

Abstract
The human gut microbiota consists of ten times more microorganisms than there are cells in our body, processes otherwise indigestible nutrients, and produces important energy precursors, essential amino acids, and vitamins. In this study, we assembled and validated a genome-scale metabolic reconstruction of Bacteroides thetaiotaomicron (iAH991), a prominent representative of the human gut microbiota, consisting of 1488 reactions, 1152 metabolites, and 991 genes. To create a comprehensive metabolic model of host-microbe interactions, we integrated iAH991 with a previously published mouse metabolic reconstruction, which was extended for intestinal transport and absorption reactions. The two metabolic models were linked through a joint compartment, the lumen, allowing metabolite exchange and providing a route for simulating different dietary regimes. The resulting model consists of 7239 reactions, 5164 metabolites, and 2769 genes. We simultaneously modeled growth of mouse and B. thetaiotaomicron on five different diets varying in fat, carbohydrate, and protein content. The integrated model captured mutually beneficial cross-feeding as well as competitive interactions. Furthermore, we identified metabolites that were exchanged between the two organisms, which were compared with published metabolomics data. This analysis resulted for the first time in a comprehensive description of the co-metabolism between a host and its commensal microbe. We also demonstrate in silico that the presence of B. thetaiotaomicron could rescue the growth phenotype of the host with an otherwise lethal enzymopathy and vice versa. This systems approach represents a powerful tool for modeling metabolic interactions between a gut microbe and its host in health and disease.
Keywords: Bacteroides thetaiotaomicron, Mus musculus, computational modeling, constraint-based modeling, host-microbe interactions, metabolism, systems biology
Introduction
The gut microbiota plays an essential role in human health and is thought to be involved in the development of numerous complex diseases.1 Advances in metagenomics have made it possible to access the diversity of the human gut microbiota and its collective genome, the gut microbiome.2,3 Increasing evidence points to a connection between the gut microbiota composition and health and disease states, such as changes in its composition in obese or dieting individuals.2 Recently, the existence of enterotypes, which characterize the gut microbial composition of human individuals, has been proposed.3,68 These enterotypes also point toward a role of the gut microbiota in idiosyncratic human response to drug and diet.3
The human gut microbiota is dominated by two bacterial phyla, Firmicutes and Bacteroidetes, as 90 to 99% of the identified phylotypes in metagenomic analysis belong to either of them4. On the genus level, the gram-negative Bacteroides of the Bacteroidetes phylum are the most abundant.3,5 A common representative of the Bacteroides is Bacteroides thetaiotaomicron. This beneficial human gut inhabitant has adapted to its environment by developing an impressive repertoire of enzymes targeting both dietary plant polysaccharides and host-derived mucosal glycans.6,7 Humans are well equipped to hydrolyze disaccharides, such as lactose and sucrose, as well as some forms of starch, but are limited in their ability to utilize other dietary polysaccharides.8 Saccharolytic gut inhabitants, such as B. thetaiotaomicron, benefit the human host by fermenting these otherwise inaccessible dietary polysaccharides to short-chain fatty acids (SCFAs), which are consumed by the host. SCFA absorption in the large intestine is estimated to contribute 5–10% to daily caloric intake in humans on an average Western diet.9 Besides serving as nutrients, SCFAs also regulate inflammatory responses.1 Other beneficial roles performed by the gut microbiota include maturation of the host immune system and intestinal epithelial cell homeostasis.1
Several experimental approaches have been applied to study the relationships between a host and its gut microbiota. A common method is the use of gnotobiotic mice, which are delivered by Caesarian section, raised germfree and then colonized with a defined microbiota.10 Gnotobiotic mice colonized selectively with B. thetaiotaomicron are particularly well studied. In combination with transcriptomic analysis, such humanized mouse models have been used to study their response to B. thetaiotaomicron,11 and the adaptation of the latter to the gut habitat and to the presence of other gut microbes.12-14
Systems biology aims to analyze the interactions between cellular components by generation of high-throughput “omics” data and computational analysis of these data. Genome-scale high-quality metabolic reconstructions are frequently used to put omics data into context.15 They are generated in a bottom-up manner and represent a biochemical, genetic and genomic (BiGG) knowledge bases for target organisms. Metabolic reconstructions can be converted into mathematical models and used to simulate the organism’s phenotypic behavior given some environmental constraints (e.g., medium compositions).16 The reconstruction process requires intensive manual curation effort,16 though recent progress has made it possible to automate some steps of the reconstruction process.17
High-throughput “omics” methods, such as metagenomics, metabolomics and metaproteomics, have been used to study the complex ecosystem residing in the mammalian gut.18-20 The metabolic interactions between a host and gut microbes have not yet been modeled using bottom-up systems biology methods. Here, we reconstructed and analyzed the first integrated stoichiometric model of murine and B. thetaiotaomicron metabolism.
Results
In this study, we manually assembled a metabolic reconstruction for B. thetaiotaomicron and expanded the published mouse reconstruction with an intestinal absorption module. After integration into a single model, we computed the tradeoff between the growth of mouse and of B. thetaiotaomicron on five different dietary regimes. Furthermore, we investigated nutrient competition and mutually beneficial cross-feeding in the integrated model. In silico metabolite exchange and secretion profiles were computed and compared with in vivo metabolomics data. Finally, we performed a genome-wide in silico gene deletion study for the integrated mouse and B. thetaiotaomicron model.
Genome-scale metabolic reconstruction of the human symbiont, B. thetaiotaomicron
We assembled a draft genome-scale metabolic reconstruction for B. thetaiotaomicron from the Model Seed pipeline.17 Subsequent extensive manual curation (Fig. S1, see Supplementary text for detailed description of the reconstruction process and content) ensured that the reconstruction-derived models were able (1) to produce all known biomass precursors on defined anaerobic minimal medium, (2) to metabolize all major reported carbon sources (Table S1) and (3) to secrete known by-products (Table S2). The model predicted gene essentiality with an accuracy of 86.6% (Supplementary text). We compared predicted quantitative growth rates with experimentally reported ones and found moderate overlap, which was, at least in part, due to missing experimental information on substrate uptake rates (Table S3a). The model also released known secretion products in ratios that compared well with experimental data (Table S3b). The final manually curated and validated reconstruction of B. thetaiotaomicron contains 1213 metabolic and transport reactions, 275 exchange and demand reactions, 1152 unique metabolites, and 991 genes (Table 1). The reconstruction was deemed iAH991, where i stands for in silico, AH are the initials of the lead reconstructor, and 991 the number of captured genes. Information from 150 primary and review papers was incorporated into the final reconstruction, ensuring that it captures the known biochemistry and physiology of B. thetaiotaomicron.
Table 1. Summary of the metabolic reconstructions employed in this study. BΘ, B. thetaiotaomicron.
| Bacteroides thetaiotaomicron | Mus musculus | Integrated model | |
|---|---|---|---|
| Acronym |
iAH991 |
iSS1393 |
iexGFMM_BΘ |
| Total reactions |
1488 |
4091 |
7239 |
| Metabolic and transport reactions |
1213 |
3505 |
5443 |
| Exchange and sink reactions |
275 |
585 |
Internal: 728 External: 1068 |
| Compartment specific metabolites |
1152 |
2950 |
5164 |
| Genes |
991 |
1393 (1778 transcripts) |
2769 |
| Gene associated reactions |
1068 |
2357 |
3428 |
| Reversible reactions |
45% |
n. d. |
n. d. |
| Irreversible reactions |
55% |
n. d. |
n. d. |
| Blocked reactions |
87 |
784 |
1219 |
| Number of compartments |
2 |
8 |
12 |
| Usable carbohydrates as sole carbon sources |
63 |
n. d. |
n. d. |
| Growth on minimal medium (MM) |
Yes, on anaerobic glucose MM67 |
Yes, on aerobic amino acid MM supplemented with glucose, fatty acids and ions21 |
n. d. |
| Oxygen requirement in silico |
no |
yes |
Mouse: yes BΘ: no |
| Genes predicted to be essential on minimal medium | 204 | 54 | n.d. |
Expansion of the genome-scale metabolic reconstruction of Mus musculus
The published mouse metabolic reconstruction, iMM1415, represents the metabolic capabilities found in any mouse cell.21 It can be employed to assess the overall growth capability of a mouse given some nutrient uptake rates. To enable the simulation of different dietary regimes as well as of mucus-type O-glycan secretion,7 of bilirubin transport, and of menaquinone uptake,22 we expanded iMM1415 with intestinal transport and absorption reactions based on literature. Altogether, we added 371 transport and exchange reactions, 27 metabolites, and 29 genes. When performing quality control, we identified and removed 67 genes in iMM1415 that were artifacts and were not associated with reactions. We revised the genes associated with the reactions and removed 23 incorrectly included human genes, while adding further 39 missing mouse homologous genes. Due to these changes, 54 transcripts were predicted to be essential on amino acid minimal medium supplemented with glucose in the updated reconstruction, compared with 53 transcripts in iMM1415.21 The additional essential transcript encodes a phosphoserine phosphatase. The updated and expanded mouse reconstruction (deemed iSS1393) consisted of 4091 reactions, 2950 metabolites, 1393 genes, and 1778 transcripts.
An integrated model of mouse: B. thetaiotaomicron interaction
To model metabolic interactions, we linked the two metabolic models through metabolites in a common compartment, the intestinal lumen, which allowed metabolite exchange between mouse and B. thetaiotaomicron, while providing a route for simulating different dietary regimes (Fig. 1A). The resulting B. thetaiotaomicron-associated mouse model was called iexGFMM_BΘ, where exGFMM stands for ex-germfree Mus musculus. It consists of 2769 genes, 5164 (non-unique) metabolites, and 7239 reactions (Table 1) distributed over 12 compartments: two B. thetaiotaomicron compartments, eight mouse compartments, the lumen and a compartment for secretion into mouse body fluids (Fig. 1A). Furthermore, to avoid biologically implausible solutions, we added further constraints to iexGFMM_BΘ linking the flux through to one of the organism’s reactions with the respective biomass reaction (see Materials and Methods and Figs. S2-S4).

Figure 1. Simultaneous optimization of mouse and B. thetaiotaomicron growth rate using an integrated model of host and gut symbiont metabolism. (A) Schematic representation of iexGFMM_BΘ and the possible metabolite exchange between iAH991 and iSS1393. Compartments: [u], lumen; [b], body fluids; [c], cytoplasm; [e], extracellular. The numbers of exchanged and secreted metabolites on a Western diet are shown. Green arrows indicate metabolites B. thetaiotaomicron provides to the mouse model. Purple arrows indicate mouse metabolites provided to B. thetaiotaomicron. (B) Compositions of diets simulated in this study. CHO = carbohydrate. (C) Predicted growth rates for mouse and B. thetaiotaomicron in iexGFMM_BΘ, and in the individual models are listed for the five dietary regimes employed in this study. Note that the computed mouse growth rates were not realistic, since the biomass reaction summed the required fractions of biomass precursors for a new cell but not for an entire new mouse. (D) Trade-off between the two organisms’ maximal achievable growth rates in iexGFMM_BΘ (Pareto optimality curve). Due to the constraints imposed by the ATP maintenance reactions in iexGFMM_BΘ, fixing either biomass reaction at very low growth rates produces an infeasible solution.
The B. thetaiotaomicron-associated mouse model captures mutualistic growth
We investigated mutual growth dependencies in five different dietary regimes varying in protein, carbohydrate, and fat content (Fig. 1). In all dietary regimes, at low growth rates of B. thetaiotaomicron, the mouse profited significantly from the presence of B. thetaiotaomicron in the lumen. However, as B. thetaiotaomicron’s growth rate increased, competition for nutrients increased and subsequently, the maximal mouse growth rate decreased rapidly (Fig. 1D). Based on iexGFMM_BΘ’s predictions, the ideal growth rate of B. thetaiotaomicron for optimal mouse growth rate was between 0.05−0.29 h−1 depending on the diet (Fig. 1C). In vivo, it has been estimated that E. coli divides about 1.2 times per day in mono-associated mice (equal to a growth rate of 0.05 h−1), and in conventional mice, the average microbial doubling time is 3.3−5.7 divisions per day.23 To our knowledge, in vivo growth rates for B. thetaiotaomicron in mice have not been reported. Since iexGFMM_BΘ corresponds to a mouse colonized with only one microbe, the predicted B. thetaiotaomicron growth rates compares favorably with experimental data.
Presence of B. thetaiotaomicron significantly influences the growth phenotype of mouse
We predicted and compared the growth optima of mouse and B. thetaiotaomicron under different diets (Fig. 1B–D). On a carbohydrate-rich diet, optimal growth rate of both B. thetaiotaomicron and mouse was achieved. While lower than maximal growth rates of B. thetaiotaomicron and mouse were reached on a high-protein diet, a lower growth rate of B. thetaiotaomicron was necessary for an optimal growth rate of the mouse model, suggesting a reduced contribution from B. thetaiotaomicron. On a high fat diet, a higher growth rate of B. thetaiotaomicron was needed for the mouse model to reach an optimal growth rate. On a ketogenic diet, consisting almost entirely of fatty acids, the growth rate of both mouse and B. thetaiotaomicron was poorest. To determine growth-limiting nutrients for each model, we repeated the analysis with iSS1393 and iAH991 individually (Fig. 1C). The high-carbohydrate diet was a very good source of nutrients for the B. thetaiotaomicron model, which agrees with B. thetaiotaomicron being saccharolytic and efficient at utilizing dietary polysaccharides.24 On its own, iAH991 was unable to grow on a high-protein diet, in accordance with the Bacteroides genus’ known incapability to utilize proteins as sole carbon source and the particularly low proteolytic capacity of B. thetaiotaomicron.25 iAH991 was also incapable of growing on high-fat or ketogenic diet. Accordingly, B. thetaiotaomicron has no β-oxidation pathways annotated in its genome.6 For the germfree mouse model, the growth rate was highest on a high-protein diet (75% protein) and lowest on the ketogenic diet (4.5% protein) (Fig. 1C), revealing that amino acid content was growth-limiting for the mouse model. This observation agrees with the known requirement for several essential amino acids in mice.26 In iexGFMM_BΘ, the requirement for essential amino acids was satisfied by B. thetaiotaomicron at low, non-competition inducing microbial growth rates.
Metabolic dependency between mouse and B. thetaiotaomicron
We investigated the metabolite exchanges between mouse and B. thetaiotaomicron, by determining the minimal and maximal possible exchange flux values, while simulating growth on high-protein, high-fat and Western diets. For comparison, we also performed this analysis using the in silico germfree mouse model. When comparing the germfree with the microbe associated mouse model, we identified 266 nutrients, whose associated exchange fluxes changed in at least one diet (Fig. S5; Table S4). The exchanged metabolites belonged to a variety of subsystems, including energy, fatty acid, cholesterol, amino acid, and nucleotide metabolism (Table S4). We predict that the mouse supplies nine metabolites to B. thetaiotaomicron, whereas B. thetaiotaomicron provided 52 metabolites to the mouse (Fig. S5; Table S4). Metabolites provided by B. thetaiotaomicron to mouse included essential amino acids, nucleotides, and SCFAs, i.e., acetate and propionate (Table S4). Metabolic products of mouse consumed by B. thetaiotaomicron included hyaluronan and mucin-type O-gluycans, which are well known to be utilized by the microbe.27 In total, the mouse and B. thetaiotaomicron secreted 30 and 89 metabolites, respectively, into the lumen (Fig. S5). The mouse model secreted 226 compounds into the body fluids compartment, including a number of glycolipids and hormones (Table S4). The mouse model could not metabolize ten dietary carbohydrates in the absence of B. thetaiotaomicron (Fig. S5). When B. thetaiotaomicron was present, these polysaccharides were metabolized leading to the liberation of simple sugars, such as galactose and mannose, which were then used by the mouse model (Table S4).
Identification of competitive dietary components
To identify dietary components, for which the two organisms compete, we compared the minimal and maximal possible exchange flux values for the exchange reactions between B. thetaiotaomicron/mouse and the lumen. We considered three scenarios: at optimal mouse growth rate (diamonds in Fig. 1D), at optimal B. thetaiotaomicron growth rate (triangles in Fig. 1D), and during nutrient competition (squares in Fig. 1D). The two organisms had 32 metabolite exchange reactions in common, for which they could compete (Fig. S5). The analysis was performed on high-protein diet, high-fat diet, and Western diet. Under these conditions, 22, 24, and 25 dietary metabolite exchange reactions, respectively, had flux values above the cutoff (0.01 mmol.gDw−1.hr−1) and showed at least a 2-fold flux change between two or three scenarios (Table S5). On high-fat and Western diet, the production of ten and 11 amino acids, respectively, by B. thetaiotaomicron was required to enable maximal mouse growth rate. These amino acids included all nine essential mouse amino acids, and cysteine, as well as tyrosine on Western diet. For eight non-essential amino acids, the possible flux range was more flexible, spanning from consumption to production by B. thetaiotaomicron, highlighting that their biosynthesis by the mouse did not influence the maximal possible mouse growth rate. In the competition phase, the B. thetaiotaomicron model supplied lower fluxes of threonine and methionine to the mouse, making them major competition-inducing nutrients (Table S5).
When the growth rate of B. thetaiotaomicron was optimized, the microbe did not synthesize several essential mouse amino acids but took them up from the lumen. This change affected the maximal possible growth rate of the mouse (Table S5). Additionally, on Western diet, B. thetaiotaomicron was forced to consume some of the glucose, maltose, lactose, and sucrose provided by the diet to achieve maximal growth rate, which negatively affected the maximal possible mouse growth rate (Fig. 1D). In contrast, on high-fat diet, the flux rate for mouse-derived hyaluronan was increased (data not shown). B. thetaiotaomicron in turn used hyaluronan as an energy and carbon source and thus the competition for dietary simple sugars was reduced. On a high-protein diet, the high influx of amino acids removed competition for dietary carbohydrates. The mouse required the presence of B. thetaiotaomicron for the synthesis of six essential amino acids for optimal growth. Phenylalanine, histidine and tryptophan did not need to be provided by B. thetaiotaomicron under these conditions (Table S5).
In silico metabolome profile in the B. thetaiotaomicron-associated mouse model
Metabolomics analysis of biofluids and tissue from conventional or gnotobiotic and germfree animals has demonstrated that germ-free animals display a large range of differences to conventional animals, including effects on body metabolism, electrolysis, and nutrient requirements.10 We computed the in silico metabolome profile of germfree and B. thetaiotaomicron-associated mouse while simulating growth on high-protein, high-fat, and Western diet. Numerous exchange reactions for metabolites known to be modulated by the gut microbiota, such as amino acids, fatty acids and energy metabolites, were found to change in flux between the germfree and microbe-associated model simulations. B. thetaiotaomicron secreted acetate, propionate, succinate, lactate, fumarate, and phenylacetate into the lumen. In general, the germfree mouse was predicted to have higher secretion fluxes for metabolites containing carbon, nitrogen, and sulfur, as its maximal possible growth rate was limited by availability of essential amino acids in absence of the microbe. Several important vitamins, such as folate, tetrahydrofolate, and niacin, were only found in the body fluid metabolome in the presence of the microbe. Changes in the in silico metabolome were similar in the three diets. Notable exceptions were (1) ammonia, which was only secreted by B. thetaiotaomicron into the lumen on high-protein diet, (2) urea secretion into body fluids (higher in germfree on high-protein diet, but lower in germfree on high-fat and Western diet), and (3) nitric oxide secretion into body fluids (higher in germfree mice on a high-protein and Western diet, but lower in germfree mice on a high-fat diet) (Table S4).
We compared the in silico metabolome profile with five metabolomics studies on conventional and germfree mice20,28-31 (Table 2). While the in vivo measurements in lumen correspond to the lumen compartment in our model setup, the “biofluid” compartment represents serum, blood, and urine (Fig. 1A). The in silico metabolome profile contained 266 metabolites, of which 52 overlapped with experimental data. Our predictions agreed, in general, much better with metabolome changes in the lumen, especially for metabolites of the bile acid, fatty acid, and cholesterol metabolism. Moreover, 42 metabolites were measured experimentally but are not currently captured by our model.
Table 2. Comparison of in silico metabolite profiles between iexGFMM_BΘ and the germfree mouse model with in vivo measured metabolite concentration differences between conventional and germfree mouse20,28-31.
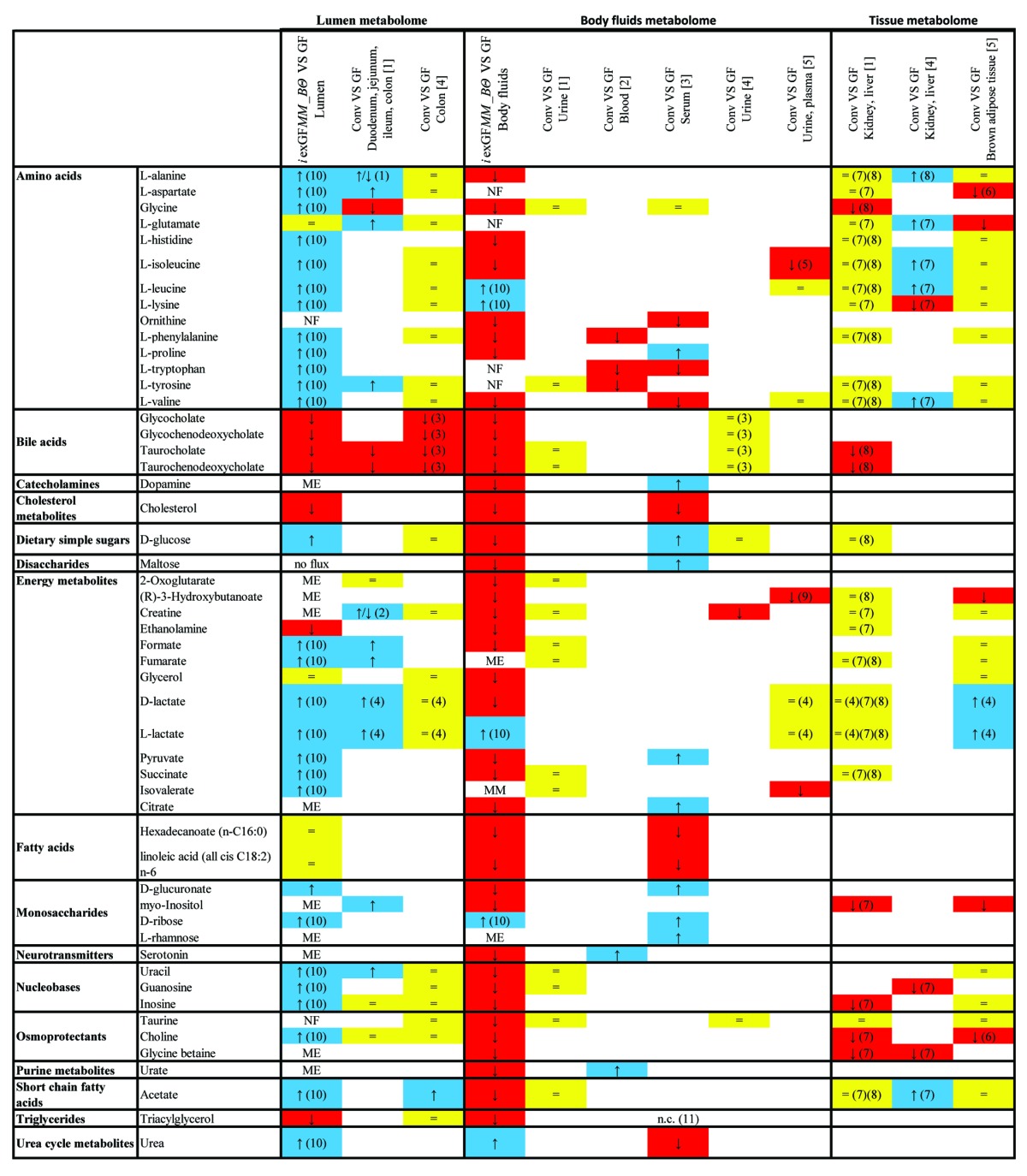
The in silico compartments were body fluids [b] and lumen [u]. Agreements between in silico and in vivo data are presented as follows: higher flux/ concentration (blue, arrow up), lower flux/ concentration (red, arrow down), equal flux/ concentration (yellow), authors do not report detection of this metabolite in this compartment (white), zero flux (white, NF), missing exchange reaction in the model or constraints preventing flux (white, ME) and metabolite not in model (white, MM). Abbreviations: (1) lower levels in ileum and higher levels in duodenum of germfree mice; (2) lower levels in colon and higher levels in jejunum of germfree mice; (3) types of measured bile acids not specified; (4) not specified if D- or L-lactate was measured; (5) female animals only; (6) male animals only; (7) observed in kidney; (8) observed in liver; (9) observed in plasma; (10) flux only when B. thetaiotaomicron is present; and 11. not comparable (the metabolite information in iSS1393 was not specific enough).
B. thetaiotaomicron rescues lethal mouse gene knockouts and vice versa
We aimed to determine how the presence of mouse and B. thetaiotaomicron affected the essentiality of genes when both organisms metabolically interact. When performing an in silico single transcript deletion study on Western diet, 50 transcripts were essential in the germfree mouse (iSS1393). When the same analysis was performed for iexGFMM_BΘ only 29 mouse knockouts resulted in a lethal growth phenotype of the mouse. Thus, the presence of B. thetaiotaomicron rescued the growth phenotypes for 21 single knockouts by compensating for the lost enzymatic functions. As expected, the deletion of B. thetaiotaomicron genes did not result in any lethal growth phenotypes in mouse. However, 184 gene deletions in B. thetaiotaomicron reduced the maximal possible mouse rate growth by more than 50%. In iAH991, 160 genes were essential for maximal possible microbial growth on Western diet, but only 156 B. thetaiotaomicron genes were in silico essential in iexGFMM_BΘ. The rescued gene knockouts were: BT_0554 (glutamine-fructose-6-phosphate transaminase, EC 2.6.1.16), BT_0558 (mannose-1-phosphate guanylyltransferase, EC 2.7.7.22), BT_1224 (GDP-D-mannose dehydratase, EC 4.2.1.47), and BT_1225 (GDP-L-fucose synthase, EC 1.1.1.271). The deletion of mouse genes did not cause any lethality in B. thetaiotaomicron, but reduced the maximal achievable microbial growth rate by up to 50% in 42 cases and by at least 50% in 31 cases.
Metabolic activity of B. thetaiotaomicron rescues on the mouse growth phenotypes in three in silico IEM models
To obtain further insight into the 21 rescued mouse growth phenotypes, we retrieved information for known enzymopathies associated with the genes by mapping a recently assembled compendium of human inborn errors of metabolism (IEMs)32 onto the mouse model. Of the 50 lethal growth phenotypes, ten corresponded to known IEMs. Five IEMs are known to be lethal in mouse (Table S6), one of which is also lethal in humans (D-bifunctional protein deficiency, OMIM: 261515), and two vary in lethality and severity of symptoms in humans (desmosterolosis, OMIM: 602398; dihydrofolate reductase deficiency, OMIM: 126060). For the remaining five in silico lethal IEMs, we could not find information on growth phenotype in mice, but in human they exhibit non-lethal phenotypes. Three of the 21 rescued growth phenotypes overlapped with IEMs, being AICA-ribosiduria (OMIM: 608688), ribose 5-phosphate isomerase deficiency (OMIM: 608611), and orotic aciduria (OMIM: 258900). The enzyme-deficient iexGFMM_BΘ models had growth rates equal to the wildtype iexGFMM_BΘ model.
How can B. thetaiotaomicron compensate the lethal effects of these diseases? We analyzed the effect of the three simulated IEMs on the achievable fluxes in iexGFMM_BΘ. For orotic aciduria, the fluxes of 18 reactions in the B. thetaiotaomicron model and 11 reactions in the mouse model, all involved in nucleotide metabolism and nucleoside transport, increased more than 3-fold compared with the wildtype iexGFMM_BΘ (Table S7a). In particular, the flux values for uridine secretion by B. thetaiotaomicron into the lumen was over 5-fold increased. The mouse luminal uptake reaction for uridine showed a corresponding increase in flux value. Lumen transport reaction fluxes for cytidine and deoxycytidine were also more than 4-fold higher. B. thetaiotaomicron reactions increased in flux range included B. thetaiotaomicron’s orotidine monophosphate decarboxylase (EC 4.1.1.23, BiGG ID: OMPDC) and orotate: pyrophosphoryl transferase (EC 2.4.2.10, BiGG ID: ORPT). The supplementation of the germfree orotic aciduria mouse model with uracil, uridine, deoxyuridine, cytidine, or deoxycytidine rendered the enzyme deficiency non-lethal (data not shown). However, supplementation of other nucleosides or nucleobases did not restore the growth phenotype.
When AICA-ribosiduria was simulated, 28 B. thetaiotaomicron reactions and 12 mouse reactions showed at least a 3-fold increase in flux values (Table S7b), which were in involved in nucleotide metabolism and transport/exchange. The most notable change was the 8-fold flux increase through the inosine exchange reaction between the two models, followed by a > 6-fold increase in guanosine exchange, and a > 4-fold increase in deoxyadenosine exchange. Simulation of the ribose-5-phosphate isomerase deficiency produced a > 3-fold increase in D-ribose transport flux and increased fluxes through further 15 metabolic reactions involved in ribonucleoside metabolism (Table S7c). B. thetaiotaomicron rescued the in silico mouse growth phenotype of AICA-ribosiduria by supplying purine nucleosides, and of ribose-5-phosphate isomerase deficiency through supply of D-ribose liberated from ribonucleosides.
Discussion
In this study, we developed a constraint-based modeling framework permitting to investigate systematically the metabolic interactions between gut microbes and their host, and demonstrated using mouse and B. thetaiotaomicron. Our results suggest that(1) our model accurately captures known growth dependencies between mouse and B. thetaiotaomicron, (2) amino acids are major competition-inducing nutrients; (3) the model can predict changes in biofluid metabolome composition due to microbe association; (4) the association with mouse increases B. thetaiotaomicron’s growth fitness; and (5) gut microbes provide important metabolites to enzyme deficient hosts turning a lethal genotype into a viable one. The presented modeling approach can be used to further our understanding of how the gut microbiota contributes to human health.
A constraint-based reconstruction and analysis (COBRA) approach has been used to investigate symbiotic relationships between microbes,33 to discover previously unknown putative commensal or mutualistic microbial growth dependencies,34,35 and to analyze host-pathogen interactions.36 Here, we expanded the COBRA approach to a modeling framework (Fig. 1A), which permits the systematically investigation of complex metabolic host-microbe interactions. This framework can be easily extended to model the host’s association with multiple microbes. For instance, metabolic reconstructions are available for at least ten gut microbes.15,37,38 The reconstruction process has been partially automated yielding draft metabolic networks in a short time frame.17 However, as illustrated in the current work, these networks still require substantial manual refinement. The number of draft metabolic reconstructions for human gut microbes is likely to increase significantly in near future due to the increasing number of gut microbial genomes published by the Human Microbiome Project.39 Thus, the presented scalable, tractable modeling framework for studying symbiotic and competitive interactions between the gut microbiota and its host, including human,40,41 will open new avenues for studying in silico host-microbiota interactions and their role in human health. The resulting hypothesis may drive experiments, which then could in turn inform the model. Using this combined computational-experimental approach of hypothesis generation and testing has the promise to accelerate new discoveries.
The integrated model of B. thetaiotaomicron and mouse captured known growth dependencies, such as the gut microbe’s ability to extract energy from dietary polysaccharides, which would otherwise be worthless to the host8 (Fig. 1D). Acetate and propionate production by B. thetaiotaomicron25 and the consumption by the host9 were also captured accurately by iexGFMM_BΘ (Fig. 2; Table S4). Moreover, the mouse model could not metabolize ten dietary carbohydrates in absence of B. thetaiotaomicron (Fig. S5), including raffinose, an oligosaccharide that accumulates in colonocytes of germfree mice.20 The B. thetaiotaomicron model could ferment indigestible oligosaccharides, such as raffinose,42 and complex carbohydrates, such as pullulan,24 arabinogalactan, and pectins27 to SCFAs. These examples illustrate the predictive capacity of the presented modeling framework and of the integrated mouse-B. thetaiotaomicron model in particular for studying microbial contributions to host nutrition. Moreover, we demonstrated that this contribution could be investigated in the light of different nutritional regimes.

Figure 2. Metabolite dependency of mouse and B. thetaiotaomicron in iexGFMM_BΘ. On a Western diet, changes in flux rates in iexGFMM_BΘ, when maximizing for mouse growth rate, were compared with those in the maximally growing germfree mouse model and depicted by arrows (up: increased flux, down: decreased flux compared with germfree mouse). For simplicity not all observed changes and related pathways are shown. Metabolites with increased exchange flux compared with the germfree model are shown in green, while metabolites with reduced exchange flux are shown in red. Metabolites that only secreted in iexGFMM_BΘ are shown in blue. Orange arrows indicate metabolites that B. thetaiotaomicron provides to the mouse. Purple arrows indicate mouse metabolites provided to B. thetaiotaomicron. CHO, carbohydrate; Bas, bile acids; TCA cycle, tricarboxylic acid cycle; = B. thetaiotaomicron’s polysaccharide- degrading enzyme repertoire.
We systematically elucidated the metabolic dependencies and interactions between mouse and B. thetaiotaomicron in silico. We found that B. thetaiotaomicron supplied essential and non-essential amino acids to the mouse model in addition to their availability in the diets, thus, boosting the maximum possible growth rate of the mouse model (Fig. 2; Table S5). While amino acid absorption occurs mainly in the small intestine,43 where B. thetaiotaomicron is normally present at lower numbers than in the colon,44 it is still conceivable that B. thetaiotaomicron is capable of providing the host with amino acids in vivo. For example, up to 20% of lysine is provided by gut microbes.43 Our simulation suggested that essential amino acids are major competition-inducing nutrients. Slow growing B. thetaiotaomicron synthesized amino acids to the benefit of the mouse. However, when B. thetaiotaomicron grew maximally (“bacterial overgrowth”), it depleted dietary amino acids, as well as sugars, significantly reducing the maximal achievable mouse growth rate. While the predicted competition for amino acids may be obvious, it highlights the balance that has to be achieved by the host and its microbiota to ensure a viable, sustainable interaction. It also emphasizes the fine line between commensalism and parasitism. In fact, many commensal gut microbes are known to be pathogenic once they escape the gut. The question of what defines a stable microbial community within the gut is subject to current research and we believe that metabolic modeling will play an important role in addressing this challenge.
The human metabolome database45 reports 5039 distinct metabolites in human biofluids. It can be expected that a significant number of these metabolites are of microbial origin and that the gut microbiota contributes substantially. We predicted numerous metabolites, which were only present in the in silico biofluid metabolome when the mouse model was associated with B. thetaiotaomicron (Table S4). One example being phenylacetate, a product of aromatic amino acid fermentation, which is likely deleterious to human health and has been recently found to be increased in volunteers consuming a high-protein, low-carbohydrate diet.46 We predicted similar flux values for phenylacetate production on all three diets (data not shown), since B. thetaiotaomicron can synthesize aromatic amino acids de novo, independent from dietary protein input. Another example is tetrahydrofolate, which is produced from B. thetaiotaomicron-derived folate. Nitric oxide secretion by mouse into body fluids was increased in silico, on a high-fat diet when B. thetaiotaomicron was present, but decreased on a high-protein or Western diet. Nitric oxide is important for the motility of the gastrointestinal tract, and both excess and deficiency in nitric oxide generation have been associated with diseases.47 Overall, our model predicted changes in biofluid metabolome composition due to microbe association (Fig. 2, Table 2), but the prediction accuracy varied with the compared biofluid. The moderate agreement with the reported plasma metabolome can be partially explained by the fact that the mouse reconstruction represents the global mouse metabolism rather than the metabolism occurring in individual organs. Also, the published studies compared germfree mice with conventional animals possessing a complete microbiota, while we only modeled the presence of B. thetaiotaomicron. For example, consistent with B. thetaiotaomicron’s metabolic inability to synthesize indole-3-propionic acid,31 we did not predict any changes in secretion flux for this metabolite in the microbe-associated mouse. A more comprehensive host-microbiota computational model is needed to capture more accurately the metabolic diversity of the gut microbes. In addition, the simulated diets may not match the diets fed to animals in the experimental studies. Considering these simplifications in our integrated model, the observed agreements with the metabolome prediction are quite remarkable and indicate the predictive potential of the presented modeling framework for host-microbe interactions.
We predict that four lethal growth phenotypes of B. thetaiotaomicron can be rescued by the presence of the murine host. Three of these genes (BT_0558, BT_1224, and BT_1225) were involved in the GDP-fucose biosynthesis pathway. Bacteroides species are well known for their L-fucose containing cell surface and mutants with deficient L-fucose synthesis cannot colonize the mouse intestine under competitive conditions.48 Bacteroides species are also capable of incorporating host-derived L-fucose into their capsule polysaccharides using L-fucose-1-phosphate as an intermediate.48 Accordingly, the mouse model provided L-fucose, enabling B. thetaiotaomicron to synthesize L-fucose containing polysaccharides. The rescue of the growth phenotype was also predicted when L-fucose was added to the in silico growth medium, which is consistent with experimental evidence of B. fragilis being able to incorporate medium-derived L-fucose into its capsule polysaccharides.48 For the fourth rescued growth phenotype (glutamine-fructose-6-phosphate transaminase, BT_0554), no information could be found for B. thetaiotaomicron, but in E. coli, the gene knockout causes a glucosamine requirement.49 When supplying either glucosamine or N-acetylglucosamine to the in silico diet, the growth capability of the knockout was restored. Components of host-derived glycans, such as L-fucose and N-acetylglucosamine, are well known to serve as carbon sources for microbes colonizing the intestine, e.g., E. coli.50 Similarly, B. thetaiotaomicron has been shown to upregulate a L-fucose utilization operon in the mouse intestine, allowing the microbe to use mucus-derived L-fucose.11 We predict that host supply of these important monosaccharides compensates for otherwise lethal gene deletions in gut microbes.
In silico, B. thetaiotaomicron was able to compensate for mouse gene deletions, including three inborn errors of metabolism (IEMs). One of these was orotic aciduria, a defect in the uridine monophosphate synthase, which has orotate:pyrophosphoryl transferase and orotidine monophosphate decarboxylase activities. In these patients, the uridine monophosphate synthesis pathway is blocked and orotate accumulates. B. thetaiotaomicron rescued the in silico enzyme-deficient mouse model by providing uridine and other nucleosides. Subsequent analysis revealed that nutritional supplementation with one of the nucleosides could also restore the growth phenotype of the germfree knockout mouse model. Consistently, the positive effect of uridine replacement therapy in orotic aciduria patients is well-documented.51-53 In contrast, has been shown that human patients do not respond to uracil treatment.52 Recently, it was discovered that the orthologous gene to the rat intestinal uracil transporter is defective by humans, likely resulting in poor uracil absorption in the human intestine.54 This new finding explains the discrepancy between our predictions and clinical failure of uracil medication. The possible treatment with cytidine has been demonstrated in cell culture,51 but since uridine is less expensive, it may be preferred in clinical applications.51 To our knowledge, treatment with deoxynucleosides has not been tested in vivo. It is well established that the gut microbiota is an important source of amino acids and vitamins.43,55 We predict that the gut microbiota also provides the host with nucleosides, which could explain the severe but non-lethality in orotic aciduria patients. AICA-ribosiduria and ribose 5-phosphate deficiency have each been described in a single patient53,56 and no treatment methods are available. A computational system biology approach could be employed to suggest candidate dietary supplementations that could alleviate such rare inherited diseases.
While the IEMs modeled in this study are individually rare and to our knowledge, the effects of gut microbial activity on the disease phenotype have not been studied, the etiology of common diseases, such as obesity, inflammatory bowel disease, and circulatory diseases, has been directly linked to the gut microbiota.1 Moreover, it has been recently shown that enzymes carrying out specific metabolic functions are enriched or depleted in the metagenomes of obesity-associated microbiomes.57 Computational modeling of human-gut microbiota co-metabolism will elucidate metabolic strengths and shortcomings that particular genera possess based on their enzymatic repertoire. The impact of particular genera on the caloric load consumed by the human host can also be investigated. We believe that the presented modeling framework will contribute to the ongoing discussion on the relationship between diet, gut microbiota composition, and host metabolism, and the influence of the gut microbiota on human health. Moreover, with the increasing availability of omics data detailing the gut function, the presented modeling framework will enable contextualization and integrative analysis of diverse omic data types beyond correlative analysis and may lead to the discovery of previously unknown relationships between gut microbiota and human in health and disease states.
Methods
Genome-scale metabolic reconstruction of B. thetaiotaomicron
The draft metabolic reconstruction was generated by importing the genome sequence of B. thetaiotaomicron VPI-5482 (obtained from NCBI, ftp.ncbi.nih.gov, January 2011) into the RAST prokaryotic genome annotation server.58 We used the web-based resource Model Seed17 to obtain a draft reconstruction, deemed BΘ_Seed_v1, which was manually curated based on literature using our previously described reconstruction protocol.16 The reaction directionality was assigned in accordance to the directionality listed in the BiGG database.59 The curation and validation process is depicted in Figure S1. Details on the reconstruction and curation process can be found in the Supplementary text. Simulation conditions and constraints for all growth capacity and model behavior tests are listed in Table S8. The complete list of reactions and metabolites included in iAH991 with the associated genes, notes, and references can be found in spreadsheet format in Table S10a−g. Reactions that underwent changes in GPR association are listed in Table S10g.
Expanded genome-scale metabolic reconstruction of Mus musculus
A previously published mouse reconstruction21 was expanded by intestinal transport reactions based on literature and was improved by correcting for duplicate or nonfunctional reactions and erroneous GPRs. This updated mouse reconstruction was named iSS1393. We then determined gene essentiality, while simulating growth on amino acid minimal medium supplemented with glucose and fatty acids and compared the results with the previous mouse reconstruction.21 All reactions, metabolites and associated genes included in iSS1393 as well as a description of the added intestinal transport and absorption module can be found in Table S11a and b.
Construction of the integrated metabolic model of B. thetaiotaomicron and Mus musculus
The integrated model (iexGFMM_BΘ) was constructed by joining the two metabolic models, iAH991 and iSS1393 (Fig. 1A). The integrated model contained a separate extracellular space [e] for each model and the lumen [u] as a common environment. Metabolites from simulated growth medium entered the lumen and could be consumed by either model. The B. thetaiotaomicron model secreted metabolic end products into the lumen. For the mouse model an additional compartment “body fluids” [b] was defined, which served as an outlet for metabolites produced by the mouse (i.e., in urine, blood, lymph). The uptake of metabolites from the lumen was unidirectional for the mouse model except selected transport reactions known or assumed to be reversible. Secretion by the host cytosol [c] into host body fluids [b] was also unidirectional, with the exception of oxygen transport, which could be transported in both directions representing the oxygen uptake through the lungs. Reactions and metabolites in the mouse model started with the letters MM, while B. thetaiotaomicron was assigned the prefix BT. The complete list of reactions and metabolites included in iexGFMM_BΘ can be found in spreadsheet format in Table S12. The germfree mouse model obtained by setting the bounds on all reactions in iAH991 to 0 mmol gDw−1.hr−1.
Addition of further constraints to iexGFMM_BΘ
To model the metabolic dependency between the two organisms in a realistic manner, additional constraints were implemented, such that the flux of reaction i (e.g., from the mouse) and of reaction rxnC (e.g., mouse biomass reaction) were proportional (vi: vrxnC). We bound the ratio between reaction i and reaction rxnC using a factor c, such that for irreversible reactions: vi − c × vrxnC ≥ u, and for reversible reactions: vi − c × vrxnC ≥ u in the forward direction and vi + c × vrxnC ≤ u in the reverse direction. The parameter u ≥ 0 allowed a small reaction flux when vrxnC = 0 (Fig. S2). This parameter takes into account the required flux for “housekeeping,” or maintenance, of cellular function when the cell is not dividing. A sensitivity analysis for different factors c was performed (Supplementary text, Figure S4). For all simulations, c was set to be 400 and u = 0.01 mmol gDw−1.hr−1, respectively. These constraints maintain the linear, scalable, and tractable nature of the COBRA approach. These constraints are in analogy to the coupling constraints developed for non-metabolic networks.60 Constraints for simulation of Western diet, which was used for sensitivity analysis of coupling constraints, are listed in Table S9.
Definition of diets
Five dietary conditions were defined: Western, high-protein, high-carbohydrate, high-fat, and ketogenic (extreme high fat) diet (Fig. 1D). The simulated diets consisted of the same compounds, but varied in protein, fat, and carbohydrate content. The simulated “Western” diet was based on daily amounts typically consumed by an average American citizen.61 The uptake rates for the simulated media were calculated as follows: (1) carbohydrate content was defined as 50% mono- and disaccharides and 50% polysaccharides; fat content was defined as equal fractions of glycerol, cholesterol, saturated fatty acids, and unsaturated fatty acids; and protein was assumed to consist of equal fraction for all 20 amino acids. (2) The uptake rates were scaled to the number of carbon atoms, with glucose as reference, to account for differences in molecule size. (3) The uptake rates were multiplied with the corresponding percentages for each diet. In total, each diet contained carbon sources corresponding to 20 mmol.gDw−1.hr−1 of carbon skeletons with six carbon atoms. Thus, each diet provided the same components and the same net sum of carbon skeletons, only with varying amounts of protein, fat and carbohydrate. Details on simulation constraints and the composition of the simulated diets are reported in Table S9.
Pareto optimality analysis
We investigated the growth dependencies between the two organisms in the integrated model using Pareto optimality analysis,62,63 a bi-objective linear programming approach to analyze the trade-off between two reactions i and j. Briefly, we determined Pareto optimum curves by determining the minimal and maximal flux through reaction i, then fixing the flux through i at different steps spanning minimal to maximal flux, and then maximizing the flux through reaction j for each step. The procedure was repeated with reaction i and j exchanged.
Analysis of metabolite exchange and secretion in iexGFMM_BΘ
Cross-feeding and metabolite exchange in iexGFMM_BΘ was analyzed using flux variability analysis (FVA),64 which minimizes (mini) and maximizes (maxi) the flux through each reaction i. At three different points of the Pareto optimality curve (see Fig. 1D, diamonds, squares, triangles), we performed FVA on the exchange and transport reactions resulting in metabolite exchange profiles, which were compared for identified competition-inducing nutrients. By convention, for exchange reactions negative flux values represent uptake, while positive flux values correspond to secretion. The flux span of reaction i was defined as ||maxi-mini||. The analysis was performed on high-protein diet, high-fat diet, and Western diet. The cutoff for exchange reactions to be included in the analysis was an absolute flux value of 0.01 mmol.gDw−1.hr−1.
In silico metabolome analysis
The metabolite exchange profile was determined for the in silico germfree mouse model. The differences in metabolite exchange and secretion fluxes between B. thetaiotaomicron-associated and germfree mouse model were determined. The analysis was performed on high-protein diet, high-fat diet and Western diet. We only considered exchange reactions with absolute flux values greater than 0.01 mmol.gDw−1.hr−1 in at least one direction and with at least 10% difference in fluxes between germfree and B. thetaiotaomicron-associated mouse model. We defined the in silico “metabolome” as the flux through internal and external exchange reactions between B. thetaiotaomicron and lumen (“gut”), between mouse and lumen, between lumen and outside (“faeces”), and between mouse and a separate outlet (“body fluids”). Metabolite concentrations reported in five in vivo metabolomic studies of conventional and germfree mice20,28-31 were compared with the computed flux differences. Differences in the levels of triglyceride, phosphatidylcholine, and sphingomyelin species30 were not considered, since the mouse reconstruction used for iexGFMM_BΘ did not specify fatty acid residues.
Mapping human inborn errors of metabolism (IEMs) onto iSS1393 and iexGFMM_BΘ
Recently, we assembled a compendium of inborn errors of metabolism, which accounts for 250 human genes and 235 hereditary human diseases,32 many of which have known mouse models (http://www.omim.org/). Using the HomoloGene database (http://www.ncbi.nlm.nih.gov/homologene) and the mouse genome informatics database (http://informatics.jax.org/), the human genes were mapped onto the corresponding mouse homologs. We identified 243 homologs in the mouse genome associated with 222 IEMs that were captured by iSS1393.
Performing a genome-scale single gene deletion study for iexGFMM_BΘ, iAH991 and iSS1393
An in silico single gene deletion study was performed for all transcripts in iSS1393 and all genes in iAH991. The analysis was also performed using iexGFMM_BΘ. In all cases, growth on Western diet was simulated (Table S9). An in silico knockout model was obtained by setting the bounds of the corresponding reaction(s), as defined by the GPR association, to 0 mmol.gDw−1.hr−1. To prevent infeasible LP problems, the lower constraints on the ATP maintenance reactions (ATPM, BTATPM, MMATPM) were set to 0 mmol.gDw−1.hr−1. We then optimized for the respective biomass reaction. Essential genes were those that resulted in a zero flux through the biomass reaction. For the three rescued IEMs, we determined the flux span for each reaction. We only considered reactions with flux values greater than 0.01 mmol.gDw−1.hr−1 in at least one direction and with a fold change of at least three between germfree and B. thetaiotaomicron-associated mouse model. We confirmed potential dietary supplements using the enzyme-deficient germfree mouse model and added the metabolite to the in silico Western diet. Similarly, we examined B. thetaiotaomicron gene knockouts, which were lethal in iAH991, but not in iexGFMM_BΘ, and added candidate metabolites subsequently to the in silico Western diet of the enzyme-deficient B. thetaiotaomicron.
For the reconstruction process, we employed a reconstruction environment tool, rBioNet.65 All simulations were performed using the Matlab-based COBRA Toolbox.66 Tomopt (Tomlab, Inc.) was employed as linear programming solver. We used Matlab (Mathworks, Inc.) as programming environment.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was funded by a Marie Curie International Reintegration grant (No. 249261) within the 7th European Community Framework Program. SS was funded by an Icelandic Research Fund grant (No. 100406022). The authors thank Dr. S. Gudmundsson for valuable discussions.
Glossary
Abbreviations:
- AICA-ribosiduria
5-amino-4-imidazolecarboxamide (AICA)-ribosiduria
- BiGG
biochemical, genetic and genomic
- COBRA
constraint-based reconstruction and analysis
- CPS
capsular polysaccharide
- FVA
flux variability analysis
- GPR
gene-protein-reaction
- IEM
inborn error of metabolism
- NO
nitric oxide
- OMIM
online Mendelian inheritance in man
- RAST
rapid annotations based on subsystem technology
- SCFA
short-chain fatty acid
Supplemental Material
Supplemental materials may be downloaded here:
http://www.landesbioscience.com/journals/gutmicrobes/article/22370/
Footnotes
Previously published online: www.landesbioscience.com/journals/gutmicrobes/article/22370
References
- 1.Kinross JM, Darzi AW, Nicholson JK. Gut microbiome-host interactions in health and disease. Genome Med. 2011;3:14. doi: 10.1186/gm228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tilg H, Kaser A. Gut microbiome, obesity, and metabolic dysfunction. J Clin Invest. 2011;121:2126–32. doi: 10.1172/JCI58109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. MetaHIT Consortium Enterotypes of the human gut microbiome. Nature. 2011;473:174–80. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–8. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007;14:169–81. doi: 10.1093/dnares/dsm018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, et al. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science. 2003;299:2074–6. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- 7.Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 2008;4:447–57. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr. 2002;22:283–307. doi: 10.1146/annurev.nutr.22.011602.092259. [DOI] [PubMed] [Google Scholar]
- 9.McNeil NI. The contribution of the large intestine to energy supplies in man. Am J Clin Nutr. 1984;39:338–42. doi: 10.1093/ajcn/39.2.338. [DOI] [PubMed] [Google Scholar]
- 10.Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol. 2007;19:59–69. doi: 10.1016/j.smim.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, et al. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science. 2005;307:1955–9. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- 12.Sonnenburg JL, Chen CT, Gordon JI. Genomic and metabolic studies of the impact of probiotics on a model gut symbiont and host. PLoS Biol. 2006;4:e413. doi: 10.1371/journal.pbio.0040413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci U S A. 2009;106:5859–64. doi: 10.1073/pnas.0901529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samuel BS, Gordon JI. A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism. Proc Natl Acad Sci U S A. 2006;103:10011–6. doi: 10.1073/pnas.0602187103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oberhardt MA, Palsson BO, Papin JA. Applications of genome-scale metabolic reconstructions. Mol Syst Biol. 2009;5:320. doi: 10.1038/msb.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiele I, Palsson BO. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010;5:93–121. doi: 10.1038/nprot.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 2010;28:977–82. doi: 10.1038/nbt.1672. [DOI] [PubMed] [Google Scholar]
- 18.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–9. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verberkmoes NC, Russell AL, Shah M, Godzik A, Rosenquist M, Halfvarson J, et al. Shotgun metaproteomics of the human distal gut microbiota. ISME J. 2009;3:179–89. doi: 10.1038/ismej.2008.108. [DOI] [PubMed] [Google Scholar]
- 20.Claus SP, Tsang TM, Wang Y, Cloarec O, Skordi E, Martin FP, et al. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol Syst Biol. 2008;4:219. doi: 10.1038/msb.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sigurdsson MI, Jamshidi N, Steingrimsson E, Thiele I, Palsson BO. A detailed genome-wide reconstruction of mouse metabolism based on human Recon 1. BMC Syst Biol. 2010;4:140. doi: 10.1186/1752-0509-4-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shearer MJ, Newman P. Metabolism and cell biology of vitamin K. Thromb Haemost. 2008;100:530–47. [PubMed] [Google Scholar]
- 23.Gibbons RJ, Kapsimalis B. Estimates of the overall rate of growth of the intestinal microflora of hamsters, guinea pigs, and mice. J Bacteriol. 1967;93:510–2. doi: 10.1128/jb.93.1.510-512.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salyers AA, Vercellotti JR, West SEH, Wilkins TD. Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl Environ Microbiol. 1977;33:319–22. doi: 10.1128/aem.33.2.319-322.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dworkin M, Falkow S. The prokaryotes: a handbook on the biology of bacteria. New York; [London]: Springer, 2006. [Google Scholar]
- 26.Fox JG. The mouse in biomedical research. Amsterdam; Boston: Elsevier, AP, 2007. [Google Scholar]
- 27.Martens EC, Lowe EC, Chiang H, Pudlo NA, Wu M, McNulty NP, et al. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 2011;9:e1001221. doi: 10.1371/journal.pbio.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Claus SP, Ellero SL, Berger B, Krause L, Bruttin A, Molina J, et al. Colonization-induced host-gut microbial metabolic interaction. MBio. 2011;2:e00271–10. doi: 10.1128/mBio.00271-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mestdagh R, Dumas ME, Rezzi S, Kochhar S, Holmes E, Claus SP, et al. Gut Microbiota Modulate the Metabolism of Brown Adipose Tissue in Mice. J Proteome Res. 2011 doi: 10.1021/pr200938v. [DOI] [PubMed] [Google Scholar]
- 30.Velagapudi VR, Hezaveh R, Reigstad CS, Gopalacharyulu P, Yetukuri L, Islam S, et al. The gut microbiota modulates host energy and lipid metabolism in mice. J Lipid Res. 2010;51:1101–12. doi: 10.1194/jlr.M002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A. 2009;106:3698–703. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sahoo S, Franzson L, Jonsson JJ, Thiele I. A compendium of inborn errors of metabolism mapped onto the human metabolic network. Mol Biosyst. 2012;8:2545–58. doi: 10.1039/c2mb25075f. [DOI] [PubMed] [Google Scholar]
- 33.Stolyar S, Van Dien S, Hillesland KL, Pinel N, Lie TJ, Leigh JA, et al. Metabolic modeling of a mutualistic microbial community. Mol Syst Biol. 2007;3:92. doi: 10.1038/msb4100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klitgord N, Segrè D. Environments that induce synthetic microbial ecosystems. PLoS Comput Biol. 2010;6:e1001002. doi: 10.1371/journal.pcbi.1001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freilich S, Zarecki R, Eilam O, Segal ES, Henry CS, Kupiec M, et al. Competitive and cooperative metabolic interactions in bacterial communities. Nat Commun. 2011;2:589. doi: 10.1038/ncomms1597. [DOI] [PubMed] [Google Scholar]
- 36.Bordbar A, Lewis NE, Schellenberger J, Palsson BO, Jamshidi N. Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol Syst Biol. 2010;6:422. doi: 10.1038/msb.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thiele I, Hyduke DR, Steeb B, Fankam G, Allen DK, Bazzani S, et al. A community effort towards a knowledge-base and mathematical model of the human pathogen Salmonella Typhimurium LT2. BMC Syst Biol. 2011;5:8. doi: 10.1186/1752-0509-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thiele I, Vo TD, Price ND, Palsson BO. Expanded metabolic reconstruction of Helicobacter pylori (iIT341 GSM/GPR): an in silico genome-scale characterization of single- and double-deletion mutants. J Bacteriol. 2005;187:5818–30. doi: 10.1128/JB.187.16.5818-5830.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, et al. NIH HMP Working Group The NIH Human Microbiome Project. Genome Res. 2009;19:2317–23. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thiele I, al. e. A community-generated reconstruction of the human metabolic network and its use for the analysis of omics data. Submitted.
- 41.Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD, et al. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci U S A. 2007;104:1777–82. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krieg NR. Bergey's manual of systematic bacteriology, 2nd ed., volume 4. New York: Springer, 2010. [Google Scholar]
- 43.Metges CC. Contribution of microbial amino acids to amino acid homeostasis of the host. J Nutr. 2000;130:1857S–64S. doi: 10.1093/jn/130.7.1857S. [DOI] [PubMed] [Google Scholar]
- 44.Hayashi H, Takahashi R, Nishi T, Sakamoto M, Benno Y. Molecular analysis of jejunal, ileal, caecal and recto-sigmoidal human colonic microbiota using 16S rRNA gene libraries and terminal restriction fragment length polymorphism. J Med Microbiol. 2005;54:1093–101. doi: 10.1099/jmm.0.45935-0. [DOI] [PubMed] [Google Scholar]
- 45.Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B, et al. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009;37(Database issue):D603–10. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Russell WR, Gratz SW, Duncan SH, Holtrop G, Ince J, Scobbie L, et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am J Clin Nutr. 2011;93:1062–72. doi: 10.3945/ajcn.110.002188. [DOI] [PubMed] [Google Scholar]
- 47.Shah V, Lyford G, Gores G, Farrugia G. Nitric oxide in gastrointestinal health and disease. Gastroenterology. 2004;126:903–13. doi: 10.1053/j.gastro.2003.11.046. [DOI] [PubMed] [Google Scholar]
- 48.Coyne MJ, Reinap B, Lee MM, Comstock LE. Human symbionts use a host-like pathway for surface fucosylation. Science. 2005;307:1778–81. doi: 10.1126/science.1106469. [DOI] [PubMed] [Google Scholar]
- 49.Wu G, Sun Y, Qu W, Huang Y, Lu L, Li L, et al. Application of GFAT as a novel selection marker to mediate gene expression. PLoS One. 2011;14:e17082. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang DE, Smalley DJ, Tucker DL, Leatham MP, Norris WE, Stevenson SJ, et al. Carbon nutrition of Escherichia coli in the mouse intestine. Proc Natl Acad Sci U S A. 2004;101:7427–32. doi: 10.1073/pnas.0307888101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Becroft DM, Phillips LI. Hereditary Orotic Aciduria and Megaloblastic Anaemia: A Second Case, with Response to Uridine. Br Med J. 1965;1:547–52. doi: 10.1136/bmj.1.5434.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becroft DM, Phillips LI, Simmonds A. Hereditary orotic aciduria: long-term therapy with uridine and a trial of uracil. J Pediatr. 1969;75:885–91. doi: 10.1016/S0022-3476(69)80318-5. [DOI] [PubMed] [Google Scholar]
- 53.Fernandes J. Inborn metabolic diseases: diagnosis and treatment. Heidelberg: Springer, 2006. [Google Scholar]
- 54.Yamamoto S, Inoue K, Murata T, Kamigaso S, Yasujima T, Maeda JY, et al. Identification and functional characterization of the first nucleobase transporter in mammals: implication in the species difference in the intestinal absorption mechanism of nucleobases and their analogs between higher primates and other mammals. J Biol Chem. 2010;285:6522–31. doi: 10.1074/jbc.M109.032961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hill MJ. Intestinal flora and endogenous vitamin synthesis. Eur J Cancer Prev. 1997;6(Suppl 1):S43–5. doi: 10.1097/00008469-199703001-00009. [DOI] [PubMed] [Google Scholar]
- 56.Wamelink MMC, Gruning NM, Jansen EEW, Bleumlein K, Lehrach H, Jakobs C, et al. The Difference between Rare and Exceptionally Rare: Molecular Characterisation of Ribose 5-Phosphate Isomerase Deficiency. J Inherit Metab Dis. 2010;33:S70–S. doi: 10.1007/s00109-010-0634-1. [DOI] [PubMed] [Google Scholar]
- 57.Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci U S A. 2012;109:594–9. doi: 10.1073/pnas.1116053109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schellenberger J, Park JO, Conrad TM, Palsson BO. BiGG: a Biochemical Genetic and Genomic knowledgebase of large scale metabolic reconstructions. BMC Bioinformatics. 2010;11:213. doi: 10.1186/1471-2105-11-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thiele I, Fleming RM, Bordbar A, Schellenberger J, Palsson BO. Functional characterization of alternate optimal solutions of Escherichia coli’s transcriptional and translational machinery. Biophys J. 2010;98:2072–81. doi: 10.1016/j.bpj.2010.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Freedman MR, King J, Kennedy E. Popular diets: a scientific review. Obes Res. 2001;9(Suppl 1):1S–40S. doi: 10.1038/oby.2001.113. [DOI] [PubMed] [Google Scholar]
- 62.Nagrath D, Avila-Elchiver M, Berthiaume F, Tilles AW, Messac A, Yarmush ML. Integrated energy and flux balance based multiobjective framework for large-scale metabolic networks. Ann Biomed Eng. 2007;35:863–85. doi: 10.1007/s10439-007-9283-0. [DOI] [PubMed] [Google Scholar]
- 63.Oberhardt MA, Goldberg JB, Hogardt M, Papin JA. Metabolic network analysis of Pseudomonas aeruginosa during chronic cystic fibrosis lung infection. J Bacteriol. 2010;192:5534–48. doi: 10.1128/JB.00900-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gudmundsson S, Thiele I. Computationally efficient flux variability analysis. BMC Bioinformatics. 2010;11:489. doi: 10.1186/1471-2105-11-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thorleifsson SG, Thiele I. rBioNet: A COBRA toolbox extension for reconstructing high-quality biochemical networks. Bioinformatics. 2011;27:2009–10. doi: 10.1093/bioinformatics/btr308. [DOI] [PubMed] [Google Scholar]
- 66.Schellenberger J, Que R, Fleming RM, Thiele I, Orth JD, Feist AM, et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat Protoc. 2011;6:1290–307. doi: 10.1038/nprot.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Varel VH, Bryant MP. Nutritional features of Bacteroides fragilis subsp. fragilis. Appl Microbiol. 1974;28:251–7. doi: 10.1128/am.28.2.251-257.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–8. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.