

Abstract
Background
The currently available antifungal drugs suffer from toxicity, greatest potential drug interactions with other drugs, insufficient pharmacokinetics properties, and development of resistance. Thus, development of new antifungal agents with optimum pharmacokinetics and less toxicity is urgent task. In the search for new azole antifungals, we have been previously described azolylchromanone oxime ethers as rigid analogs of oxiconazole. In continuation of our work, we incorporated phenylhydrazone moiety instead of oxime ether fragment in azolylchromanone derivatives.
Methods
The 3-azolyl-4-chromanone phenylhydrazones were synthesized via ring closure of 2-azolyl-2'-hydroxyacetophenones and subsequent reaction with phenylhydrazine. The biological activity of title compounds was evaluated against different pathogenic fungi including Candida albicans, Saccharomyces cerevisiae, Aspergillus niger, and Microsporum gypseum. Docking study, in silico toxicity risks and drug-likeness predictions were used to better define of title compounds as antifungal agents.
Results
The in vitro antifungal activity of compounds based on MIC values revealed that all compounds showed good antifungal activity against C. albicans, S. cerevisiae and M. gypseum at concentrations less than 16 μg/mL. Among the test compounds, 2-methyl-3-imidazolyl derivative 3b showed the highest values of drug-likeness and drug-score.
Conclusion
The 3-azolyl-4-chromanone phenylhydrazones considered as analogs of 3-azolyl-4-chromanone oxime ethers basically designed as antifungal agents. The antifungal activity of title compounds was comparable to that of standard drug fluconazole. The drug-likeness data of synthesized compounds make them promising leads for future development of antifungal agents.
Keywords: Azole antifungals; Antifungal activity; 1 H-imidazole; 1,2,4-triazole; Chroman-4-one; Hydrazone
Introduction
Recently, the incidence of life-threatening fungal diseases has dramatically increased due to the rising number of immunocompromised patients, increased use of anti-cancer drugs, and immunosuppressive therapy for organ transplantation [1]. On the other hand, the currently available antifungal drugs suffer from toxicity, greatest potential drug interactions with other drugs, insufficient pharmacokinetics properties, and development of resistance [2]. Consequently, it is urgent to develop new broad-spectrum antifungal agents with optimum pharmacokinetics and less toxicity.
Several antifungal drugs, including azoles, polyenes, echinocandins and allylamines have been developed to overcome the fungal infections [3]. However, in comparison to the large number of antibacterial drugs the number of clinically available antifungal agents is rather low. Azole antifungals such as ketoconazole, fluconazole, voriconazole, and posaconazole, which inhibit fungal cytochrome P-450 dependent 14α-demethylase, are among the most investigated compounds with respect to their clinical impact and individual mechanism of action [4]. These antifungal drugs contain imidazole or triazole heterocycles and act through a mechanism in which the heterocyclic nitrogen atom (N-3 of imidazole or N-4 of triazole) binds to the heme iron atom in cytochrome P-450 enzymes. Particularly, the triazole derivatives have a greater affinity for fungal cytochrome P-450 enzymes than for mammalian ones contributing to a favorable safety profile [5].
In the search for new azole antifungals, we have been described azolylchromanone oxime ethers (Figure 1) as rigid analogs of oxiconazole [6-8]. The chroman scaffold also found in some natural flavonoids possessing antifungal activity [9-11]. In continuation of our work, we incorporated phenylhydrazone moiety instead of oxime ether fragment in azolylchromanone derivatives (Figure 1). This modification was made on the basis of zinoconazole, a well-known azole antifungal possessing phenylhydrazone substructure (Figure 2). Thus we describe here, the synthesis and antifungal activity of 3-azolyl-4-chromanone phenylhydrazones. Furthermore, docking study and in silico toxicity risks and drug-likeness predictions were used to better define of title compounds as antifungal agents.
Figure 1.
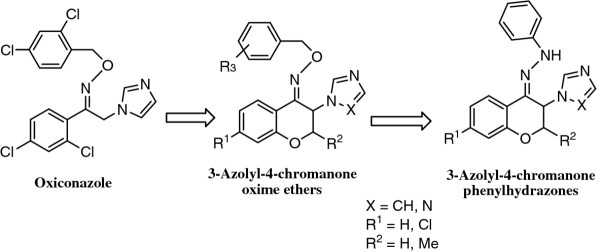
Structures of azolylchromanone oxime ethers as rigid analogs of oxiconazole and designed compounds 3-azolyl-4-chromanone phenylhydrazones.
Figure 2.

Zinoconazole, a well-known azole antifungal bearing phenylhydrazone substructure.
Material and methods
Chemistry
All chemical reagents and solvents were purchased from Merck AG Chemical (Germany) and used without further purification. The progress of reactions and the purity of compounds were checked by TLC analysis on silica gel 60 F254 plates (Merck) and visualization was done with UV light (254 nm). Melting points were determined in open glass capillaries using Bibby Stuart Scientific SMP3 apparatus (Stuart Scientific, UK) and are uncorrected. The IR spectra were recorded on FT-IR (PerkinElmer, USA) spectrophotometer using KBr disks. 1 H-NMR spectra were recorded using a Bruker 400 (Rheinstatten, Germany) spectrometer, and chemical shifts are expressed as δ (ppm) with tetramethylsilane as internal standard. The MS spectra were obtained with a Finnigan MAT TSQ-70 spectrometer (Finnigan MAT, Germany).
General procedure for the synthesis of 3-azolyl-4-chromanone derivatives 2a-c
Compounds 2a-c was synthesized from 2-azolyl-2'-hydroxyacetophenones 1a-c according to the literature methods [6-8].
General procedure for the synthesis of phenyhydrazone derivatives 3a-c
Phenylhydrazine hydrochloride was added to a solution of compound 2 (0.5 mmol) in methanol (4 mL) and the solution was stirred at room temperature. The completion of the reaction was checked by TLC. After consumption of compound 2 (3–7 days), water was added and the mixture was left in refrigerator overnight. The precipitated solid was separated by filtration and washed with water. The product was re-crystallized from methanol.
2,3-Dihydro-3-(1H-1,2,4-triazol-1-yl)-4H-1-benzopyran-4-one phenylhydrazone (3a)
Yield 55 %; IR (KBr, cm-1) 3418, 3238, 3128, 1703, 1603, 1561, 1497, 1478, 1459, 1278, 1261, 1216, 1138, 1055, 831, 763, 691, 678. 1 H NMR (400 MHz, DMSO-d6) δ 4.44 (m, 1 H), 4.82-4.87 (m, 1 H), 5.99 (dd, 1 H, J = 10.8 and 6.6 Hz), 6.79-6.90 (m, 2 H), 7.04 (t, 2 H, J = 7.2 Hz), 7.12-7.26 (m, 3 H), 7.66 (dt, 1 H, J = 7.4 and 2.0 Hz), 7.83 (dd, 1 H, J = 7.6 and 2.0 Hz), 8.43 (s, 1 H), 8.64 (s, 1 H), 10.05 (s, 1H). Anal. Calcd for C17H15N5O: C, 66.87; H, 4.95; N, 22.94. Found: C, 66.98; H, 5.14; N, 22.71.
trans-2,3-Dihydro-3-(1H-imidazol-1-yl)-2-methyl-4H-1-benzopyran-4-one phenylhydrazone (3b)
Yield 62%; m.p. 228–230°C; IR (KBr, cm-1) 3444, 3246, 1603, 1567, 1498, 1457, 1295, 1255, 1220, 1124, 834, 750, 696. 1 H NMR (400 MHz, DMSO-d6) δ 1.16 (d, 3 H, J = 6.4 Hz), 5.19 (dq, 1 H, J = 12.4 and 6.4 Hz), 5.88 (d, 1 H, J = 12.8 Hz), 6.75 (d, 2 H, J = 7.2 Hz), 6.83 (t, 1 H, J = 8.0 Hz), 6.92 (t, 2 H, J = 8.0 Hz), 7.01 (d, 1 H, J = 8.0 Hz), 7.11 (t, 1 H, J = 7.6 Hz), 7.14-7.36 (m, 3 H), 7.68 (t, 1 H, J = 8.4 Hz), 8.07 (d, 1 H, J = 7.6 Hz), 10.34 (s, 1 H). Anal. Calcd for C19H18N4O: C, 71.68; H, 5.70; N, 17.60. Found: C, 71.50; H, 5.79; N, 17.86.
7-Chloro-2,3-dihydro-3-(1H-imidazol-1-yl)-4H-1-benzopyran-4-one phenylhydrazone (3c)
Yield 58%; m.p. 192–194°C; IR (KBr, cm-1) 3444, 1602, 1560, 1497, 1418, 1252, 1163, 1082, 814, 754, 696. MS (m/z, %) 338 (M+, 25), 270 (100), 207 (10), 154 (15), 105 (28), 77 (78), 67 (37). Anal. Calcd for C18H15ClN4O: C, 63.81; H, 4.46; N, 16.54. Found: C, 63.66; H, 4.28; N, 16.57.
Antifungal activity
The antifungal activities of title compounds 3a-c and 2b were tested against several representative pathogenic fungi including Candida albicans PTCC 5027 and Saccharomyces cerevisiae PTCC 5177 (as yeast), Microsporum gypseum PTCC 5070 (as a dermatophyte) and Aspergillus niger PTCC 5012 (as a mould) using agar dilution method [12]. Stock solutions of tested compounds were prepared in DMSO and serially diluted. Sabouraud Dextrose Agar was used for fungal growth. Inocula containing approximately 105 CFUs/mL of fungi were prepared from broth cultures in log phase growth. Fungal plates were made in triplicate and incubated at 30°C about 24–48 h for yeast, about 72 h for moulds and about 168 h for dermatophytes. The MIC value was defined as lowest concentration of the antifungal agent at which the test strain does not demonstrate visible growth. The reported MICs are representative of at least two independent experiments.
Docking study
Crystal structure of cytochrome P450 14α-sterol demethylase (Cyp51) from Mycobacterium tuberculosis in complex with fluconazole (ID 1EA1) was provided from the Protein Data Bank and prepared for docking calculations using AutoDock Tools 1.5.4 with default settings. Docking was performed using AutoDock vina,and standard parameters were used. Water molecules were not included. The binding site was defined to any residues with 10 Å distant from the co-crystallized fluconazole in the complex crystal structure of the enzyme [13].
In silico drug-likeness and toxicity predictions
OSIRIS Property Explorer [14] was used to estimate the risks of side effects, such as mutagenic, tumorigenic, irritant and reproductive effects, as well as drug-relevant properties including cLogP, LogS (solubility), MW, drug-likeness and overall drug-score.
Results and discussion
Chemistry
The synthetic route to phenylhydrazone derivatives 3a-c was illustrated in Scheme 1. The starting 2-azolyl-2'-hydroxyacetophenones 1a-c was prepared from corresponding 2-bromo-2'-hydroxyacetophenones according to the literature method [15]. Cyclization of compounds 1a-c with paraformaldehyde or acetaldehyde in refluxing acetic acid gave 3-azolyl-4-chromanones 2a-c[6-8]. The synthesis of hydrazone derivative 3a was first attempted by reaction of 3-triazolyl-4-chromanone 2a with phenylhydrazine hydrochloride in refluxing methanol in the presence of Na2CO3 as a base, which resulted in poor yield with mixture of unknown degraded products. Since the condensation reactions of amines and hydrazines with ketones are commonly catalyzed by acids or bases, thus the alternative condition was using hydrochloride salt of phenylhydrazine in the absence of a base. Accordingly, the reaction of compound 2a-c with phenylhydrazine hydrochloride in methanol at room temperature afforded final compounds 3a-c in a mild condition. It seems that the acid released by hydrochloride salt of phenylhydrazine is adequate for catalyzing the condensation reaction.
Scheme 1.
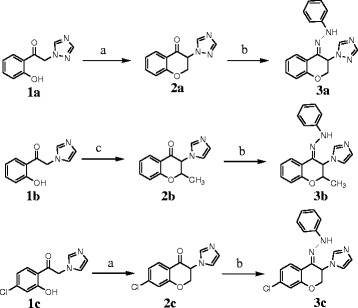
Synthetic routes to phenylhydrazone derivatives 3a-c. (a) paraformaldehyde, AcOH, reflux; (b) phenylhyrazine hydrochloride, MeOH, r.t.; (c) acetaldehyde, AcOH, 90°C.
Theoretically, the reactivity of chromanone derivatives toward N-nucleophiles like substituted hydrazine related to two sites; carbonyl group and C-2 position (Scheme 2). The attack of hydrazine to the C-2 position of chroman ring could result in ring opening and subsequent pyrazoline formation.
Scheme 2.

Possible reactivity of 3-triazolylchroman-4-one derivative 2a toward phenylhydrazine.
Previously, the reaction of chromanone and flavanone derivatives with substituted hydrazines has been investigated in different conditions [16,17]. Kállay et al. have been reported that the flavanone hydrazones are predominantly obtained under acidic conditions while alkaline conditions give the pyrazolines and the 2-hydroxychalcone derivatives due to the ring cleavage of the hetero ring of chromanone [16]. Similarly, in our experiment with 3-azolyl-4-chromanones, the hydrazone derivatives were predominantly obtained under mild acidic conditions, but in the presence of weak base Na2CO3, possibly produced a mixture of corresponding hydrazone, acyclic N-substituted hydrazine and pyrazoline derivatives which was difficult to separate and characterize (Scheme 2).
The structures of compounds 3a-c were properly characterized by IR, 1 H NMR or Mass spectroscopy as well as elemental analyses. For example, elemental analysis proved the molecular formula of compound 3b as C19H18N4O. The IR spectra of compound 3b did not show any absorption due to the presence of C = O group and a broad band was found in 3444 cm-1 due to the presence of a N − H bond. The 1 H NMR spectrum of 3b showed a singlet at 10.34 for NH, a double at 8.07 (J = 7.6 Hz) and a triplet at 7.68 (J = 8.4 Hz) for H-5 and H-7 of chroman ring, respectively. The resonance for imidazole protons was observed as a multiplet at the range of 7.14-7.39 ppm. The H-6 and H-8 of chroman ring resonated as a triplet at 7.11 and a doublet at 7.01 ppm. Five protons related to the phenyl group appeared at 6.92, 6.83 and 6.75 ppm. The aliphatic protons of chroman (H-3 and H-2) resonated as a doublet at 5.88 and a doublet of quartet at 5.19 ppm. A doublet appeared at 1.16 ppm, assigned to the methyl protons.
Antifungal activity
The antifungal activities of title compounds 3a-c were tested against several representative pathogenic fungi. The resulting MICs (minimum inhibitory concentrations) of hydrazone compounds were compared with those of fluconazole as a reference antifungal agent and intermediate compound 2b (Table 1). The results revealed that 2-methyl-3-imidazolylchroman-4-one (2b) showed no activity against Saccharomyces cerevisiae, Aspergillus niger and Microsporum gypseum (MIC >64 μg/mL). Its activity against Candida albicans was weak. Compounds 3a,b exhibited good growth inhibitory activity against different fungal strains with MIC values of 8–16 μg/mL. The 7-chloro-3-imidazolyl derivative 3c also showed good antifungal activity against Candida albicans, Saccharomyces cerevisiae and Microsporum gypseum (MIC = 16 μg/mL), but its activity against A. niger was moderate. The MIC values of fluconazole against test strains were 8–32 μg/mL. Thus, the antifungal activity of title compounds was comparable to that of standard drug fluconazole. By comparing the MIC values of hydrazone derivative 3b and its ketone counterpart 2b, it is revealed that the introduction of phenylhydrazone on chroman ring improves the antifungal activity against all strains.
Table 1.
In vitro antifungal activity of compounds 3a-c in comparison with 2b and fluconazole
|
Compound |
Tested fungi (MICs in μg /mL) |
|||
|---|---|---|---|---|
| C. albicans | S. cerevisiae | A. niger | M. gypseum | |
|
2b |
64 |
>64 |
>64 |
>64 |
|
3a |
16 |
16 |
16 |
8 |
|
3b |
8 |
16 |
16 |
16 |
|
3c |
16 |
16 |
64 |
16 |
| Fluconazole | 8 | 32 | 32 | 32 |
Previously, we have described azolylchromanone oxime ethers as conformationally restricted analogs of oxiconazole (Figure 1). Many of these derivatives exhibit high activity against different fungal strains [6-8]. In this work, we replaced O-benzyloxime ether fragment in azolylchromanone derivatives by phenylhydrazone moiety. The comparison of MIC values of triazole derivative 3a with its O-benzyloxime ether counterpart demonstrates that replacement of O-benzyloximino moiety with hyrazone can increase antifungal potency. However, in the cases of 3b and 3c, their O-benzyloximino analogs have a better profile of antifungal activity.
Docking study
Azole antifungals such as fluconazole inhibit fungal cytochrome P-450 dependent 14α-demethylase [4]. With the aim of rationalizing the antifungal activity data obtained, docking study was performed for the selected derivatives 2b and 3a in order to investigate the possible interactions with cytochrome P450 14α-sterol demethylase from Mycobacterium tuberculosis (Mycobacterium P450 DM). Because no experimental data are available for the target enzyme Candida P450 DM in the Protein Data Bank, the crystallographic structure of the complex between Cytochrome P450 14α-sterol demethylase from Mycobacterium tuberculosis (Mycobacterium P450 DM) and fluconazole (ID 1EA1) was used for the docking study [18].
Figure 3 shows the docked reference compound fluconazole in the active site of the same enzyme for validation of our docking protocol and confirmation of the biological data. Figures 4 and 5 show complex between the enzyme's active site and compounds 2b and 3a, respectively. The aromatic part of chroman ring of the both compounds is situated in a lipophilic region and interacts with Phe255, Met99, Phe83 and Met79. Oxygen atom (sp3 hybridized) of the chroman ring is oriented towards the polar region of the enzyme constituted by Arg96 and Arg95. The phenyl attached to the hydrazone moiety is inserted into a hydrophobic pocket above the heme ring formed by ceiling residues Met79, Phe78, Phe255, Val434, Leu321 and Tyr76. The azole ring of both compounds 2b and 3a are positioned above the porphyrine plan, with a ring nitrogen atom coordinated to the heme iron. The distance between nitrogen atom of compounds 2b and 3a and the heme iron is 2.6 Å and 2.8 Å, respectively.
Figure 3.

Computed binding geometry of the reference compound fluconazole (ball and stick) in the active site of Mycobacterium P450DM.
Figure 4.

Compound 2b (ball and stick) in the active site of Mycobacterium P450 DM. For clarity, only the amino acids with 10 Ǻ distant from the compound 2b are shown.
Figure 5.

Compound 3a (ball and stick) in the active site of Mycobacterium P450 DM. For clarity, only the amino acids with 10 Ǻ distant from the compound 3a are shown.
It has been previously reported that hydrophobic interactions with Val434 is responsible for the potent inhibitory activity of antifungals against Aspergillus niger whereas hydrophobic interactions with Leu321, Ile323, Leu324 may be responsible for an enhanced inhibitory activity against Candida albicans and other Candida spp. [18]. As shown in Figures 4 and 5, these interactions only can be seen with compound 3a prototype bearing lipophilic phenylhydrazone side chain and not with compound 2b; explaining the major potency differences observed in obtained results between 2b and compounds 3ac.
In silico drug-likeness and toxicity predictions
Currently, several approaches have been developed to assess drug-likeness of bioactive compounds based on topological descriptors, fingerprints of molecular structure or other properties such as molecular weight, water solubility and cLogP[19]. In this work, open-source program OSIRIS Property Explorer [14] was used to predict the fragment-based drug-likeness of title compounds and comparing them with fluconazole. OSIRIS program involves the database of traded drugs and commercially available compounds (Fluka) assumable as non drug-like dataset to assess the occurrence frequency of each fragment in the individual structure. The program estimated the risks of side effects, such as mutagenic, tumorigenic, irritant and reproductive effects, as well as drug-relevant properties including cLogP, LogS (solubility), MW and drug-likeness. Moreover, the overall drug-score was estimated by combining outcome of cLogP, LogS, MW, toxicity risks and drug-likeness. Drug-score is a measure of compound's potential to meet the criteria of a possible drug candidate [14].
The in silico drug-relevant properties obtained by OSIRIS Property Explorer are given in Table 2. Interestingly, the potential drug-likeness values of compounds 3a-c were significantly higher than that of fluconazole, which showed a negative value of −1.13. As shown in Table 2, compounds 3a-c had high in silico tumorigenic and medium mutagenic toxicity risks due to the phenylhydrazone moiety. However, the in silico prediction by OSIRIS Property Explorer showed that the introduction of chlorine on phenylhydrazone can reduce the risk of tumorigenic and mutagenic toxicities. Generally, the drug-score values of compounds 3a-c (0.2-0.34) were less than that of fluconazole (0.46). Among the final compounds, 2-methyl-3-imidazolyl derivative 3b showed the highest values of drug-likeness and drug-score.
Table 2.
Toxicity risks and physicochemical properties of compounds 3a-c in comparison with fluconazole, predicted by OSIRIS Property Explorer
|
Compound |
Toxicity risks |
Physicochemical properties |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Mutagenic | Tumorigenic | Irritant | Reproductive effect | CLogP | Slobility | MW | Drug likeness | Drug-score | |
|
3a |
(±) |
(+) |
(−) |
(±) |
4.18 |
−3.07 |
305 |
1.33 |
0.26 |
|
3b |
(±) |
(+) |
(−) |
(−) |
4.46 |
−2.78 |
318 |
2.81 |
0.34 |
|
3c |
(±) |
(+) |
(−) |
(±) |
4.79 |
−3.14 |
338 |
0.17 |
0.2 |
| Flu | (−) | (−) | (−) | (±) | −0.21 | −2.17 | 306 | −1.13 | 0.46 |
Conclusion
In conclusion, a series of 3-azolyl-4-chromanone phenylhydrazones were synthesized via ring closure of 2-azolyl-2'-hydroxyacetophenones and subsequent reaction with phenylhydrazine. These compounds considered as analogs of 3-azolyl-4-chromanone oxime ethers basically designed as antifungal agents. The in vitro antifungal activity of compounds based on MIC values revealed that all compounds showed good antifungal activity against Candida albicans, Saccharomyces cerevisiae and Microsporum gypseum at concentrations less than 16 μg/mL. The antifungal activity of title compounds was comparable to that of standard drug fluconazole. The biological data and docking study demonstrated that the introduction of phenylhydrazone moiety on chroman ring improves the antifungal activity. The 2-methyl-3-imidazolyl derivative 3b which showed the highest drug-likeness and drug-score values serves as a lead for future development of antifungal agents.
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
AA participated in the synthesis of compounds. MF performed antifungal activity evaluation. HI participated in the molecular modeling study and performed the docking analysis. SE participated in the design of compounds, supervision of synthetic and computational parts, identifying of the structures of target compounds, accomplishing drug-likeness predictions and manuscript preparation. All authors read and approved the final manuscript.
Contributor Information
Adile Ayati, Email: sd_emami@yahoo.com.
Mehraban Falahati, Email: sd_emami@yahoo.com.
Hamid Irannejad, Email: sd_emami@yahoo.com.
Saeed Emami, Email: sd_emami@yahoo.com.
Acknowledgements
This work was supported by grants from Research Council of Mazandaran University of Medical Sciences and Iran National Science Foundation (INSF).
References
- Pappas PG The role of azoles in the treatment of invasive mycoses: review of the Infectious Diseases Society of America guidelines Curr Opin Infect Dis 201124S1–S13.21200179 [Google Scholar]
- Nosanchuk JD. Current status and future of antifungal therapy for systemic mycoses. Recent Patents on Anti-Infective Drug Discovery. 2006;1:75–84. doi: 10.2174/157489106775244109. [DOI] [PubMed] [Google Scholar]
- Odds FC. Antifungal agents: their diversity and increasing sophistication. Mycologist. 2003;17(Part 2):51–55. [Google Scholar]
- Kathiravan MK, Salake AB, Chothe AS, Dudhe PB, Watode RP, Mukta MS, Gadhwe S. The biology and chemistry of antifungal agents: a review. Bioorg Med Chem. 2012. http://dx.doi.org/10.1016/j.bmc.2012.04.045. [DOI] [PubMed]
- Shalini K, Kumar N, Drabu S, Sharma PK. Advances in synthetic approach to and antifungal activity of triazoles. Beilstein J Org Chem. 2011;7:668–677. doi: 10.3762/bjoc.7.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emami S, Falahati M, Banifatemi A, Moshiri K, Shafiee A. Stereoselective synthesis and in vitro antifungal evaluation of (E)- and (Z)-imidazolylchromanone oxime ethers. Arch Pharm Pharm Med Chem. 2002;335:318–324. doi: 10.1002/1521-4184(200209)335:7<318::AID-ARDP318>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Emami S, Falahati M, Banifatemi A, Amanlou M, Shafiee A. (E)- and (Z)-1,2,4-triazolylchromanone oxime ethers as conformationally constrained antifungals. Bioorg Med Chem. 2004;12:3971–3976. doi: 10.1016/j.bmc.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Emami S, Falahati M, Banifatemi A, Shafiee A. Stereoselective synthesis and antifungal activity of (Z)-trans-3-azolyl-2-methylchromanone oxime ethers. Bioorg Med Chem. 2004;12:5881–5889. doi: 10.1016/j.bmc.2004.08.030. [DOI] [PubMed] [Google Scholar]
- Weidenbörner M, Jha HC. Antifungal spectrum of flavone and flavanone tested against 34 different fungi. Mycol Res. 1997;101:733–736. doi: 10.1017/S0953756296003322. [DOI] [Google Scholar]
- Pretorius JC. Flavonoids: a review of its commercial application potential as anti-infective agents. Curr Med Chem Anti-Infective Agents. 2003;2:335–353. doi: 10.2174/1568012033482971. [DOI] [Google Scholar]
- Wei DG, Yang GF, Wan J, Zhan CG. Binding model construction of antifungal 2-Aryl-4-chromanones. J Agric Food Chem. 2005;53:1604–1611. doi: 10.1021/jf048313r. [DOI] [PubMed] [Google Scholar]
- Massa S, Di Santo R, Retico A, Simonetti N, Fabrizi G, Lamba D. Antifungal agents. 1. Synthesis and antifungal activities of estrogen-like imidazole and triazole derivatives. Eur J Med Chem. 1992;27:495–502. doi: 10.1016/0223-5234(92)90183-2. [DOI] [Google Scholar]
- Banfi E, Scialino G, Zampieri D, Mamolo MG, Vio L, Ferrone M, Fermeglia M, Paneni MS, Pric S. Antifungal and antimycobacterial activity of new imidazole and triazole derivatives. A combined experimental and computational approach. J Antimicrob Chemother. 2006;58:76–84. doi: 10.1093/jac/dkl182. [DOI] [PubMed] [Google Scholar]
- Organic Chemistry Portal. 2012. Available at http://www.organic-chemistry.org/prog/peo/. Accessed May 22.
- Emami S, Foroumadi A, Falahati M, Lotfali E, Rajabalian S, Ebrahimi SA, Farahyar S, Shafiee A. 2-Hydroxyphenacyl azoles and related azolium derivatives as antifungal agents. Bioorg Med Chem Lett. 2008;18:141–146. doi: 10.1016/j.bmcl.2007.10.111. [DOI] [PubMed] [Google Scholar]
- Kállay F, Janzsó G, Koczor I. The reactions of flavanone with substituted hydrazines. Tetrahedron. 1967;23:4317–4321. doi: 10.1016/S0040-4020(01)88830-X. [DOI] [Google Scholar]
- Léavai A. Synthesis of 2-pyrazolines by the reactions of α, β-unsaturated aldehydes, ketones, and esters with diazoalkanes, nitrile imines, and hydrazines. J Heterocyclic Chem. 2002;39:1–13. doi: 10.1002/jhet.5570390101. [DOI] [Google Scholar]
- Patel PD, Patel MR, Kocsis B, Kocsis E, Graham SM, Warren AR, Nicholson SM, Billack B, Fronczek FR, Talele TT. Design, synthesis and determination of antifungal activity of 5(6)-substituted benzotriazoles. Eur J Med Chem. 2010;45:2214–2222. doi: 10.1016/j.ejmech.2010.01.062. [DOI] [PubMed] [Google Scholar]
- Tetko IV. Computing chemistry on the web. Drug Discov Today. 2005;10:1497–1500. doi: 10.1016/S1359-6446(05)03584-1. [DOI] [PubMed] [Google Scholar]