

Abstract
Caveolin-1 is a key regulator of pulmonary endothelial barrier function. Here, we tested the hypothesis that caveolin-1 expression is required for ventilator-induced lung injury (VILI). Caveolin-1 gene-disrupted (Cav-1-/-) and age-, sex-, and strain-matched wild-type (WT) control mice were ventilated using two protocols: volume-controlled with protective (8 mL/kg) versus injurious (21 mL/Kg) tidal volume for up to 6 hours; and pressure-controlled with protective (airway pressure = 12 cm H2O) versus injurious (30 cm H2O) ventilation to induce lung injury. Lung microvascular permeability (whole-lung 125I-albumin accumulation, lung capillary filtration coefficient [Kf, c]) and inflammatory markers (bronchoalveolar lavage [BAL] cytokine levels and neutrophil counts) were measured. We also evaluated histologic sections from lungs, and the time course of Src kinase activation and caveolin-1 phosphorylation. VILI induced a 1.7-fold increase in lung 125I-albumin accumulation, fourfold increase in Kf, c, significantly increased levels of cytokines CXCL1 and interleukin-6, and promoted BAL neutrophilia in WT mice. Lung injury by these criteria was significantly reduced in Cav-1-/- mice but fully restored by i.v. injection of liposome/Cav-1 cDNA complexes that rescued expression of Cav-1 in lung microvessels. As thrombin is known to play a significant role in mediating stretch-induced vascular injury, we observed in cultured mouse lung microvascular endothelial cells (MLECs) thrombin-induced albumin hyperpermeability and phosphorylation of p44/42 MAP kinase in WT but not in Cav-1-/- MLECs. Thus, caveolin-1 expression is required for mechanical stretch-induced lung inflammation and endothelial hyperpermeability in vitro and in vivo.
Keywords: high tidal volume mechanical ventilation, lung inflammation, thrombin, caveolae, albumin permeability
Acute Lung Injury (ALI) is a major complication of systemic inflammation due to sepsis, pneumonia, multiple trauma, and gastric acid aspiration.[1,2] It is characterized by increased-permeability pulmonary edema resulting from damage to the alveolar-capillary membrane upon exposure to noxious stimuli such as bacterial products, endogenous vasoactive substances, and also to mechanical distention during artificial ventilation.[1] Disruption of endothelial cell (EC) junctions by destabilizing homotypic VE cadherin adhesive interactions[3] or by provoking contraction of the actin cytoskeleton and cellular retraction are thought to be primary mechanisms causing lung edema formation.[4] It was previously demonstrated that caveolin-1 (Cav-1), a 22 kDa structural protein of caveolar endocytic vesicles, also functions as a scaffold for various cell-signaling and junctional proteins.[5,6] Cav-1 expression has been shown not only to be required for caveolae formation and trafficking,[7] but also to play a critical role in regulating junctional integrity.[7] Several cell signaling pathways regulated by Cav-1 have been implicated in the mechanisms by which vasoactive permeability- enhancing agents increase actin polymerization/cytoskeletal remodeling and decrease cell-cell adhesion, most notably Rho, Rac, Src, Ca2+, NO, and MAP kinases.[7] In this context, mice lacking Cav-1 (Cav-1-/-) show tolerance to lung injury induced by lipopolysaccharide and oxidants,[8,9] defective neutrophil function,[10] and endothelial Ca++ influx.[11] We thus hypothesized that Cav-1 expression is required for endothelial cell inflammatory and stretch-induced signaling that mediates increased endothelial permeability during ventilator-induced ALI.
To approach this hypothesis, we subjected Cav-1-/- mice to injurious mechanical ventilation (ventilator-induced lung injury-VILI). Mechanical stretch applied to lung endothelium and epithelium by ventilation with excessive tidal volumes leads to Ca++ influx, activation of protein kinases, transcription of proinflammatory genes, and formation of actin stress fibers.[12–14] Thus, EC distention can increase endothelial permeability in a manner comparable to vasoactive agents. In a previous study, Hoetzel et al. reported increased susceptibility of Cav-1-/- mice to positive-pressure mechanical ventilation[15] by augmenting the production of carbon monoxide. We used a higher level of mechanical ventilation (approximately twofold greater than normal tidal volume ventilation) in order to induce distention on the endothelium similar to what one might expect in ventilated patients with lung injury where healthy alveoli receive the bulk of the delivered gas volume. For confirmation, we employed a pressure-controlled ventilation mode and measured water permeability in ex vivo-perfused mouse lungs following in situ injurious ventilation. Finally, we tested cell-signaling and permeability responses of ECs isolated from wild-type (WT) and Cav-1-/- mice with and without challenge with thrombin to determine the role of endothelial Cav-1-/- expression in the mechanism of pulmonary edema in ALI.
MATERIALS AND METHODS
Mice
Animal studies were approved by the University of Illinois Animal Care and Use Committee. Two independent strains of caveolin-1 knockout mice (Cav1-/-; Cav1tm1Mls and Cav1B6129/SJ2) were purchased from The Jackson Laboratory (Jax; Bar Harbor, Me.). Adult male Black Swiss and B6129/SJ2 mice were purchased from Taconic (Hudson, N.Y.) and Jax for use as strain-matched controls.
Antibodies
Mouse pTyr14-caveolin-1 antibody was purchased from Transduction Laboratories (Franklin Lakes, N.J.). Rabbit anti-phospho-Src family kinase and anti-p44/42 antibody were purchased from Cell Signaling Technology (Danvers, Mass.). All other antibodies were purchased from Sigma Chemical Co., St. Louis, Mo.
Reagents
Enhanced chemiluminesence kit (ECL) and BCA protein quantification reagent were purchased from Pierce (Rockford Ill). Bioplex custom assays were purchased from Biorad, Hercules, Calif. All other reagents were purchased from Sigma Chemical Co., St. Louis, Mo.
Liposome preparation and in vivo gene delivery
Rescue studies were made in mouse lungs from Cav-1-/- mice by liposome-mediated plasmid DNA transfection.[16,19] Myc-tagged WT-Cav-1 cDNA in pcDNA6 plasmid vector were used for caveolin-1 repletion studies. In vivo gene delivery/transfection efficiency was assessed by Western blotting for Myc-tagged Cav-1 and eBFP plasmid (empty vector) transfection and confocal microscopy was used to visualize liposome-mediated lung vascular cDNA transfection in lung sections. The liposome was prepared as previously described and intravenously injected in mice.[16] Briefly, the mixture consisting of dimethyldioctadecylammonium bromide and cholesterol (1:1 molar ratio) was dried using a Rotavaporator (Brinkmann, Westbury, N.Y.) and dissolved in 5% glucose. The complex consisting of the transgene (50 μg per mouse) and liposomes was combined at a ratio of 1 μg of DNA to 8 nmol/L of liposomes. Successful expression of caveolin-1 was confirmed by Western blot of lung homogenates and by detection of transfected eBFP in lung vessels by fluorescence confocal microscopy of 100 μm fresh-frozen lung sections.
Mechanical ventilation protocols
Sevoflurane-anesthetized mice (1.2 MAC) were ventilated via tracheostomy with FiO2 = 0.25, tidal volume 21 mL/Kg, respiratory rate 60 breaths/min, Positive End-Expiratory Pressure (PEEP = 0 injurious ventilation) or 8 mL/Kg, respiratory rate 120 breaths/min, PEEP =2 (protective ventilation) for two or six hours using a Harvard ventilator. For experiments using pressure-controlled ventilation, peak airway pressure was set at either 12 cm H2O, respiratory rate 120 breaths/min (protective ventilation) or 30 cm H2O and respiratory rate of 60 breaths/min (injurious ventilation).
Lung albumin accumulation
125I-BSA (1 μCi) was injected via a PE-10 catheter in the internal jugular vein one hour before the end of the protocol. For catheter insertion, a paramedian neck incision was performed and the jugular vein was separated from the surrounding tissue with a blunt curved forceps. Two 6-0 silk sutures were placed around the vessel, which was ligated by tying the proximal suture. Mild traction was applied to the distal suture and the vessel wall was incised in order to insert the catheter past the distal suture, which was then tied in order to secure the catheter in place. After 60 minutes, a venous blood sample was drawn from the catheter and the lungs were excised, rinsed in PBS and weighed. Radioactivity of lung tissue and blood was measured with a γ-counter (Packard Instruments, Downers Grove, Ill). The lungs were then placed in a drying oven for 48 hours at 60°C and the dry weight was obtained. Results are expressed as lung counts per min/blood counts per min per ml/dry lung weight in grams.
Capillary filtration coefficient Kf,c
Kf,c was measured in isolated lung preparations explanted from mice after two hours of injurious or control ventilation. The procedure is described in detail by Gorovoy et al.[17] Following thoracotomy, a catheter was placed in the pulmonary artery and secured with a suture. The left atrium was excised. The lungs were then perfused to clear all blood from the pulmonary circulation using HEPES-buffered RPMI at a constant flow of 2 ml/min. The heart and lungs were explanted and suspended from a force displacement transducer for weight determination. Pulmonary arterial pressure was measured through the pulmonary artery catheter which was connected to a pressure transducer. To measure Kf, c, a constant pressure pulse of 10 cm H2O (DR) was administered to the lung through a fluid reservoir connected to the main inflow tubing. Four such five-minute pulses were given 20 minutes apart, starting after a 20-minute equilibration period. A regression line was fit through the weight-time tracing of the last three minutes of this maneuver. Kf, c was calculated as the slope of the weight-time tracing/ΔR/dry lung weight in g.
Lung tissue preparation and Western blot
Lungs were flushed free of blood with PBS through the pulmonary artery, extracted lobe by lobe, and snap frozen in liquid nitrogen. One lobe was placed in 1 ml of lysis buffer containing 0.1% SDS, 0.5% Na deoxycholate, 1% Triton-×100, and phosphatase inhibitors I and II diluted 1:100. Specimens were cooled on ice for 30 minutes before homogenization with a Polytron homogenizer at maximal speed for 30 seconds, sonicated (3 × 5 s), and sedimented by centrifugation at 10,000 g for 10 minutes at 4°C. Total protein in each specimen was spectrophotometrically quantified using a BCA kit and the supplied albumin standards. Twenty μg of protein were loaded on 10% SDS-polyacrylamide gels and transferred onto nitrocellulose membranes. Membranes were blocked with 5% fat-free milk and probed with appropriate concentrations of primary antibodies at 4°C overnight. Secondary antibodies conjugated to horseradish peroxidase (HRP) were applied for one hour at room temperature. Protein bands were visualized on film after adding HRP substrate to blots. Densitometry was performed using NIH ImageJ software.
Determination of bronchoalveolar lavage cytokine levels and neutrophil counts
To obtain BAL specimens, three 1-ml aliquots of PBS were injected into the tracheal tube and withdrawn. Specimens were centrifuged at 1,000 ×g at 4°C for five minutes and the supernatant was stored at -80°C. The cell pellets were resuspended and transferred to Cytospin glass slides, centrifuged, fixed with methanol, and stained with the Giemsa-Wright stain. The neutrophil fraction was expressed as the percentage of neutrophils among 500 BAL cells counted. BAL supernatants were analyzed for interleukin-6 and CXCL1 using the Biorad Bioplex system according to the manufacturer's instructions.
Lung tissue histology
Lungs were inflated on a 20 cm H2O column of 4% paraformaldehyde for tissue fixation and to prevent alveolar collapse for one hour. The lungs were then removed and stored in 4% paraformaldehyde until processing. Following tissue dehydration, the tissues were embedded in paraffin, sectioned at a thickness of 5 μM, placed on slides, and stained with hematoxylin-eosin.
Cell-culture and in vitro permeability assay
Cells were isolated and cultured as described.[14] For the permeability assay, cells (2 × 104 cells per well) were plated on 6.5-mm-diameter polyethylene terephthalate microporous filters (Transwell, 1.0 μm pore size, Millipore, Billerica, Mass., USA) and grown for four days at 37°C in 5% CO2. The upper and lower chambers contained 200 and 800 μL growth medium, respectively. On the day of the experiment, growth medium was replaced with medium supplemented with 1% bovine albumin w/o serum. After two hours, the medium from the upper chamber was removed and cells were incubated with medium containing thrombin (1 U/mL, Calbiochem, San Diego, Calif., USA). After the 15-minute incubation period, thrombin-containing medium was removed and replaced with medium containing 1 mg/mL of Fluorescein-Isothiocyanate-labelled bovine serum albumin (Sigma-Aldrich) in addition to thrombin 1U/mL. The fluorescence of each sample from the lower chamber was measured on a fluorescence microplate reader (Tecan GENios, Männedorf, Switzerland) using excitation and emission filters set at 480 and 520 nm, respectively.
Statistical analysis
Data are expressed as sample mean ± standard deviation. Sample means of VILI groups were compared to the baseline data obtained at protective ventilator settings for each strain as well as to each other using one-way ANOVA. The SNK test was applied for post-hoc comparisons involving more than two groups.
RESULTS
Time-course of caveolin-1 phosphorylation in mechanically ventilated lungs
Since Cav-1 phosphorylation has been previously shown to be associated with increased endothelial permeability responses to vasoactive agents,[9] we investigated the time-course of Cav-1 phosphorylation following application of high tidal volumes to mouse lungs by immunoblotting (Fig. 1). We observed a rapid increase in Cav-1 Tyr14 phosphorylation, in addition to concomitant activation of Src kinase, which is known to be the primary kinase responsible of Cav-1 phosphorylation.[18]
Figure 1.
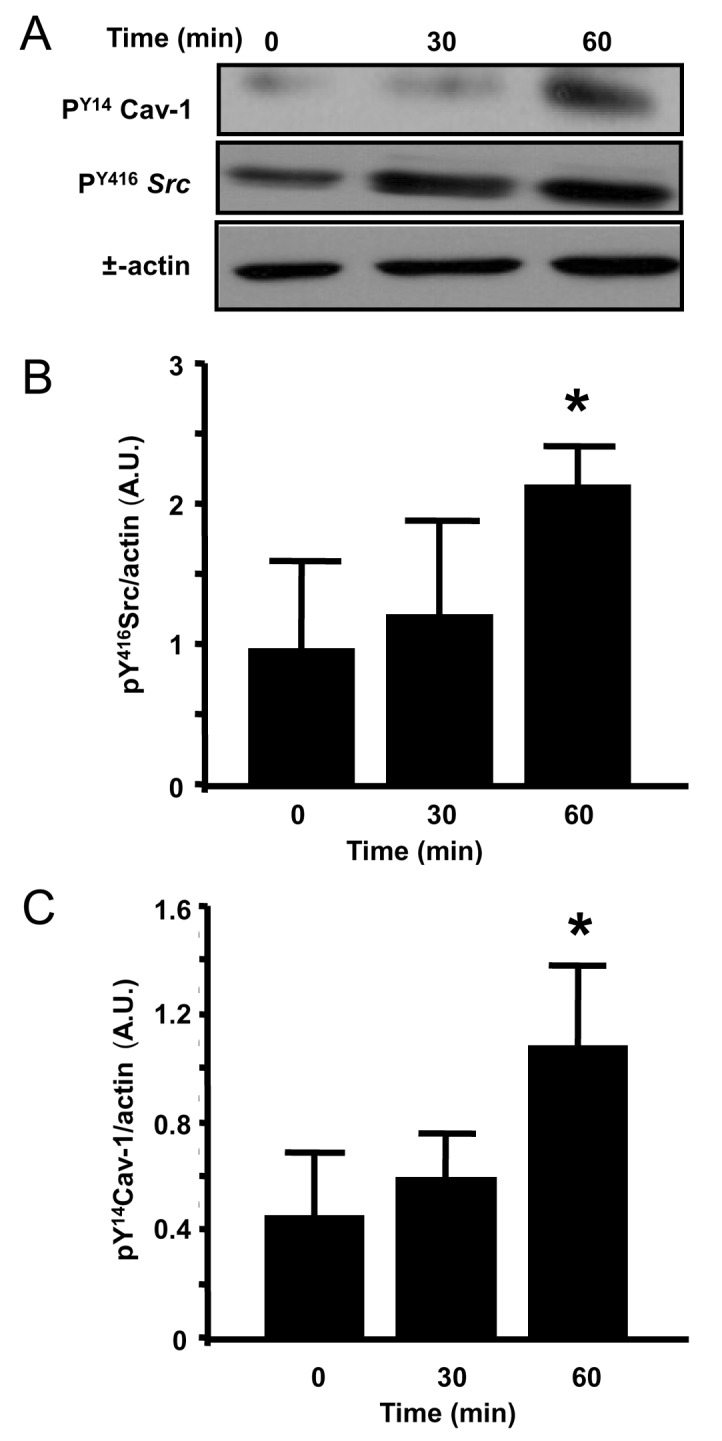
Time course of Src and caveolin-1 phosphorylation: The time course of pY416Src and pY14 caveolin-1 was followed in WT mice undergoing ventilation with 8 mL/Kg for 30 min (B) and 21 mL/Kg for 30 or 60 min by western blot analysis (A). Sustained Src activation was evident within 30 min of 21 ml/Kg (n = 4 time course experiments). Band density is reported relative to ctr in arbitrary units (C).
Decreased endothelial permeability induced by VILI in Cav-1-/- mouse lungs
To determine pulmonary microvascular permeability responses and edema formation following VILI, we measured lung uptake of 125I-BSA and microvascular filtration coefficient Kf, c in WT and Cav-1-/- mice (Fig. 2). For 125I-BSA determination, mice were ventilated for two hours with a tidal volume of 21 mL/Kg and 60 bpm. In mice receiving 8 mL/Kg (protective ventilation), 125I-BSA uptake was higher in lungs from Cav-1-/- mice compared to WT (2.25 ± 0.21 vs. 1.66 ± 0.23 cpm/mL/dry g, P < 0.05, n = 5). Ventilation with 21 mL/Kg for 2 hours increased lung albumin accumulation by 1.7-fold in WT mice (from 1.66 ± 0.23 to 2.85 ± 0.496 cpm/mL/dry g, P < 0.05, n = 4-5). However, no increase in lung 125I-BSA was seen in Cav-1-/- mice after 2 hours of 21 mL/Kg compared to 8 mL/Kg (from 2.26 ± 0.5 to 2.25 ± 0.21 cpm/mL/dry g).
Figure 2.
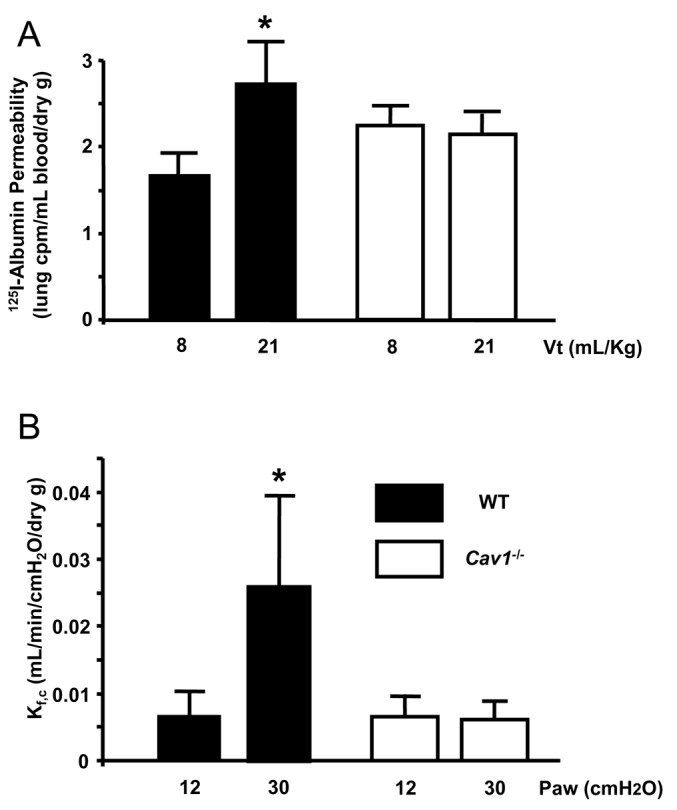
Microvascular permeability is reduced in lungs from Cav-1-/- mice subjected to injurious ventilation. (A) 125I-Bovine Serum Albumin accumulation (125I-BSA) in mouse lungs as a marker of protein leak during VILI was assessed by injecting 125I- BSA intravenously and measuring lung radioactivity in counts/min/dry g. Mice were ventilated for two hours with 8 or 21 mL/Kg. In WT lungs, injurious ventilation increased 125I-BSA 1.7-fold compared to baseline (P < 0.05, n = 4-5) but this was abrogated in Cav-1-/-. (B) Capillary filtration coefficient Kf, c, a measure of pulmonary microvascular permeability to water, was measured in explanted perfused mouse lungs following in situ ventilation with 12 cm H2 O or 30 cm H2O for two hours. After two hours of VILI, Kf, c increased four-fold in WT mouse lungs (P < 0.05, n = 4) but did not change in Cav-1-/- mice.
Pulmonary microvascular permeability. To corroborate the above findings using a different experimental strategy, we ventilated WT and Cav-1-/- mice with a pressure-cycled ventilator using injurious ventilation (airway pressure 30 cmH 2 O) or protective ventilation (airway pressure 12 cmH 2 O) for two hours as described in “MATERIALS AND METHODS”. After the end of the ventilation period, the lungs were explanted and perfused with buffer in order to measure the capillary filtration coefficient, Kf, c (Fig. 2). Values in mice ventilated with 12 cm H2O did not differ between WT and Cav-1-/- mice (0.0065 ± 0.0039 vs. 0.0064 ± 0.0039 mL/min/cm H2O/dry g, n = 4/group, P > 0.05). Ventilation with 30 cm H2O induced a fourfold increase in Kf, c in WT mice (to 0.025 ± 0.013 mL/min/cm H2O/dry g; n = 4; P < 0.05 compared to 12 cm H2O) whereas no change in Kf, c was observed in Cav-1-/- mice (0.0060 ± 0.0028 ml/min/cm H2O/dry g; n = 4; P > 0.05 vs. Cav-1-/- exposed to 12 cm H20).
Decreased lung injury in Cav-1-/- mice assessed histologically
We obtained lung tissue sections to determine whether histopathological alterations were of reflective and consistent with changes in permeability. Hematoxylin-Eosin staining of WT unventilated mice showed normal anatomy (not shown). After two hours of VILI using the volume-controlled settings of 21 mL/Kg, there was substantial congestion of pulmonary capillaries with erythrocytes, focal intra-alveolar hemorrhage, and mononuclear cell infiltration compared to mice ventilated with 8 mL/Kg for two hours (Fig. 3). In Cav-1-/- mice, we observed the previously described abnormalities in lung micromorphology (alveolar septal thickening and hypercellularity)[19] but no additional changes were observed following two hours of high tidal volume mechanical ventilation (n = 3/group).
Figure 3.
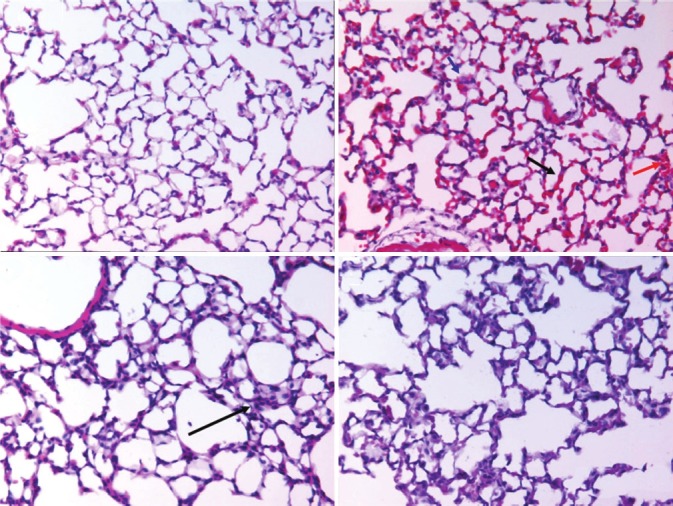
Lung tissue histology: Lung tissue sections from WT mice ventilated at 8 ml/Kg show normal anatomy (A). At 21 ml/Kg for two hours (B) WT lungs appear congested (black arrow), with areas of alveolar hemorrhage (red arrow) and mononuclear infiltrates (green arrow). In Cav-1-/- mice ventilated for two hours with 8 ml/Kg (C), we observed the previously reported baseline alterations (alveolar septal thickening and hypercellularity, yellow arrow) (C) but no or very mild additional changes after two hours of 21 ml/Kg ventilation (D) (n = 3, ×400).
Decreased inflammatory cytokines and neutrophil infiltration in Cav-1-/-
Levels of CXCL1 and IL-6. Cytokines CXCL1/KC and interleukin-6 (IL-6) were measured in WT and Cav-1-/- as markers of the inflammatory response and are expressed in log10 of the BAL concentration in pg/mL (Fig. 4). For these experiments, we assessed cytokine levels at two hours and six hours after high tidal volume ventilation, while control mice were ventilated with protective settings for 30 min. In WT mice, CXCL1 increased from 1.1 ± 0.09 in lungs ventilated with 8 mL/Kg to 1.8 ± 0.17 after two hours of 21 mL/Kg ventilation and to 3.1 ± 0.49 at six hours (n = 3-4, P < 0.05). In Cav-1-/-, this increase was blunted (from 1.1 ± 0.02 with normal tidal volume ventilation to 1.5 ± 0.35 after two hours and to 1.9 ± 0.26 after six hours; n = 3-4, P < 0.05). At six hours of 21 mL/Kg, WT mice had significantly higher levels of CXCL1 in the BAL than Cav-1-/- mice (3.1 ± 0.49 and 1.9 ± 0.26 respectively, n = 3, P < 0.05) but not at 2 hours (1.8 ± 0.17 and 1.5 ± 0.35 respectively, n = 4). Similar observations were made regarding IL-6. In WT mice, IL-6 levels increased from 0.3 ± 0.17 in lungs ventilated 30 minutes with 8 ml/Kg) to 1.5 ± 0.44 (two hours 21 mL/Kg) and to 3.49 ± 0.51 (six hours 21 ml/Kg) (P < 0.05 for comparisons of 21 mL/Kg groups to 8 mL/Kg, n = 3-4). In Cav-1-/- mice, IL-6 levels increased from 0.3 ± 0.23 (30 minutes of 8 ml/Kg) to 1.3 ± 0.78 (two hours of 21 mL/Kg) and 2.5 ± 0.05 (six hours 21 mL/Kg) (n = 3-4, P < 0.05 for comparisons with 8 mL/Kg). IL-6 levels did not differ significantly between groups at two hours (1.5 ± 0.44 for WT and 1.3 ± 0.78 for Cav-1-/- , n = 4, P > 0.05). At six hours however, WT mouse lung BAL contained significantly greater IL-6 compared to Cav-1-/- BAL (3.49 ± 0.51 and 2.5 ± 0.05; n = 3, P < 0.05).
Figure 4.
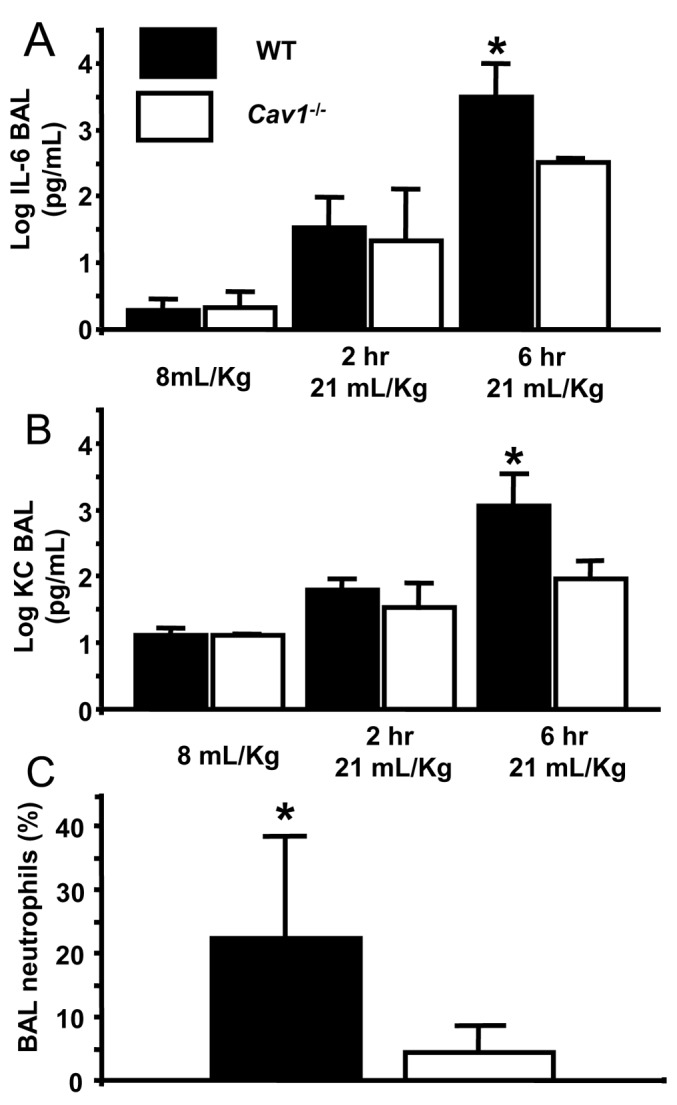
Attenuated inflammation in the broncho-alveolar lavage of Cav-1-/- mice. (A and B) Levels of cytokines CXCl1/IL-6 measured in WT and Cav-1-/- are expressed in log10 of the BAL concentration in pg/mL. In WT mice, VILI induced significant induction of IL-6 (B) and CXCL1 (A) (n = 3-4, *=P<0.05 for comparisons with control). In Cav-1-/-, this increase was blunted (n = 3-4, P < 0.05 for comparisons with control). At 6 hours of VILI, WT mice had significantly higher levels of CXCL1 in the BAL than Cav-1-/- mice (n = 3, P < 0.05) and IL-6 (n = 3, P < 0.05). (C) The neutrophil fraction in the BAL of WT mice was significantly higher than Cav-1-/- at 6 hours (n = 5-6, P < 0.05).
%PMN in BAL. BAL neutrophilia was present in WT mice at six hours but not in Cav-1-/- (22 ± 16 and 4.5 ± 4.6% respectively; n = 5-6, P < 0.05) (Fig. 4C).
The above data suggest blunted biotrauma and inflammation in the absence of Cav-1.
Rescue of Cav-1 expression in Cav-1-/- mouse lung microvessels restores injury response to mechanical ventilation
To establish the direct relationship between the absence of Cav-1 and the observed protected phenotype, we rescued Cav-1 expression in Cav-1-/- mice and subjected these to injurious ventilation with 21 mL/Kg (Fig. 5). Liposome-mediated delivery and transduction of eBFP vector or WT myc-tagged-Cav-1 cDNA in lung microvessels[16] rescued Cav-1 expression in lungs of Cav-1-/- mice, as shown by fluorescence microscopy of lung tissue sections (Fig. 5A) and immunoblotting total lung homogenates (Fig. 5B). Mice subsequently underwent injurious ventilation and then 125I-BSA activity was measured in BAL. As expected, BSA permeability following mechanical ventilation was significantly reduced in Cav-1-/- mice compared to WT mice ventilated with high tidal volume. Interestingly, rescue of Cav-1 expression restored the hyperpermeability response observed in WT mice following lung hyperinflation (Fig. 5C).
Figure 5.
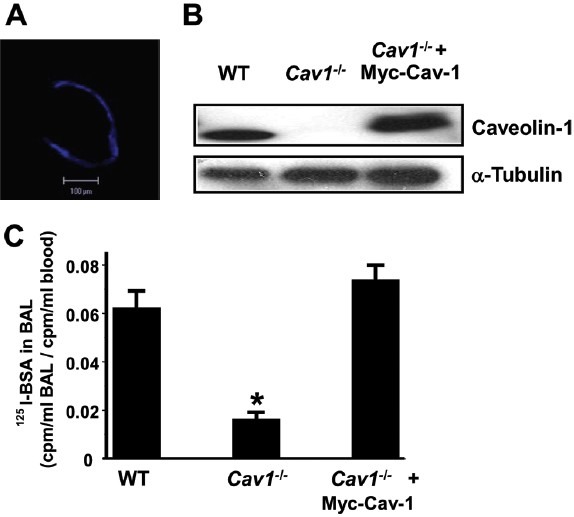
Rescue of Cav-1 expression in Cav1-/- mouse lungs using liposomes. Liposome-mediated gene delivery was used to restore Cav-1 expression in mouse lungs. (A) Incorporation of pEBFP (blue fluorescent protein) in the lung circulation 24hrs after transfection. (B) Rescue of Cav-1 protein expression in Cav1-/- mouse lungs 48hrs after transfection. (C) Restoration of the VILI transvascular 125I-albumin hyperpermeability response in Cav1-/- mouse lungs 48 hrs after liposome transfection of Cav-1 cDNA (n = 3/group).
Cav-1-/- endothelial cells are resistant to thrombin-induced increase in endothelial permeability
Endothelial monolayer permeability studies. To determine the role of Cav-1 expression in the endothelial response to vasoactive and permeability enhancing agents, pulmonary microvascular endothelial cells isolated from WT and Cav-1-/- mice were cultured on microporous filters and permeability was measured as the rate of fluorescent albumin flux across the endothelial monolayer. Upon thrombin challenge (1 U/mL), we observed an increase in albumin permeability in WT cells (Fig. 6A). However, in cells obtained from Cav-1-/- mice, no increase albumin permeability in response to thrombin as noted (Fig. 6A).
Figure 6.
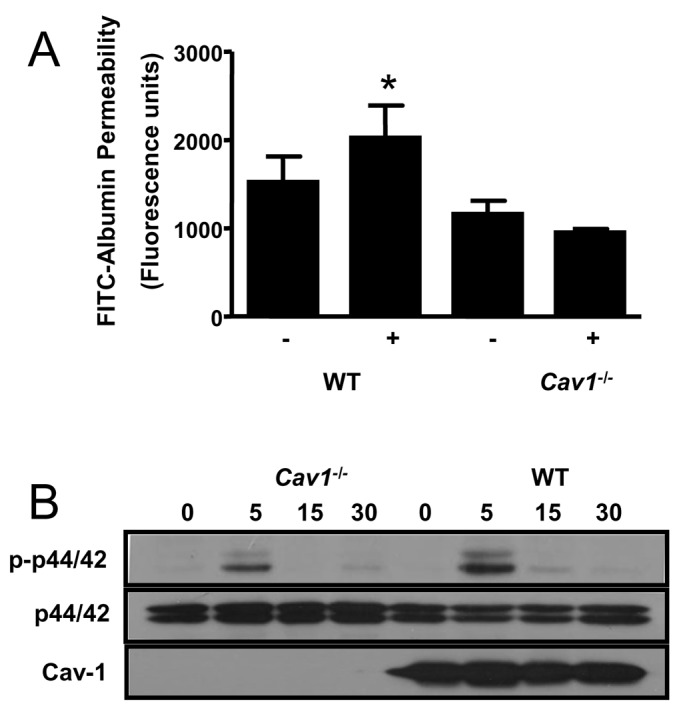
Requirement for Cav-1 for thrombin-induced increase in permeability and signaling in cultured pulmonary endothelial cells. (A) Pulmonary microvascular endothelial cells harvested from mouse lungs were cultured on microporous filters and monolayer permeability to fluorescein-isothiocyanate-labeled bovine serum albumin was measured following a 15-min incubation with thrombin 1 U/mL. Thrombin elicited an increase in albumin permeability in cells expressing Cav-1, but not in cells derived from Cav-1-/- mice. (B) Incubation of cultured endothelial cells with thrombin induced a time-dependent increase in p44/42 phosphorylation by immunoblotting, which was attenuated in cells lacking Cav-1. (A and B) representative of three experiments with three wells/treatment.
Cell-signaling studies. Mitogen-activated protein kinases are important signaling intermediates of the cellular stress response. We thus examined the role of Cav-1 expression on thrombin-induced p44/42 expression and phosphorylation. In WT ECs, robust activation of p44/42 was evident within 5 min following addition of thrombin to the cell culture medium (Fig. 6B). In the absence of Cav-1-/-, however, p44/42 activation was markedly attenuated.
DISCUSSION
In this study, we demonstrate that Cav-1-/- is required for increased endothelial permeability in ALI induced by mechanical ventilation. In addition to being the major coat protein of caveolar vesicles, Cav-1 binds and inhibits several components of key cell signaling pathways[20] (p44/42, Src, eNOS) and is thus an important regulator of fundamental cellular processes.[7] Several lines of evidence point to a possible involvement of endothelial Cav-1 in the pathological increase in permeability. For example, tyrosine kinases, for which Cav-1 is a substrate, are activated in white blood cells by VILI;[21] Src is a major activator of caveolar albumin uptake triggered by activated neutrophils bound to ICAM-1,[16] and Cav-1 expression can be upregulated in response to bacterial endotoxin stimulation.[20] Here we show that lung distention activates Cav-1, an event important in the generation of lung injury.
In our experiments, Src was phosphorylated within 30 minutes of onset of high tidal volume ventilation which was accompanied by synchronous phosphorylation of Cav-1 at Tyr14. In previous work, it was shown that Cav-1 phosphorylation is a crucial step in endothelial dysfunction induced by oxidants.[8] To determine the functional significance of Cav-1 activation in vivo, we subjected Cav-1-/- mice and appropriate controls to VILI. We found that Cav-1-/- mice were protected from lung permeability increases using two different protocols: (1) by ventilating with 21 mL/Kg and measuring 121 I-BSA uptake in the lung; and (2) by ventilating with 30 cmH 2 O pressure and measuring capillary filtration coefficient. Using these complementary approaches, we found that Cav-1-/- mouse lungs were less susceptible to pulmonary endothelial hyperpermeability induced by injurious ventilation. It seems therefore evident that Cav-1 expression is required for microvascular endothelial cell injury to occur.
The presence of lung injury by our volume-cycled experimental protocol was documented in histologic sections from mouse lungs stained with hematoxylin-eosin. While mice ventilated with protective settings showed no significant histologic abnormalities, use of the injurious protocol was associated with substantial congestion of pulmonary capillaries, focal intraalveolar hemorrhage, and mononuclear cell infiltration.
To assess the degree of inflammatory response to alveolar over-distention, we measured the levels of the inducible cytokines IL-6 and CXCL1 as well as the BAL neutrophil fraction. Both cytokines were substantially elevated in WT mice ventilated with high tidal volumes; however in Cav-1-/- mice, the levels were significantly lower, indicating a blunted inflammatory response. Contrary to WT, which had a marked BAL neutrophilia, Cav-1-/- mice subjected to injurious ventilation had near-normal BAL neutrophil counts.
The observed attenuation in permeability and innate immune reaction observed in Cav-1-/- is suggestive of an important role of this protein in initiating these fundamental responses. To confirm this result, we re-established Cav-1 expression in Cav-1-/-, which were then exposed to high lung inflation volumes. We observed that rescue of Cav-1 expression restored the hyperpermeability response. This is in line with the initial findings in Cav-1-/- mice and also indicates that these were not due to the known chronic pulmonary alterations of these mice, e.g. increased interstitial matrix deposition, which could make the lung more resistant to distention. The reversal of the phenotype within 48 hours of restoring Cav-1 expression in lung endothelia supports the direct role of Cav-1 as a regulator of immediate permeability responses to acute insults.
We next sought to determine the importance of Cav-1 expression in the permeability response of ECs. To this end, we used cultured pulmonary vascular ECs isolated from WT and Cav-1-/- mice. To increase endothelial permeability in vitro, cells were incubated with thrombin, a serine protease with procoagulant properties, and an extremely potent vasoactive agent known to induce endothelial barrier disruption via ligation of protease-activated receptor-1. Endothelial cells exposed to thrombin contracted resulting in the loosening of interendothelial junctions and underlying focal contacts with the matrix, a conformation which favors leakage of plasma water, solutes, and proteins into the interstitial space. This process can, to some extent, be duplicated in vitro by culturing cells on microporous filters by measuring the rate of labeled albumin tracer flux across this endothelial monolayer. In the presence of thrombin, we observed increased EC permeability only in the presence of Cav-1, indicating that this protein somehow triggers or is required for thrombin-induced signaling events that result in increased monolayer permeability.
In this and other experimental systems, thrombin results in activation of MAP-kinases, most notably p38, p44/42, and c-Jun N-terminal kinase.[22] We observed time-dependent p44/42 activation in ECs induced by thrombin which was substantially reduced in cells lacking Cav-1, providing evidence that Cav-1 is an important regulator of thrombin signaling and possibly of other vasoactive mediators as well. Putative mechanisms of Cav-1 regulation of stress-induced endothelial pathways include control of Ca++ influx, spatial organization of signaling pathways, and regulation of nitric oxide release. Recently, a role for Cav-1 in the rearrangement processes taking place in endothelial adherence junctions, which are required for the permeability-enhancing effect of thrombin, has also been recently described.[23]
Our results are consistent with other models of lung inflammation and fibrosis which favor a role of Cav-1 in the innate immune response.[24,25] A notable exception is the study by Hoetzel et al., which, by employing an injurious ventilation model of lower intensity than ours, found that Cav-1-/- mice were actually more susceptible to lung injury.[15] This discrepancy illustrates the importance of experimental design among animal studies, in particular with respect to the dose of the applied stressor. In our experimental system, a relatively high tidal volume was delivered using two different strategies, one being volume-controlled and other via pressure-controlled ventilation, and we observed that Cav-1-/- mice were less susceptible to lung injury. This approach of ventilating experimental animals with tidal volumes that far exceed what is used in clinical practice is commonly the subject of criticism. However, in patients requiring respiratory support because of lung injury, large areas of the lung are collapsed and not accessible to ventilation, while less affected alveoli receive the bulk of delivered gas volume. Under these circumstances, distention of alveoli to an unknown extent may still occur, even if protective ventilation with low tidal volume is applied.
In summary, we present evidence that Cav-1 is required for endothelial permeability increases in response to insults relevant to acute lung injury, including mechanical stretch and thrombin signaling.
Footnotes
Source of Support: This work was supported by NIH R01 HL71626 and P01 HL60678 (RDM), and American Thoracic Society/Acute Respiratory Distress Syndrome Foundation/Sepsis Alliance Partnership Research Grant (NAM).
Conflict of Interest: None declared.
REFERENCES
- 1.Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. 2011;6:147–63. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donahoe M. Acute respiratory distress syndrome: A clinical review. Pulm Circ. 2011;1:192–211. doi: 10.4103/2045-8932.83454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vestweber D. VE-cadherin: The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler Thromb Vasc Biol. 2008;28:223–32. doi: 10.1161/ATVBAHA.107.158014. [DOI] [PubMed] [Google Scholar]
- 4.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 5.Galbiati F, Volonte D, Brown AM, Weinstein DE, Ben-Ze›ev A, Pestell RG, et al. Caveolin-1 expression inhibits Wnt/beta-catenin/Lef-1 signaling by recruiting beta-catenin to caveolae membrane domains. J Biol Chem. 2000;275:23368–77. doi: 10.1074/jbc.M002020200. [DOI] [PubMed] [Google Scholar]
- 6.Song L, Ge S, Pachter JS. Caveolin-1 regulates expression of junction-associated proteins in brain microvascular endothelial cells. Blood. 2007;109:1515–23. doi: 10.1182/blood-2006-07-034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–79. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- 8.Garrean S, Gao XP, Brovkovych V, Shimizu J, Zhao YY, Vogel SM, et al. Caveolin-1 regulates NF-kappaB activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J Immunol. 2006;77:4853–60. doi: 10.4049/jimmunol.177.7.4853. [DOI] [PubMed] [Google Scholar]
- 9.Sun Y, Hu G, Zhang X, Minshall RD. Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ Res. 2009;105:676–85. doi: 10.1161/CIRCRESAHA.109.201673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu G, Ye RD, Dinauer MC, Malik AB, Minshall RD. Neutrophil caveolin-1 expression contributes to mechanism of lung inflammation and injury. Am J Physiol Lung Cell Mol Physiol. 2008;294:L178–86. doi: 10.1152/ajplung.00263.2007. [DOI] [PubMed] [Google Scholar]
- 11.Kwiatek AM, Minshall RD, Cool DR, Skidgel RA, Malik AB, Tiruppathi C. Caveolin-1 regulates store-operated Ca2+ influx by binding of its scaffolding domain to transient receptor endothelial cells. Mol Pharmacol. 2006;70:1174–83. doi: 10.1124/mol.105.021741. [DOI] [PubMed] [Google Scholar]
- 12.Han B, Lodyga M, Liu M. Ventilator-induced lung injury: Role of protein-protein interaction in mechanosensation. Proc Am Thorac Soc. 2005;2:181–7. doi: 10.1513/pats.200501-008AC. [DOI] [PubMed] [Google Scholar]
- 13.dos Santos CC, Slutsky AS. The contribution of biophysical lung injury to the development of biotrauma. Annu Rev Physiol. 2006;68:585–618. doi: 10.1146/annurev.physiol.68.072304.113443. [DOI] [PubMed] [Google Scholar]
- 14.Sverdlov M, Shinin V, Place AT, Castellon M, Minshall RD. Filamin A regulates caveolae internalization and trafficking in endothelial cells. Mol Biol Cell. 2009;20:4531–40. doi: 10.1091/mbc.E08-10-0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoetzel A, Schmidt R, Vallbracht S, Goebel U, Dolinay T, Kim HP, et al. Carbon monoxide prevents ventilator-induced lung injury via caveolin-1. Crit Care Med. 2009;37:1708–15. doi: 10.1097/CCM.0b013e31819efa31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu G, Vogel SM, Schwartz DE, Malik AB, Minshall RD. Intercellular adhesion molecule-1-dependent neutrophil adhesion to endothelial cells induces caveolae-mediated pulmonary vascular hyperpermeability. Circ Res. 2008;102:e120–31. doi: 10.1161/CIRCRESAHA.107.167486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorovoy M, Neamu R, Niu J, Vogel S, Predescu D, Miyoshi J, et al. RhoGDI-1 modulation of the activity of monomeric RhoGTPase RhoA regulates endothelial barrier function in mouse lungs. Circ Res. 2007;101:50–8. doi: 10.1161/CIRCRESAHA.106.145847. [DOI] [PubMed] [Google Scholar]
- 18.Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68:673–82. doi: 10.1016/0092-8674(92)90143-z. [DOI] [PubMed] [Google Scholar]
- 19.Maniatis NA, Shinin V, Schraufnagel DE, Okada S, Vogel SM, Malik AB, et al. Increased pulmonary vascular resistance and defective pulmonary artery filling in caveolin-1-/- mice. Am J Physiol Lung Cell Mol Physiol. 2008;294:L865–73. doi: 10.1152/ajplung.00079.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minshall RD, Sessa WC, Stan RV, Anderson RG, Malik AB. Caveolin regulation of endothelial function. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1179–83. doi: 10.1152/ajplung.00242.2003. [DOI] [PubMed] [Google Scholar]
- 21.Yiming MT, Parthasarathi K, Issekutz AC, Bhattacharya S. Sequence of endothelial signaling during lung expansion. Am J Respir Cell Mol Biol. 2005;33:549–54. doi: 10.1165/rcmb.2005-0133OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 23.Kronstein R, Seebach J, Grossklaus S, Minten C, Engelhardt B, Drab M, et al. Caveolin-1 opens endothelial cell junctions by targeting catenins. Cardiovasc Res. 2012;93:130–40. doi: 10.1093/cvr/cvr256. [DOI] [PubMed] [Google Scholar]
- 24.Shivshankar P, Brampton C, Miyasato S, Kasper M, Thannickal VJ, Jourdan Le Saux C. Caveolin-1 Deficiency Protects from Pulmonary Fibrosis by Modulating Epithelial Cell Senescence in Mice. Am J Respir Cell Mol Biol. 2012;47:28–36. doi: 10.1165/rcmb.2011-0349OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin Y, Lee SJ, Minshall RD, Choi AM. Caveolin-1: A critical regulator of lung injury. Am J Physiol Lung Cell Mol Physiol. 2011;300:L151–60. doi: 10.1152/ajplung.00170.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]