

Abstract
The RNA helicase p68 (DDX5) is an established co-activator of the p53 tumour suppressor that itself plays a pivotal role in orchestrating the cellular response to DNA damage. Although several factors influence the biological outcome of p53 activation, the mechanisms governing the choice between cell cycle arrest and apoptosis remain to be elucidated. In the present study we show that, while p68 is critical for p53-mediated transactivation of the cell cycle arrest gene p21WAF1/CIP1, it is dispensable for induction of several pro-apoptotic genes in response to DNA damage. Moreover, p68 depletion results in a striking inhibition of recruitment of p53 and RNA Pol II to the p21 promoter but not to the Bax or PUMA promoters, providing an explanation for the selective effect on p21 induction. Importantly, these findings are mirrored in a novel inducible p68 knockout mouse model in which p68 depletion results in a selective inhibition of p21 induction in several tissues. Moreover, in bone marrow, p68 depletion results in an increased sensitivity to γ-irradiation, consistent with an increased level of apoptosis. These data highlight a novel function of p68 as a modulator of the decision between p53-mediated growth arrest and apoptosis in vitro and in vivo.
Keywords: p68 (DDX5) RNA helicase, p53, DNA damage, cell cycle arrest, apoptosis
Introduction
The p53 tumour suppressor protein is a tightly regulated transcription factor that plays a pivotal role in orchestrating the cellular response to tumorigenic agents such as activated oncogenes or DNA damage (1). The outcome of inducing and activating p53 varies from the instigation of a reversible growth arrest that permits repair and recovery, to the exclusion of the cell from the cycling population by senescence (permanent arrest) or its elimination by apoptosis. Several mechanisms/agents have been identified as mediators of the decision between growth arrest and programmed cell death and two alternative models, involving either selective binding of p53 to specific pro-arrest or pro-apoptotic promoters or selective activation (context) of bound p53 through modifications or recruitment of co-factors at specific promoters, have been proposed to achieve the flexibility of the p53 response (2). Furthermore, it is well established that there is considerable tissue-specific heterogeneity in the p53 response to stress in vivo; the combination of factors present in different tissue/cell environments has been likened to a “barcode” that governs p53 activity (3) and is likely to be critical in the choice between cell cycle arrest and apoptosis. However there is relatively little known about the factors underpinning this selectivity in vivo.
p68 (DDX5) is a prototypic member of the DEAD box family of RNA helicases that can unwind RNA in an ATP-dependent manner (4). p68 is a multi-functional protein known to be involved in several cellular processes requiring manipulation of RNA structures, including processing of pre-mRNA, rRNA, and miRNA, and has been shown to be aberrantly expressed/modified in a range of cancers (5). Studies from several groups, including our own, have demonstrated that p68 also plays an important, and apparently RNA helicase-independent, role as a co-activator of several transcription factors that are critical to cancer development, including Estrogen Receptor alpha (6), Androgen Receptor (7), and the tumour suppressor p53 (8). p68 is recruited to the promoters of responsive genes under conditions in which these transcription factors are activated, consistent with a role in transcription initiation (9).
We have previously demonstrated that p68 is important for the p53 response to DNA damage (8). In the present study we show that, while p68 is critical for p53-mediated transactivation of the pro-survival cell cycle arrest gene CDKN1 (p21WAF1/CIP1 - herein referred to as p21) and for the G1/S cell cycle checkpoint, it is dispensable for the expression of several pro-apoptotic genes, or the induction of apoptosis, in cell lines in response to DNA damage. We also demonstrate that siRNA depletion of p68 results in a striking inhibition of recruitment of p53 and RNA Pol II to the p21 promoter but not to the Bax or PUMA promoters, providing a means by which the selective inhibition of p21 induction could be achieved. Finally, using our novel inducible p68 knockout mouse model, we show that p68 is required for p53-dependent induction of p21 in several, although not all, tissues in response to γ-irradiation and does not appear to be required for induction of pro-apoptotic genes. Taken together, our data highlight an important function of p68 in selectively regulating p53-dependent p21 expression and thus potentially influencing the decision between p53-mediated growth arrest and apoptosis in vitro and in vivo. Importantly, such a role for p68 in vivo may provide a mechanism by which elevated p68 in cancer cells could undermine p53-dependent cell death.
Results
p68 is required for the DNA damage-induced G1/S cell cycle checkpoint but has no effect on the induction of apoptosis
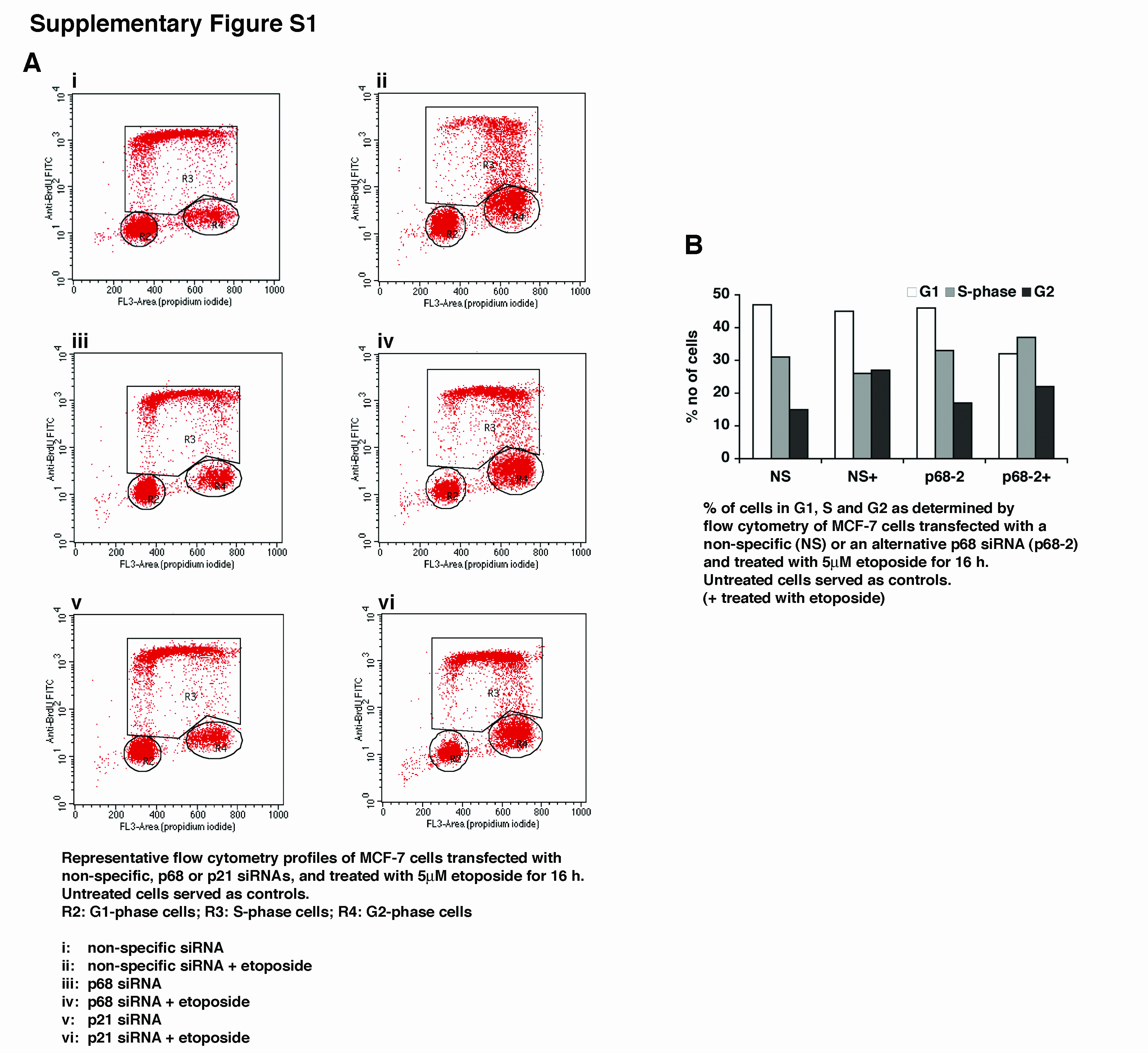
Our previous work demonstrated that p68 is critical for p53-dependent p21 induction in response to DNA damage (8). We therefore considered whether p68 depletion affects DNA damage-induced, p53-dependent, cell cycle arrest. To this end MCF-7 cells (wt p53) were transfected with either non-specific, p68 or p21 siRNAs, treated with the chemotherapeutic drug etoposide to induce cell cycle arrest and analysed by flow cytometry. As Figure 1A shows, in the absence of etoposide, approximately 50% of the cells are in G1, 40% in S and 10% in G2 and the cell cycle profiles are similar regardless of p68 or p21 depletion. In cells transfected with a non-specific siRNA, etoposide treatment does not cause a significant change in the G1 population but results in a reduction in the proportion of S-phase cells and an increase in the proportion of G2 cells, consistent with the induction of a G2/M arrest under these conditions of DNA damage. In contrast, in cells transfected with the p68 siRNA, while the etoposide-induced G2/M arrest is maintained, and indeed enhanced, the proportion of G1 cells is significantly reduced suggesting that depletion of p68 selectively results in a failure of the G1/S checkpoint, allowing cells to progress through S to G2/M. Strikingly this defect is similar to that observed in cells transfected with a p21 siRNA, consistent with the idea that the G1/S checkpoint failure in cells depleted of p68 is due to their failure to induce p21. Flow cytometry profiles are shown in Supplementary Figure S1A.
Figure 1. p68 is critical for DNA-damage induced G1/S cell cycle arrest and p21 expression but is not required for induction of pro-apoptotic genes.

MCF-7 cells were transfected with non-specific (NS), p68 or p21 siRNAs and treated with etoposide prior to analysis by flow cytometry or RNA extraction for qRT-PCR (TaqMan). Untreated cells served as controls. +: cells treated with 5μM etoposide for 16 h.
A: Percentage of cells in G1, S and G2 as determined by flow cytometry after BrdU treatment and propidium iodide staining. B-E: Levels of mRNAs as indicated shown relative to respective mRNA cells transfected with a non-specific siRNA and not treated with etoposide. In each case this is shown as 1. F: corresponding western blots.
In all graphs the values shown are the averages from 3 independent experiments +/− s.e.m. and statistically significant differences are indicated. Where graphs are marked by * instead of bars for clarity, p values were <0.01.
Analysis of p68, p53 and p21 mRNA levels (Figure 1B-D) confirmed the p68/p21 knockdown and showed no effect on p53 mRNA. Strikingly p68 depletion resulted in a significant reduction of both basal and etoposide-induced p21 mRNA levels while there was no reduction in the expression of Bax mRNA in cells treated with p68 or p21 siRNA (Figure 1E); instead there was a minor but reproducible and statistically significant increase in etoposide-induced Bax mRNA levels. Corresponding western blots showing p68, p53, p21 and Bax protein levels are shown in Figure 1F. A small increase in p68 mRNA was observed upon etoposide treatment (Figure 1B-note different scales in graphs); however no corresponding increase was seen in p68 protein. These observations suggest that p68 siRNA knockdown does not affect the expression of all p53 target genes. In order to investigate this further we examined the induction of GADD45, which is a mediator of the G2/M checkpoint (10) and of several other pro-apoptotic genes. Interestingly, p68 or p21 knockdown resulted in a significant increase in the induction of GADD45 (consistent with the observed increase in the population of G2 cells) and the pro-apoptotic genes PUMA, Noxa, Fas and PIG3 (Supplementary Figure S2). Interestingly, in the case of GADD45, PUMA and Noxa, the baseline (uninduced) mRNA levels are also increased upon p68 or p21 knockdown. These findings suggest that p68 is selectively required for p21 expression and that, under certain conditions, may indeed protect against apoptosis.
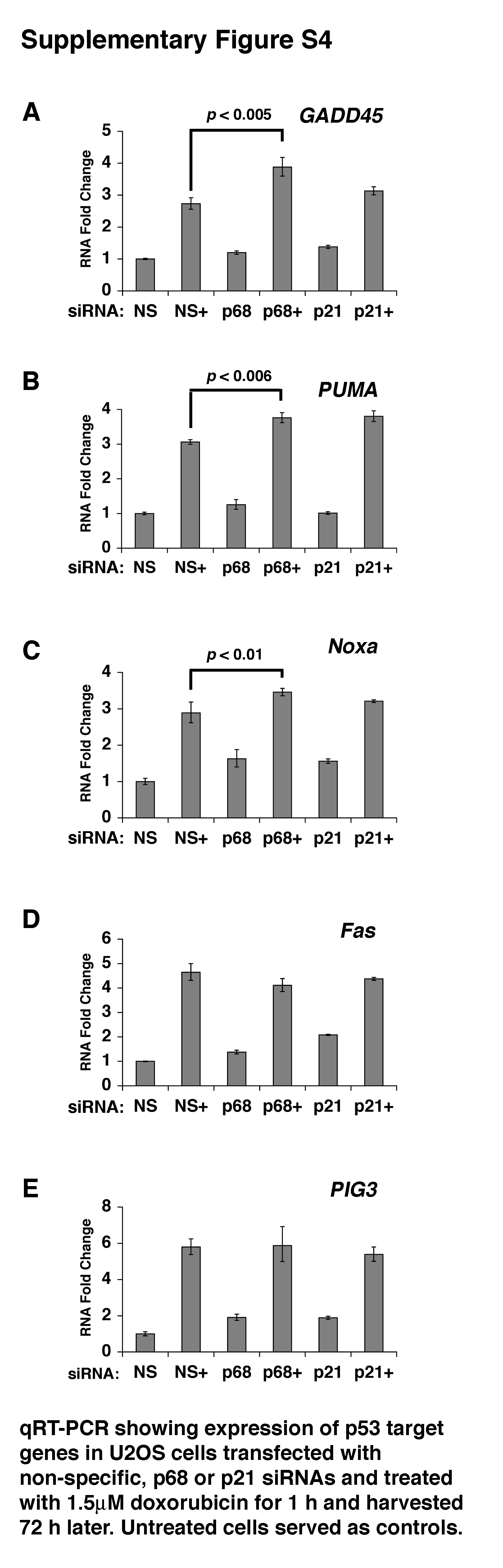
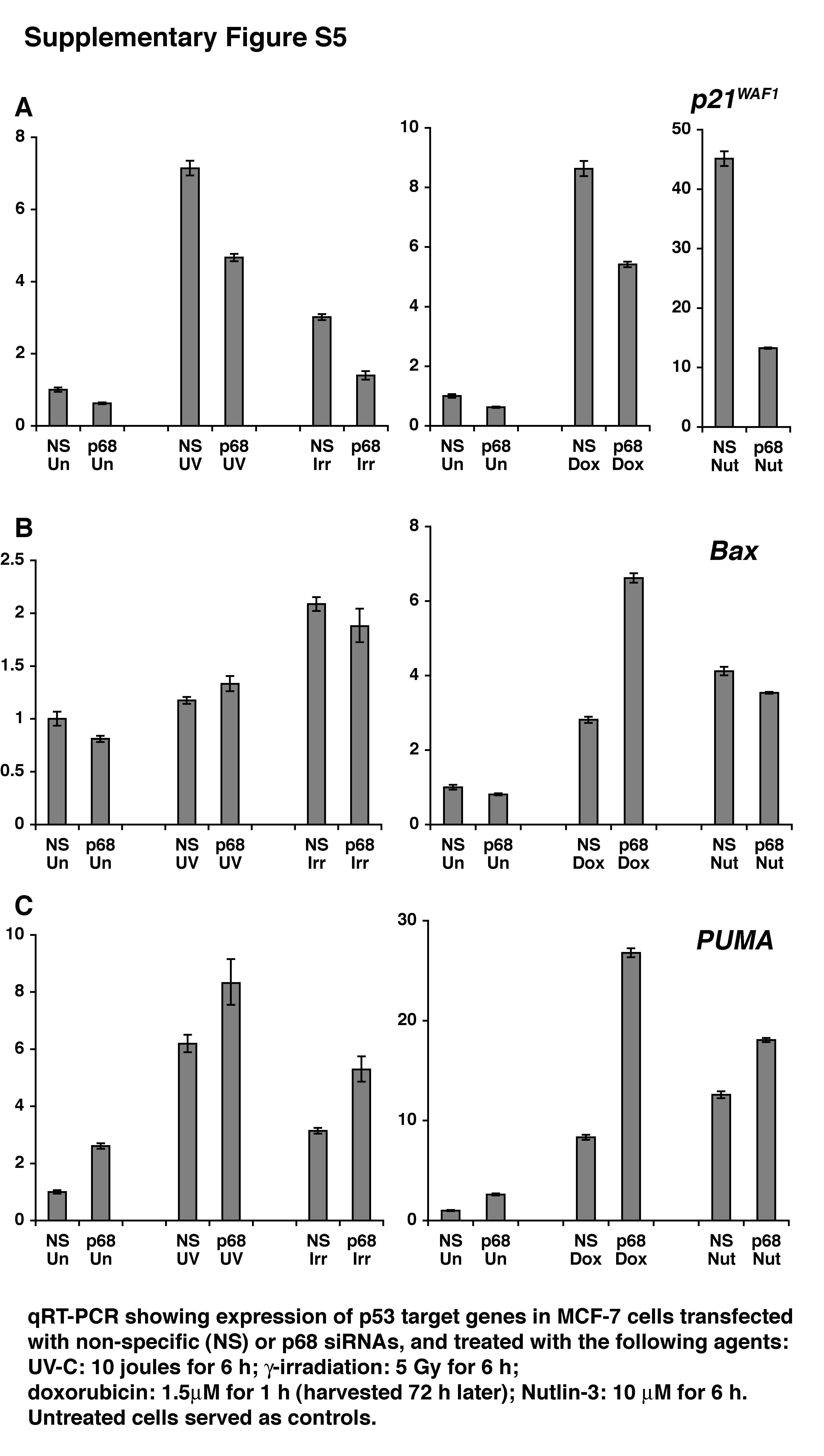
To test this further we used an established U2OS cell line model of apoptosis: treatment of these cells with 1.5μM doxorubicin for 1 h results in a marked induction of apoptosis (measured 72 h later-Figure 2A). Flow cytometry profiles are shown in Supplementary Figure S3A. Analysis of p68, p53 and p21 mRNA and protein levels (Figure 2B-F) confirmed the p68/p21 knockdown and, as in the MCF-7 cells (above), showed that p68 depletion resulted in a significant reduction of both basal and doxorubicin-induced p21 but no difference in Bax induction. Under these conditions p68 siRNA knockdown did not significantly alter the induction of apoptosis by doxorubicin while p21 knockdown resulted in a marked enhancement of apoptosis induction (Figure 2A), consistent with the idea that p21 is protective against apoptosis. (Note: p21 siRNA results in almost complete abrogation of p21 as opposed to the ~50% reduction observed with the p68 siRNA). We observed a minor but statistically significant increase GADD45, PUMA and Noxa induction but no significant effect on the induction of Fas or PIG3 (Supplementary Figure S4). For both cell cycle arrest and apoptosis experiments we obtained similar results using a second p68 siRNA directed against a different region in the p68 gene (Supplementary Figures S1B, S3B). Importantly, treatment of both MCF-7 and U2OS cells with other DNA damaging agents and Nutlin-3 (which induces p53 activity in the absence of DNA damage) gave similar results (Supplementary Figures S5, S6).
Figure 2. p68 is not required for DNA damage-induced apoptosis or the induction of Bax.

U2OS cells were transfected with non-specific (NS), p68 or p21 siRNAs and treated with doxorubicin prior to analysis by flow cytometry or RNA extraction for qRT-PCR (TaqMan). Untreated cells served as controls. +: cells treated with 1.5μM doxorubicin.
A: Percentage of apoptotic cells as determined by Annexin V staining and flow cytometry.
B-E: Levels of mRNAs as indicated shown relative to respective mRNA cells transfected with a non-specific siRNA and not treated with doxorubicin. In each case this is shown as 1. F: corresponding western blots.
In all graphs the values shown are the averages from 3 independent experiments +/− s.e.m. and statistically significant differences (p=<0.05) are indicated.
Taken together, our data show that p68 is not required for the induction of pro-apoptotic genes or apoptosis by DNA damage and suggest that, at least under certain conditions, it may protect against apoptosis. In contrast, p68 is critical for induction of p21 and the G1/S checkpoint, suggesting that p68 levels may play an important role in influencing the decision between cell cycle arrest and apoptosis in response to DNA damage.
In a previous study (8) we observed that p68 siRNA knockdown also inhibited induction of Fas and PIG3 upon etoposide treatment. However, with the siRNA transfection reagents available at that time, the earlier experiments required two sequential transfections to achieve efficient p68 knockdown. These may have resulted in additional transfection-induced stress, which could explain the different results. In the current study (using a single siRNA transfection and different transfection reagents) all DNA damaging agents and all doses/times (including 100μM etoposide for 4 hr as in the previous study) gave similar results (Supplementary Figures S5, S6 and S7), i.e. p68 was required for p21 but not pro-apoptotic gene induction, indicating that these are not dependent on the DNA damage or dose used.
p68 is important for recruitment of p53 and RNA polymerase II to the p21 promoter but has minor effects on recruitment to the Bax or PUMA promoters
We had previously shown that p68 is recruited to the p21 promoter in a p53 and DNA damage-dependent manner (8). By chromatin immunoprecipitation, using a p68-specific antibody, we confirmed that p68 is recruited to the p21 as well as the Bax and PUMA promoters and that its recruitment to the p21 and PUMA promoters is enhanced by DNA damage (Figure 3A). Since p68 has been shown to be important for recruitment of other transcriptional regulators to certain promoters (11) we reasoned that one mechanism by which p68 could selectively modulate induction of certain p53-target genes in response to DNA damage might be through differentially regulating the recruitment of p53, and/or of other components of the transcriptional machinery, to specific promoters. We therefore compared the recruitment of p53 and RNA polymerase II (RNA Pol II) to the p21, Bax and PUMA promoters in MCF-7 cells, under conditions of p68 siRNA knockdown, in the presence and absence of etoposide treatment (5μM for 16 hr-Figure 3B, C). While p68 depletion had little effect on the baseline recruitment (in the absence of DNA damage) it resulted in a striking reduction (>50%) in the recruitment of both p53 and RNA Pol II to the p21 promoter following DNA damage. In contrast, there was only a minor reduction in recruitment of p53 or RNA Pol II to the Bax and PUMA promoters upon p68 knockdown. Similar results were obtained after treatment with 100μM etoposide for 2 hr (Supplementary Figure S8). These findings demonstrate that p68 is crucial for recruitment of p53 and RNA Pol II to the p21 promoter, but not to the Bax and PUMA promoters, following genotoxic stress thus providing a way in which p68 may selectively regulate p53- and DNA damage-dependent induction of p21.
Figure 3. p68 is required for recruitment of p53 and RNA Pol II to the p21 promoter but not to the Bax and PUMA promoters.

MCF-7 cells were transfected with non-specific (NS) or p68 siRNAs and treated with etoposide prior to chromatin immunoprecipitation and qPCR (SYBR Green). Untreated cells served as controls. +: cells treated with 5μM etoposide for 16 h.
Recruitment of (A) p68, (B) p53 and (C) RNA Pol II to the p21, Bax and PUMA promoters is shown as percentage relative to input DNA.
In all graphs the values shown are the averages from 3 independent experiments +/− s.e.m. and statistically significant differences (p = <0.05) are indicated.
p68 is critical for γ-irradiation-induced p21 expression in some but not all tissues in a conditional p68 knockout mouse model
Given our findings in cell lines, we wished to investigate whether p68 also influences the decision between cell cycle arrest and apoptosis in vivo. Since constitutive p68 knockout results in embryonic lethality (~E11.5) (12) we generated a conditional, tamoxifen-inducible (Cre-ERT2), p68 knockout (p68KO) mouse, to allow us to inducibly knock out p68 expression in adult mice, and investigated the p53 response to γ-irradiation. As shown in Supplementary Figure S9, a significant Cre-dependent knockout of p68 was achieved in a wide range of tissues. Within specific tissues we observed a strong bias in the extent of p68 depletion towards certain sub-populations of cells. For example, in liver there was a complete p68 knockout in hepatocytes but apparently normal p68 expression in Kupffer cells while, in the spleen, p68 was depleted in most haemopoietic cells with a significant proportion of stromal cells retaining p68 expression.
In order to investigate the importance of p68 in the p53 response to DNA damage in vivo, p68KO (Cre+) and Cre− controls were treated with tamoxifen to induce Cre and then subjected to 2 Gy γ-irradiation (a standard dose for patients receiving radiotherapy). Expression of p53, and of p21 and cleaved caspase-3 (as indicators of cell cycle arrest and apoptosis respectively), was examined as described previously (13, 14) in spleen, liver, bone marrow and large intestine (Figures 4, 5), tissues in which the p53 response to irradiation has been established previously (14-17). Tissues from mice that had not been irradiated were similarly stained as controls and showed no significant p53, p21 or caspase-3 staining (Figure 4A, C).
Figure 4. The p53 DNA damage response in spleen and liver from p68 knockout mice.

Immunohistochemistry showing p68, p53, p21 and caspase-3 expression in: spleen (A, B) and liver (C, D) from untreated and γ-irradiated control and p68 knockout mice. Tissues were harvested 2 h after irradiation. Images are representative of 6 control and 6 p68KO mice. Bar= 50μm.
Figure 5. The p53 DNA damage response in bone marrow and large intestine from p68 knockout mice.

Immunohistochemistry showing p68, p53, p21 and caspase-3 expression in: bone marrow (A, B) and large intestine (C, D) from untreated and γ-irradiated control and p68 knockout mice. Tissues were harvested 2 h after irradiation. Images are representative of 6 control and 6 p68KO mice. Bar= 50μm.
In the irradiated spleen (Figure 4B), p53 staining was observed in the nuclei of cells in both the red pulp and white pulp areas; the proportion of cells staining positive for p53 was similar in the control and p68KO mice, confirming that depletion of p68 does not affect p53 stabilisation. However, there was a striking reduction in the proportion of cells staining for p21 in the p68KO mice, as compared with the control mice (Figure 4A, B) indicating that, as observed in our cell line models (Figures 1, 2), p68 is important for the induction of p21. In contrast, the induction of cleaved caspase-3 was similar in the control and p68KO mice, consistent with our finding that p68 siRNA knockdown does not affect the induction of pro-apoptotic genes in cell lines (Supplementary Figures S1, S3, S5, S6). In the liver, p53 was barely detectable in hepatocytes after irradiation (Figure 4D), as described previously (15, 16). Nevertheless, in the control (Cre) mice there was a significant increase of p21 in hepatocytes following irradiation, indicating activation of p53. High p21 induction in the context of low levels of p53 in the liver has been described previously (17); moreover, we confirmed that irradiation-induced p21 expression in hepatocytes is p53-dependent by demonstrating that there is no p21 induction in p53-null mice (Supplementary Figure S10A). Strikingly, in the p68KO mice, there was no induction of p21 in the hepatocytes (from which p68 was completely depleted) while induction in the Kuppfer cells (which retain p68) appeared to be normal. In both control and p68KO mice there was little induction of cleaved caspase-3; this is consistent with earlier findings showing little apoptosis induction in the liver by irradiation (15-17). Our data thus demonstrate that, in the spleen and liver, p68 depletion results in the abrogation of DNA damage-induced p21 expression but has no measureable effect on induction of apoptosis.
The data from bone marrow and large intestine are more complex, suggesting tissue specificity in p68 function. A significant, p68 knockout is obtained; however p68KO results in hypoplastic bone marrow and alterations in tissue organisation in the large intestine, even in the absence of irradiation as shown by histologic examination (Figure 5). Moreover, there was significant expression of p53 and p21 in the p68KO mice in the absence of radiation (Figure 5A, C); this however, was not enhanced upon irradiation (Figure 5B, D), suggesting that the p53/p21 expression is due to other, DNA-damage independent, cellular stresses induced by lack of p68 in these tissues. In contrast in the control mice there was no p53 or p21 staining in the non-irradiated mice (Figure 5A, C) with a significant induction upon irradiation (Figure 5B, D). In bone marrow, although p68 knockout resulted in an increase in capase-3 independently of irradiation (Figure 5A), there was a further enhancement upon irradiation (Figure 5B) suggesting that, in this tissue, lack of p68 favours the induction of apoptosis. No significant caspase-3 staining was observed in large intestine (Figure 5C, D). As expected, p21 and caspase-3 induction in bone marrow was p53-dependent since no expression was observed in p53-null mice (Supplementary Figure S10B).
The effect of p68 status on radiosensitivity of haemopoietic stem cells
In order to examine the effect of p68 depletion on the sensitivity of bone marrow haemopoietic stem cells to γ-irradiation, suspensions of femoral bone marrow obtained from individual mice were γ-irradiated with doses of 1, 2 or 3 Gy (or sham-irradiated) and immediately plated in the established clonogenic CFU-A assay (18), which produces mature cells derived from members of the haemopoietic stem cell compartment (short term repopulating haemopoietic stem cells) and is a well-established technique for assessing effects on the survival of irradiated cells. p68 knockout in these cells was confirmed by immunocytochemistry (Supplementary Figure S11). As shown in Figure 6 and Supplementary Table S1, haemopoietic cells from the p68KO mice were significantly more sensitive to γ-irradiation than their control counterparts, consistent with our observation that in the bone marrow of γ-irradiated p68KO mice there was a slightly increased number of cells staining positive for cleaved caspase-3 (Figure 5A, B), as compared with control mice, and would be indicative of increased apoptosis. This idea is also supported by our findings in cell lines, which demonstrated that DNA damage in the context of p68 siRNA knockdown resulted in an enhanced induction of several pro-apoptotic genes (Supplementary Figures 2, 4). It is established that radiosensitivity of haemopoietic cells is p53-dependent (19, 20); therefore our data are consistent with lack of p68 favouring the induction of p53-dependent apoptosis in this context, possibly through loss of protection by p21.
Figure 6. Bone marrow cells from p68 knockout mice are more sensitive to γ-irradiation than those from control mice.

Survival of multipotential haemopoietic cells (from clonogenic CFU-A assays) after exposure to 1-3 Gy γ-irradiation. Bone marrow cells were obtained from 4 individual control mice (■-solid line) and 8 individual p68KO mice (●-dashed line).
Discussion
The biological outcome of p53 induction is not only dependent on the particular stress but also on the cell/tissue environment and the many factors that act as co-regulators of p53 function (1, 3). Our data demonstrate that p68 plays an important role in the p53 response to DNA damage both in cell lines and in vivo. In cell lines, our siRNA knockdown experiments demonstrate that p68 is crucial for both basal and DNA damage-induced expression of p21. Interestingly, in the absence of DNA damage there was no obvious difference in the cell cycle profiles between control cells and those in which p68 or p21 had been depleted by siRNA. However, in response to etoposide treatment, there was a striking reduction in the G1 population of cells lacking p68 under conditions of DNA damage, suggesting a failure of the G1/S checkpoint. Moreover, the effect of p68 siRNA knockdown on the response to DNA damage was similar to that observed with a p21 siRNA, indicating that the defect observed upon depletion of p68 may be due to the lack of p21 induction.
In contrast, p68 appears to be dispensible for expression of pro-apoptotic genes; indeed p68 siRNA knockdown results in an increased expression of several pro-apoptotic genes. These observations are supported by our data showing that p68 is not required for the induction of apoptosis in a cell line model and that bone marrow cells from p68KO mice are more sensitive to irradiation than their control counterparts. These findings suggest that p68 is required for p53-dependent p21 expression and that, following genotoxic stress, p68 favours cell survival.
Other factors have been found to modulate p53 transactivation to favour the induction of cell cycle arrest. iASPP (21) and YB1 selectively inhibit the ability of p53 to induce expression of pro-apoptotic genes but have no effect on p21 transactivation. In contrast, Brn-3a (22, 23) and Hzf (24) repress the induction of pro-apoptotic genes while enhancing induction of cell cycle arrest genes. p68 appears to have a somewhat different modulatory effect on p53 function: in our cell line models p68 siRNA results in the abrogation of p21 expression while the enhancement of induction of pro-apoptotic genes appears to exhibit cell- and/or DNA damage-specific effects, suggesting that p68 might repress the expression of pro-apoptotic genes only in certain contexts.
Our chromatin immunoprecipitation data provide a possible mechanism by which the selective effect of p68 depletion on p21 induction could be achieved. Although p68 is recruited to the p21, Bax and PUMA promoters, p68 siRNA knockdown results in a striking reduction in the recruitment of p53 and RNA Pol II to the p21 promoter upon DNA damage, but has only minor effects on their recruitment to the Bax and PUMA promoters, suggesting that p68 may be important for the DNA damage response at selected p53-dependent promoters. Similarly Hzf and Brn-3a selectively enhance recruitment of p53 to the p21 promoter (22, 24). One possibility for this selectivity is that promoter-specific sequence or structural elements, partner proteins present in certain cell types or indeed p53 post-translational modifications (25), may influence the requirement for p68 at these promoters and may explain the tissue dependent effects we observe in vivo. In this respect the previously reported differences in the mechanism of activation and the composition of transcriptional initiation complexes at the p21 and pro-apoptotic Fas promoters are particularly relevant (26, 27). Interestingly, the p21 promoter has been shown to be ‘pre-loaded’ with p53 and paused RNA Pol II in the absence of stress (26), but to be intrinsically inefficient for reinitiation (27). Our data show that p68 depletion had a negligible effect on p53 and RNA Pol II recruitment to the p21 promoter in the absence of DNA damage but a striking effect on their recruitment after DNA damage, suggesting that p68 may play an additional role in the enhancement of transcriptional initiation in response to stress and providing a possible explanation for the observed effect of p68 depletion on cell cycle profiles only in the presence of DNA damage.
The results from our p68 knockout mouse support our model from the cell line data but demonstrate tissue specificity in the requirement of p68 for p53-dependent p21 induction. In the liver and spleen p68 is required for p21 induction in response to irradiation. Although there was little p53 induction in the liver there was a strong p53-dependent p21 induction in the control mice, which was absent in the mice lacking p68, underscoring the requirement for p68 and indicating that only low levels of p53 induction are required to induce significant levels of p21. High p21 induction by relatively low p53 levels has been previously reported (28, 29). The results from the bone marrow and large intestine are more difficult to interpret since organisation/cell populations are altered in these tissues in the p68KO mice and basal levels of p53 and p21 are high in the absence of DNA damage and were not induced upon irradiation, suggesting that lack of p68 induces other, DNA damage-independent, cellular stresses. This is perhaps not surprising given the demonstrated multiple functions of p68 (5). Interestingly, however, although there was a higher level of caspase-3 staining in non-irradiated p68KO mice compared with controls, this was further increased upon irradiation, consistent with our finding that bone marrow cells from these mice are more sensitive to irradiation. Taken together our data indicate that the requirement of p68 for p53-dependent p21 expression is tissue- and context-dependent. Interestingly, the p53 response itself shows tissue and context specificity in terms of the induction of cell cycle arrest or pro-apoptotic genes by irradiation (14, 17, 30).
Our findings thus demonstrate that p68 is a selective regulator of the p53 DNA damage response and that it can modulate the decision between cell cycle arrest and apoptosis in a tissue- and context-dependent manner. This is particularly interesting since several studies have shown that p68 is aberrantly expressed and/or modified in a wide range of cancers (7, 31-33). p68 levels and function in cancer tissues could therefore modulate p53-dependent tissue responses to radiotherapy or chemotherapy by determining whether cells survive or die. This in turn could be important in the development of better therapeutic strategies for cancer treatment, or choice of treatment depending on p53 and p68 status, that strike the correct balance between cell cycle arrest/DNA repair and apoptosis to achieve the optimal therapeutic effect and minimise deleterious side effects.
Materials and Methods
Antibodies
p68: PAb204 (Millipore, Temecula, USA) and 2907 (polyclonal against the C-terminal 15 residues of p68); p53: DO1 (Santa Cruz Biotechnology, Santa Cruz, USA) and CM1 (polyclonal-recognises human and mouse p53), p21: H164 (Santa Cruz Biotechnology-western blotting) and Ab-5 (Oncogene Science, Cambridge, USA-immunohistochemistry); Bax: N-20 (Santa Cruz Biotechnology); RNA Pol II: Ab817 (Abcam, Cambridge, USA); cleaved caspase-3: 9664 (Cell Signalling, Hitchin, UK); actin: A2066 (Sigma-Aldrich, Poole, UK). Vectastain ABC kits (Vector Labs) were used for immunohistochemistry.
Transfections/siRNA transfections
siRNA transfections were performed using LipofectamineTM RNAiMax (MCF-7) and LipofectamineTM 2000 (U2OS) (Invitrogen) and 30pmols of p68, p21 or non-silencing siRNAs [Dharmacon (Lafayette, USA)-Supplementary Table S2A].
RNA extraction and qRT-PCR
Total cell RNA was extracted using the RNeasy kit (Qiagen, Crawley, UK). 1μg of RNA was reverse-transcribed using M-MLV Reverse Transcriptase and random primers (Invitrogen). Quantitative PCR was performed using TaqMan Universal PCR Master Mix and gene expression assays (Applied Biosystems, Foster City, USA) or specifically-designed primers (Eurofins, Ebersberg, Germany)-Supplementary Table S2B-C. β-actin was used as control; fold changes were calculated using the ΔΔCt method in Microsoft Excel.
Chromatin Immunoprecipitation
p53-, p68- or RNA Pol II-bound chromatin was immunoprecipitated using 3μg of DO-1, PAb204 or Ab817 antibody respectively, as described previously (8). qPCR was performed using SYBR Green QuantiTect Mastermix (Qiagen, Crawley, UK). Promoter occupancy was calculated as % of input DNA using the ΔΔCt method in Microsoft Excel. Primer sequences and PCR cycles are in Supplementary Table S2D.
Cell cycle arrest and apoptosis analysis-Flow cytometry
Analysis of cells for cell cycle arrest was performed after BrdU treatment and propidium iodide staining as described previously (34). Apoptotic cells were detected by AnnexinV-FITC (Cambridge Bioscience, Cambridge, UK).
Induction of p68 knockout in mice and γ-irradiation
The gene-targeting strategy (Supplementary Figure S12A), and generation of founder mice was performed by TaconicArtemis (Koln, Germany). Induction of Cre and irradiation were as described in Supplementary Figure S8 and correct Cre-mediated excision of the p68 gene was confirmed by PCR- see Supplementary Figure S12B). All experiments on mice were performed in accordance with Home Office guidelines under project licence PPL60/2841.
Preparation of mouse tissues for western blotting and immunohistochemistry
For western blotting, tissues were ground into powder under liquid nitrogen and homogenized in lysis buffer containing 50mM Tris-HCl (pH6.8), 2% SDS, 10% glycerol, 3.575M β-mercaptoethanol, 1mM EDTA and 0.5mM DTT. For immunohistochemistry, tissues were fixed by immersion in 10% buffered formalin and processed to paraffin wax. 4μm sections were stained as described previously (7).
Irradiation of bone marrow cells and Clonogenic CFU-A assay
Suspensions of femoral bone marrow obtained from individual mice were γ-irradiated (or sham-irradiated) using a Bio International 637 Cesium irradiator. Immediately after irradiation cells were plated in 45-mm Petri dishes containing 2ml α-MEM supplemented with 20% pre-tested horse serum (Invitrogen/Life Technologies) and conditioned media from the AF1, 19T and L929 cell lines batch tested to produce maximal colony stimulating activities as previously described (18).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Colin Henderson for helpful discussions. This work was supported by grants from Cancer Research UK (C8745/A11216) and the Association for International Cancer Research (06-613).
Footnotes
Conflict of interest The authors declare no conflict of interest.
References
- 1.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 2.Espinosa JM. Mechanisms of regulatory diversity within the p53 transcriptional network. Oncogene. 2008;27:4013–4023. doi: 10.1038/onc.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol. 2008;9:702–712. doi: 10.1038/nrm2451. [DOI] [PubMed] [Google Scholar]
- 4.Linder P. Dead-box proteins: a family affair--active and passive players in RNP-remodeling. Nucleic Acids Res. 2006;34:4168–4180. doi: 10.1093/nar/gkl468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuller-Pace FV, Moore HC. RNA helicases p68 and p72: multifunctional proteins with important implications for cancer development. Future Oncol. 2011;7:239–251. doi: 10.2217/fon.11.1. [DOI] [PubMed] [Google Scholar]
- 6.Endoh H, Maruyama K, Masuhiro Y, Kobayashi Y, Goto M, Tai H, et al. Purification and identification of p68 RNA helicase acting as a transcriptional coactivator specific for the activation function 1 of human estrogen receptor alpha. Mol Cell Biol. 1999;19:5363–5372. doi: 10.1128/mcb.19.8.5363. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Clark EL, Coulson A, Dalgliesh C, Rajan P, Nicol SM, Fleming S, et al. The RNA helicase p68 is a novel androgen receptor coactivator involved in splicing and is overexpressed in prostate cancer. Cancer Res. 2008;68:7938–7946. doi: 10.1158/0008-5472.CAN-08-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bates GJ, Nicol SM, Wilson BJ, Jacobs AM, Bourdon JC, Wardrop J, et al. The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor. EMBO J. 2005;24:543–553. doi: 10.1038/sj.emboj.7600550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, et al. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 10.Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu L, et al. GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci U S A. 1999;96:3706–3711. doi: 10.1073/pnas.96.7.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caretti G, Schiltz RL, Dilworth FJ, Di Padova M, Zhao P, Ogryzko V, et al. The RNA helicases p68/p72 and the noncoding RNA SRA are coregulators of MyoD and skeletal muscle differentiation. Dev Cell. 2006;11:547–560. doi: 10.1016/j.devcel.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Fukuda T, Yamagata K, Fujiyama S, Matsumoto T, Koshida I, Yoshimura K, et al. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nat Cell Biol. 2007;9:604–611. doi: 10.1038/ncb1577. [DOI] [PubMed] [Google Scholar]
- 13.Wallace M, Coates PJ, Wright EG, Ball KL. Differential post-translational modification of the tumour suppressor proteins Rb and p53 modulate the rates of radiation-induced apoptosis in vivo. Oncogene. 2001;20:3597–3608. doi: 10.1038/sj.onc.1204496. [DOI] [PubMed] [Google Scholar]
- 14.Coates PJ, Lorimore SA, Lindsay KJ, Wright EG. Tissue-specific p53 responses to ionizing radiation and their genetic modification: the key to tissue-specific tumour susceptibility? J Pathol. 2003;201:377–388. doi: 10.1002/path.1456. [DOI] [PubMed] [Google Scholar]
- 15.Midgley CA, Owens B, Briscoe CV, Thomas DB, Lane DP, Hall PA. Coupling between gamma irradiation, p53 induction and the apoptotic response depends upon cell type in vivo. J Cell Sci. 1995;108:1843–1848. doi: 10.1242/jcs.108.5.1843. [DOI] [PubMed] [Google Scholar]
- 16.MacCallum DE, Hupp TR, Midgley CA, Stuart D, Campbell SJ, Harper A, et al. The p53 response to ionising radiation in adult and developing murine tissues. Oncogene. 1996;13:2575–2587. [PubMed] [Google Scholar]
- 17.Fei P, Bernhard EJ, El-Deiry WS. Tissue-specific induction of p53 targets in vivo. Cancer Res. 2002;62:7316–7327. [PubMed] [Google Scholar]
- 18.Lorimore SA, Pragnell IB, Eckmann L, Wright EG. Synergistic interactions allow colony formation in vitro by murine haemopoietic stem cells. Leuk Res. 1990;14:481–489. doi: 10.1016/0145-2126(90)90036-9. [DOI] [PubMed] [Google Scholar]
- 19.Lotem J, Sachs L. Hematopoietic cells from mice deficient in wild-type p53 are more resistant to induction of apoptosis by some agents. Blood. 1993;82:1092–1096. [PubMed] [Google Scholar]
- 20.Lorimore SA, Goodhead DT, Wright EG. The effect of p53 status on the radiosensitivity of haemopoietic stem cells. Cell Death Differ. 1995;2:233–234. [PubMed] [Google Scholar]
- 21.Bergamaschi D, Samuels Y, O’Neil NJ, Trigiante G, Crook T, Hsieh JK, et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet. 2003;33:162–167. doi: 10.1038/ng1070. [DOI] [PubMed] [Google Scholar]
- 22.Budram-Mahadeo V, Morris PJ, Latchman DS. The Brn-3a transcription factor inhibits the pro-apoptotic effect of p53 and enhances cell cycle arrest by differentially regulating the activity of the p53 target genes encoding Bax and p21(CIP1/Waf1) Oncogene. 2002;21:6123–6131. doi: 10.1038/sj.onc.1205842. [DOI] [PubMed] [Google Scholar]
- 23.Hudson CD, Morris PJ, Latchman DS, Budhram-Mahadeo VS. Brn-3a transcription factor blocks p53-mediated activation of proapoptotic target genes Noxa and Bax in vitro and in vivo to determine cell fate. J Biol Chem. 2005;280:11851–11858. doi: 10.1074/jbc.M408679200. [DOI] [PubMed] [Google Scholar]
- 24.Das S, Raj L, Zhao B, Kimura Y, Bernstein A, Aaronson SA, et al. Hzf Determines cell survival upon genotoxic stress by modulating p53 transactivation. Cell. 2007;130:624–637. doi: 10.1016/j.cell.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aylon Y, Oren M. Living with p53, dying of p53. Cell. 2007;130:597–600. doi: 10.1016/j.cell.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Espinosa JM, Verdun RE, Emerson BM. p53 functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol Cell. 2003;12:1015–1027. doi: 10.1016/s1097-2765(03)00359-9. [DOI] [PubMed] [Google Scholar]
- 27.Morachis JM, Murawsky CM, Emerson BM. Regulation of the p53 transcriptional response by structurally diverse core promoters. Genes Dev. 2010;24:135–147. doi: 10.1101/gad.1856710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu X, Burbidge SA, Griffin S, Smith HM. Discordance between accumulated p53 protein level and its transcriptional activity in response to u.v. radiation. Oncogene. 1996;13:413–418. [PubMed] [Google Scholar]
- 29.Chene P, Fuchs J, Bohn J, Garcia-Echeverria C, Furet P, Fabbro D. A small synthetic peptide, which inhibits the p53-hdm2 interaction, stimulates the p53 pathway in tumour cell lines. J Mol Biol. 2000;299:245–253. doi: 10.1006/jmbi.2000.3738. [DOI] [PubMed] [Google Scholar]
- 30.Bouvard V, Zaitchouk T, Vacher M, Duthu A, Canivet M, Choisy-Rossi C, et al. Tissue and cell-specific expression of the p53-target genes: bax, fas, mdm2 and waf1/p21, before and following ionising irradiation in mice. Oncogene. 2000;19:649–660. doi: 10.1038/sj.onc.1203366. [DOI] [PubMed] [Google Scholar]
- 31.Causevic M, Hislop RG, Kernohan NM, Carey FA, Kay RA, Steele RJ, et al. Overexpression and poly-ubiquitylation of the DEAD-box RNA helicase p68 in colorectal tumours. Oncogene. 2001;20:7734–7743. doi: 10.1038/sj.onc.1204976. [DOI] [PubMed] [Google Scholar]
- 32.Yang L, Lin C, Liu ZR. Phosphorylations of DEAD box p68 RNA helicase are associated with cancer development and cell proliferation. Mol Cancer Res. 2005;3:355–363. doi: 10.1158/1541-7786.MCR-05-0022. [DOI] [PubMed] [Google Scholar]
- 33.Wortham NC, Ahamed E, Nicol SM, Thomas RS, Periyasamy M, Jiang J, et al. The DEAD-box protein p72 regulates ERalpha-/oestrogen-dependent transcription and cell growth, and is associated with improved survival in ERalpha-positive breast cancer. Oncogene. 2009;28:4053–4064. doi: 10.1038/onc.2009.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoy CA, Carswell C, Schimke RT. Bromodeoxyuridine/DNA analysis of replication in CHO cells after exposure to UV light. Mutat Res. 1993;290:217–230. doi: 10.1016/0027-5107(93)90162-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.