

Abstract
Sepsis, sepsis-induced hyperinflammation and subsequent sepsis-associated immunosuppression (SAIS) are important causes of death. Here we show in humans that the loss of the major reactive oxygen species (ROS) scavenger, glutathione (GSH), during SAIS directly correlates with an increase in the expression of activating transcription factor 3 (ATF3). In endotoxin-stimulated monocytes, ROS stress strongly superinduced NF-E2–related factor 2 (NRF2)–dependent ATF3. In vivo, this ROS-mediated superinduction of ATF3 protected against endotoxic shock by inhibiting innate cytokines, as Atf3−/− mice remained susceptible to endotoxic shock even under conditions of ROS stress. Although it protected against endotoxic shock, this ROS-mediated superinduction of ATF3 caused high susceptibility to bacterial and fungal infections through the suppression of interleukin 6 (IL-6). As a result, Atf3−/− mice were protected against bacterial and fungal infections, even under conditions of ROS stress, whereas Atf3−/−Il6−/− mice were highly susceptible to these infections. Moreover, in a model of SAIS, secondary infections caused considerably less mortality in Atf3−/− mice than in wild-type mice, indicating that ROS-induced ATF3 crucially determines susceptibility to secondary infections during SAIS.
Sepsis and sepsis-associated multiorgan failure are major causes of death worldwide1–3. During sepsis, some people die early from bacteremia or from sepsis-induced hyperinflammation, an uncontrolled overactivation of the innate immune system with initiation of the complement system and high amounts of circulating proinflammatory cytokines4,5. Notably, the neutralization of inflammatory mediators with glucocorticosteroids6, endotoxin-specific antibodies7, tumor necrosis factor (TNF) inhibitors8,9 or interleukin-1 (IL-1) receptor antagonists10 does not improve the overall survival of people with sepsis. Some affected individuals die from secondary infections during the phase of SAIS3,11,12, which is characterized by cutaneous anergy to recall skin antigens12, neutrophil paralysis13, lymphopenia14, lower amounts of circulating proinflammatory cytokines (TNF, IL-6)15 and increased IL-10 levels16,17.
Systems analyses of endotoxin-stimulated macrophages have identified ATF3 as a rapidly induced transcription factor of endotoxin-stimulated macrophages and have found that ATF3 strongly represses IL-6 transcription18,19. ATF3 also reduces TNF- and IL-6–dependent airway hyper-reactivity and interferon-γ production in cytomegalovirus-infected mice20,21. Because ROS deplete GSH18,22 during SAIS, and GSH depletion enhances endotoxin-induced ATF3 expression, we investigated the role of ROS-induced ATF3 expression and ATF3-mediated IL-6 suppression in lipopolysaccharide (LPS)-induced shock, in bacterial or fungal sepsis, and during SAIS.
RESULTS
During sepsis, low glutathione correlates with high ATF3
During sepsis, sepsis-induced hyperinflammation and SAIS, ROS stress severely depletes GSH, which is the primary intracellular ROS scavenger23,24. We found that GSH levels were 600 μmol l−1 in the blood samples of patients with acute sepsis admitted to the intensive care unit (ICU; Supplementary Table 1), compared with 1,000 μmol l−1 in healthy individuals (Fig. 1a). GSH further decreased to 400 μmol l−1 by the time the patients left the ICU (Fig. 1a). Data from cell lines suggest that ROS-induced GSH depletion enhances the expression of stress-regulated ATF3 in response to Toll-like receptor (TLR) ligands18,19,22. Therefore, we investigated whether GSH depletion during SAIS induces ATF3. In acute sepsis, ATF3 mRNA levels were tenfold lower in leukocytes of affected subjects than in those of healthy controls (Fig. 1b). Low ATF3 correlated with high IL-6 levels (Fig. 1). Until patients left the ICU (Supplementary Table 1), ATF3 mRNA and protein levels significantly increased during SAIS (Fig. 1b,c). The increased ATF3 directly correlated with the decreased GSH (r2 = −0.49; P < 0.05; Supplementary Fig. 1a,b). This inverse correlation between ATF3 and GSH may be physiologically relevant, because ATF3 suppresses TNF, IL12B and IL6 transcription18. Accordingly, IL-6 serum levels were high during acute sepsis and significantly lower during SAIS (Fig. 1d).
Figure 1.

Declining glutathione levels correlate with increasing ATF3 and decreasing IL-6 levels during SAIS. We collected blood from subjects with sepsis (n = 16, subjects 1–16; two subjects died during sepsis, and four did not provide postsepsis samples; see Supplementary Table 1) who met the inclusion criteria during acute sepsis and after acute sepsis when leaving the ICU, and from four healthy individuals, as described in Online Methods50. (a) GSH concentrations in whole blood, analyzed by HPLC. (b) Increase in ATF3 expression in blood cells of subjects with sepsis, measured with real-time quantitative PCR (subjects with acute sepsis versus healthy controls, P < 0.001; subjects after sepsis versus healthy controls, P < 0.05). (c) Western blot of ATF3 in PBMCs from a different group of subjects with sepsis (n = 8, subjects 17–24; two subjects died during sepsis, and two did not provide second samples; see Supplementary Table 1). S, acute sepsis; PS, post-sepsis. Graph shows ATF3 normalized to β-actin using the ImageJ software program. (d) IL-6 measured by ELISA (n = 16; error bars represent means ± s.d.). *P < 0.05; **P < 0.01; ***P < 0.001 (ANOVA). ND, not detectable.
ROS stress superinduces ATF3 and abrogates IL-6 production
To model sepsis-induced GSH depletion in vitro, we depleted GSH in CD14+ human monocytes using dimethylfumarate (DMF). DMF diminished intracellular GSH to 40–50% of its concentration in controls (Fig. 2a), levels similar to those measured during SAIS in human blood. Flow cytometry revealed that GSH depletion by DMF increased intracellular ROS to the same extent as did H2O2 treatment (Fig. 2a). N-acetylcysteine (NAC) restored intracellular GSH and returned ROS to normal levels (Fig. 2a). As in mouse macrophages18, LPS stimulation of CD14+ human monocytes increased ATF3 mRNA expression two- to threefold, with a maximum at 2 h (Fig. 2b). To selectively mimic ROS stress during SAIS, we depleted GSH in CD14+ human monocytes, stimulated them with LPS and measured ATF3 mRNA and protein levels 2 h later. After GSH depletion, LPS superinduced ATF3 mRNA about fivefold (Fig. 2c). This resulted in 12-fold greater ATF3 mRNA expression compared to unstimulated cells (Fig. 2b,c), equivalent to the tenfold ATF3 mRNA increase measured during SAIS in vivo (Fig. 1b). NAC restored normal ROS levels (Fig. 2a) and prevented superinduction of ATF3 mRNA in GSH-depleted monocytes (Fig. 2c). Because ATF3 negatively regulates IL-6 and IL-12 p40 (ref. 18), cytokines that are crucial in both LPS-induced shock and pathogen defense, we determined whether the ATF3 superinduction caused by GSH depletion affected the expression of IL6 or IL12B mRNAs in CD14+ monocytes or their protein production in peripheral blood mononuclear cells (PBMCs). GSH depletion suppressed the LPS-induced IL6 or IL12B mRNA by 80–90%; NAC fully reversed this suppression (Fig. 2d). GSH depletion also suppressed IL-6 or IL-12 p40 protein production (Supplementary Fig. 1c). In addition, DMF impaired LPS-induced TNF expression (Supplementary Fig. 1c), and NAC reversed ATF3 superinduction and restored the LPS-induced expression and production of ATF3-regulated cytokines (Fig. 2d and Supplementary Fig. 1c). To determine whether ATF3 superinduction caused the ROS-mediated suppression of IL-6, IL-12 p40 and TNF, we selectively impaired ATF3 translation using ATF3-specific short interfering RNA (siRNA; Fig. 2e). We depleted GSH in ATF3-knockdown monocytes or control siRNA–transfected monocytes, stimulated them with LPS and measured IL-6 production. In monocytes transfected with control siRNAs, GSH depletion caused ATF3 superinduction and suppressed IL-6 production to <10% of the level seen in untreated monocytes (Fig. 2f). The DMF-treated ATF3-knockdown monocytes produced fourfold more IL-6 than DMF-treated monocytes transfected with the control siRNA. Thus, ROS-induced ATF3 protein is required to inhibit LPS-triggered IL-6 production by human monocytes. The ATF3 knockdown did not affect IL-12 p40 or TNF (data not shown).
Figure 2.
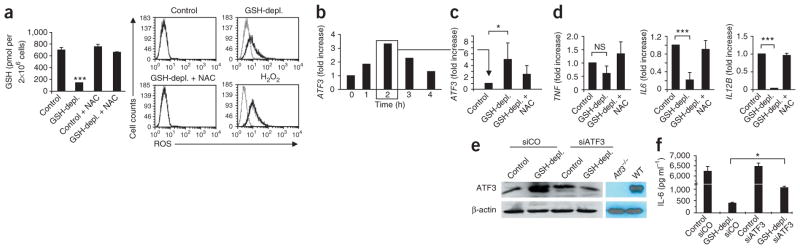
ROS-mediated superinduction of ATF3 suppresses IL-6. (a) Left, intracellular content of GSH measured by colorimetric assay in human CD14+ monocytes (n = 6 healthy donors) after incubation with GSH-depleting DMF (GSH-depl.), NAC or both (GSH-depl. + NAC), as described in Online Methods. Control, untreated monocytes. Data from six donors are expressed as means ± s.d. Right, flow-cytometry analysis of intracellular ROS levels of the THP-1 monocytic cell line after GSH depletion with DMF (GSH-depl.), H2O2 treatment or NAC compensation. Dark lines, ROS-treated cells; gray lines, sham-treated cells; data are from one representative experiment. The x axis indicates the mean fluorescence intensity of ROS staining, and the y axis indicates the cell counts. (b) ATF3 mRNA expression measured by quantitative PCR in monocytes stimulated for the indicated times with LPS; data are from one representative experiment (of three total). (c,d) mRNAs measured by quantitative PCR 2 h after monocytes with the indicated pretreatment were stimulated with LPS. Data are means ± s.d. of three (c) or six (d) different donors. (e,f) ATF3 protein measured by western blotting (e), and cytokine production after LPS stimulation measured by ELISA (f), after the indicated treatments. Control, monocytes not treated with DMF; siATF3, ATF3 mRNA knocked down by specific siRNA; siCo, control siRNA. Right two lanes in e show cell lysates of macrophages from wild-type (WT) or Atf3−/− mice. One representative experiment of three is shown in e. Data from three experiments are means ± s.d. in f. NS, not significant; *P < 0.05; ***P < 0.001 (ANOVA).
ROS-mediated ATF3 superinduction requires NRF2
To determine the functional relevance of ROS-induced increases in ATF3 during SAIS after bacterial sepsis or sepsis-induced hyper-inflammation, we studied GSH depletion in C57BL/6 (wild-type) mice or congenic C57BL/6 × Atf3−/− mice. DMF diminished GSH levels in macrophages of wild-type or Atf3−/− mice by 70%, and NAC prevented this GSH depletion (Supplementary Fig. 2a). DMF and H2O2 induced similar ROS increases in wild-type and Atf3−/− macrophages, and these increases were fully reversed by NAC (Supplementary Fig. 2b). LPS increased Atf3 mRNA levels two- to threefold in mouse macrophages18 (and data not shown), and GSH depletion again significantly (P < 0.05) superinduced Atf3 mRNA levels in response to LPS (Fig. 3a,b).
Figure 3.

ROS-mediated superinduction of ATF3 requires NRF2 signaling. (a,b) Quantification of Atf3 expression in GSH-depleted peritoneal macrophages from wild-type mice (n = 6) by quantitative PCR (a) or western blot analysis (b) after incubation with DMF (GSH-depl.) or H2O2 and stimulation with LPS. Control, LPS stimulation alone. Data are means ± s.d. (c) Quantification of NRF2 by western blotting in BMDCs after DMF-induced GSH depletion. The positive controls were BMDCs treated with tert-butylhydroquinone (tBHQ) or butylated hydroxyanisole (BHA); the negative control (Co) was BMDCs treated with DMSO. Data are representative of two independent experiments. (d) ARE1 sequences amplified by PCR after chromatin immunoprecipitation (ChIP) with an antibody to NRF2 in untreated (Co) or GSH-depleted THP-1 cells. (e) Luciferase activity (normalized to β-galactosidase activity) in mouse RAW 264.7 cells transfected with a wild-type (pATF3-3.6k-luc) or mutant ATF3 promoter–luciferase construct (pATF3-3.6k-luc-mut) and treated with tBHQ or DMF. The normalized activity in a DMSO-treated sample (Co) was set as 1. Data are means ± s.d. (n = 3). (f) ATF3 measured by western blotting (normalized to β-actin) in control BMDCs (wild-type mice, WT) and GSH-depleted BMDCs (Nrf2−/− mice) stimulated with LPS. Data are means ± s.d. (n = 3). (g) ELISAs showing cytokine production by peritoneal WT or Atf3−/− mouse macrophages incubated with DMSO (Control), DMF or DMF plus NAC, and stimulated with LPS. Data are means ± s.d. (n = 4). NS, not significant; *P < 0.05; **P < 0.01; ***P < 0.001 (t test (f) or ANOVA).
NRF2, which is a ROS-sensitive transcription factor, is rapidly degraded in the cytoplasm under normal oxidative conditions25. During ROS stress, NRF2 accumulates in the nucleus and binds antioxidant response elements (AREs) of various promoter regions, including the ATF3 promoter26. GSH depletion dose-dependently increased nuclear NRF2 protein, as did the NRF2 activators tBHQ and BHA (Fig. 3c). GSH depletion also increased NRF2 binding to ARE1 in the ATF3 promoter (Fig. 3d). The enhanced NRF2 binding was functional, as shown in mouse macrophage RAW lines and in human astrocytes transfected with various ATF3 promoter–luciferase constructs. Notably, NRF2 bound intact but not mutated ARE1. NRF2 binding induced luciferase approximately twofold after GSH depletion or tBHQ treatment (Fig. 3e and Supplementary Fig. 2c). Western blots from mouse bone marrow–derived dendritic cells (BMDCs) highlighted the functional relevance of this binding. In the absence of ROS stress, LPS moderately induced ATF3 (Fig. 3f). GSH depletion superinduced LPS-triggered ATF3 in wild-type BMDCs, but not in Nrf2-deficient BMDCs (Fig. 3f). Therefore, GSH-mediated ATF3 superinduction required the nuclear binding of NRF2 to the ARE1 promoter region of ATF3.
ROS-mediated ATF3 superinduction abrogates IL-6 production
GSH depletion abrogated LPS-induced Il6 and Il12b mRNA in mouse macrophages, and NAC fully reversed this suppression (Supplementary Fig. 2d). In wild-type macrophages, GSH depletion suppressed the LPS-induced production of IL-6, IL-12 p40 or IL-1β by ≥90% and of TNF by 50%, and NAC again reversed this suppression (Fig. 3g). In the absence of GSH depletion, Atf3−/− macrophages produced amounts of innate cytokines similar to wild-type macrophages. In contrast, the Atf3−/− macrophages were largely resistant to ROS-induced IL-6 suppression (Fig. 3g). This demonstrates that the ROS-mediated superinduction of ATF3 directly silences IL-6. In contrast, ATF3 suppressed IL-12 p40 and TNF only partially, and did not affect IL-1β production (Fig. 3g). These results were confirmed in BMDCs (Supplementary Fig. 3a) and were independent of apoptosis or necrosis induction (Supplementary Fig. 3b).
ROS stress establishes resistance to endotoxin-induced shock
Sepsis and SAIS are complex diseases that affect multiple molecular signaling cascades2,3,12. To study the functional relevance of ROS-mediated GSH depletion in vivo, which is a hallmark of SAIS12, we diminished GSH by feeding mice DMF (Supplementary Fig. 4a). DMF-treated mice had similar GSH blood concentrations as humans or mice with SAIS (Fig. 1a and Supplementary Figs. 1 and 4). GSH depletion also significantly enhanced endotoxin-induced Atf3 mRNA expression in peritoneal macrophages in vivo (Fig. 4a). Because low ATF3 sensitizes mice to endotoxic (LPS-induced) shock18, ATF3 superinduction by GSH depletion should protect mice against LPS-induced shock. Indeed, in vivo depletion of GSH by DMF treatment (Supplementary Fig. 4a) protected >80% of mice against LPS-induced shock in conditions that rapidly killed 50% of controls (Fig. 4b,c). This LPS resistance strictly required GSH depletion, as NAC treatment to prevent GSH depletion restored susceptibility to LPS-induced shock (Supplementary Fig. 4a,b). DMF-mediated protection from LPS-induced shock correlated with substantial suppression of Il6, Tnf and Il12b mRNA expression in the spleen and of the corresponding protein expression in serum (Fig. 4d and Supplementary Fig. 4c,d). Together, these data suggest that GSH depletion protects against LPS-induced shock and that this protection is at least partially mediated through the superinduction of ATF3. Therefore, we challenged the DMF-treated Atf3−/− mice with LPS. As in wild-type mice, DMF diminished GSH in Atf3−/− mice by 50% (Supplementary Fig. 4a). However, in contrast to the DMF-treated wild-type mice, GSH depletion suppressed neither the LPS-induced Tnf expression nor the production of IL-6 or IL-12 p40 in Atf3−/− mice (Fig. 4d and Supplementary Fig. 4c,d). Moreover, GSH depletion did not protect the Atf3−/− mice against LPS-induced shock (Fig. 4b,c). Therefore, ROS-induced ATF3 directly suppressed the innate immune response to endotoxins and prevented endotoxin-induced shock.
Figure 4.
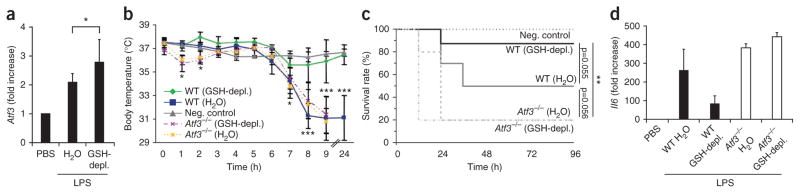
ROS-mediated protection from endotoxin-induced shock is ATF3-dependent. (a) Atf3 expression in mouse peritoneal macrophages measured by quantitative PCR, after GSH depletion in wild-type mice (GSH-depl.) as described in the Online Methods, and LPS challenge intraperitoneally with LPS or PBS (control). Data are means ± s.d. from two independent experiments (n = 6 mice per group per experiment). (b,c) Body temperatures (b) and survival rates (c) of WT and Atf3−/− mice treated as in a. Data are means ± s.d. from five independent experiments (n = 4 mice per group per experiment). (d) Quantitative PCR showing cytokine mRNA levels in spleens of mice treated as in a, normalized to expression in healthy, PBS-injected controls. Data are means ± s.d. from two independent experiments (n = 4 mice per group per experiment). *P < 0.05; **P < 0.01; ***P < 0.001 (ANOVA and log-rank statistics).
ROS-induced ATF3 abrogates innate control of bacterial sepsis
Although ROS-induced GSH depletion protected against LPS-induced shock via NRF2-dependent superinduction of ATF3, the attenuation of the cytokine storm by ATF3 may render mice and humans more vulnerable to bacterial and fungal infections. To address this possibility, we studied fungal and Escherichia coli sepsis and bacterial peritonitis (cecal ligation and puncture, CLP), three important model diseases of human sepsis27. In conditions that killed 20% of water-treated controls, CLP killed 80% of DMF-treated mice (Fig. 5a). The ROS-mediated induction of ATF3 directly caused this susceptibility to bacteremia, as 80% of the Atf3−/− mice were even protected against CLP conditions that killed 100% of wild-type mice (Fig. 5b). Moreover, after CLP, the peritoneal lavage of Atf3−/− mice contained only 10% of the bacteria found in Atf3 wild-type mice (Fig. 5c). In addition, Atf3−/− mice were largely protected from E. coli– or Aspergillus fumigatus–induced sepsis (Supplementary Fig. 5a–d), indicating that ATF3 activation attenuated the control of systemic bacterial or fungal infection. To determine whether ROS-mediated ATF3 induction causes increased susceptibility to bacterial sepsis, we depleted GSH in wild-type and Atf3−/− mice using DMF and then performed CLP in conditions that killed approximately 50% of undepleted wild-type mice (Fig. 5d). After GSH depletion, this CLP condition killed all wild-type mice (Fig. 5d). In contrast, GSH depletion did not attenuate the protection against CLP in Atf3−/− mice (Fig. 5d). In line with this, GSH depletion enhanced the CLP-induced expression of ATF3 protein in peritoneal macrophages of wild-type mice (Supplementary Fig. 6a) and suppressed the CLP-induced expression of IL-6 and IL-12 p40 serum proteins in wild-type mice. Atf3−/− mice had significantly higher IL-6 and IL-12 p40 serum levels than wild-type mice (P < 0.05), but only slightly higher TNF levels (Supplementary Fig. 6b).
Figure 5.
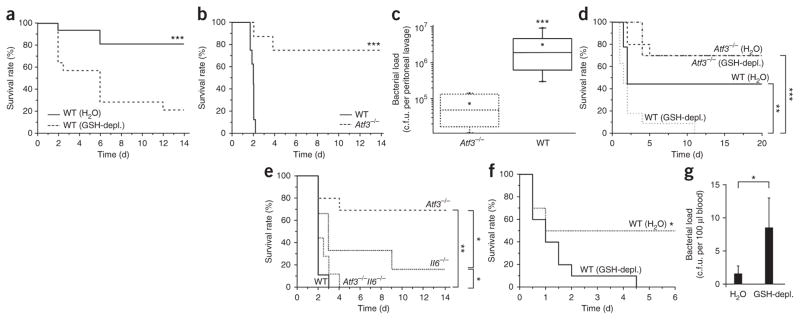
ROS-induced ATF3 causes high susceptibility to bacterial peritonitis and E. coli sepsis by suppressing IL-6. (a) Mortality of wild-type (WT) mice (littermates) fed either DMF (GSH-depl.) or water for 10 d and subjected to mild CLP. Data represent four independent experiments (n = 8 mice per group per experiment). (b) Mortality of WT or Atf3−/− mice after severe CLP. Data represent three independent experiments (n = 6–8 mice per group per experiment). (c) Bacterial load measured by peritonial lavage in WT or Atf3−/− mice 24 h after CLP. Data represent two independent experiments (n = 5 mice per group per experiment). (d) Mortality after intermediate CLP of WT or Atf3−/− mice treated with water or DMF, as in a. Data represent two independent experiments (n = 9 or 10 mice per group per experiment). (e) Mortality of WT, Atf3−/−, Il6−/−, and Atf3−/−Il6−/− mice after CLP. Data represent two independent experiments (n = 9 or 10 mice per group per experiment). (f,g) Mortality of WT mice (littermates) fed DMF (GSH-depl.) or water for 10 d then challenged with E. coli intraperitoneally (1 × 1010 c.f.u.) and treated with antibiotics as described in Online Methods. Data represent two independent experiments (n = 5–6 mice per group per experiment). (g) Bacterial load in blood of mice in f, 24 h after E. coli injection (n = 6 mice per group; data are means ± s.d.). *P < 0.05; **P < 0.01; ***P < 0.001 (t test (c) or log-rank statistic).
Control of bacterial or fungal infections requires IL-6 (ref. 28). To test whether ATF3 also attenuates IL-6– or TNF-independent inflammation, we analyzed GPI-induced arthritis, which requires IL-1 but not IL-6 (ref. 29). This inflammation was ATF3-independent, as arthritis was similar in the Atf3−/− and wild-type mice (Supplementary Fig. 7).
ATF3-mediated IL-6 suppression sensitizes for bacterial sepsis
ROS-induced ATF3 severely suppressed serum IL-6 during bacteremia. To determine whether the ATF3-mediated suppression of IL-6 caused the susceptibility to CLP, we studied CLP in Atf3−/−Il6−/− double-knockout mice (Fig. 5e). In CLP conditions that killed 100% of the wild-type mice, 70% of the Atf3−/− mice survived (Fig. 5e). However, 100% of the Atf3−/−Il6−/− mice died from this CLP (Fig. 5e), indicating that the ATF3-mediated IL-6 suppression directly caused the high susceptibility of the wild-type mice to CLP. Therefore, the endotoxin-mediated induction of ATF3 provided robust protection against LPS-induced shock while strongly enhancing susceptibility to bacteremia. ROS-mediated GSH depletion amplified both of these effects, an outcome reminiscent of SAIS.
To more closely mimic SAIS in hospitalized patients with antibiotic therapy, we induced E. coli sepsis, a frequent cause of sepsis in humans30. Antibiotics protected 100% of mice infected with 1 × 106 colony-forming units (c.f.u.) of E. coli; this dose caused 60% mortality in untreated mice (Supplementary Fig. 5a). At 1 × 1010 c.f.u. of E. coli, the antibiotics protected only 30% of the wild-type mice but 70% of the Atf3−/− mice (Supplementary Fig. 5b; P < 0.05), which demonstrates that ATF3 enhanced susceptibility to bacterial sepsis during antibiotic therapy. Moreover, GSH depletion increased E. coli–induced mortality and bacteremia in antibiotic-treated wild-type mice (Fig. 5f,g) and increased Atf3 mRNA levels but decreased serum IL-6 and IL-12 p40 protein abundance (Supplementary Fig. 5c).
ATF3 causes susceptibility to secondary infections during SAIS
SAIS causes high susceptibility to secondary infections by bacterial, viral and fungal pathogens. The causes of SAIS remain unknown, but they are correlated with high ROS levels and the depletion of GSH11,23,24. During SAIS, decreasing GSH levels were inversely correlated with ATF3 induction (Fig. 1a,b), and GSH depletion caused high, ATF3-dependent susceptibility to bacterial and fungal pathogens. These results suggest that ATF3 induction is an important contributor to SAIS and to the severe susceptibility to secondary infections. To investigate this, we first induced sublethal CLP, a well-established model of SAIS31,32, in wild-type and Atf3−/− mice. During immunosuppression, we then challenged the mice with the fungal pathogen A. fumigatus at doses that were nonpathogenic to healthy mice. During SAIS, all of the wild-type mice died from this sublethal A. fumigatus infection within 8 d; the Atf3−/− mice lived significantly longer, and 20% of the Atf3−/− mice survived this challenge (Fig. 6a). To further aggravate the GSH depletion, we treated mice with DMF during SAIS. This additional GSH depletion enhanced the susceptibility of the immunosuppressed mice to A. fumigatus (Fig. 6b), further diminished the already low GSH abundance (Fig. 6c), increased the fungal load (Fig. 6d), superinduced ATF3 protein (Fig. 6e) and decreased serum IL-6 and IL-12 p40 abundance (Fig. 6f and Supplementary Fig. 5e). Thus, the ROS-induced increase in ATF3 expression caused the susceptibility to acute bacterial and fungal infections and contributed to SAIS and the high sensitivity to secondary infections.
Figure 6.
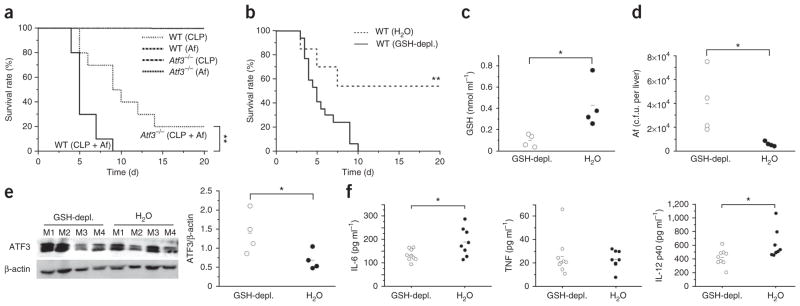
ROS-induced ATF3 crucially regulates susceptibility to secondary infections during SAIS. (a) Mortality in wild-type or Atf3−/− mice subjected to sublethal CLP and then infected intravenously with a nonlethal dose of A. fumigatus (Af; 2 × 106 conidia) 2 d later. Line at top of graph indicates survival of all controls (WT or Atf3−/− mice with sublethal CLP alone or infected with A. fumigatus alone). Data are from two independent experiments (n = 10 mice per group per experiment). (b–f) Mortality (b), GSH in blood (c), fungal load in livers (d), ATF3 in cells from peritoneal lavages (e; western blot from single mice, M1–M4), and cytokine levels in serum after 16 h (f; IL-10 not detectable) in WT mice treated with DMF (GSH-depl.) or water for 10 d, then subjected to sublethal CLP and injected with a nonlethal dose of A. fumigatus (n = 13–17 mice per group). ATF3 blots were normalized to β-actin using the ImageJ software program. *P < 0.05 (t test); **P < 0.01 (log-rank statistic).
DISCUSSION
Here we have shown that ROS-mediated depletion of GSH, which is the primary cellular ROS scavenger, strongly superinduces endotoxin- triggered ATF3 in human monocytes and mouse macrophages in response to TLR signals. This was demonstrated in human subjects with sepsis, septic mice and GSH-depleted mice. ROS-mediated ATF3 superinduction required nuclear translocation of the ROS-sensitive transcription factor NRF2 after GSH depletion and NRF2 binding to the ATF3 promoter. ATF3 superinduction severely impaired the endotoxin-induced production of innate cytokines (Supplementary Fig. 8), which protected against endotoxin-induced lethal shock. However, ATF3-mediated suppression of the innate cytokine storm (specifically of IL-6) abrogated the control of bacteria and fungi and caused high susceptibility to secondary infections. This indicates that IL-6 is crucial for controlling bacterial and fungal infections, whereas silencing of additional cytokines is required to attenuate LPS-induced shock.
The key role of a single transcription factor, ATF3, in determining the outcome of SAIS surprised us, because SAIS is a complex disease associated with severe impairment of multiple immune functions12–17. Until now, to our knowledge no single target molecule of therapeutic value has been identified3,12, and clinical trials targeting the ‘sepsis complex’ with a single compound have not improved overall survival3,6–10. Our data reveal that sepsis-associated ROS stress itself sensitizes monocytes and macrophages to TLR-mediated ATF3 superinduction. This ATF3 superinduction may protect against acute stresses that mimic LPS-induced shock, such as mechanical lung ventilation, acute kidney injury or allergen exposure18,21,22,33,34, certain chemicals, avian flu, or systemic TLR triggers that cause rapid lung injury and death5,35,36 but are attenuated by neutralization of innate cytokines18,37–39.
In contrast, ATF3 induction during SAIS increases susceptibility to secondary infections2,3,12, and treatment with antioxidants, such as NAC, may be beneficial. NAC improves the survival of mice with severe bacterial infection40 and peroxidative indices and clinical scores in people with sepsis41. However, NAC does not improve the overall survival of people with sepsis41. Our results support a potential benefit of NAC during sepsis, but in clinical studies NAC therapy requires clearly defined patient populations and correct dosing and timing. Several reports support this view: appropriate neutrophil recruitment by IL-33 improves bacterial clearance and survival in CLP42; restoring the production of interferon-γ by monocytes improves the outcome of sepsis in humans43; and negative regulators of TLRs, such as IRAK-M and ST2, enhance susceptibility to secondary infections44,45.
Our results are reminiscent of those in Tlr4−/− or Tnf−/− mice, yet they extend further these findings27,37,46–49. First, ATF3 regulation by ROS differentially determined the susceptibility to either endotoxins such as LPS or to pathogens such as bacteria and fungi. Second, a single ROS-regulated transcription factor had opposing effects on LPS-induced shock and on septicemia. This explains why endotoxin shock, septicemia and SAIS require different treatment protocols and why studies of treatments that do not improve the overall survival of individuals with sepsis frequently identify subpopulations who do benefit50,51. Therefore, GSH and ATF3 expression in PBMCs may be reliable parameters to monitor susceptibility to secondary infections. Moreover, because ATF3 can be regulated by GSH in vivo, our data suggest ATF3 as a powerful target for prophylactic or therapeutic intervention in endotoxic shock, bacterial sepsis or SAIS.
ONLINE METHODS
Subjects
We conducted clinical evaluations at the Medical University of Vienna, Department of Surgery, after the study was approved by the local ethics committee of the Medical University of Vienna, Vienna, Austria, and written informed consent was obtained from the subjects (EK number 580/2008). We included 16 subjects with sepsis, defined according to the criteria of the Society of Critical Care Medicine, the European Society of Intensive Care Medicine, the American College of Chest Physicians, the American Thoracic Society and the Surgical Infection Society; the affected subjects had a Sequential Organ Failure Assessment score of 9.2 ± 4 (mean ± s.d.) within 24 h of diagnosis in the ICU50. We determined the SOFA score during acute sepsis. The criteria for the definition of septic shock and exclusion criteria are provided in the Supplementary Methods. We collected 10 ml of whole blood from the subjects during acute sepsis, within 24 h after the diagnosis of sepsis and when leaving the ICU. Depending on the experiment, four to six healthy volunteers served as controls. We obtained PBMCs from a second group of eight subjects with sepsis for the ATF3 western blot analysis.
Mice
Female C57BL/6 mice aged 8–12 weeks were obtained from Charles River, and Il6−/− (Il6tm1Kopf)28 mice were obtained from the Jackson Laboratory. T.H. provided the Atf3−/− mice51. S. Werner (Eidgenössische Technische Hochschule Zürich) provided the Nrf2−/− (Nfe2l2tm1Ywk) mice25 with the permission of J.A. Johnson (University of California, San Francisco). We generated the Atf3−/−Il6−/−mice by crossing the respective single-knockout mice. We screened the mice by standard PCR using ear tissue and published primer sequences (http://jaxmice.jax.org/). The experiments were performed with the approval and supervision of the Regierungspräsidium Tübingen (HT03/07, HT04/07, HT01/11) and the University of Regensburg (54-2532.1-32/08). We used sex- and age-matched mice for all of the experiments. We kept the mice for 3–6 weeks in identical conditions to adapt the gut flora, and we used littermates where indicated.
Cell purification, generation and culture
We obtained peritoneal macrophages by lavage of the peritoneal cavity with PBS and cultured them in complete RPMI medium at 0.5 × 106 cells ml−1. We collected the PBMCs by Ficoll-Paque density gradient separation from the leukapheresis products of healthy volunteers provided by the University of Tübingen blood bank. We purified monocytes by positive selection using CD14 microbeads (Miltenyi)52. We cultured the PBMCs and monocytes in X-VIVO 15 medium (BioWhittaker) that contained 1% autologous human plasma at 1 × 106 cells ml−1. We cultured THP-1 cells from American Type Culture Collection in supplemented RPMI medium and generated mouse BMDCs as described53. Where indicated, we incubated the cells with 70 μM DMF, 1 mM NAC, 50 μM tBHQ or 50 μM BHA (all from Sigma-Aldrich) or 10 mM H2O2 (Roth) for 2–6 h and then stimulated cells with LPS (10 ng ml−1; Sigma-Aldrich).
Quantitative PCR, short interfering RNA, western blotting, luciferase reporter assay and chromatin immunoprecipitation
For the quantitative PCR analysis, RNA from the defined cell populations, whole spleens, PBMCs, or human (PAXgene RNA tubes) or mouse blood was isolated using the NucleoSpin RNA II kit (Macherey-Nagel) or the Peqlab Total RNA kit (Peqlab). After the DNase digestion, cDNA synthesis was performed using the SuperScript III First-Strand Synthesis SuperMix (Invitrogen) or the Sensiscript RT kit (Qiagen). For the relative quantification by quantitative PCR, 20 ng of each cDNA sample was analyzed in a LightCycler 480 Real-Time PCR System using the FastStart DNA Masterplus SYBR Green I kit (Roche Diagnostics). The sequences of the primer pairs are reported in the Supplementary Methods. The relative gene expression was normalized to that of the housekeeping gene G6PDH or β-actin and expressed in relative units. Where indicated, the results were normalized to the controls. For the knockdown experiments, ATF3-specific siRNA (a pool of three target-specific 19- to 25-nt siRNAs) and control siRNA were purchased from Santa Cruz Biotechnology, and the knockdown was performed according to the manufacturer’s instructions. Briefly, human CD14+ monocytes (0.5 × 106 cells) were transfected with 60 pmol siRNA (ATF3 or control) using the transfection reagent and transfection medium (Sigma-Aldrich) for 72 h. The cells were then incubated with the control medium or DMF (70 μM) and stimulated with LPS (10 ng ml−1). ATF3 expression was analyzed by western blotting, and cytokine production was analyzed using ELISA. For the western blot analyses, we prepared the cellular extracts by lysing the cells in SDS-PAGE sample loading buffer and boiling them at 95 °C for 5 min. The proteins were resolved in 4–20% gradient SDS/PAGE gels and transferred to a PVDF membrane, which was blocked with 5% milk and TBS plus Tween and hybridized overnight at 4 °C with an ATF3-specific, NRF2-specific (both Santa Cruz) or β-actin–specific (Cell Signaling) antibody in TBS plus Tween. After incubation with a secondary antibody (Cell Signaling), the blots were developed with enhanced chemiluminescence (Pierce). In some experiments, the blots were analyzed quantitatively using image analysis software (Image J). For the luciferase reporter assays, RAW cells and human astrocytes were transfected using PolyFect reagents (Qiagen) with ATF3 promoter–luciferase constructs26. All of the cells were cotransfected with the β-galactosidase expression vector (pSV–β-galactosidase). Luciferase activity was measured using the Luciferase Assay System (Promega), and β-galactosidase activity was measured using O-nitrophenyl β-galactopyranoside. Chromatin immunoprecipitation was performed using the ChIP kit (Upstate Biotechnology). ARE1 in the ATF3 promoter was amplified by PCR using primers listed in the Supplementary Methods.
Glutathione assay, flow cytometry and ELISA
We measured the intracellular and plasma glutathione levels in mouse and human cells using a colorimetric assay (Sigma-Aldrich)54. The content of glutathione in whole blood was determined using HPLC (in the laboratory of A. Bayer). To measure apoptosis in cell culture, we used an Annexin V/PI staining kit (BD). The ELISAs were performed according to the manufacturers’ instructions (BD and eBioscience). In some experiments, mouse serum was analyzed with a Cytometric Bead Array according to the manufacturer’s instructions (BD).
Lipopolysaccharide-induced endotoxemia, and bacterial and fungal sepsis models
In some experiments, the mice (C57BL/6 and Atf3−/−) were treated orally with DMF (400 μg daily) or DMF plus NAC (1600 μg daily) for 5–10 d. For the endotoxemia experiments, the mice were challenged intraperitoneally with LPS (20 mg per kg body weight; Sigma-Aldrich), and body temperatures were recorded in the rectum with a BAT thermometer (WPI). Bacterial peritonitis was induced with an intraperitoneal injection of 106–1010 c.f.u. of E. coli ATCC25922. The mice were treated with ceftriaxone (100 mg kg−1 intraperitoneally) plus gentamicin (20 mg per kg body weight intraperitoneally) 1 h after the E. coli injection and then with ceftriaxone alone every 24 h. The CLP experiments (C57BL/6, Atf3−/−, Il6−/− or Atf3−/−Il6−/− mice) were performed as described27. Where mentioned, the mice were infected intravenously with A. fumigatus (2 × 106–3 × 106 conidia). Blood samples, peritoneal lavage and livers were harvested to quantify bacterial and fungal contents. Liver cell suspensions were prepared, serial dilutions were generated and 50–100 μl of each dilution was plated on tryptic soy or Sabouraud glucose agar with antibiotic (Fisher Scientific). The plates were incubated for 1–2 d at 37 °C, and the c.f.u. were counted.
Statistical analyses
Parametric data were analyzed using the unpaired Student’s t test. For the nonparametric data, the Wilcoxon rank-sum test was used (ANOVA). For the survival studies, the log-rank test was used to determine significance. Pearson’s correlation was used to calculate the correlation. A P value of <0.05 was considered to be statistically significant; P values of <0.01 and <0.001 are indicated separately.
Additional methods
Detailed methodology is described in the Supplementary Methods.
Supplementary Material
Acknowledgments
We thank B. Pichler, N. Suttorp, T. Welte, S. Werner and P. Dotto for helpful discussions, and for reviewing the manuscript, and M. Haberbosch and S. Hemberger for technical assistance. S. Werner (Eidgenössische Technische Hochschule Zürich) provided the Nrf2−/− (Nfe2l2tm1Ywk) mice25 with the permission of J.A. Johnson (University of California, San Francisco). This work has been supported by grants from the Sander Stiftung (2005.043.2 and 2005.043.3), Deutsche Krebshilfe (no. 109037), Deutsche Forschungsgemeinschaft (SFB 685 A6 and C1, Bi 696/3-3, Bi 696/5-1), German Federal Ministry of Education and Research (BMBF FKZ 0315079 to K.G., DLR 01GN0970 to M.R.), University of Tuebingen (W.H., f-33654-87; E.G., f-1803-0-0; K.G. and M.R., IZKF-Tuebingen, collaborative research program), European Union FP7-HEALTH-2007-2.4.4-1 200515 (M.R.), The German Dermatologic Society/Arbeitsgemeinschaft Dermatologische Forschung (W.H.), a National Research Foundation of Korea grant that was funded by the Ministry of Education, Science and Technology through the Creative Research Initiative Program (grant R16-2004-001010010,2010) and a World Class University grant (no. R31-2008-000-10103-0).
Footnotes
Supplementary information is available on the Nature Medicine website.
AUTHOR CONTRIBUTIONS
W.H., B.E., E.G., N.V., A.T. and F.W. performed the experiments and analytical methods (quantitative PCR, western blots, flow cytometry, ELISA, CLP and endotoxin model) and analyzed the data. J.-H.P., K.-H.K. and K.-W.K. performed the luciferase and chromatin immunoprecipitation experiments. K.H. and C.K. collected the blood samples of subjects with sepsis. P.H. measured the bacterial and fungal loads. K.F. and M.K. performed arthritis experiments. J.B., K.G. and M.R. developed the glutathione depletion model in vitro and in vivo and the initial proof of concept. W.H., B.E., E.G., F.W., T.B. and M.R. designed the experiments. W.H., B.E., E.G., F.W., J.B., K.G., T.B. and M.R. discussed the manuscript. M.R. developed the concept. W.H., E.G., B.E. and M.R. coordinated and directed the project. T.H. provided the Atf3−/− mice. W.H., T.B. and M.R. interpreted the data and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 2.Riedemann NC, Guo RF, Ward PA. Novel strategies for the treatment of sepsis. Nat Med. 2003;9:517–524. doi: 10.1038/nm0503-517. [DOI] [PubMed] [Google Scholar]
- 3.Hotchkiss RS, Opal S. Immunotherapy for sepsis—a new approach against an ancient foe. N Engl J Med. 2010;363:87–89. doi: 10.1056/NEJMcibr1004371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bone RC, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest; The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine; 1992; 2009. p. e28. [Google Scholar]
- 5.Imai Y, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bone RC, et al. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med. 1987;317:653–658. doi: 10.1056/NEJM198709103171101. [DOI] [PubMed] [Google Scholar]
- 7.Ziegler EJ, et al. Treatment of gram-negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin. A randomized, double-blind, placebo-controlled trial. The HA-1A Sepsis Study Group. N Engl J Med. 1991;324:429–436. doi: 10.1056/NEJM199102143240701. [DOI] [PubMed] [Google Scholar]
- 8.Fisher CJ, Jr, et al. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N Engl J Med. 1996;334:1697–1702. doi: 10.1056/NEJM199606273342603. [DOI] [PubMed] [Google Scholar]
- 9.Abraham E, et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAb Sepsis Study Group. J Am Med Assoc. 1995;273:934–941. [PubMed] [Google Scholar]
- 10.Fisher CJ, Jr, et al. Initial evaluation of human recombinant interleukin-1 receptor antagonist in the treatment of sepsis syndrome: a randomized, open-label, placebo-controlled multicenter trial. Crit Care Med. 1994;22:12–21. doi: 10.1097/00003246-199401000-00008. [DOI] [PubMed] [Google Scholar]
- 11.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 12.Ward NS, Casserly B, Ayala A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med. 2008;29:617–625. doi: 10.1016/j.ccm.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muller Kobold AC, et al. Leukocyte activation in sepsis; correlations with disease state and mortality. Intensive Care Med. 2000;26:883–892. doi: 10.1007/s001340051277. [DOI] [PubMed] [Google Scholar]
- 14.Hotchkiss RS, et al. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci USA. 1999;96:14541–14546. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Osuchowski MF, Welch K, Siddiqui J, Remick DG. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J Immunol. 2006;177:1967–1974. doi: 10.4049/jimmunol.177.3.1967. [DOI] [PubMed] [Google Scholar]
- 16.Gogos CA, Drosou E, Bassaris HP, Skoutelis A. Pro- versus anti-inflammatory cytokine profile in patients with severe sepsis: a marker for prognosis and future therapeutic options. J Infect Dis. 2000;181:176–180. doi: 10.1086/315214. [DOI] [PubMed] [Google Scholar]
- 17.Steinhauser ML, et al. IL-10 is a major mediator of sepsis-induced impairment in lung antibacterial host defense. J Immunol. 1999;162:392–399. [PubMed] [Google Scholar]
- 18.Gilchrist M, et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–178. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- 19.Whitmore MM, et al. Negative regulation of TLR-signaling pathways by activating transcription factor-3. J Immunol. 2007;179:3622–3630. doi: 10.4049/jimmunol.179.6.3622. [DOI] [PubMed] [Google Scholar]
- 20.Rosenberger CM, Clark AE, Treuting PM, Johnson CD, Aderem A. ATF3 regulates MCMV infection in mice by modulating IFN-gamma expression in natural killer cells. Proc Natl Acad Sci USA. 2008;105:2544–2549. doi: 10.1073/pnas.0712182105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilchrist M, et al. Activating transcription factor 3 is a negative regulator of allergic pulmonary inflammation. J Exp Med. 2008;205:2349–2357. doi: 10.1084/jem.20072254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U. ATF3 and stress responses. Gene Expr. 1999;7:321–335. [PMC free article] [PubMed] [Google Scholar]
- 23.Biolo G, Antonione R, De Cicco M. Glutathione metabolism in sepsis. Crit Care Med. 2007;35:S591–S595. doi: 10.1097/01.CCM.0000278913.19123.13. [DOI] [PubMed] [Google Scholar]
- 24.Fläring UB, Hebert C, Wernerman J, Hammarqvist F, Rooyackers OE. Circulating and muscle glutathione turnover in human endotoxaemia. Clin Sci (Lond) 2009;117:313–319. doi: 10.1042/CS20080462. [DOI] [PubMed] [Google Scholar]
- 25.Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci USA. 2001;98:4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim KH, Jeong JY, Surh YJ, Kim KW. Expression of stress-response ATF3 is mediated by Nrf2 in astrocytes. Nucleic Acids Res. 2010;38:48–59. doi: 10.1093/nar/gkp865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Echtenacher B, Mannel DN, Hultner L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature. 1996;381:75–77. doi: 10.1038/381075a0. [DOI] [PubMed] [Google Scholar]
- 28.Kopf M, et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339–342. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- 29.Kneilling M, et al. Targeted mast cell silencing protects against joint destruction and angiogenesis in experimental arthritis in mice. Arthritis Rheum. 2007;56:1806–1816. doi: 10.1002/art.22602. [DOI] [PubMed] [Google Scholar]
- 30.Annane D, Bellissant E, Cavaillon JM. Septic shock. Lancet. 2005;365:63–78. doi: 10.1016/S0140-6736(04)17667-8. [DOI] [PubMed] [Google Scholar]
- 31.Benjamim CF, Hogaboam CM, Lukacs NW, Kunkel SL. Septic mice are susceptible to pulmonary aspergillosis. Am J Pathol. 2003;163:2605–2617. doi: 10.1016/S0002-9440(10)63615-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benjamim CF, Hogaboam CM, Kunkel SL. The chronic consequences of severe sepsis. J Leukoc Biol. 2004;75:408–412. doi: 10.1189/jlb.0503214. [DOI] [PubMed] [Google Scholar]
- 33.Akram A, et al. Activating transcription factor 3 confers protection against ventilator induced lung injury. Am J Respir Crit Care Med. 2010;182:489–500. doi: 10.1164/rccm.200906-0925OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li HF, Cheng CF, Liao WJ, Lin H, Yang RB. ATF3-mediated epigenetic regulation protects against acute kidney injury. J Am Soc Nephrol. 2010;21:1003–1013. doi: 10.1681/ASN.2009070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsung A, et al. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204:2913–2923. doi: 10.1084/jem.20070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gill R, Tsung A, Billiar T. Linking oxidative stress to inflammation: Toll-like receptors. Free Radic Biol Med. 2010;48:1121–1132. doi: 10.1016/j.freeradbiomed.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marino MW, et al. Characterization of tumor necrosis factor-deficient mice. Proc Natl Acad Sci USA. 1997;94:8093–8098. doi: 10.1073/pnas.94.15.8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229:869–871. doi: 10.1126/science.3895437. [DOI] [PubMed] [Google Scholar]
- 39.Fattori E, et al. Defective inflammatory response in interleukin 6-deficient mice. J Exp Med. 1994;180:1243–1250. doi: 10.1084/jem.180.4.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Villa P, Saccani A, Sica A, Ghezzi P. Glutathione protects mice from lethal sepsis by limiting inflammation and potentiating host defense. J Infect Dis. 2002;185:1115–1120. doi: 10.1086/340042. [DOI] [PubMed] [Google Scholar]
- 41.Ortolani O, et al. The effect of glutathione and N-acetylcysteine on lipoperoxidative damage in patients with early septic shock. Am J Respir Crit Care Med. 2000;161:1907–1911. doi: 10.1164/ajrccm.161.6.9903043. [DOI] [PubMed] [Google Scholar]
- 42.Alves-Filho JC, et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med. 2010;16:708–712. doi: 10.1038/nm.2156. [DOI] [PubMed] [Google Scholar]
- 43.Döcke WD, et al. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med. 1997;3:678–681. doi: 10.1038/nm0697-678. [DOI] [PubMed] [Google Scholar]
- 44.Deng JC, et al. Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J Clin Invest. 2006;116:2532–2542. doi: 10.1172/JCI28054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoogerwerf JJ, et al. Loss of suppression of tumorigenicity 2 (ST2) gene reverses sepsis-induced inhibition of lung host defense in mice. Am J Respir Crit Care Med. 2011;183:932–940. doi: 10.1164/rccm.201006-0934OC. [DOI] [PubMed] [Google Scholar]
- 46.Sultzer BM. Genetic control of leucocyte responses to endotoxin. Nature. 1968;219:1253–1254. doi: 10.1038/2191253a0. [DOI] [PubMed] [Google Scholar]
- 47.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 48.O’Brien AD, et al. Genetic control of susceptibility to Salmonella typhimurium in mice: role of the LPS gene. J Immunol. 1980;124:20–24. [PubMed] [Google Scholar]
- 49.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 50.Levy MM, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Intensive Care Med. 2003;29:530–538. doi: 10.1007/s00134-003-1662-x. [DOI] [PubMed] [Google Scholar]
- 51.Hartman MG, et al. Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol Cell Biol. 2004;24:5721–5732. doi: 10.1128/MCB.24.13.5721-5732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghoreschi K, et al. Interleukin-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat Med. 2003;9:40–46. doi: 10.1038/nm804. [DOI] [PubMed] [Google Scholar]
- 53.Biedermann T, et al. IL-4 instructs TH1 responses and resistance to Leishmania major in susceptible BALB/c mice. Nat Immunol. 2001;2:1054–1060. doi: 10.1038/ni725. [DOI] [PubMed] [Google Scholar]
- 54.Ghoreschi K, et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J Exp Med. 2011;208:2291–2303. doi: 10.1084/jem.20100977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.