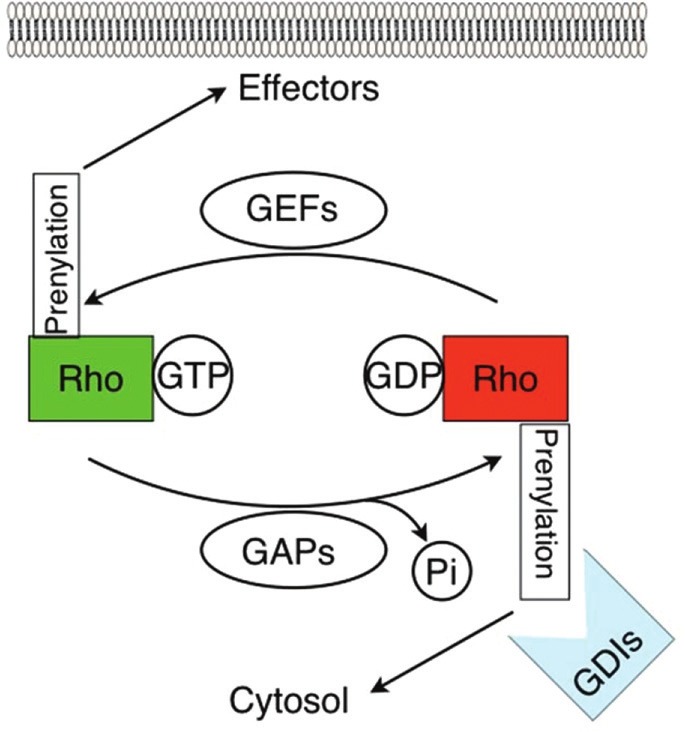
Regulation of RhoGTPase Activity. Statin-induced endothelial cell signaling and activation is mediated in part by Rho GTPases. These proteins operate as molecular switches, toggling between a GTP-bound active state (green) and a GDP-bound inactive state (red). In turn, these states may be regulated by guanine exchange factors (GEFs) and GTPase activating proteins (GAPs). Prenylation regulates GTPase function either by promoting their association with membranes where many downstream effector targets and GEFs are located, or by increasing their association with guanine dissociation inhibitors (GDIs) which favors GDP-binding and inactivation of RhoGTPases localized to the cytosol. Statin inhibition of protein prenylation leads to decreased RhoGTPase membrane association and thus decreased activation by GEFs at the membrane. However, statins also augment cytosolic GTPasebinding of Rho GTPases, with unclear functional significance, indicative of the complex effects of these drugs.
Nonetheless, in cellular and animal models of ALI a plethora of vascular-protective changes in EC phenotype can be attributed to statin-induced inhibition of mevalonate production and the resultant changes in Rho GTPase activity and localization. Moreover, there are mechanisms of statin activity described in studies examining vascular inflammation in atherosclerosis which are not known to be significant contributors to ALI pathogenesis/resolution but may be relevant in this clinical context by virtue of their associated anti-inflammatory and vascular protective effects. For example, induction of the transcription factor, KLF-2, is thought to be an important component of the statin-induced endothelial atheroprotective phenotype, but includes inhibition of proinflammatory and prothrombotic gene responses that are relevant to ALI pathogenesis.[18] Additionally, at least one mevalonate-independent mechanism of statin-mediated endothelial anti-inflammation has been identified, and is thought to occur via a direct modification of COX-2.[19,20] Here we review three aspects of endothelial protection by statins which contribute to their potential benefits in ALI, namely endothelial cytoskeletal regulation, eNOS activity, and ROS production via NADPH oxidase. This will be followed by a short summary of mechanisms arising from differential gene expression.
Endothelial cytoskeletal rearrangement and barrier protection by statins
The pulmonary microcirculation is granted access to the lung interstitium by the semipermeable monolayer of ECs. Under normal conditions, endothelial permeability is dictated by two molecular pathways, one paracellular and the other transcellular. Although much less is known with regards to the nature of the transcellular pathway and its interactions with the paracellular route, it is generally believed that the paracellular pathway is primarily responsible for microvascular barrier incompetence during the early pathologic changes characteristic of ALI.[21,22] Endothelial regulation of paracellular permeability can be understood in terms of the tensegrity model which describes paracellular junctional integrity as a balance between competing cytoskeletal contractile forces and adhesive cell-cell or cell-matrix tethering forces.[23,24]
During the increased permeability phase of ALI, EC cytoskeletal contraction results in cell rounding observed by electron microscopy and contributes to the formation of paracellular gaps.[21,22] Actin microfilaments, made up of polymerized globular actin proteins, interact with phosphorylated myosin to impart rigidity to cell structural elements. Dynamic remodeling of actin filaments in peripherally distributed, cortical bands facilitates the maintenance of endothelial junctional integrity in the basal barrier state, while inhibition of actin polymerization with cytochalasin D increases EC permeability.[25] Inflammatory stimuli may induce a dramatic rearrangement of actin with a disintegration of peripheral filaments and the augmentation of organized actin cables which traverse the intracellular space and participate in EC contraction.[24]
Actin remodeling and redistribution is regulated by various actin-binding proteins. Many actin-binding proteins such as nmMLCK, filamin, cofilin, VASP, gelsolin, and cortactin are in turn subject to regulation by Rho GTPases, making EC cytoskeletal rearrangement and the resultant increase in paracellular permeability susceptible to the effects of statins administered during ALI.[26] For example, pretreatment of confluent ECs with simvastatin significantly attenuates thrombin-induced transcellular stress fiber formation while increasing peripheral localization of actin.[27] These changes are associated with a decrease in paracellular gap formation and improvement in EC-barrier function as measured both by transendothelial electrical resistance (TER) and by FITC-dextran flux across EC monolayers.[27,28] Notably, simvastatin-mediated EC barrier protection can be reproduced in some measure by Rho inhibition via the use of silencing RNA specific for RhoA, or Y27632, a pharmacologic inhibitor of Rho kinase.[28] Moreover, siRNA-mediated knockdown of Rac1 produces similar barrier protection albeit to a lesser extent than RhoA inhibition. However, statin-induced inhibition of Rho GTPase prenylation has numerous implications for EC signaling and barrier regulation beyond direct effects on the cytoskeleton.
NADPH oxidase regulation by statins
Abundant oxidative stress is a prominent feature of ALI.[29] In patients with ALI this has been observed as increased levels of H2O2 in exhaled breath condensates,[30] oxidized proteins in bronchoalveolar lavage (BAL) fluid,[31] and a relative reduction in various antioxidants measured in both blood and BAL fluid.[32,33] At the cellular level, the redox balance of the endothelial environment plays a major role in vascular function as endotoxin-induced endothelial hyperpermeability is prevented by pretreatment with antioxidants.[34] The mechanisms by which oxidant stress induces barrier dysfunction are diverse and range from activation of redox-sensitive transcription factors to direct modification of cytoskeletal and junctional proteins.[35] Although the predominant source of oxidants in ALI are neutrophils and macrophages,[36,37] the major source of endothelial reactive oxygen species (ROS) is an endogenous form of NADPH oxidase,[38,39] the activity of which is subject to regulation by statins.
The EC NADPH oxidase complex consists of two membrane-bound components, p22phox and Nox2, as well as several cytosolic regulatory subunits including p47phox, p67phox, and Rac1.[40,41] Stimulation of EC by proinflammatory cytokines, mechanical forces, ischemia/reperfusion, or hyperoxia, leads to post-translational modification and translocation of the cytosolic components to form the activated NADPH oxidase complex at the membrane.[39] Dissociation of Rac1 from Rho-GDI in the cytosol and its subsequent activation by binding GTP is essential for NADPH oxidase complex assembly and activation (Fig. 3). As predicted via the known inhibition of Rac1 geranylgeranylation by statins, simvastatin attenuates LPS-induced EC superoxide generation by preventing the translocation of Rac1 and, consequently, p47phox to the membrane.[28] These effects are reproduced by transfection of silencing RNA for Rac1, and are reversed by the addition of GGPP. Notably, the actin cytoskeleton independently regulates NADPH oxidase activity as p47phox associates actin and actin destabilization via pretreatment of EC with cytochalasin D enhances NADPH oxidase mediated ROS production.[42] Thus, statins may also attenuate NADPH oxidase activity via direct effects on actin cytoskeletal reorganization.
Figure 3.
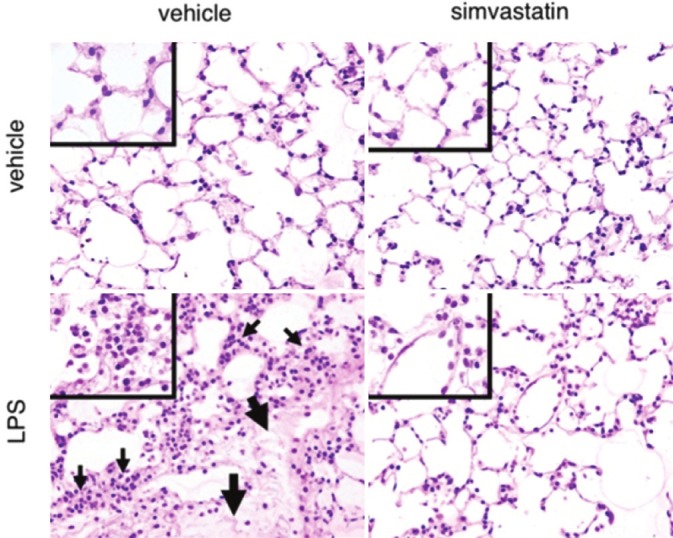
Protection by Statins in a Murine Model of ALI. Mice were administered either vehicle control or LPS (2 μg/g) and lungs were harvested for histology after 24 hours Inflammatory cell infiltration (small arrows) and interstitial edema (large arrows) were abrogated by intraperitoneal administration of simvastatin (20 mg/kg) 24 hours prior to LPS inhalation, and again at the time of inhalation. (Reproduced with permission from reference 81).
Statins and ENOS
An important consequence of increased oxidative stress during ALI is the production of reactive nitrogen species such as the reaction between superoxide anion and nitric oxide (NO) producing peroxynitrite (ONOO-). Peroxynitrite formation both induces nitrosative damage of proteins relevant to endothelial barrier function, and reduces the bioavailability of NO.[43] NO is thought to be lung protective through a number of mechanisms including free radical scavenging, local pulmonary vasodilatation, inhibition of leukocyte adhesion, protection of alveolar epithelium subjected to mechanical stress, and preservation of normal endothelial function.[44–47] In support of this idea, higher levels of endogenous NO measured in the urine of ALI patients has been correlated with a lower mortality.[48] EC NO synthesis is mediated by the endothelial isoform of nitric oxide synthase (eNOS). The effects of statins on eNOS have been described with respect to regulation of both mRNA stability and enzyme activity.[49–51]
Effects of statins on eNOS expression is in fact predicted by evidence of a functional link between Rho GTPase activity and eNOS mRNA levels.[49,52] This is suggested by increased eNOS expression in response to inhibition of Rho by either Clostridium botulinum C3 transferase or overexpression of a dominant-negative N19RhoA mutant, while Rho activation induced by Escherichia coli cytotoxic nectrotizing factor-1 is associated with decreased eNOS expression.[53] These events appear to primarily involve actin cytoskeletal remodeling since disruption of actin polymerization by cytochalasin D reproduces the increase in eNOS mRNA expression associated with Rho inhibition.[54] Separately, while a nuclear pool of β-actin may serve as a transcription factor for eNOS expression,[55] enhanced stabilization of eNOS mRNA transcripts has been linked to effects of statins on EC actin polymerization. Stability of eNOS transcripts is mediated by polyadenylation of its 3’ end via RNA polymerase II (RNAP II). Statin treatment of cultured EC induces an increase in the number of stable poly(A) eNOS transcripts. This finding has been linked to the statin-mediated phosphorylation of RNAP II resulting in increased polyadenylating processing activity for eNOS mRNA.[54] Interestingly, the increase in poly(A) transcripts by statins can be reproduced by cytochalasin D.[54] While it is unknown whether cytochalasin D treatment results in RNAP II phosphorylation, actin has been shown to be independently involved in mRNA transcription, processing, and nucleocytoplasmic transport,[56] thus representing a potential distinct mechanism underlying augmented eNOS mRNA stability by statins. Altogether these effects culminate in increased eNOS expression and NO production and are thought to be central to the benefits of statins in at least one disease model of endothelial injury as, in a murine model of ischemic stroke, the protective effects of statins were noted to be absent in eNOS-deficient mice.[57]
In addition to stabilizing eNOS mRNA expression, statins appear to increase enzyme activity of eNOS by at least two possible mechanisms. The first relates to the observation that statin treatment of ECs induces early phosphorylation of phosphatidylinositol 3-kinase (PI3-K) which in turn activates the protein kinase, Akt.[58] Akt activation and translocation to the cell membrane is necessary for subsequent eNOS activation via phosphorylation.[59] These statin-induced events are inhibited by wortmannin, a PI3-K inhibitor, mutation of the pleckstrin homology domain of Akt, necessary for membrane targeting;[60] overexpression of dominant-negative Akt,[51] and extracellular cholesterol supplementation of EC media.[58] Cholesterol depletion by statins may be linked to a second mechanism of eNOS upregulation as expression of caveolin-1, the primary component of EC plasmalemmal microdomains, is dependent on cholesterol availability, and is an important negative regulator of eNOS. Statin treatment decreases caveolin-1 expression which leads to subsequent disinhibition of eNOS activity.[50]
Differential gene expression by statins
Beyond the direct vascular-protective effects relevant to ALI pathophysiology, statins also modulate the expression of a number of EC genes known to mediate ALI pathobiology. While the mechanisms by which these effects occur and interact are incompletely understood, the cumulative pattern of gene expression strengthens the assertion that statins induce a multi-faceted vascular-protective EC phenotype in response to inflammatory stimuli.[27] For example, statins downregulate the G protein-coupled angiotensin II type-1 (AT-1) receptor gene in EC.[61] Activation of AT-1 by its ligand, angiotensin II, leads to protein kinase C and Rac1 activation, and is linked to increased superoxide generation by NADPH oxidase.[62,63] The mechanism of AT-1 down regulation by statins is cholesterol-independent, reproducible by specific inhibition of geranylgeranyl-transferase, and involves decreased mRNA stability with no change in the rate of transcription.[64] The importance of this gene in the effect of statins on endothelial function is highlighted by the findings of the Endothelial Protection, AT1 Blockade, and Cholesterol-Dependent Oxidative Stress (EPAS) trial.[65] In this study endothelial function was measured in human patients using a calculation termed the endothelial expression quotient which is based on the mRNA expression of specific genes, including eNOS and NADPH oxidase. The expression quotient associated with statin use was found to be highly consistent with that achieved via AT1 receptor blockade using the drug irbesartan.
Additionally, a number of genes involved in coagulation are differentially expressed in EC treated with statins. These include thrombomodulin, tissue factor, plasminogen activator inhibitor-1 (PAI-1), and the thrombin receptor protease-activated receptor-1 (PAR-1).[27,66,67] Increased thrombomodulin expression in response to statins has been linked to inhibition of Rac1 and Cdc42 geranylgeranylation and to NO production, while the attenuation of tissue factor and PAI-1 expression is associated with inhibition of RhoA geranylgeranylation.[68–70] Altogether, these coagulation gene changes culminate in EC resistance to the effects of thrombin, an inflammatory agonist which induces EC activation, contraction, and increased permeability.[27,28,71–73]
Finally, two other genes worth noting that are profoundly regulated by statins in EC are caldesmon and integrin-β4. Caldesmon is a cytoskeletal actin binding protein which mediates actin rearrangement associated with EC barrier incompetence during exposure to inflammatory conditions.[74,75] Expression of caldesmon is decreased at both the gene and protein level by more than two-fold in statin-treated EC.[27] Conversely, the expression of integrin β4 is dramatically upregulated, more than seven-fold, in statin-treated EC. While several β integrins have been implicated as mediators of ALI,[76–78] less is known about integrin β4 in this context. However, integrin β4 has been implicated in the activation of the Rho GTPases and in MAPK signaling,[79] both pathways relevant to the EC response to inflammatory conditions, and we have reported that EC inflammatory responses are augmented in response to integrin β4 silencing.[80] These findings suggest that the upregulation of EC integrin β4 is an important mechanism of the anti-inflammatory properties of statins and contribute to the potential protective effects of statins in ALI.
Statins in animal models of ALI
Abundant evidence that the vascular-protective effects of statins include many facets of endothelial function relevant to ALI has led investigators to examine their impact in integrated, animal models of inflammatory lung injury. As statins are thought to work at the level of a final common pathologic pathway during lung injury, a variety of inciting conditions which produce lung injury have been used to investigate the therapeutic potential of statins. These include endotoxin inhalation, bacteremia, bleomycin treatment, ischemia-reperfusion, high tidal volume ventilation, and burn injury with smoke inhalation.[10,81–86]
Our lab has employed a lipopolyascharide (LPS)-induced ALI model and studied mice pretreated with simvastatin (20 mg/kg) or placebo prior to the intratracheal administration of LPS.[81] Markers of lung injury including Evans blue albumin dye extravasation, BAL fluid neutrophil and albumin content, and lung tissue histology demonstrated protection from vascular leak and inflammation in mice pretreated with simvastatin (Fig. 4). Further, statin-mediated protection was associated with differential whole lung gene expression in a number of relevant ontologies (inflammation and immune response) as well as with respect to individual genes implicated in ALI pathogenesis and severity (IL-6, TLR 4). In support of our findings, Fessler and colleagues reported similar findings of reduced LPS-induced lung inflammation and vascular leak by statins despite the fact that statin-treated mice demonstrated reduced pulmonary and systemic clearance of bacteria following inhalation.[10] Notably, both the attenuation of lung inflammation and impairment of host defense by statins were noted to be fully reversed by the co-administration of mevalonic acid. Separately, in a murine model of bleomycin-induced inflammatory lung injury, pravastatin (30 mg/kg) administered intraperitoneally on a daily basis following intratracheal bleomycin exposure resulted in reduced lung histologic edema, hyaline membrane formation, and inflammatory cell infiltration up to one week following exposure.[85] BAL fluid inflammatory cell counts were similarly diminished in a dose-dependent manner by statins.
Figure 4.
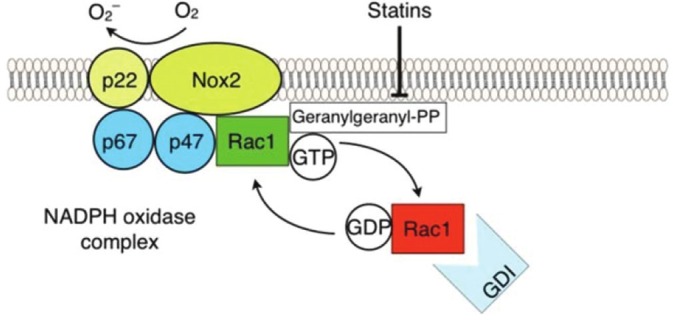
Inhibition of NADPH Oxidase by Statins. The EC NADPH complex comprises two membrane-bound components, p22phox and Nox2, and three cytosolic regulatory subunits, p47phox, p67phox, and Rac1. Statins inhibit complex formation primarily via the inhibition of Rac1 geranylgeranylation, thereby preventing its translocation and subsequent activation at the cell membrane, essential for NADPH oxidase complex assembly and activation.
Oxidative stress following ischemia-reperfusion during lung transplantation is an important cause of lung injury, and is a significant factor in primary graft failure and mortality after the procedure. The mechanisms of ROS production in these circumstances include NADPH oxidase activity and NO bioavailability both subject to modulation by statins. Naidu et al. examined the effect of statins in a rat model of ischemia-reperfusion lung injury in which lung ischemia was induced for 90 minutes followed by reperfusion for four hours.[82] Pretreatment with oral simvastatin (0.5 mg/kg) for five days prior to these maneuvers was associated with reduced lung vascular permeability as measured by extravasation of radio-iodinated bovine serum albumin. Markers of inflammation such as lung tissue myeloperoxidase content and BAL inflammatory cell counts were also reduced in the statin-treated animals. Notably, observations in human lung transplant patients support the potential utility of statins in this setting as patients taking statins for hyperlipidemia have been observed to have significantly better lung function and six-year survival following lung transplant compared to patients not on a statin.[87]
The effects of statins on mechanical stress-induced lung injury have been examined in at least two animal studies. Muller et al. subjected mice to high tidal volume mechanical ventilation (VT=12 ml/kg) with half of these animals pretreated with simvastatin (20 mg/kg).[83] Oxygenation, as assessed by PaO2/FiO2 at constant PEEP, was significantly higher in statin-treated animals after six hours of mechanical ventilation. Endothelial permeability was measured by BAL fluid content of human serum albumin (HSA) administered in advance intravascularly. HSA BAL/plasma ratios were reduced by statin treatment in both spontaneously breathing and mechanically ventilated mice. Finally, histology demonstrated a normal distribution of caveolae in the endothelium of statin-treated mice indicating intact transcellular-mediated barrier permeability compared to a reduction in untreated mice. In a separate study by Siempos and colleagues, isolated lung preparations from rabbits were subjected to either high pressure (23 cm H2O) or low pressure (11 cm H2O) ventilation. The influence of oral atorvastatin (20 mg/ kg/ day) for three days prior to sacrifice was then examined with respect to vascular permeability via determination of an ultrafiltration coefficient (Kfc).[84] At baseline, there was no difference in Kfc between any of the lung preparations. Although high pressure ventilation for one hour was associated with increased Kfc compared to lungs subjected to low pressure, there was no difference in Kfc between lungs subjected to high and low pressure amongst the statin-treated animals. These findings were corroborated by measurement of lung weight gain. Interestingly, mean pulmonary artery pressure during high pressure ventilation was 10-fold higher in lungs from untreated animals compared to those pretreated with a statin. Lungs were also twice as compliant in the statin-treated groups resulting in a greater increase in tidal volume (normalized to initial lung weight) during pressure control ventilation.
Finally, burn injury and smoke inhalation are important precipitants of lung injury in trauma patients. Belli et al. investigated the effect of statins in a rat model of burn injury and cotton smoke inhalation.[86] The degree of lung injury was measured in this study solely on the basis of histologic scoring for inflammation and leukocyte tissue infiltration which both measured significantly lower when animals were treated with orogastrically administered simvastatin (25 mg/kg/day) for 48 hours prior to burn injury and smoke inhalation events.
Clinical studies of statins in ALI
The effects of statins on endothelial barrier function and lung inflammation in relevant animal models serves as the basis for their potential use as therapeutic agents in patients with ALI. However, the HMG-CoA-dependent prenylation pathway is essential to numerous processes in a variety of cell types, many of which may be significant players in the pathogenesis of both ALI and the comorbidities that influence its outcome. Thus, as animal models of ALI are known to lack the ability to reproduce all of these key pathologic components of the human condition,[88] the effects of statins in clinical studies of ALI patients, either positive or negative, must be interpreted independently. Additionally, both the choice of statin and the timing of intervention, which are subject to less restriction in animal studies, are limited when studying the human condition. The available forms of statins differ pharmacodynamically based on the degree of their individual hydrophilic or lipophilic properties (Fig. 5). The lipophilic statins are more widely distributed after administration and are therefore associated with a greater number of side effects, favoring the use of the more hydrophilic forms for study in critically ill populations with lung injury. However, the wider distribution of lipophilic statins is also more likely to allow them to directly modualte lung endothelial function. There is data to support this notion as simvastatin has been observed to be more effective at improving endothelium-dependent vasodilation in diabetic patients than its hydrophilic counterpart, rosuvastatin.[89]
Figure 5.
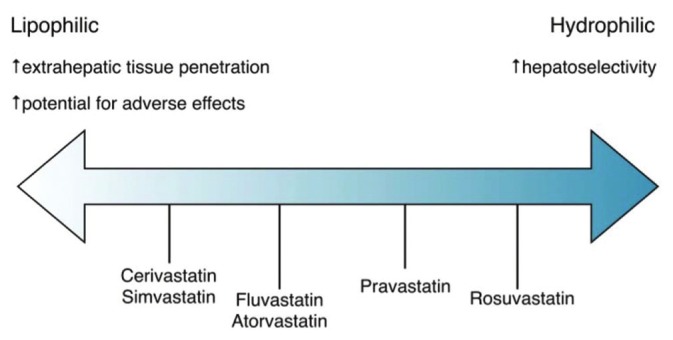
Variability of Statin Lipophilicity. The solubilities of various statins are represented along a spectrum from the most lipophilic to the most hydrophilic. While the more lipophilic statins are associated with a higher propensity for adverse effects (e.g., myositis and myopathy) by virtue of their increased extrahepatic tissue penetration relative to the more hydrophilic statins, they may also be more likely to confer endothelial-protective effects relevant to ALI for this same reason.
Another potential problem associated with clinical studies of statins in ALI is the fact that intervention with statins in human studies is necessarily delayed by the time required for clinical diagnostic criteria to be met following an event precipitating lung injury. The luxury of knowing precisely when this inciting event has occurred is largely confined to the animal model, where statin administration often coincides with or occurs prior to the initiation of injury. This favors the ability of statins to modify the endothelial barrier dysfunction and increased lung vascular permeability which occurs early in the disease process. In contrast, the degree of ongoing endothelial barrier dysfunction versus progression to cellular infiltration and formation of airspace granulation tissue at the time of identification and enrollment of human ALI patients into clinical trials is not known. What little data exists regarding this question suggests that the proliferative stages of lung injury have already begun at a time normally considered early in the clinical course of illness.[90] Therefore, the benefits of statins in human ALI trials may ultimately be dependent in part on the specific statin studied or on strategies aimed at identifying patients early in their clinical course or even, ideally, prior to the onset of ALI.
Nonetheless, an early clinical study of the effects of statins in ALI investigated patients who had previously been prescribed statins for cardiovascular disease. This retrospective cohort study of 178 consecutive patients admitted to an ICU who met the consensus definition of ALI identified 45 patients who were receiving a statin at the time of hospitalization.[91] Statin-treated patients were found to have had a lower baseline level of organ dysfunction and sepsis upon admission to the ICU compared to those who had never received a statin. However, statin use did not appear to affect the subsequent clinical course when considered by multivariate analysis. In a separate study, a cross-sectional analysis of a prospective cohort of 575 critically ill patients, prehospital statin use was associated with a reduced rate of sepsis at the time of enrollment.[92] In addition, the subsequent development of ALI in the next four days was also found to be reduced in the group of patients receiving statins (19.4 vs. 28.9%, P = 0.03). Multivariate logistic regression analysis confirmed both the lower rate of severe sepsis associated with statin use, and the reduced risk for development of ALI in these patients.
The effects of statins in a human model of inflammatory lung injury were reported in a double-blinded, placebo-controlled study of 30 healthy subjects exposed to 50 μg of inhaled bacterial endotoxin and divided into three groups: (1) 10 subjects had been given 40 mg of simvastatin for four days prior to endotoxin administration; (2) another 10 subjects were treated with 80 mg of simvastatin; and (3) the third group of 10 subjects received a placebo.[93] BAL fluid was taken six hours after endotoxin exposure and examined for markers of pulmonary inflammation. Patients treated with simvastatin were found to have significant reductions in BAL fluid neutrophil counts, myeloperoxidase concentrations, and TNF-α concentration. There were no differences between subjects given 40 versus 80 mg of simvastatin.
Finally, a Phase 2 clinical trial examining the effects of statins in ALI patients was completed by the Irish Critical Care Trials Group in 2010.[94] This double-blinded, placebo-controlled, single-center, randomized controlled trial studied mechanically ventilated patients admitted to an ICU who met the consensus definition of ALI, excluding those who exhibited high creatine kinase levels, elevated liver function tests, severe renal disease without renal replacement therapy, severe liver disease, or a history of prior treatment with a statin. Patients receiving 80 mg of simvastatin for up to 14 days while on mechanical ventilation were compared with those receiving placebo under the same conditions. The study was underpowered to detect any differences in clinical outcomes associated with statin use. However, there were no differences in adverse events between groups and there were no unexpected serious events associated with statin use in this population.
Collectively, these studies have paved the way for the SAILS study (Statins for Acutely Injured Lungs from Sepsis), a multicentered study sponsored by the NHLBI-ARDS network and expected to complete enrollment by the end of 2012 (http://clinicaltrials.gov/ct2/show/NCT00979121). In this multicentered, double-blinded, placebo-controlled study, patients with ALI will be randomized to receive either rosuvastatin (20 mg daily) or placebo for up to 28 days or until discharge from the hospital. Although the results of this study are eagerly awaited, there are some potential limitations of the study design that should be noted in advance including the use of rosuvastatin, the most hydrophilic statin available and thus likely to have less vascular-protective effects, submaximal dosing, and a study population that already meets ALI criteria at the time of enrollment, as opposed to a population at risk whose outcomes could potentially be more favorably affected by statin treatment.
CONCLUSION
Since their discovery as antimicrobial agents in the 1970s, inhibitors of HMG-CoA reductase have had a widespread impact on the understanding and treatment of cardiovascular disease. The evolving literature demonstrates that their impact continues to grow, expanding into the realm of disorders characterized by increased lung vascular permeability and inflammation, including ALI. While it remains to be seen whether statins will ultimately prove useful as therapeutic agents in the clinical management of ALI, or perhaps in a subgroup of ALI patients, they have already provided invaluable insights into many relevant endothelial signaling events and lung inflammatory responses which underlie this devastating condition.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Endo A. The discovery and development of HMG-CoA reductase inhibitors. J Lipid Res. 1992;33:1569–82. [PubMed] [Google Scholar]
- 2.Alberts AW, Chen J, Kuron G, Hunt V, Huff J, Hoffman C, et al. Mevinolin: A highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci U S A. 1980;77:3957–61. doi: 10.1073/pnas.77.7.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Endo A, Kuroda M, Tsujita Y. ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrinium. J Antibiot. 1976;29:1346–8. doi: 10.7164/antibiotics.29.1346. [DOI] [PubMed] [Google Scholar]
- 4.Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–9. [PubMed] [Google Scholar]
- 5.Landmesser U, Bahlmann F, Mueller M, Spiekermann S, Kirchhoff N, Schulz S, et al. Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation. 2005;111:2356–63. doi: 10.1161/01.CIR.0000164260.82417.3F. [DOI] [PubMed] [Google Scholar]
- 6.Liu PY, Liu YW, Lin LJ, Chen JH, Liao JK. Evidence for statin pleiotropy in humans: Differential effects of statins and ezetimibe on rho-associated coiled-coil containing protein kinase activity, endothelial function, and inflammation. Circulation. 2009;119:131–8. doi: 10.1161/CIRCULATIONAHA.108.813311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buhaescu I, Izzedine H. Mevalonate pathway: A review of clinical and therapeutical implications. Clin Biochem. 2007;40:575–84. doi: 10.1016/j.clinbiochem.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 8.Blum A, Shamburek R. The pleiotropic effects of statins on endothelial function, vascular inflammation, immunomodulation and thrombogenesis. Atherosclerosis. 2009;203:325–30. doi: 10.1016/j.atherosclerosis.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 9.Bu DX, Griffin G, Lichtman AH. Mechanisms for the anti-inflammatory effects of statins. Curr Opin Lipidol. 2011;22:165–70. doi: 10.1097/MOL.0b013e3283453e41. [DOI] [PubMed] [Google Scholar]
- 10.Fessler MB, Young SK, Jeyaseelan S, Lieber JG, Arndt PG, Nick JA, et al. A role for hydroxy-methylglutaryl coenzyme a reductase in pulmonary inflammation and host defense. Am J Respir Crit Care Med. 2005;171:606–15. doi: 10.1164/rccm.200406-729OC. [DOI] [PubMed] [Google Scholar]
- 11.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, S, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–93. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 12.Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: Four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol. 2005;33:319–27. doi: 10.1165/rcmb.F305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suratt BT, Parsons PE. Mechanisms of acute lung injury/acute respiratory distress syndrome. Clin Chest Med. 2006;27:579–89. doi: 10.1016/j.ccm.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 14.Amaya M, Baranova A, van Hoek ML. Protein prenylation: A new mode of host-pathogen interaction. Biochem Biophys Res Commun. 2011;416:1–6. doi: 10.1016/j.bbrc.2011.10.142. [DOI] [PubMed] [Google Scholar]
- 15.Sinensky M. Recent advances in the study of prenylated proteins. Biochim Biophys Acta. 2000;1484:93–106. doi: 10.1016/s1388-1981(00)00009-3. [DOI] [PubMed] [Google Scholar]
- 16.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–35. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 17.Jacobson JR. Statins in endothelial signaling and activation. Antioxid Redox Signal. 2009;11:811–21. doi: 10.1089/ars.2008.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parmar KM, Nambudiri V, Dai G, Larman HB, Gimbrone MA, Jr, Garcia-Cardena G. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. J Biol Chem. 2005;280:26714–9. doi: 10.1074/jbc.C500144200. [DOI] [PubMed] [Google Scholar]
- 19.Spite M, Serhan CN. Novel lipid mediators promote resolution of acute inflammation: Impact of aspirin and statins. Circ Res. 2010;107:1170–84. doi: 10.1161/CIRCRESAHA.110.223883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Planagumà A, Pfeffer MA, Rubin G, Croze R, Uddin M, Serhan CN, et al. Lovastatin decreases acute mucosal inflammation via 15-epi-lipoxin A4. Mucosal Immunol. 2010;3:270–9. doi: 10.1038/mi.2009.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majno G, Palade GE. Studies on inflammation. 1. The effect of histamine and serotonin on vascular permeability: an electron microscopic study. J Biophys Biochem Cytol. 1961;11:571–605. doi: 10.1083/jcb.11.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Majno G, Palade GE, Schoefl GI. Studies on inflammation. II. The site of action of histamine and serotonin along the vascular tree: A topographic study. J Biophys Biochem Cytol. 1961;11:607–26. doi: 10.1083/jcb.11.3.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia JG, Schaphorst KL. Regulation of endothelial cell gap formation and paracellular permeability. J Investig Med. 1995;43:117–26. [PubMed] [Google Scholar]
- 24.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 25.Shasby DM, Shasby SS, Sullivan JM, Peach MJ. Role of endothelial cell cytoskeleton in control of endothelial permeability. Circ Res. 1982;51:657–61. doi: 10.1161/01.res.51.5.657. [DOI] [PubMed] [Google Scholar]
- 26.Lang G, Chiang ET, Garcia JG. Lung vascular dysfunction and repair in acute lung injury: Role of the endothelial cytoskeleton. In: Choi A, editor. Acute Respiratory Distress Syndrome. 2nd ed. New York: Informa Healthcare USA Inc; 2010. pp. 282–86. [Google Scholar]
- 27.Jacobson JR, Dudek SM, Birukov KG, Ye SQ, Grigoryev DN, Girgis RE, et al. Cytoskeletal activation and altered gene expression in endothelial barrier regulation by simvastatin. Am J Respir Cell Mol Biol. 2004;30:662–70. doi: 10.1165/rcmb.2003-0267OC. [DOI] [PubMed] [Google Scholar]
- 28.Chen W, Pendyala S, Natarajan V, Garcia JG, Jacobson JR. Endothelial cell barrier protection by simvastatin: GTPase regulation and NADPH oxidase inhibition. Am J Physiol Lung Cell Mol Physiol. 2008;295:L575–83. doi: 10.1152/ajplung.00428.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chabot F, Mitchell JA, Gutteridge JM, Evans TW. Reactive oxygen species in acute lung injury. Eur Respir J. 1998;11:745–57. [PubMed] [Google Scholar]
- 30.Kietzmann D, Kahl R, Muller M, Burchardi H, Kettler D. Hydrogen peroxide in expired breath condensate of patients with acute respiratory failure and with ARDS. Intensive Care Med. 1993;19:78–81. doi: 10.1007/BF01708366. [DOI] [PubMed] [Google Scholar]
- 31.Lenz AG, Jorens PG, Meyer B, De Backer W, Van Overveld F, Bossaert L, et al. Oxidatively modified proteins in bronchoalveolar lavage fluid of patients with ARDS and patients at-risk for ARDS. Eur Respir J. 1999;13:169–74. doi: 10.1034/j.1399-3003.1999.13a31.x. [DOI] [PubMed] [Google Scholar]
- 32.Metnitz PG, Bartens C, Fischer M, Fridrich P, Steltzer H, Druml W. Antioxidant status in patients with acute respiratory distress syndrome. Intensive Care Med. 1999;25:180–5. doi: 10.1007/s001340050813. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt R, Luboeinski T, Markart P, Ruppert C, Daum C, Grimminger F, et al. Alveolar antioxidant status in patients with acute respiratory distress syndrome. Eur Respir J. 2004;24:994–9. doi: 10.1183/09031936.04.00120703. [DOI] [PubMed] [Google Scholar]
- 34.Dimmeler S, Brinkmann S, Neugebauer E. Endotoxin-induced changes of endothelial cell viability and permeability: Protective effect of a 21-aminosteroid. Eur J Pharmacol. 1995;287:257–61. doi: 10.1016/0014-2999(95)00499-8. [DOI] [PubMed] [Google Scholar]
- 35.Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. Am J Physiol Cell Physiol. 2001;280:C719–41. doi: 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- 36.Brigham KL. Role of free radicals in lung injury. Chest. 1986;89:859–63. doi: 10.1378/chest.89.6.859. [DOI] [PubMed] [Google Scholar]
- 37.Rinaldo JE. Mediation of ARDS by leukocytes.Clinical evidence and implications for therapy. Chest. 1986;89:590–3. doi: 10.1378/chest.89.4.590. [DOI] [PubMed] [Google Scholar]
- 38.Park HS, Chun JN, Jung HY, Choi C, Bae YS. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc Res. 2006;72:447–55. doi: 10.1016/j.cardiores.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 39.Pendyala S, Usatyuk PV, Gorshkova IA, Garcia JG, Natarajan V. Regulation of NADPH oxidase in vascular endothelium: The role of phospholipases, protein kinases, and cytoskeletal proteins. Antioxid Redox Signal. 2009;11:841–60. doi: 10.1089/ars.2008.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones SA, O’Donnell VB, Wood JD, Broughton JP, Hughes EJ, Jones OT. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am J Physiol. 1996;271:H1626–34. doi: 10.1152/ajpheart.1996.271.4.H1626. [DOI] [PubMed] [Google Scholar]
- 41.Li JM, Shah AM. Differential NADPH- versus NADH-dependent superoxide production by phagocyte-type endothelial cell NADPH oxidase. Cardiovasc Res. 2001;52:477–86. doi: 10.1016/s0008-6363(01)00407-2. [DOI] [PubMed] [Google Scholar]
- 42.Usatyuk PV, Romer LH, He D, Parinandi NL, Kleinberg ME, Zhan S, et al. Regulation of hyperoxia-induced NADPH oxidase activation in human lung endothelial cells by the actin cytoskeleton and cortactin. J Biol Chem. 2007;282:23284–95. doi: 10.1074/jbc.M700535200. [DOI] [PubMed] [Google Scholar]
- 43.Knepler JL, Jr, Taher LN, Gupta MP, Patterson C, Pavalko F, Ober MD, et al. Peroxynitrite causes endothelial cell monolayer barrier dysfunction. Am J Physiol Cell Physiol. 2001;281:C1064–75. doi: 10.1152/ajpcell.2001.281.3.C1064. [DOI] [PubMed] [Google Scholar]
- 44.Muzaffar S, Jeremy JY, Angelini GD, Stuart-Smith K, Shukla N. Role of the endothelium and nitric oxide synthases in modulating superoxide formation induced by endotoxin and cytokines in porcine pulmonary arteries. Thorax. 2003;58:598–604. doi: 10.1136/thorax.58.7.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cobb JP, Danner RL. Nitric oxide and septic shock. JAMA. 1996;275:1192–6. [PubMed] [Google Scholar]
- 46.Gessner C, Hammerschmidt S, Kuhn H, Lange T, Engelmann L, Schauer J, et al. Exhaled breath condensate nitrite and its relation to tidal volume in acute lung injury. Chest. 2003;124:1046–52. doi: 10.1378/chest.124.3.1046. [DOI] [PubMed] [Google Scholar]
- 47.Wort SJ, Evans TW. The role of the endothelium in modulating vascular control in sepsis and related conditions. Br Med Bull. 1999;55:30–48. doi: 10.1258/0007142991902286. [DOI] [PubMed] [Google Scholar]
- 48.McClintock DE, Ware LB, Eisner MD, Wickersham N, Thompson BT, Matthay MA. Higher urine nitric oxide is associated with improved outcomes in patients with acute lung injury. Am J Respir Crit Care Med. 2007;175:256–62. doi: 10.1164/rccm.200607-947OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–35. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 50.Feron O, Dessy C, Desager JP, Balligand JL. Hydroxy-methylglutaryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation. 2001;103:113–8. doi: 10.1161/01.cir.103.1.113. [DOI] [PubMed] [Google Scholar]
- 51.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–10. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laufs U, Endres M, Stagliano N, Amin-Hanjani S, Chui DS, Yang SX, et al. Neuroprotection mediated by changes in the endothelial actin cytoskeleton. J Clin Invest. 2000;106:15–24. doi: 10.1172/JCI9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–71. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 54.Kosmidou I, Moore JP, Weber M, Searles CD. Statin treatment and 3’ polyadenylation of eNOS mRNA. Arterioscler Thromb Vasc Biol. 2007;27:2642–9. doi: 10.1161/ATVBAHA.107.154492. [DOI] [PubMed] [Google Scholar]
- 55.Ou H, Shen YH, Utama B, Wang J, Wang X, Coselli J, et al. Effect of nuclear actin on endothelial nitric oxide synthase expression. Arterioscler Thromb Vasc Biol. 2005;25:2509–14. doi: 10.1161/01.ATV.0000189306.99112.4c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pederson T, Aebi U. Nuclear actin extends, with no contraction in sight. Mol Biol Cell. 2005;16:5055–60. doi: 10.1091/mbc.E05-07-0656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, et al. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1998;95:8880–5. doi: 10.1073/pnas.95.15.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Skaletz-Rorowski A, Lutchman M, Kureishi Y, Lefer DJ, Faust JR, Walsh K. HMG-CoA reductase inhibitors promote cholesterol-dependent Akt/PKB translocation to membrane domains in endothelial cells. Cardiovasc Res. 2003;57:253–64. doi: 10.1016/s0008-6363(02)00618-1. [DOI] [PubMed] [Google Scholar]
- 59.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andjelkovic M, Jakubowicz T, Cron P, Ming XF, Han JW, Hemmings BA. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc Natl Acad Sci U S A. 1996;93:5699–704. doi: 10.1073/pnas.93.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang D, Hirase T, Inoue T, Node K. Atorvastatin inhibits angiotensin II-induced T-type Ca2+ channel expression in endothelial cells. Biochem Biophys Res Commun. 2006;347:394–400. doi: 10.1016/j.bbrc.2006.06.084. [DOI] [PubMed] [Google Scholar]
- 62.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: Upstream mediators. Circ Res. 2002;91:406–13. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- 63.Warnholtz A, Nickenig G, Schulz E, Macharzina R, Bräsen JH, Skatchkov M, et al. Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: Evidence for involvement of the renin-angiotensin system. Circulation. 1999;99:2027–33. doi: 10.1161/01.cir.99.15.2027. [DOI] [PubMed] [Google Scholar]
- 64.Wassmann S, Laufs U, Bäumer AT, Müller K, Konkol C, Sauer H, et al. Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: Involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol Pharmacol. 2001;59:646–54. doi: 10.1124/mol.59.3.646. [DOI] [PubMed] [Google Scholar]
- 65.Morawietz H, Erbs S, Holtz J, Schubert A, Krekler M, Goettsch W, et al. Endothelial Protection, AT1 blockade and Cholesterol-Dependent Oxidative Stress: The EPAS trial. Circulation. 2006;114:I296–301. doi: 10.1161/CIRCULATIONAHA.105.001313. [DOI] [PubMed] [Google Scholar]
- 66.Eto M, Kozai T, Cosentino F, Joch H, Luscher TF. Statin prevents tissue factor expression in human endothelial cells: role of Rho/Rho-kinase and Akt pathways. Circulation. 2002;105:1756–9. doi: 10.1161/01.cir.0000015465.73933.3b. [DOI] [PubMed] [Google Scholar]
- 67.Bourcier T, Libby P. HMG CoA reductase inhibitors reduce plasminogen activator inhibitor-1 expression by human vascular smooth muscle and endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:556–62. doi: 10.1161/01.atv.20.2.556. [DOI] [PubMed] [Google Scholar]
- 68.Lin SJ, Chen YH, Lin FY, Hsieh LY, Wang SH, Lin CY, et al. Pravastatin induces thrombomodulin expression in TNFalpha-treated human aortic endothelial cells by inhibiting Rac1 and Cdc42 translocation and activity. J Cell Biochem. 2007;101:642–53. doi: 10.1002/jcb.21206. [DOI] [PubMed] [Google Scholar]
- 69.Masamura K, Oida K, Kanehara H, Suzuki J, Horie S, Ishii H, et al. Pitavastatin-induced thrombomodulin expression by endothelial cells acts via inhibition of small G proteins of the Rho family. Arterioscler Thromb Vasc Biol. 2003;23:512–7. doi: 10.1161/01.ATV.0000060461.64771.F0. [DOI] [PubMed] [Google Scholar]
- 70.Shi J, Wang J, Zheng H, Ling W, Joseph J, Li D, et al. Statins increase thrombomodulin expression and function in human endothelial cells by a nitric oxide-dependent mechanism and counteract tumor necrosis factor alpha-induced thrombomodulin downregulation. Blood Coagul Fibrinolysis. 2003;14:575–85. doi: 10.1097/00001721-200309000-00010. [DOI] [PubMed] [Google Scholar]
- 71.Esmon C. Thrombomodulin as a model of molecular mechanisms that modulate protease specificity and function at the vessel surface. FASEB J. 1995;9:946–55. doi: 10.1096/fasebj.9.10.7615164. [DOI] [PubMed] [Google Scholar]
- 72.Fay WP, Garg N, Sunkar M. Vascular functions of the plasminogen activation system. Arterioscler Thromb Vasc Biol. 2007;27:1231–7. doi: 10.1161/ATVBAHA.107.140046. [DOI] [PubMed] [Google Scholar]
- 73.van ’t Veer C, Mann KG. Regulation of tissue factor initiated thrombin generation by the stoichiometric inhibitors tissue factor pathway inhibitor, antithrombin-III, and heparin cofactor-II. J Biol Chem. 1997;272:4367–77. doi: 10.1074/jbc.272.7.4367. [DOI] [PubMed] [Google Scholar]
- 74.Bogatcheva NV, Birukova A, Borbiev T, Kolosova I, Liu F, Garcia JG, et al. Caldesmon is a cytoskeletal target for PKC in endothelium. J Cell Biochem. 2006;99:1593–605. doi: 10.1002/jcb.20823. [DOI] [PubMed] [Google Scholar]
- 75.Stasek JE, Jr, Patterson CE, Garcia JG. Protein kinase C phosphorylates caldesmon77 and vimentin and enhances albumin permeability across cultured bovine pulmonary artery endothelial cell monolayers. J Cell Physiol. 1992;153:62–75. doi: 10.1002/jcp.1041530110. [DOI] [PubMed] [Google Scholar]
- 76.Song Y, Pittet JF, Huang X, He H, Lynch SV, Violette SM, et al. Role of integrin alphav beta6 in acute lung injury induced by Pseudomonas aeruginosa. Infect Immun. 2008;76:2325–32. doi: 10.1128/IAI.01431-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Su G, Hodnett M, Wu N, Atakilit A, Kosinski C, Godzich M, et al. Integrin alphavbeta5 regulates lung vascular permeability and pulmonary endothelial barrier function. Am J Respir Cell Mol Biol. 2007;36:377–86. doi: 10.1165/rcmb.2006-0238OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wary KK, Vogel SM, Garrean S, Zhao YD, Malik AB. Requirement of alpha(4)beta(1) and alpha(5)beta(1) integrin expression in bone-marrow-derived progenitor cells in preventing endotoxin-induced lung vascular injury and edema in mice. Stem Cells. 2009;27:3112–20. doi: 10.1002/stem.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Giancotti FG, Tarone G. Positional control of cell fate through joint integrin/receptor protein kinase signaling. Annu Rev Cell Dev Biol. 2003;19:173–206. doi: 10.1146/annurev.cellbio.19.031103.133334. [DOI] [PubMed] [Google Scholar]
- 80.Chen W, Garcia JG, Jacobson JR. Integrin beta4 attenuates SHP-2 and MAPK signaling and reduces human lung endothelial inflammatory responses. J Cell Biochem. 2010;110:718–24. doi: 10.1002/jcb.22582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jacobson JR, Barnard JW, Grigoryev DN, Ma SF, Tuder RM, Garcia JG. Simvastatin attenuates vascular leak and inflammation in murine inflammatory lung injury. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1026–32. doi: 10.1152/ajplung.00354.2004. [DOI] [PubMed] [Google Scholar]
- 82.Naidu BV, Woolley SM, Farivar AS, Thomas R, Fraga C, Mulligan MS. Simvastatin ameliorates injury in an experimental model of lung ischemia-reperfusion. J Thorac Cardiovasc Surg. 2003;126:482–9. doi: 10.1016/s0022-5223(03)00699-8. [DOI] [PubMed] [Google Scholar]
- 83.Müller HC, Hellwig K, Rosseau S, Tschernig T, Schmiedl A, Gutbier B, et al. Simvastatin attenuates ventilator-induced lung injury in mice. Crit Care. 2010;14:R143. doi: 10.1186/cc9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Siempos II, Maniatis NA, Kopterides P, Magkou C, Glynos C, Roussos C, et al. Pretreatment with atorvastatin attenuates lung injury caused by high-stretch mechanical ventilation in an isolated rabbit lung model. Crit Care Med. 2010;38:1321–8. doi: 10.1097/CCM.0b013e3181d9dad6. [DOI] [PubMed] [Google Scholar]
- 85.Kim JW, Rhee CK, Kim TJ, Kim YH, Lee SH, Yoon HK, et al. Effect of pravastatin on bleomycin-induced acute lung injury and pulmonary fibrosis. Clin Exp Pharmacol Physiol. 2010;37:1055–63. doi: 10.1111/j.1440-1681.2010.05431.x. [DOI] [PubMed] [Google Scholar]
- 86.Belli S, Basaran O, Ozdemir BH, Türkoğlu S, Karabay G, Kut A, et al. Protective role of simvastatin on lung damage caused by burn and cotton smoke inhalation in rats. J Surg Res. 2011;167:e283–90. doi: 10.1016/j.jss.2010.01.035. [DOI] [PubMed] [Google Scholar]
- 87.Johnson BA, Iacono AT, Zeevi A, McCurry KR, Duncan SR. Statin use is associated with improved function and survival of lung allografts. Am J Respir Crit Care Med. 2003;167:1271–8. doi: 10.1164/rccm.200205-410OC. [DOI] [PubMed] [Google Scholar]
- 88.Proudfoot AG, McAuley DF, Griffiths MJ, Hind M. Human models of acute lung injury. Dis Model Mech. 2011;4:145–53. doi: 10.1242/dmm.006213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bellia A, Rizza S, Galli A, Fabiano R, Donadel G, Lombardo MF, et al. Early vascular and metabolic effects of rosuvastatin compared with simvastatin in patients with type 2 diabetes. Atherosclerosis. 2010;210:199–201. doi: 10.1016/j.atherosclerosis.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 90.Polunovsky VA, Chen B, Henke C, Snover D, Wendt C, Ingbar DH, et al. Role of mesenchymal cell death in lung remodeling after injury. J Clin Invest. 1993;92:388–97. doi: 10.1172/JCI116578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kor DJ, Iscimen R, Yilmaz M, Brown MJ, Brown DR, Gajic O. Statin administration did not influence the progression of lung injury or associated organ failures in a cohort of patients with acute lung injury. Intensive Care Med. 2009;35:1039–46. doi: 10.1007/s00134-009-1421-8. [DOI] [PubMed] [Google Scholar]
- 92.O’Neal HR, Jr, Koyama T, Koehler EA, Siew E, Curtis BR, Fremont RD, et al. Prehospital statin and aspirin use and the prevalence of severe sepsis and acute lung injury/acute respiratory distress syndrome. Crit Care Med. 2011;39:1343–50. doi: 10.1097/CCM.0b013e3182120992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shyamsundar M, McKeown ST, O’Kane CM, Craig TR, Brown V, Thickett DR, et al. Simvastatin decreases lipopolysaccharide-induced pulmonary inflammation in healthy volunteers. Am J Respir Crit Care Med. 2009;179:1107–14. doi: 10.1164/rccm.200810-1584OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Craig TR, Duffy MJ, Shyamsundar M, McDowell C, O’Kane CM, Elborn JS, et al. A randomized clinical trial of hydroxymethylglutaryl- coenzyme a reductase inhibition for acute lung injury (The HARP Study) Am J Respir Crit Care Med. 2011;183:620–6. doi: 10.1164/rccm.201003-0423OC. [DOI] [PubMed] [Google Scholar]