

Abstract
Most animal cells express mixtures of the three subtypes of inositol 1,4,5-trisphosphate receptor (IP3R) encoded by vertebrate genomes. Activation of each subtype by different agonists has not hitherto been examined in cells expressing defined homogenous populations of IP3R. Here we measure Ca2+ release evoked by synthetic analogues of IP3 using a Ca2+ indicator within the lumen of the endoplasmic reticulum of permeabilized DT40 cells stably expressing single subtypes of mammalian IP3R. Phosphorylation of (1,4,5)IP3 to (1,3,4,5)IP4 reduced potency by ∼100-fold. Relative to (1,4,5)IP3, the potencies of IP3 analogues modified at the 1-position (malachite green (1,4,5)IP3), 2-position (2-deoxy(1,4,5)IP3) or 3-position (3-deoxy(1,4,5)IP3, (1,3,4,5)IP4) were similar for each IP3R subtype. The potency of an analogue, (1,4,6)IP3, in which the orientations of the 2- and 3-hydroxyl groups were inverted, was also reduced similarly for all three IP3R subtypes. Most analogues of IP3 interact similarly with the three IP3R subtypes, but the decrease in potency accompanying removal of the 1-phosphate from (1,4,5)IP3 was least for IP3R3. Addition of a large chromophore (malachite green) to the 1-phosphate of (1,4,5)IP3 only modestly reduced potency suggesting that similar analogues could be used to measure (1,4,5)IP3 binding optically. These data provide the first structure-activity analyses of key IP3 analogues using homogenous populations of each mammalian IP3R subtype. They demonstrate broadly similar structure-activity relationships for all mammalian IP3R subtypes and establish the potential utility of (1,4,5)IP3 analogues with chromophores attached to the 1-position.
Introduction
Most animal cells express inositol 1,4,5-trisphosphate receptors (IP3R), which fulfil an essential role in linking the many cell-surface receptors that stimulate IP3 formation to release of Ca2+ from the endoplasmic reticulum [1]. Vertebrates have genes for three IP3R subunits, while invertebrates have only a single IP3R gene. All functional IP3R are tetrameric assemblies of these subunits. The similar primary sequences of the IP3R subunits suggest that all IP3R are likely to share similar structures, although we presently have only a limited understanding of the structure of the entire IP3R [1], [2]. Each subunit has an N-terminal region to which IP3 binds. This region comprises the N-terminal suppressor domain (SD, residues 1–223) and the IP3-binding core (IBC, residues 224–604 in IP3R1, Figure 1A), which is alone sufficient to bind IP3 with appropriate selectivity [3]. The SD both modulates the affinity of the IBC for agonists and provides an essential link between IP3 binding and opening of the pore [4], [5], [6], [7]. A large cytoplasmic region separates the N-terminal from the six transmembrane domains. The last pair of these, together with the intervening luminal loop, form the Ca2+-permeable pore [8] (Figure 1A). Each subunit terminates in a short C-terminal tail, which has also been implicated in the regulation of gating [9]. The diversity provided by three genes is further increased by multiple splice variants of at least two of the three IP3R subtypes (IP3R1 and IP3R2), by formation of homo- or hetero-tetrameric assemblies of IP3R subunits, by association with an enormous diversity of modulatory proteins and by post-translational modifications [10]. At present, we have only a limited understanding of the functional significance of this complexity for IP3-evoked Ca2+ signals in native tissues.
Figure 1. Structure of the N-terminal of the IP3 receptor and structures of the ligands used.

(A) Key regions of a single IP3R subunit (numbering for rat IP3R1) are shown highlighting N-terminal domains and the six C-terminal transmembrane domains (TMD) that form the pore. A high-resolution structure of the N-terminal (NT, residues 1–604) with (1,4,5)IP3 bound is shown (Protein Data Bank, 3UJO). The NT comprises the suppressor domain (SD) and IP3-binding core (IBC). The essential 4- and 5-phosphate groups of (1,4,5)IP3 interact with residues in the β-domain and α-domain of the IBC, respectively. (B) Structures of the ligands used.
The broadly similar structures of the three IP3R subunits are matched by many shared functional properties, most notably co-regulation of all IP3R by IP3 and Ca2+ [10], [11]. Nevertheless, there are differences in the patterns of expression of IP3R in different tissues [12], [13], in their subcellular distributions [14], [15], sensitivities to IP3 [16], modulation by accessory proteins and additional signals [17], [18], [19], and in the functional consequences of IP3R ablation [20], [21]. Heterogeneous populations of IP3R in most cells make it difficult to establish clearly the characteristics of each IP3R subtype and to define their functional roles. A better knowledge of the ligand recognition properties of the three IP3R3 subtypes is needed if ligands selective for IP3R subtypes are to be developed to help resolve these problems. All known high-affinity agonists of IP3R retain structures equivalent to the 4,5-bisphosphate and 6-hydroxyl groups of (1,4,5)IP3 (Figure 1B) [22]. The only exception is a low-affinity analogue of adenophostin A (3″-dephospho-adenophostin A) in which interactions between the adenine moiety and IP3R appear partially to compensate for loss of a phosphate (equivalent to the 5-phosphate of (1,4,5)IP3) within the critical bisphosphate moiety [23]. Here we use a selection of synthetic analogues of IP3 that preserve the key structures of the high-affinity agonists to assess their activity at each IP3R subtype.
Materials and Methods
Materials
Thapsigargin was from Alomone Laboratories (Jerusalem, Israel). The structures of the ligands used and their abbreviations are shown in Figure 1B. (1,4,5)IP3 was from Alexis Biochemicals (Nottingham, U.K.). 3-deoxy(1,4,5)IP3, (1,3,4)IP3 and (1,3,4,5)IP4 were from Calbiochem (Nottingham, U.K.). (1,4,6)IP3 from both Alexis Biochemicals and synthesized as reported previously [24] was used. Malachite green IP3 (MG(1,4,5)IP3) was synthesized using the methods described by Inoue et al. [25]. (4,5)IP2 [26], 2-deoxy(1,4,5)IP3 [27] and synthetic (1,3,4,5)IP4 [28] were synthesized as previously described. All synthesized ligands were purified by ion-exchange chromatography, fully characterized by the usual spectroscopic methods and accurately quantified by total phosphate assay. 3H-IP3 (185 Bq/mmol) was from PerkinElmer (Bucks, U.K.).
Anti-peptide antisera to peptides conserved in all mammalian IP3R subtypes (AbC, residues 62–75 of rat IP3R1) or unique to IP3R1 (Ab1, residues 2724–2739) or IP3R2 (Ab2, residues 2685–2701) were described previously [29]. A monoclonal antibody that recognizes N-terminal residues (22–230) of IP3R3 (Ab3) was from BD Transduction Laboratories (Oxford, U.K.). Anti-β-actin antibody was from AbCam (Cambridge, U.K.). Donkey secondary antibodies (anti-rabbit or anti-mouse) conjugated to horseradish peroxidase were from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). Sources of other materials are provided in earlier publications [30], [31].
Cell Culture
DT40 cells lacking genes for all three IP3R subtypes (DT40-KO cells) [32] and the same cells stably expressing rat IP3R1 (GenBank accession number GQ233032.1) [33], mouse IP3R2 (GU980658.1) [31] or rat IP3R3 (GQ233031.1) [34] were cultured in RPMI 1640 medium supplemented with 10% foetal bovine serum, 1% heat-inactivated chicken serum, 2 mM glutamine and 50 µM 2-mercaptoethanol at 37°C in 95% air and 5% CO2. Cells were passaged every 2 days when they reached a density of ∼1.5×106 cells/mL.
Immunoblotting
Cells (∼7×107) were centrifuged (650 g, 5 min), resuspended in Hepes-buffered saline (HBS), re-centrifuged, and the pellet was solubilized in 1 mL of medium containing 140 mM NaCl, 5 mM NaF, 10 mM Tris, 1 mM Na4P2O7, 0.4 mM Na3VO4, 1% Triton X-100 and a protease inhibitor tablet (1 tablet/10 mL, Roche Diagnostics, Mannheim, Germany). HBS had the following composition: 135 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 1.5 mM CaCl2, 11.6 mM Hepes, 11.5 mM glucose, pH 7.3. The solubilized cells were sonicated on ice (Trans Scientific 1420 sonicator, 50–60 Hz, 3×10 s), incubated with gentle rotation for 1 h at 2°C and then centrifuged (6000 g, 10 min). A sample of the supernatant (13 µL) was mixed with dl-dithiothreitol (2 µL, 100 mM) and NuPAGE LDS sample buffer (5 µL, Invitrogen, Paisley, U.K.). After heating (70°C, 10 min), 20-µL samples were loaded onto NuPAGE 3–8% Tris acetate gels for SDS-PAGE using Novex Tris acetate buffer (Invitrogen). Broad range spectrum marker and MagicMark molecular weight markers (Invitrogen) were used to monitor protein migration during SDS-PAGE and for calibration, respectively. After transfer to a PVDF membrane using an iBlot dry-blotting system (Invitrogen), the membrane was blocked by incubation (1 h) with Tris-buffered saline (TBS) containing 5% non-fat dry milk. It was then incubated with primary antiserum (1∶1000 and 1∶10,000 for IP3R and β-actin antibodies, respectively) in TBS containing 2.5% w/v BSA for 12 h. TBS had the following composition: 140 mM NaCl, 20 mM Tris, 0.1% v/v Tween, pH 7.6. After incubation with primary antibodies, the blots were washed in TBS (3×5 min), incubated with horseradish peroxidase-conjugated secondary antibodies (1∶1000, donkey anti-rabbit or donkey anti-mouse) for 1 h in TBS supplemented with 2.5% BSA, and washed again (3×5 min). Bands were detected using Supersignal West Pico chemiluminescent substrate (ThermoScientific, Rockford, IL, U.S.A.) and quantified using GeneTools software (Syngene, Frederick, MD, U.S.A.).
Measurement of IP3-evoked Ca2+ Release
A low-affinity Ca2+ indicator (Mag fluo-4) trapped within the ER lumen was used to measure IP3-evoked Ca2+ release from permeabilized DT40 cells stably expressing mammalian IP3R [35]. Cells (50 mL, 106 cells/mL) were loaded with Mag fluo-4 AM (20 µM) in HBS supplemented with pluronic F127 (0.02% w/v) for 1 h at 20°C in the dark with gentle shaking. The cells were centrifuged (650 g, 5 min), resuspended in Ca2+-free cytosol like medium (CLM) and permeabilized by addition of saponin (10 µg/mL, ∼4 min, 37°C). Ca2+-free CLM had the following composition: 140 mM KCl, 2 mM NaCl, 1 mM EGTA, 2 mM MgCl2, 20 mM Pipes, pH 7. After washing (650 g, 2 min), permeabilized cells were resuspended in CLM without Mg2+, but supplemented with CaCl2 (375 µM) to give a final free [Ca2+] of ∼220 nM (after addition of 1.5 mM MgATP) and with carbonyl cyanide 4-trifluoromethoxy-phenyl hydrazone (FCCP, 10 µM) to inhibit mitochondrial Ca2+ uptake. Cells were distributed (5×105 cells/well) into half area 96-well, black-walled, poly-lysine-coated plates and centrifuged (300 g, 2 min). Fluorescence (excitation and emission wavelengths of 485 nm and 520 nm, respectively) was recorded at 1-s intervals using a FlexStation™ 3 fluorescence plate reader (Molecular Devices, Berkshire, U.K.) at 20°C. Addition of MgATP (1.5 mM) allowed Ca2+ uptake into the intracellular stores, and after 150 s, IP3 (or its analogues) was added together with thapsigargin (1 µM) to prevent further Ca2+ uptake. IP3-evoked Ca2+ release is expressed as a fraction of that released by addition of ionomycin (1 µM) [35].
3H-IP3 Binding to Native Type 1 IP3 Receptors
Mouse cerebellar membranes (5 mg protein) in a final volume of 500 µL of CLM with a free [Ca2+] of 220 nM were incubated with 3H-IP3 (1.5 nM) and appropriate concentrations of competing ligand at 4°C [4]. After 5 min, during which equilibrium was attained, the reactions were terminated by centrifugation (2000 g). The supernatant was removed and the pellet was washed (500 µL CLM) and then solubilized (200 µL CLM with 1% v/v Triton-X-100) before determining its radioactivity using Ecoscint A scintillation cocktail (National Diagnostics, Atlanta, GA, USA, 1 mL). Total 3H-IP3 binding was ∼3000 disintegrations/min (d.p.m.) and non-specific binding was ∼300 d.p.m. (determined by addition of 1 µM IP3 or by extrapolation of IP3 competition curves to infinite IP3 concentration). Results were fitted to Hill equations (GraphPad Prism, version 5, GraphPad Software Inc., CA, U.S.A.) from which half-maximal inhibitory concentration (IC50) and thereby KD values were calculated.
Statistical Analysis
Concentration-effect relationships were fitted to Hill equations using Graph Pad Prism, from which Hill coefficients (h), the fraction of the intracellular Ca2+ stores released by maximally effective concentrations of agonist, and pEC50 values were calculated. For clarity, some results are reported as EC50 values, but all statistical comparisons used pEC50 values. Each experiment included an analysis of the effects of (1,4,5)IP3 to allow paired comparisons of pEC50 values for IP3 and each analogue (ΔpEC50). Results are expressed as means ± SEM for n independent experiments, with each experiment performed in triplicate. Statistical comparisons used paired Student’s t test or ANOVA followed by Bonferroni test, with P<0.05 is considered significant.
Homology Modelling and Ligand Docking
Sequence alignments were performed using MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/). Structural homology models were built in Modeller 9.10 [36] using templates of crystal structures of the IBC (Protein Data Bank: 1N4K) and SD (Protein Data Bank: 1XZZ). The geometric qualities of the models were evaluated with the Molprobity server [37]. Finally, the N-terminal regions of mouse IP3R2 and rat IP3R3 comprising the modelled SD and IBC structures of each were reconstructed by aligning the individual domains against cognate regions of the NT of rat IP3R1 (Protein Data Bank: 3UJ4) [7] using UCSF Chimera 1.6.1 [38].
We used docking of IP3 analogues into a rigid IBC and subsequent superposition of the structure onto a model of the entire IP3R solely to assess whether full-length IP3R was likely to bind the analogues without steric clashes. Docking of IP3 analogues was performed using the IBC (Protein Data Bank: 1N4K) [3] and AutoDock Vina 1.1.2 [39]. Prior to docking, bound (1,4,5)IP3 and all water molecules (except those within 5 Å of the IP3-binding site) were removed. The search space comprised a grid of 20×20×20 points, each separated by 0.375 Å, around the IP3-binding site. Ligands (except (1,4,5)IP3) were drawn and energy-minimized with MM2 force field using ChemBioOffice 2008 (http://www.cambridgesoft.com). Polar hydrogens and the Gasteiger partial atomic charges were then added to the protein and ligands using AutoDockTools (http://autodock.scripps.edu/resources/adt), and the prepared structures were used as input files for docking. Only the best pose is considered for each ligand. For MG(1,4,5)IP3, the ligand was superposed onto (1,4,5)IP3 within the NT monomer (Protein Data Bank: 3UJ0A) and the complex (with (1,4,5)IP3-removed) was energy-minimized using MMFF94 Forcefield [40]. PyMol was used to present docked structures (http://pymol.sourceforge.net/).
Results
Expression of Mammalian IP3 Receptor Subtypes in DT40 Cells
Immunoblotting with IP3R subtype-selective antisera (Ab1-3) established that each of the stable DT40 cell lines specifically expressed only a single IP3R subtype (Figure 2A). An antiserum that recognizes a peptide sequence present in all mammalian IP3R subtypes (AbC) [29] was used to quantify relative levels of IP3R expression in the three cell lines (Figure 2A). The results demonstrate that, relative to IP3R3 (100%), levels of expression of IP3R1 and IP3R2 were 71±8% and 48±5%, respectively (Figure 2A). Because the density of IP3R may affect the sensitivity of intracellular Ca2+ stores to IP3
[6] and it is impracticable to generate cell lines expressing identical levels of each IP3R subtype, comparisons of the relative potencies of IP3 analogues for each IP3R subtype are expressed relative to the potency of (1,4,5)IP3 in the same cell line 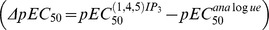 (see Methods).
(see Methods).
Figure 2. Functional expression of recombinant IP3 receptor subtypes in DT40 cells.

(A) Western blots from DT40 cells stably expressing each of the IP3R subtypes. Each lane was loaded with lysate derived from ∼1.4×106 cells stably expressing IP3R1, IP3R2 or IP3R3, or from DT40-KO cells (KO). The antisera used are selective for IP3R1-3 (Ab1-3) or they interact equally with all three IP3R subtypes (AbC) [29] (see Materials). An antiserum to β-actin was used to confirm the equivalent loading of lanes. Quantitative analysis of AbC immunoreactivity from 3 similar gels was used to establish expression levels of the three IP3R subtypes relative to IP3R3 (means ± SEM, n = 3) (lower panel). (B) A luminal Ca2+ indicator was used to record Ca2+ uptake into the intracellular stores of permeabilized DT40 cells expressing IP3R1 after addition of MgATP. Subsequent addition of (1,4,5)IP3 (at the concentrations shown) with thapsigargin (1 µM) allowed (1,4,5)IP3-evoked Ca2+ release to be quantified. The typical experiment shows traces averaged from 3 wells in a single plate. For clarity only a selection of the (1,4,5)IP3 concentrations are shown. The same methods were used to quantify the effects of all analogues. (C) Concentration-dependent effects of (1,4,5)IP3 on Ca2+ release from the intracellular stores of cells expressing IP3R1 (black), IP3R2 (blue) or IP3R3 (red). The same colour-codes are used in all subsequent figures. Results are means ± SEM from the number of independent experiments given in Table 1. Here, and in many subsequent figures, some error bars are smaller than the symbols.
IP3-evoked Ca2+ Release by Subtypes of IP3 Receptor
A low-affinity luminal Ca2+ indicator was used to report the free Ca2+ concentration within the endoplasmic reticulum of permeabilized DT40 cells (Figure 2B). IP3 failed to evoke Ca2+ release in DT40-KO cells, which lack IP3R (not shown) [34], but it was effective in DT40 cells stably expressing each of the three IP3R subtypes (Figure 2B and C) [31]. The results demonstrate that in each cell line, IP3 caused a concentration-dependent release of 60–75% of the intracellular Ca2+ stores, with Hill coefficients (h) of about 1, and pEC50 values of 7.06±0.05, 6.84±0.06 and 6.38±0.05 for IP3R1, IP3R2 and IP3R3, respectively (Table 1 and Figure 2C). Notwithstanding the moderate differences in IP3R expression in the different DT40 cell lines (Figure 2A), the relative sensitivities of IP3R1-3 to IP3 in these assays (IP3R1∼IP3R2>IP3R3) are consistent with a general consensus that IP3R3 is the least sensitive of the IP3R subtypes [16].
Table 1. Effects of IP3 analogues on Ca2+ release by subtypes of IP3 receptor.
| IP3R1 | IP3R2 | IP3R3 | |||||||||||||
| EC50 (nM) | pEC50 | h | Ca2+release (%) | n | EC50 (nM) | pEC50 | h | Ca2+release (%) | n | EC50 (nM) | pEC50 | h | Ca2+release (%) | n | |
| (1,4,5)IP3 | 87 | 7.06±0.05 | 0.99±0.05 | 75±1 | 31 | 145 | 6.84±0.06 | 1.26±0.09 | 61±2 | 34 | 417 | 6.38±0.05 | 1.26±0.07 | 64±2 | 30 |
| (4,5)IP2 | 8128 | 5.09±0.12 | 1.11±0.15 | 69±4 | 4 | 6310 | 5.20±0.20 | 1.67±0.27 | 56±4 | 5 | 11482 | 4.94±0.07 | 1.28±0.03* | 83±2 | 3 |
| (1,3,4,5)IP4 | 11749 | 4.93±0.05 | 1.09±0.16 | 66±6 | 3 | 5495 | 5.26±0.09 | 2.26±0.18 | 61±5 | 3 | 114815b | 3.94±0.03 | 1.08±0.22 | 36±1a | 3 |
| 2-deoxy(1,4,5)IP3 | 123 | 6.91±0.15 | 0.84±0.07 | 82±3 | 4 | 115 | 6.94±0.11 | 1.71±0.37 | 65±2 | 6 | 324 | 6.49±0.02 | 1.36±0.10 | 74±3 | 3 |
| (1,4,6)IP3 | 4365 | 5.36±0.19 | 0.98±0.10 | 81±4 | 6 | 12589 | 4.90±0.16 | 1.06±0.17 | 68±2 | 6 | 15849 | 4.80±0.12 | 2.58±0.61* | 46±1 | 6 |
| 3-deoxy(1,4,5)IP3 | 3311 | 5.48±0.09 | 1.75±0.24* | 61±2 | 6 | 5888 | 5.23±0.07 | 1.78±0.34 | 57±6 | 6 | 15849 | 4.80±0.13 | 2.59±0.68 | 53±7 | 6 |
| MG(1,4,5)IP3 | 447 | 6.35±0.12 | 0.95±0.10 | 73±4 | 9 | 708 | 6.15±0.09 | 1.04±0.08 | 57±3 | 9 | 1738 | 5.76±0.11 | 1.18±0.11 | 64±4 | 8 |
| (1,3,4)IP3 | ND | ND | ND | 39±8a | 3 | ND | ND | ND | 18±6a | 3 | ND | ND | ND | 2±1a | 3 |
The EC50, pEC50, Hill coefficient (h) and fraction of the intracellular Ca2+ stores released by a maximally effective concentration of each analogue are shown for each IP3R subtype. All results (except EC50) are shown as means ± SEM from n independent experiments. The (1,3,4,5)IP4 was provided by Calbiochem. ND, not determined.
Ca2+ release evoked with 100 µM of the analogue (where the highest attainable concentrations of ligand failed to saturate the response).
EC50 estimated by assuming that maximally effective concentrations of (1,3,4,5)IP4 and (1,4,5)IP3 release the same fraction of the Ca2+ stores.
Denotes Hill slopes that are significantly different (P<0.05) from 1. Statistical comparisons of maximal Ca2+ release and pEC50 values used paired comparisons (Δmax or ΔpEC50), which are presented in Table 2.
Interactions of IP3 Metabolites with IP3 Receptor Subtypes
Inositol 1,3,4,5-tetrakisphosphate ((1,3,4,5)IP4) is the immediate product of (1,4,5)IP3 phosphorylation by IP3 3-kinase [41] (Figure 3A). Although (1,3,4,5)IP4 stimulated Ca2+ release via each of the three IP3R subtypes (Figure 3B), it was ∼100-fold less potent than IP3. Indeed in DT40-IP3R3 cells, which are the least sensitive to IP3 (Figure 2C), even 100 µM (1,3,4,5)IP4 failed to release the entire IP3-sensitive Ca2+ store (Figures 3B and C, and Table 1). The purity of (1,3,4,5)IP4 supplied by Calbiochem is only ∼98%. We were therefore concerned that its effects on Ca2+ release might be mediated by contaminating (1,4,5)IP3. However, similar experiments using DT40-IP3R1 cells and synthetic (1,3,4,5)IP4, where the synthesis provides no opportunity for contamination by (1,4,5)IP3 [28], established that the two sources of (1,3,4,5)IP4 were equipotent. Relative to (1,4,5)IP3, ΔpEC50 values were 2.09±0.07 and 1.97±0.04 (n = 3) for commercial and synthetic (1,3,4,5)IP4, respectively. Synthetic (1,3,4,5)IP4 (100 µM) had no significant effect on the Ca2+ content of the intracellular stores of permeabilized DT40 cells lacking IP3R (DT40-KO cells) (not shown). We conclude that (1,3,4,5)IP4 itself stimulates Ca2+ release via all IP3R, but only at concentrations ∼100-fold higher than with (1,4,5)IP3 (Figures 3B and C). The much reduced potency of (1,3,4,5)IP4 is consistent with loss of the 3-hydroxyl group reducing potency (Table 1 and see below) and with docking analyses, which suggest that although the 3-phosphate can be accommodated, the orientations of the phosphate groups are slightly distorted relative to (1,4,5)IP3 bound to the IBC (Figure 3F).
Figure 3. (1,3,4,5)IP4 is a low-affinity full agonist of IP3 receptors.

(A) (1,4,5)IP3 can be dephosphorylated to (1,4)IP2 or phosphorylated to (1,3,4,5)IP4, which can then be dephosphorylated to (1,3,4)IP3. (B) Effects of (1,3,4,5)IP4 (Calbiochem) on Ca2+ release via each of the three IP3R subtypes. (C) Paired comparisons of the effects of (1,4,5)IP3 and (1,3,4,5)IP4 are shown for each IP3R subtype. (D) Typical results from DT40-IP3R1 cells stimulated with (1,4,5)IP3 alone (black trace) or in the presence of (1,3,4,5)IP4 (5 µM, added 35 s before IP3; red trace). (E) Summary results show the effect of (1,3,4,5)IP4 on the concentration-dependent effects of (1,4,5)IP3 on Ca2+ release. Results (B, C and E) are means ± SEM from the number of independent experiments given in Table 1. (F) (1,3,4,5)IP4 docked into the IBC of IP3R1 adopts a conformation in which the underlying (1,4,5)IP3 scaffold is only slightly distorted from that of (1,4,5)IP3 within the crystal structure of the IBC (right panel) [3]. Docking of the NT into a 3D reconstruction of the entire IP3R1 [2] has suggested a likely arrangement of the NT within the tetrameric IP3R [see 7] (left panel). Within this proposed arrangement, the IBC can still bind (1,3,4,5)IP4 without obvious steric clashes.
It has been suggested that (1,3,4,5)IP4 potentiates the Ca2+ release evoked by (2,4,5)IP3 at IP3R1 [42], although in other cells there was no evidence for this interaction [43]. We re-examined this phenomenon by first adding (1,3,4,5)IP4 to permeabilized DT40-IP3R1 cells at a concentration (5 µM) near its threshold for evoking Ca2+ release, and then assessed subsequent responses to (1,4,5)IP3 (Figure 3D). The results, which show that (1,3,4,5)IP4 has no effect on the response to any concentration of (1,4,5)IP3 (Figure 3E), provide no support for the suggestion that (1,3,4,5)IP4 potentiates IP3-evoked Ca2+ release [42]. Furthermore, the lack of inhibition of IP3-evoked Ca2+ release by (1,3,4,5)IP4 demonstrates that it is not a partial agonist of IP3R. We conclude that (1,3,4,5)IP4 is a low-affinity full agonist of all three IP3R subtypes.
Dephosphorylation of (1,3,4,5)IP4 by IP3 5-phosphatase produces (1,3,4)IP3 (Figure 3A), which can accumulate to concentrations considerably exceeding that of (1,4,5)IP3 during sustained stimulation [44]. (1,3,4)IP3, even at a concentration of 100 µM, failed to stimulate release of the entire (1,4,5)IP3-sensitive Ca2+ store in any of the three cell lines (Table 1). (1,3,4)IP3 is, therefore, at least 1000-fold less potent than (1,4,5)IP3. Comparing the fraction of the Ca2+ stores released by 100 µM (1,3,4)IP3 in cells expressing each of the three IP3R subtypes indicates that the response was greatest in DT40-IP3R1 cells and smallest in DT40-IP3R3 cells (Table 1). The effects of (1,3,4)IP3 on the three IP3R subtypes (IP3R1> IP3R2>> IP3R3) therefore match the rank order of potency of (1,4,5)IP3 (Table 1), consistent perhaps with minor contamination (<0.1%) of the (1,3,4)IP3 with (1,4,5)IP3 or (1,3,6)IP3 accounting for the activity. These results suggest that (1,3,4)IP3 is itself unlikely to bind to IP3R. This is consistent with a requirement for a vicinal 4,5-bisphosphate structure in all known inositol phosphate ligands of IP3R [45]. It is impossible for us to verify this independently by equilibrium competition 3H-IP3 binding because DT40 cells are the only homogenous source of IP3R subtypes presently available to us, but the low density of IP3Rs would require impracticably large numbers of cells for binding studies.
Type 3 IP3 Receptors are Slightly Less Sensitive to Loss of the 1-phosphate from (1,4,5)IP3
Removal of the 1-phosphate from (1,4,5)IP3 to give (4,5)IP2 caused a similar ∼83-fold decrease in potency for IP3R1 and IP3R2 (Figures 4A and B, Tables 1 and 2). Contamination of (4,5)IP2 with (1,4,5)IP3 cannot explain this activity because (4,5)IP2 was prepared by total synthesis and purified by ion-exchange chromatography [26]. This is consistent with previous functional analyses of native IP3R [45] and with the ability of (4,5)IP2 to compete with 3H-(1,4,5)IP3 for binding to these IP3R subtypes heterologously expressed in Sf9 cells [46].
Figure 4. IP3 receptor subtypes differ slightly in their requirements for a 1-phosphate group in (1,4,5)IP3.

(A) Effects of (4,5)IP2 on Ca2+ release via each IP3R subtype. (B) Paired comparisons of the effects of (1,4,5)IP3 and (4,5)IP2 are shown for each IP3R subtype. (C) The 1-phosphate group of (1,4,5)IP3 interacts directly with R568 and via water (blue sphere) with K569 within the α-domain of the IBC [3]. The primary sequences of this region (residues 561-580) are similar for each IP3R subtype. (D) Structural homology models of the NT of mouse IP3R2 and rat IP3R3 were built using structures of the SD (Protein Data Bank: 1XZZ) and IBC (Protein Data Bank: 1N4K) from IP3R1 and these were then aligned using the published structure of the NT of rat IP3R1 (Protein Data Bank: 3UJ0).
Table 2. Relative potencies of IP3 analogues at different IP3 receptor subtypes.
| IP3R1 | IP3R2 | IP3R3 | |||||||
| ΔpEC50 | Δmax (%) | (1,4,5)IP3max (%) | ΔpEC50 | Δmax (%) | (1,4,5)IP3max (%) | ΔpEC50 | Δmax (%) | (1,4,5)IP3max (%) | |
| (4,5)IP2 | 1.92±0.15 | 13±5 | 81±3 | 1.92±0.05 | 8±3 | 65±4 | 1.38±0.06* | −1±2 | 77±4 |
| 2-deoxy(1,4,5)IP3 | 0.09±0.10 | −1±2 | 81±3 | 0.19±0.14 | −3±3 | 63±4 | −0.17±0.08 | 0±2 | 77±4 |
| (1,4,6)IP3 | 1.57±0.09 | −2±5 | 76±5 | 1.67±0.15 | −3±6 | 64±5 | 1.45±0.09 | 8±6 | 55±7 |
| 3-deoxy(1,4,5)IP3 | 1.61±0.12 | 21±2* | 80±4 | 1.64±0.21 | 12±4* | 68±3 | 1.66±0.17 | 20±5* | 66±3 |
| (1,3,4,5)IP4 a | 2.09±0.07 | 17±5a | 77±1 | 2.18±0.04 | 7±0* | 67±6 | 2.38±0.07 | NDa | 77±4 |
| MG(1,4,5)IP3 | 0.69±0.10 | 4±5 | 77±3 | 0.7±0.14 | 11±3* | 64±3 | 0.64±0.15 | 4±4 | 61±3 |
From paired comparisons with (1,4,5)IP3, the relative potency 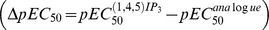 and differences in maximal Ca2+ release
and differences in maximal Ca2+ release 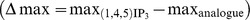 are shown for each analogue at each IP3R subtype. Results show means ± SEM, with n provided in Table 1. To allow direct comparison with responses evoked by (1,4,5)IP3, maximal Ca2+ release evoked by (1,4,5)IP3 in experiments paired with the analogues are also shown ((1,4,5)IP3 max).
are shown for each analogue at each IP3R subtype. Results show means ± SEM, with n provided in Table 1. To allow direct comparison with responses evoked by (1,4,5)IP3, maximal Ca2+ release evoked by (1,4,5)IP3 in experiments paired with the analogues are also shown ((1,4,5)IP3 max).
With (1,3,4,5)IP4 it was impossible to attain concentrations (>100 µM) that evoked a maximal response for all IP3R subtypes; in these cases the maximal responses were not subject to statistical analysis. The relative potency of (1,3,4,5)IP4 at IP3R subtypes was estimated by assuming it was a full agonist that released the same fraction of the intracellular Ca2+ stores as a maximally effective concentration of (1,4,5)IP3 in parallel analyses. ND, not determined.
Denotes values significantly different (P<0.05) from IP3R1 (for ΔpEC50 values) or from (1,4,5)IP3 in paired comparisons with the analogue (for Δmax values).
However, the difference in potency of (1,4,5)IP3 and (4,5)IP2 at IP3R3 was only ∼24-fold (ΔpEC50 = 1.38±0.06) (Figure 4B, Table 2). Our results suggest that although removal of the 1-phosphate from (1,4,5)IP3 reduces potency at all IP3R subtypes, IP3R3 is least affected. Within the IBC of IP3R1, the 1-phosphate of (1,4,5)IP3 interacts directly with R568 and, via water, with K569 [3] (Figure 4C). These residues and their immediate neighbours are conserved in IP3R2 and IP3R3 (Figure 4C). The only residues known to interact directly with (1,4,5)IP3 are within the IBC (Figure 1A) and primary sequences of the IBC are highly conserved between IP3R subtypes. It is therefore unsurprising that homology models based on the IBC of IP3R1 [3] suggest almost indistinguishable structures for the IBCs from IP3R2 and IP3R3 (Figure 4D). Furthermore, the IBCs from the three IP3R subtypes have the same affinity for (1,4,5)IP3 [16]. However, both full-length IP3R and the NT from different IP3R subtypes differ in their affinities for (1,4,5)IP3. These observations demonstrate that interactions between the IBC and other parts of the IP3R, notably the suppressor domain (SD, residues 1–223), influence ligand binding [4], [16]. The complexity of these interactions between the IBC and other domains together with the need for only very modest conformational differences between subtypes to account for the small differences in ligand potency make it difficult to define the residues responsible for modulating the interactions between the IBC and the 1-phosphate group of (1,4,5)IP3 in IP3R3.
Removal of the 2- and 3-hydroxyl Groups from (1,4,5)IP3 or Inverting their Orientations Similarly Affect Interactions with all IP3 Receptor Subtypes
The structure of (1,4,5)IP3 bound to the IBC of IP3R1 shows the 2-hydroxyl group of (1,4,5)IP3 exposed to solvent (Figure 1A) [3] and previous structure-activity studies of native IP3R suggested that removal of the 2-hydroxyl moiety from (1,4,5)IP3 to give 2-deoxy(1,4,5)IP3 minimally affects activity [4], [46]. Our results confirm that observation for all three IP3R subtypes: 2-deoxy(1,4,5)IP3 and (1,4,5)IP3 are equipotent at all three IP3R subtypes (Figures 5A and B, Tables 1 and 2).
Figure 5. The 2- and 3-hydroxy groups of (1,4,5)IP3 interact similarly with each IP3 receptor subtype.

(A) Effects of 2-deoxy-(1,4,5)IP3 on Ca2+ release via each IP3R subtype. (B) Paired comparisons of the effects of (1,4,5)IP3 and 2-deoxy-(1,4,5)IP3 are shown for each IP3R subtype. (C and D) Similar analyses of 3-deoxy-(1,4,5)IP3. Results (A-D) are means ± SEM from the number of independent experiments given in Table 1.
Removal of the 3-hydroxyl of (1,4,5)IP3 (3-deoxy(1,4,5)IP3) caused a ∼40-fold decrease in potency (ΔpEC50 ∼1.6) that was similar for all three IP3R subtypes (Figures 5C and D, Tables 1 and 2). This decrease is larger than that observed for either Ca2+ release from cells expressing largely IP3R1 (∼3-fold) [47] or for binding to heterologously expressed IP3R1-3 (7- 20-fold) [46].
(1,4,6)IP3 is an analogue of (1,4,5)IP3 in which the orientations of the 2- and 3-hydroxyl groups are inverted (Figures 1B and 6A). (1,4,6)IP3 was less potent than (1,4,5)IP3 and the loss of potency was similar for all three IP3R subtypes (Figures 6B and C, Tables 1 and 2). The loss of potency probably results from inverting the 3-hydroxyl group from its equatorial position in (1,4,5)IP3 to an axial position in (1,4,6)IP3 (Figure 6A) because l-scyllo(1,2,4)IP3, which differs from (1,4,5)IP3 only in its inverted orientation of the 2-hydroxyl group, was reported to have the same affinity as (1,4,5)IP3 for all three IP3R subtypes [46].
Figure 6. Interactions of (1,4,6)IP3 with IP3 receptor subtypes.

(A) Structures of (1,4,5)IP3 and (1,4,6)IP3 drawn to show how they differ only in the relative orientations of the 2- and 3-hydroxyl groups of (1,4,5)IP3. (B) Effects of (1,4,6)IP3 on Ca2+ release via each IP3R subtype. (C) Paired comparisons of the effects of (1,4,5)IP3 and (1,4,6)IP3 are shown for each IP3R subtype. Results (B and C) are means ± SEM from the number of independent experiments given in Table 1.
MG-(1,4,5)IP3 is a Full Agonist of IP3 Receptors with Slightly Lesser Affinity than (1,4,5)IP3
MG(1,4,5)IP3 in which 4-carboxy-malachite green is attached via an aminopropyl linkage to the 1-phosphate of (1,4,5)IP3 (Figure 1B) was originally synthesized to explore its potential as a ligand of IP3R that might allow chromophore-assisted laser inactivation (CALI) of IP3R [25], [48]. The first study, using surface plasmon resonance to assess binding to an N-terminal fragment of IP3R1 (residues 1–885), surprisingly suggested that MG(1,4,5)IP3 had ∼170-fold greater affinity than (1,4,5)IP3, whereas fluorescein similarly attached to the 1-position of (1,4,5)IP3 had no significant effect on affinity [25]. The results were important because they suggested that MG(1,4,5)IP3 might be the ligand with the highest known affinity for IP3R, and they were unexpected because disrupting interaction of the 1-phosphate group of (1,4,5)IP3 with the IP3R would be expected to reduce affinity (Figure 4 and Table 1) [45]. A subsequent study of Ca2+ release from permeabilized smooth muscle cells concluded that the EC50 for MG(1,4,5)IP3 was ∼7-fold higher than that for (1,4,5)IP3 [25]. The disparity between the reported very high affinity of MG(1,4,5)IP3 for the N-terminal of IP3R1 and its modest potency in functional assays of smooth muscle has not been explained. We considered two possibilities. MG(1,4,5)IP3 may be a high-affinity partial agonist or it may differ massively in its affinity for IP3R1 and the endogenous IP3R of smooth muscle.
Our results indicate that MG(1,4,5)IP3 is ∼5-fold less potent than (1,4,5)IP3 at each IP3R subtype (Figures 7A and B, Tables 1 and 2). This is consistent with functional assays of smooth muscle [25] and with our results from native IP3R (largely IP3R2) in rat hepatocytes, where ΔpEC50 was 0.57±0.03 for (1,4,5)IP3 (pEC50 = 6.81±0.02) and MG(1,4,5)IP3 (pEC50 = 6.24±0.01) (Taylor CW, unpublished data). These results establish that MG(1,4,5)IP3 interacts similarly with all three IP3R subtypes and that it is less potent than (1,4,5)IP3. In aqueous solution, the triphenylmethane component of MG(1,4,5)IP3 exists as a mixture of several inter-converting species, whose relative proportions are sensitive to pH [49]. At pH 7.4, a colourless triphenylmethanol form with a tetrahedral structure is likely to co-exist with the coloured propeller-shaped form shown in Figures 1B and 7F. Because the earlier surface plasma resonance experiments [25], [48] and our analyses were performed at similar pH, we assume that the proportions of the two forms were similar in each analysis.
Figure 7. MG(1,4,5)-IP3 is a full agonist of all IP3 receptor subtypes.

(A) Effects of MG(1,4,5)IP3 on Ca2+ release via each IP3R subtype. (B) Paired comparisons of the effects of (1,4,5)IP3 and MG(1,4,5)IP3 are shown for each IP3R subtype. Results (A and B) are means ± S.E.M. from the number of independent experiments given in Table 1. (C) Equilibrium-competition binding of 3H-IP3 (1.5 nM) to cerebellar membranes in CLM containing 220 nM free [Ca2+] in the presence of the indicated concentrations of (1,4,5)IP3 or MG(1,4,5)IP3. (D) Typical results showing effects of MG(1,4,5)IP3 (30 nM, added 35 s before (1,4,5)IP3) on Ca2+ release from DT40-IP3R cells. (E) Summary results (means ± SEM from 3 independent experiments) show concentration-dependent effects of (1,4,5)IP3 alone or in the presence of MG(1,4,5)IP3 (300 nM). (F) MG(1,4,5)IP3 docked into the IBC of IP3R1 (top; (1,4,5)IP3 shown in gray) shows that it can be accommodated within the likely structure of the tetrameric IP3R without steric clashes (bottom).
In equilibrium-competition binding assays in CLM using cerebellar membranes, which predominantly express IP3R1 [14], (1,4,5)IP3 bound with an affinity that was ∼7-fold greater than that of MG(1,4,5)IP3 (ΔpKD = 0.86±0.08, n = 3): pKD = 8.29±0.03 and 7.43±0.08, for (1,4,5)IP3 and MG(1,4,5)IP3, respectively (Figure 7C and Table 3). The ∼7-fold lesser affinity of MG(1,4,5)IP3 for IP3R1 relative to (1,4,5)IP3 and its similarly reduced potency (∼5-fold, Table 2) are consistent with the large malachite green structure perturbing interaction of the 1-phosphate of (1,4,5)IP3 with the IBC, and they suggest that MG(1,4,5)IP3 is a full agonist of IP3R. The latter conclusion is further substantiated by the results shown in Figures 7D and E. These show that the Ca2+ release evoked by (1,4,5)IP3 is unaffected by the presence of 300 nM MG(1,4,5)IP3, which itself released 34±4% of the Ca2+ stores. The pEC50 was 7.41±0.09 and 7.01±0.37 (n = 3) for (1,4,5)IP3 alone and in the presence of MG(1,4,5)IP3, respectively (Figure 7E). A weak partial agonist, by contrast, would be expected, at concentrations sufficient to evoke a response, to occupy enough IP3R to shift the concentration-dependence of the response to (1,4,5)IP3 to higher concentrations [4]. These results establish that MG(1,4,5)IP3 is not a partial agonist of IP3R1. We conclude that MG(1,4,5)IP3 is a full agonist of IP3R with an affinity that is ∼7-fold less than that of (1,4,5)IP3.
Table 3. Interactions of MG(1,4,5)IP3 with type 1 IP3 receptors.
| DT40-IP3R1 cells | Cerebellum | |||||
| EC50 (nM) | pEC50 (/M) | Maximal Ca2+ release (%) | KD (nM) | pKD (/M) | pKD-pEC50 | |
| (1,4,5)IP3 | 91 | 7.04±0.1 | 77±2 | 5.13 | 8.29±0.03 | 1.25±0.29 |
| MG(1,4,5)IP3 | 447 | 6.35±0.12 | 73±4 | 37.2 | 7.43±0.08 | 1.08±0.35 |
Results from functional assays of Ca2+ release from DT40-IP3R1 cells (Figure 7B) and of equilibrium competition binding to cerebellar membranes (Figure 7C) compare the pEC50 and pKD values for (1,4,5)IP3 and MG(1,4,5)IP3. Results (except EC50 and KD) show means ± SEM from 3–9 independent experiments. The final column shows pKD-pEC50 for each ligand.
Discussion
The primary sequence of the IBC, which is responsible for recognition of (1,4,5)IP3 by IP3R, is well conserved between IP3R subtypes, and in isolation the IBC from each IP3R subtype binds IP3 with similar affinity [16]. Although there are high-resolution structures of this region for only IP3R1 [3], [7], [50], [51], homology modelling suggests that the IBC structures of the three IP3R subtypes are very similar (Figure 4D). It is, therefore, unsurprising that structure-activity relationships for the three IP3R subtypes, most derived from comparisons of cells expressing mixtures of IP3R subtypes, are similar [45], [52], [53]. Nevertheless, residues outside the IBC, notably the SD (Figure 1A), reduce the affinity of the IBC to differing extents for different IP3R subtypes [16]. Such differences and the minor differences in the primary sequence of the IBC between IP3R subtypes leave open the possibility that it may be possible to develop subtype-selective ligands of IP3R. Hitherto, the only systematic comparison of the ligand recognition properties of homogenous populations of IP3R subtypes examined only ligand binding to mammalian IP3R expressed in insect Sf9 cells [46]. We have now extended the analysis to functional assays of mammalian IP3R expressed in a null background, namely DT40 cells in which the genes for all endogenous IP3R have been disrupted [32] (Figure 2).
All high-affinity inositol phosphate agonists of IP3R have structures equivalent to the 4,5-bisphosphate and 6-hydroxyl groups of (1,4,5)IP3 [22], [45], [46], and with the exception of (1,3,4)IP3 (which was effectively inactive), all ligands examined herein retain these groups (Figure 1B). Most of the ligands examined were either inactive ((1,3,4)IP3) or their potency relative to (1,4,5)IP3 suggested a lack of selectivity for IP3R subtypes (MG(1,4,5)IP3, (1,4,6)IP3, (1,3,4,5)IP4, 2-deoxy(1,4,5)IP3 and 3-deoxy(1,4,5)IP3) (Figures 3, 5, 6 and 7, Tables 1 and 2). The potencies of each of these agonists for each IP3R subtype are consistent with known structure-activity relationships [45], [46] and with the structure of (1,4,5)IP3 bound to the IBC [3] (Figure 1A). Loss of the 2-hydroxyl minimally affects activity, loss of either the 3-hydroxyl or 1-phosphate or inverting the orientation of the 3-hydroxyl causes the potency to decrease substantially, and addition of malachite green to the 1-phosphate of (1,4,5)IP3 causes a modest (∼7-fold) decrease in potency (Tables 1 and 2). We conclude that the structure-activity relationships for all three mammalian IP3R are broadly similar.
MG(1,4,5)IP3 was reported to bind with unexpectedly high affinity to an N-terminal fragment of IP3R1 [25], but our results, in line with subsequent analyses of smooth muscle cells from the same group [25], suggest that MG(1,4,5)IP3 interacts similarly with all IP3R subtypes and that it is a full agonist with an affinity that is ∼7-fold lower than that of (1,4,5)IP3 (Figure 7 and Tables 2 and 3). This is consistent with evidence suggesting that even substantial additions to the 1-phosphate of (1,4,5)IP3 (eg, fluorescein and 3-aminopropyl) are well-tolerated [25], [48], [54]. It is also consistent with our docking studies, which suggest that even these very large additions to the 1-phosphate moiety of (1,4,5)IP3 do not create steric clashes within the tetrameric IP3R (Figure 7F).
The necessity of the 4,5-bisphosphate and 6-hydroxyl moieties [55], and the considerable loss of affinity associated with modifying the 3-hydroxyl group of (1,4,5)IP3 (Figures 3C, D and 5C, D) restrict opportunities to tag (1,4,5)IP3 to modifications of the 1- and 2-positions. Our previous work has shown that modification of the 2-position is compatible with high-affinity binding to IP3R, but it reduces efficacy [4], [56]. Our demonstration that MG(1,4,5)IP3 is a high-affinity full agonist of IP3R (Figure 7) therefore identifies an opportunity to develop fluorescent, or otherwise modified, ligands of IP3R that might be expected to come close to mimicking (1,4,5)IP3 in their interactions with IP3R.
Only one synthetic ligand discriminated modestly between IP3R subtypes. (4,5)IP2 lacks the 1-phosphate group of the endogenous ligand, (1,4,5)IP3 (Figure 1B). In keeping with considerable published evidence [45], [46], this causes a substantial decrease in potency at all IP3R subtypes (Figure 4, Tables 1 and 2). The loss of potency is, however, significantly less pronounced (∼24-fold) for IP3R3 than for the other IP3R subtypes (∼80-fold) (Table 2). The difference is unlikely to be due to residues with the IBC itself because the IBCs of all three IP3R subtypes bind (1,4,5)IP3 with indistinguishable affinity [16] and the residues that interact with the 1-phosphate group are conserved between IP3R (Figure 4C). We instead suggest that subtype-selective interactions of the IBC with other domains, perhaps the SD, subtly modify interaction of the 1-phosphate group of (1,4,5)IP3 with two basic residues within the IBC (Figure 4C).
Using homogenous populations of mammalian IP3R and a variety of (1,4,5)IP3 analogues, we have shown that the three IP3R subtypes have very similar ligand recognition properties. However, the decrease in affinity caused by loss of the 1-phosphate group is less for IP3R3. Finally, we have shown that MG(1,4,5)IP3 is a full agonist of IP3R with only modestly reduced affinity, suggesting that attachment of fluorescent tags to the 1-phosphate of (1,4,5)IP3 [57] is a feasible strategy for producing modified analogues of (1,4,5)IP3 that closely mimic the native messenger.
Funding Statement
This work was supported by grants from the Wellcome Trust to CWT [085295], and BVLP and AMR [082837], and from Biotechnology and Biological Sciences Research Council to CWT [BB/H009736/1]. HS is supported by a research studentship from the Jameel Family Trust. TR was a research fellow of Pembroke College, Cambridge. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Taylor CW, Tovey SC (2010) IP3 receptors: toward understanding their activation. Cold Spring Harb Persp Biol 2: a004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ludtke SJ, Tran TP, Ngo QT, Moiseenkova-Bell VY, Chiu W, et al. (2011) Flexible architecture of IP3R1 by cryo-EM. Structure 19: 1192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bosanac I, Alattia J-R, Mal TK, Chan J, Talarico S, et al. (2002) Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature 420: 696–700. [DOI] [PubMed] [Google Scholar]
- 4. Rossi AM, Riley AM, Tovey SC, Rahman T, Dellis O, et al. (2009) Synthetic partial agonists reveal key steps in IP3 receptor activation. Nat Chem Biol 5: 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chan J, Yamazaki H, Ishiyama N, Seo MD, Mal TK, et al. (2010) Structural studies of inositol 1,4,5-trisphosphate receptor: coupling ligand binding to channel gating. J Biol Chem 285: 36092–36099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamazaki H, Chan J, Ikura M, Michikawa T, Mikoshiba K (2010) Tyr-167/Trp-168 in type1/3 inositol 1,4,5-trisphosphate receptor mediates functional coupling between ligand binding and channel opening. J Biol Chem 285: 36081–36091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seo M-D, Velamakanni S, Ishiyama N, Stathopulos PB, Rossi AM, et al. (2012) Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature 483: 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ramos-Franco J, Galvan D, Mignery GA, Fill M (1999) Location of the permeation pathway in the recombinant type-1 inositol 1,4,5-trisphosphate receptor. J Gen Physiol 114: 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uchida K, Miyauchi H, Furuichi T, Michikawa T, Mikoshiba K (2003) Critical regions for activation gating of the inositol 1,4,5-trisphosphate receptor. J Biol Chem 278: 16551–16560. [DOI] [PubMed] [Google Scholar]
- 10. Foskett JK, White C, Cheung KH, Mak DO (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87: 593–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marchant JS, Taylor CW (1997) Cooperative activation of IP3 receptors by sequential binding of IP3 and Ca2+ safeguards against spontaneous activity. Curr Biol 7: 510–518. [DOI] [PubMed] [Google Scholar]
- 12. Fujino I, Yamada N, Miyawaki A, Hasegawa M, Furuichi T, et al. (1995) Differential expression of type 2 and type 3 inositol 1,4,5-trisphosphate receptor mRNAs in various mouse tissues: in situ hybridization study. Cell Tissue Res 280: 201–210. [DOI] [PubMed] [Google Scholar]
- 13. Yamamoto-Hino M, Miyawaki A, Kawano H, Sugiyama T, Furuichi T, et al. (1995) Immunohistochemical study of inositol 1,4,5-trisphosphate receptor type 3 in rat central nervous system. Neuroreport 6: 273–276. [DOI] [PubMed] [Google Scholar]
- 14. Taylor CW, Genazzani AA, Morris SA (1999) Expression of inositol trisphosphate receptors. Cell Calcium 26: 237–251. [DOI] [PubMed] [Google Scholar]
- 15. Vermassen E, Parys JB, Mauger J-P (2004) Subcellular distribution of the inositol 1,4,5-trisphosphate receptors: functional relevance and molecular determinants. Biol Cell 96: 3–17. [DOI] [PubMed] [Google Scholar]
- 16. Iwai M, Michikawa T, Bosanac I, Ikura M, Mikoshiba K (2007) Molecular basis of the isoform-specific ligand-binding affinity of inositol 1,4,5-trisphosphate receptors. J Biol Chem 282: 12755–12764. [DOI] [PubMed] [Google Scholar]
- 17. Patterson RL, Boehning D, Snyder SH (2004) Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu Rev Biochem 73: 437–465. [DOI] [PubMed] [Google Scholar]
- 18. Ando H, Mizutani A, Matsu-ura T, Mikoshiba K (2003) IRBIT, a novel inositol 1,4,5-trisphosphate (IP3) receptor-binding protein, is released from the IP3 receptor upon IP3 binding to the receptor. J Biol Chem 278: 10602–10612. [DOI] [PubMed] [Google Scholar]
- 19. Higo T, Hattori M, Nakamura T, Natsume T, Michikawa T, et al. (2005) Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell 120: 85–98. [DOI] [PubMed] [Google Scholar]
- 20. Futatsugi A, Nakamura T, Yamada MK, Ebisui E, Nakamura K, et al. (2005) IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science 309: 2232–2234. [DOI] [PubMed] [Google Scholar]
- 21. Matsumoto M, Nakagawa T, Inoue T, Nagata E, Tanaka K, et al. (1996) Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature 379: 168–171. [DOI] [PubMed] [Google Scholar]
- 22. Potter BVL, Lampe D (1995) Chemistry of inositol lipid mediated cellular signaling. Angew Chem Int Ed Eng 34: 1933–1972. [Google Scholar]
- 23.Sureshan KM, Riley AM, Rossi AM, Tovey SC, Dedos SG, et al. (2009) Activation of IP3 receptors by synthetic bisphosphate ligands. Chem Comm: 1204–1206. [DOI] [PMC free article] [PubMed]
- 24. Mills SJ, Potter BVL (1996) Synthesis of d- and l-myo-inositol 1,4,6-trisphosphate, regioisomers of a ubiquitous second messenger. J Org Chem 61: 8980–8987. [DOI] [PubMed] [Google Scholar]
- 25. Inoue T, Kikuchi K, Hirose K, Iino M, Nagano T (1999) Synthesis and evaluation of 1-position-modified inositol 1,4,5-trisphosphate analogs. Bioorg Med Chem Lett 9: 1967–1702. [DOI] [PubMed] [Google Scholar]
- 26. Sureshan KM, Riley AM, Thomas MP, Tovey SC, Taylor CW, et al. (2012) Contribution of phosphates and adenine to the potency of adenophostins at the IP3 receptor: synthesis of all possible bisphosphates of adenophostin A. J Med Chem. 55: 1706–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Poinas A, Backers K, Riley AM, Mills SJ, Moreau C, et al. (2005) Study of the interaction of the catalytic domain of Ins(1,4,5)P3 3-kinase A with inositol phosphate analogues. ChemBioChem 6: 1449–1457. [DOI] [PubMed] [Google Scholar]
- 28. Riley AM, Mahon MF, Potter BVL (1997) Rapid synthesis of the enantiomers of myo-inositol-1,3,4,5-tetrakisphosphate by direct chiral desymmetrization of myo-inositol orthoformate. Angew Chem Int Ed Eng 36: 1472–1474. [Google Scholar]
- 29. Cardy TJA, Traynor D, Taylor CW (1997) Differential regulation of types 1 and 3 inositol trisphosphate receptors by cytosolic Ca2+ . Biochem J 328: 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rossi A, Sureshan KM, Riley AM, Potter BVL, Taylor CW (2010) Selective determinants of inositol 1,4,5-trisphosphate and adenophostin A interactions with type 1 inositol 1,4,5-trisphosphate receptors. Br J Pharmacol 161: 1070–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tovey SC, Dedos SG, Rahman T, Taylor EJA, Pantazaka E, et al. (2010) Regulation of inositol 1,4,5-trisphosphate receptors by cAMP independent of cAMP-dependent protein kinase. J Biol Chem 285: 12979–12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sugawara H, Kurosaki M, Takata M, Kurosaki T (1997) Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J 16: 3078–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pantazaka E, Taylor CW (2011) Differential distribution, clustering and lateral diffusion of subtypes of inositol 1,4,5-trisphosphate receptor. J Biol Chem 286: 23378–23387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rahman TU, Skupin A, Falcke M, Taylor CW (2009) Clustering of IP3 receptors by IP3 retunes their regulation by IP3 and Ca2+ . Nature 458: 655–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tovey SC, Sun Y, Taylor CW (2006) Rapid functional assays of intracellular Ca2+ channels. Nature Prot 1: 259–263. [DOI] [PubMed] [Google Scholar]
- 36. Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, et al. (2006) Comparative protein structure modeling using Modeller. Curr Prot Bioinform 15: 5.6.1–5.6.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, et al. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35: W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, et al. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612. [DOI] [PubMed] [Google Scholar]
- 39. Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Merlino A, Benitez D, Campillo NE, Paez JA, Tinoco LW, et al. (2012) Amidines bearing benzofuroxan or benzimidazole 1,3-dioxide core scaffolds as Trypanosoma cruzi-inhibitors: structural basis for their interactions with cruzipain. Med Chem Comm 3: 90–101. [Google Scholar]
- 41. Irvine RF, Schell MJ (2001) Back in the water: the return of the inositol phosphates. Nat Rev Mol Cell Biol 2: 327–338. [DOI] [PubMed] [Google Scholar]
- 42. Loomis-Husselbee JW, Walker CD, Bottomley JR, Cullen PJ, Irvine RF, et al. (1998) Modulation of Ins(2,4,5)P 3-stimulated Ca2+ mobilization by Ins(1,3,4,5)P 4: enhancement by activated G-proteins, and evidence for the involvement of a GAP1 protein, a putative Ins(1,3,4,5)P 4 receptor. Biochem J 331: 947–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bird GSJ, Putney JW Jr (1996) Effect of inositol 1,3,4,5-trisphosphate on inositol trisphosphate-activated Ca2+ signaling in mouse lacrimal cells. J Biol Chem 271: 6766–6770. [DOI] [PubMed] [Google Scholar]
- 44. Burgess GM, McKinney JS, Irvine RF, Putney JW (1985) Inositol 1,4,5-trisphosphate and inositol 1,3,4-trisphosphate formation in Ca2+-mobilizing-hormone-activated cells. Biochem J 232: 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wilcox RA, Primrose WU, Nahorski SR, Challiss RAJ (1998) New developments in the molecular pharmacology of the myo-inositol 1,4,5-trisphosphate receptor. Trends Pharmacol Sci 19: 467–475. [DOI] [PubMed] [Google Scholar]
- 46. Nerou EP, Riley AM, Potter BVL, Taylor CW (2001) Selective recognition of inositol phosphates by subtypes of inositol trisphosphate receptor. Biochem J 355: 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kozikowski AP, Ognyanov VI, Fauq AH, Nahorski SR, Wilcox RA (1993) Synthesis of 1d-3-deoxy-, 1d-2,3-dideoxy, and 1d-2,3,6-trideoxy-myo-inositol 1,4,5-trisphosphate from quebrachitol, their binding affinities, and calcium release activity. J Am Chem Soc 115: 4429–4434. [Google Scholar]
- 48. Inoue T, Kikuchi K, Hirose K, Iino M, Nagano T (2001) Small molecule-based laser inactivation of inositol 1,4,5-trisphosphate receptor. Chem Biol 8: 9–15. [DOI] [PubMed] [Google Scholar]
- 49. Hagiwara T, Motomizu S (1994) Equilibrium and kinetic studies on the formation of the triphenylmethanols from triphenylmethane dyes. Bull Chem Soc Jpn 67: 390–397. [Google Scholar]
- 50. Bosanac I, Yamazaki H, Matsu-ura T, Michikawa M, Mikoshiba K, et al. (2005) Crystal structure of the ligand binding suppressor domain of type 1 inositol 1,4,5-trisphosphate receptor. Mol Cell 17: 193–203. [DOI] [PubMed] [Google Scholar]
- 51. Lin CC, Baek K, Lu Z (2011) Apo and InsP3-bound crystal structures of the ligand-binding domain of an InsP3 receptor. Nat Struct Mol Biol 18: 1172–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. DeLisle S, Radenberg T, Wintermantel MR, Tietz C, Parys JB, et al. (1994) Second messenger specificity of the inositol trisphosphate receptor: reappraisal based on novel inositol phosphates. Am J Physiol 266: C429–C436. [DOI] [PubMed] [Google Scholar]
- 53. Lu P-J, Gou D-M, Shieh W-R, Chen C-S (1994) Molecular interactions of endogenous d-myo-inositol phosphates with the intracellular d-myo-inositol 1,4,5-trisphosphate recognition site. Biochemistry 33: 11586–11597. [DOI] [PubMed] [Google Scholar]
- 54. Nakanishi W, Kikuchi K, Inoue T, Hirose K, Iino M, et al. (2002) Hydrophobic modifications at 1-phosphate of inositol 1,4,5-trisphosphate analogues enhance receptor binding. Bioorg Med Chem Lett 12: 911–913. [DOI] [PubMed] [Google Scholar]
- 55. Wilcox RA, Fauq A, Kozikowski AP, Nahorski SR (1997) Defining the minimal structural requirements for partial agonism at the type I myo-inositol 1,4,5-trisphosphate receptor. FEBS Lett 402: 241–245. [DOI] [PubMed] [Google Scholar]
- 56. Marchant JS, Chang Y-T, Chung S-K, Irvine RF, Taylor CW (1997) Rapid kinetic measurements of 45Ca2+ mobilization reveal that Ins(2,4,5)P 3 is a partial agonist of hepatic InsP 3 receptors. Biochem J 321: 573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lampe D, Mills SJ, Potter BVL (1992) Total synthesis of the second messenger analogue d-myo-inositol 1-phosphorothioate,4,5-bisphosphate: optical resolution of dl-1-O-allyl-2,3,6-tri-O-benzyl-myo-inositol and fluorescent labelling of myo-inositol 1,4,5-trisphosphate. J Chem Soc Perkin Trans: 2899–2906.