

Abstract
As a signaling scaffold, p62/SQSTM1 plays important roles in cell signaling and degradation of misfolded proteins. While localization of p62 to mitochondria has been reported, a description of its function once there, remains unclear. Here, we report p62 is localized to mitochondria in non-stressed situations and demonstrate that deficiency in p62 exacerbates defects in mitochondrial membrane potential and energetics leading to mitochondrial dysfunction. We report on the relationship between mitochondrial protein import and p62. In a p62 null background, mitochondrial import of the mitochondrial transcription factor TFAM is disrupted. When p62 is returned, mitochondrial function is restored to more normal levels. We identify for the first time that p62 localization plays a role in regulating mitochondrial morphology, genome integrity and mitochondrial import of a key transcription factor. We present evidence that these responses to the presence of p62 extend beyond the protein’s immediate influence on membrane potential.
Keywords: mitochondria integrity, oxidative stress, p62, TFAM, mitochondrial import
1. Introduction
Sequestosome 1/p62 was identified as a ubiquitin binding protein [1], and has been localized to aggresomes of various neurodegenerative diseases [2]. p62 plays critical roles in neurodegeneration, cancer, and obesity through regulation of cell signaling, protein degradation and NF-κB activation [3–5]. p62 contains several interaction motifs that endow the protein with scaffolding abilities [3] and is important in both the UPS and autophagy degradation pathways. In the UPS, p62 interacts with polyubiquitinated proteins through its C-terminal UBA domain allowing shuttling to the proteasome for degradation [6]. Mediation of autophagy occurs by p62 polymerizing through an N-terminal PB1 domain and interaction with the autophagic marker protein LC3 [7,8]. p62 itself is degraded by autophagy with autophagic defects causing accumulation of p62 in response to stress stimuli [9]. Inability to clear p62-containing aggregates increases ROS levels, DNA damage, tumorigenesis and cell death in the absence of autophagy [10,11].
Growing lines of evidence correlate p62 with clustering of damaged mitochondria. p62 is recruited to depolarized mitochondria in PINK1/parkin-expressing cells, two proteins linked to the pathology of Parkinson’s disease (PD) [12]. Efficient mitochondrial function and turnover depends on continuous structural remodeling through fusion and fission [13]. Daughter mitochondria produced by fission can either maintain intact membrane potential or become depolarized. Depolarized mitochondria may restore their membrane potential and return to normal fusion/fission equilibrium or remain as non-fusing mitochondria to be eliminated by autophagy. It has been proposed that fission acts as an autophagic checkpoint [14]. Depolarized mitochondria change their morphology to a more fragmented form and show perinuclear clustering (“mito-aggresomes”). Completion of the mitophagy pathway appears to be dependent on the recruitment of both p62 and HDAC6 [12,15]. Although there are conflicting reports as to p62’s role in mitophagy, it is known to be indispensable in the polymerization and transportation of mitochondria to aggregates [16,17]. This diverse suite of p62 functions is known to take place under stress or pathological conditions. However, whether p62 is localized to mitochondria and what its function might be under physiological conditions remains unclear.
The structural state of mitochondria is directly related to the organelle’s functional status [18]. Mitochondrial fragmentation correlates with bioenergetic defects and elevated oxidative stress leading to increased mtDNA mutations [19–21]. Meanwhile, mitochondrial fusion is required for mtDNA stability in skeletal muscle and protects against neurodegeneration in the cerebellum [22]. Disruption of fusion leads to loss of membrane potential and decreased cellular respiration [19]. Defects in mitochondrial dynamics causing dysfunction have been linked to multiple neurodegenerative diseases [23].
The mitochondrial genome encodes rRNAs, tRNAs and proteins important for cell respiration and ATP generation [24,25]. mtDNA is more prone to oxidative damage than nuclear DNA due to mitochondria being the major source of ROS, a lack of histone protection in mtDNA and reduced mitochondrial DNA repair ability. mtDNA damage results in decreased membrane potential (Δψm), increased apoptotic cell death [26], and is a hallmark of neurodegenerative diseases [27,28]. Depletion of mtDNA can result in mitochondrial change, such as fragmentation and reduction in number of cristae [29]. Thus, maintenance of the mitochondrial genome is crucial for cell survival.
Our previous studies revealed that p62 protected cells from oxidative damage and promoted cell survival while defects in p62 resulted in oxidative damage to nuclear DNA in association with various neurodegenerative diseases [30,31]. In the current study, the relationship between p62 and mitochondrial dynamics was investigated using p62−/− tissues and cells. p62−/− mice possess an AD-like phenotype [32] and exhibit mitochondrial morphology and mtDNA damage associated with neurodegenerative diseases. Our goal was to elucidate the relationships between p62, mtDNA stability and biosynthesis. We also wished to examine how p62 might affect mitochondria morphology and function. We show for the first time p62 plays a role in maintaining functional mitochondria energetics and is integral for increased mtDNA stability.
2. Materials and Methods
2.1 Reagents and antibodies
All chemicals in this project were obtained from Sigma (St. Louis, MO). Lipofectamine 2000 transfection reagent and ATP synthase antibody were from Life Technologies (Carlsbad, CA). p62 antibody was from ABCAM (Cambridge, MA). All other antibodies were from Santa Cruz (Santa Cruz, CA).
2.2 Cell culture and transfection
WT and p62−/− Mouse Embryonic Fibroblast (MEF) cells were cultured in DMEM media with 10% fetal bovine serum and pen/strep in a 37°C incubator in 5% CO2. Cells were transfected using Lipofectamine 2000 transfection reagent in OPTIMEM media as directed in reagent insert for a total of 48 hours prior to harvest.
2.3 Western blot and analysis
The cell lysate or isolated mitochondria was subject to SDS-PAGE in polyacrylamide gels. Samples were Western blotted with primary antibody from sources described above and HRP-tagged secondary antibody from GE Healthcare Life Sciences (Pittsburgh, PA) and processed with ECL detection reagent. Following exposure of the labeled membrane to Hyperfilm-ECL detection film, the Un-Scan-it Gel and Graph Digitizing software (Silk Scientific, Orem, UT) was used to scan and quantify the signal from the Western blot, and data were analyzed statistically (Win-SAS, Microsoft, Seattle, WA).
2.4 Mitochondria isolation
Following trypsinization, MEF cells were collected by centrifugation and mitochondria isolated essentially as described by Wieckowski, et al. 2009 [33]. Briefly, washed cells were homogenized on ice with a Teflon pestle followed by centrifugation twice at 600×g for 5 min. The Post Nuclear Pellet (PNP) was collected from the pellets and the supernatant was centrifuged at 7000×g for 10 min. The cytosolic fraction (Cyto) was obtained from the supernatant while the crude mitochondria pellet (C. Mito) was collected in the pellet. The pellet was washed with MRB Buffer (250mM Mannitol, 5 mM HEPES, pH 7.4 and 0.5 mM EDTA) before being layered over a Percol gradient. The gradient was centrifuged at 95,000×g for 30 min with a dense band containing purified mitochondria localized at the bottom of the tube. This band was collected and washed with MRB buffer before being suspended in a small volume of MRB Buffer containing protease inhibitors. Protein concentration was determined by Bradford assay (Bio-Rad Laboratories, Hercules, CA) and subjected to SDS-PAGE and Western blot or used in further experiments as mitochondrial lysates.
2.5 Immuno-Electron Microscopy
Immuno-TEM was performed essentially as described by Liu, et al. 2004 [34]. Briefly, mitochondria were isolated as by Percol gradient centrifugation, collected in homogenization buffer and fixed by addition of 4% formaldehyde, 0.4% glutaraldehyde, 4mM CaCl2 in 0.1M cacodylate buffer, pH 7.3. Mitochondrial pellets were collected and washed with 0.1M cacodylate/2mM CaCl2 prior to embedment in LR White (EMS). Sections (~80nm) were collected to 300 mesh nickel grids and etched with saturated sodium periodate (Sigma) prior to blocking with 4% Acetylated BSA in Tris buffered saline. Grids were incubated with SQSTM1/p62 antibody (1:100) in 1% Ac-BSA/TBS overnight at 4°C followed by goat-anti-rabbit conjugated with 20nm colloidal gold (EMS) for 1 hour at room temperature. Sections were postfixed in 2% glutaraldehyde, rinsed in distilled water and contrasted with 2% uranyl acetate and lead citrate.
2.6 Immunocytochemistry
For immunocytochemical analysis, MEF cells or other cells as indicated were grown on coverslips in 24-well plates in DMEM, and where indicated, transfected with myc- p62 plasmid. Cells were stained with MitoTracker Red (Life Technologies, Grand Island, NY) prior to fixation with warm 4% PFA in PBS and permeabilized with 0.1% TX-100 for 10 min. The cells were subsequently blocked in 3% nonfat dry milk in PBS and incubated with primary antibody overnight at 4°C, washed and incubated with secondary antibody coupled to FITC (Invitrogen, Carlsbad, CA) in blocking buffer for 2 h. After washing in PBS, the coverslips were mounted on slides using Vectashield Hardset Mounting media (Vector Laboratories, Burlingame, CA), and analyzed using a Zeiss Axiovert fluorescent microscope with Zeiss PLAN-Apochromatic 63X 1.4 Oil DIC lens. Images were collected and magnified with NIS-Elements AR software (Nikon, Melville, NY). Representative images are shown for all fluorescent applications from an approximate pool of 100 cells analyzed. Mitochondrial morphologies were quantitated in a blinded study as described in figure legends
2.7 mtDNA copy number and oxidative damage of mtDNA
Genomic DNA was isolated from brain tissue using the DNeasy Tissue Kit (Qiagen, Valencia, CA). Following elution with ddH2O, purified DNA was stored at −80°C. Relative Quantitative real time PCR was employed to determine mtDNA copy number. mtDNA was amplified with mtDNA primer: MDF: 5′-CCTATCACCCTTGCCATCAT-3′ and MDR: 5′-GAGGCTGTTGCTTGTGTGAC-3′. Nuclear DNA was amplified by nuclear DNA primer: NDF: 5′-ACATCTGTTGCTCCGGCTCTCATT-3′ and NDR: 5′-GCAAGCTCAAAGGGCAAGGCTAAA-3′. RT-QPCR was conducted using an ABI 7500 Real Time PCR system (Applied Biosystems, Carlsbad, CA) using Power SYBR Green PCR Master Mix. Relative mtDNA levels were calculated by the ΔΔCt method as described (Acevedo-Torres et al, DNA Repair 8:126). Larger 10 Kb fragments were amplified by GeneAmp XL PCR kit (Applied Biosystems, Carlsbad, CA) using the primer set: mt5733F: 5′-CCAGTCCATGCAGGAGCATC-3′ and mt15733R: 5′-CGAGAAGAGGGGCATTGGTG-3′. A small 91bp fragment was amplified by primer set: Mt13597F: 5′-CCCAGCTACTACCATCATTCAAGT-3′ and Mt13688R: 5′-GATGGTTTGGGAGATTGGTTGATGT-3′. Aliquots of the 10 Kb PCR products were resolved on 0.8% agarose gels, while 91 bp PCR products were resolved on a 2% agarose gel. Density of bands were scanned and quantified. Relative amplifications of 10 Kb fragments were normalized to the 91 bp small fragments. The average lesion frequency per 10kb (λ) was calculated as λ=ln AD/AO.
2.8 ATP Assay
ATP production was measured using the ATP Determination Kit (Life Technologies, Grand Island, NY). Briefly, MEF cells were detached from tissue culture plates by trypsin treatment and collected by brief centrifugation followed by PBS wash. Cell pellets were resuspended in isolation buffer (5mM HEPES, pH 7.2; 225mM Mannitol, 75mM Sucrose, 1mM EGTA, and protease inhibitors) followed by 5 times syringe using a 23 gauge needle. Samples were centrifuged at 1,500×g for 5 min prior to supernatants being used for ATP assay following the manufacturer’s directions and measured with a luminometer. Each sample was measured in triplicate.
2.9 ROS Assay
ROS levels were measured using the fluorogenic dye H2DCFDA (Life Technologies, Grand Island, NY). MEF cells were grown in 24 well tissue culture plates. Growth media was removed from attached cells and replaced with fresh DMEM media prior to treatment directly into the media with 10μM H2DCFDA. Fluorescence was measured immediately post stain addition (T=0 min) and again at T=30 min. H2DCFDA interacts with ROS produced in living cells and released into the culture media showing an increase in fluorescence when excited at 485nm and measured emission at 528nm.
2.10 Membrane potential assay
Briefly, cells were grown in DMEM media and harvested following trypsinization. Cells were washed and resuspended in media containing 5 μM JC-1 followed by incubation at 37°C for 30 minutes. Cells were pelleted and washed with PBS, resuspended in PBS and counted with an Accuri C6 Flow Cytometer. JC-1 fluorescence was measure in FL1 (530nm) and FL2 (585nm) channels with 50,000 events captured.
2.11 Statistical analyses
Data were expressed as the mean ± SEM of different group. Possible differences between group means and statistical significance between WT and p62−/− MEF cells or mice were analyzed using one way ANOVAs (SAS v9.2, SAS Institute Inc.). For significant differences, alpha was set at 0.05.
3. Results and Discussion
3.1 p62 localizes to mitochondria under physiological conditions
Mitochondrial dysfunction is a major characteristic of neurodegenerative disease, cancer and obesity [35,36]. p62 is known to be mitochondrially associated under stress conditions [12], including oxidative stress [37]. Correlatively, our research has shown that absence of p62 results in higher oxidative damage in mouse cells and tissues [31]. We reasoned that these two observations were interrelated, and thus sought to examine if p62 plays an undiscovered role in mitochondrial function and oxidative metabolism. Using embryonic fibroblasts, we first investigated whether p62 localized to mitochondria under physiological conditions in our model system. Mitochondrial fractions were isolated from WT and p62−/− MEF cells and subjected to Western blot analysis. Mitochondrial and organelle marker proteins were used to determine the purity of mitochondrial fractions (Fig. 1A). Mitochondrially associated proteins were detected predominantly in mitochondrial fractions while cellular markers were effectively limited to pre-fractionated lysate or cytoplasmic fractions. One notable exception was the ER protein calnexin found in crude mitochondrial fractions of both WT and p62−/− samples. This is likely due to the intimate association of mitochondria with ER [38,39]. However, calnexin was absent from further purified mitochondrial fractions. Endogenous p62 distribution was also examined and a pool of endogenous protein was found to be localized to purified mitochondria of WT MEF cells under physiological conditions while the absence of p62 from p62−/− cells was also confirmed (Fig. 1A).
Fig. 1. Mitochondrial localization of p62 in MEF cells.

(A) Mitochondrial fraction was isolated from WT and p62−/− MEF cells by Percoll gradient and centrifugation. Whole cell lysate (WCL), post-nuclear supernatant (PNS), cytoplasm fraction (Cyto) or mitochondrial fraction (Mito) was subjected to 10% SDS-PAGE and immunoblotted with antibodies to p62 or protein markers. TOM40 was used as outer membrane (OMM) marker and ATP synthase as inner membrane (IMM) marker for mitochondria. The cellular compartment markers are calnexin (endoplasmic reticulum), GAPDH (cytoplasm), syntaxin 6 (trans-Golgi network) and LC3-II (autophagosome). (B) FITC-labeled p62 (arrows) associates with MitoTracker Red stained mitochondria in WT MEF cells. (C) Purified mitochondria from WT MEF cells were prepared for TEM and stained with colloidal gold labeled p62 antibody. a – negative control containing only colloidal gold secondary antibody; b–d - granules of p62 are localized to the OMM as well as with internal cristae.
Mitochondrial localization of p62 was visualized by immunofluorescence and immuno-TEM. WT MEF cells were treated with MitoTracker Red prior to fixation and immunostaining for endogenous p62. Using FITC-labeled secondary antibody, unstimulated pools of p62 were found associated with mitochondria (Fig. 1B). Colocalization was quantitatively estimated using the NIS Elements software (Nikon). A Mander’s Overlap Coefficient value (40) of 0.749 was obtained suggesting colocalization of p62 with the mitochondria. Further, mitochondria isolated from WT MEFs followed by immuno-TEM with p62 antibody (Fig. 1C) showed labeling not only localized to the Outer Mitochondrial Membrane (OMM) but was also observed associated with the Inner Mitochondrial Membrane (IMM). These data indicate that p62 could be found inside, as well as on the surface of mitochondria. These results are consistent with recent reports that portions of mitochondrially localized p62 are protected from proteinase K digestion [37]. It is possible p62 localization spans all mitochondrial compartments and plays a similar role as the ATPase ATAD3A which specifically functions to regulate mitochondrial dynamics [41].
3.2 Localization of p62 to the mitochondria affects mitochondrial morphology
Since p62 localized to mitochondria and mitochondrial shape orchestrates mitochondrial function [18], we asked if p62 localization could affect mitochondrial morphology. Using a variety of cell types, plus or minus p62, we examined mitochondrial structure by MitoTracker Red staining. We found the morphology of mitochondria, irrespective of cell type, became fragmented in the absence of p62 (Fig. 2A). Mitochondrial structure was individually quantitated showing an increase in fragmented morphology in the absence of p62 while normal “tubular-like” mitochondrial structure was seen in WT cells consistently across cell types.
Fig. 2. p62 regulates mitochondrial morphology autonomous of cell type.
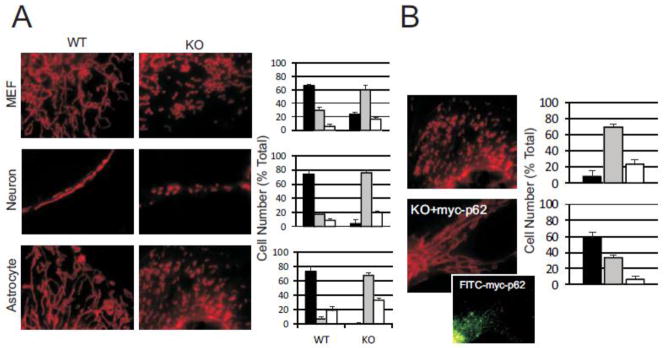
(A) p62−/− cell types were compared to WT for mitochondrial morphology and visualized by MitoTracker Red staining. Morphology was classified as Tubular (black bar), Fragmented (grey bar) or Intermediate (white bar) and cells counted in each class. Mitochondria from a minimum of 100 cells were classified for each cell type. (B) p62−/− MEF cells were transfected with myc-p62 to analyze reversion of mitochondrial phenotype. For transfected cells, only those exhibiting expression of FITC-myc-p62 were used in the analysis. Inset shows FITC-myc-p62 immunofluorescence for represented cell. Mitochondria were classified and counted as in (A). Data for all graphs are mean +/− s.e.m., P< 0.05.
We next reasoned that if removal of p62 results in mitochondrial fragmentation, restoring p62 to p62−/− cells could rescue mitochondrial morphology. p62−/− MEFs were transfected with myc-tagged p62, mitochondria stained with MitoTracker Red and immuno-stained for tagged p62. Only cells showing immunofluorescence for FITC-myc, indicating successful introduction of exogenous p62, were used in the analysis. When myc-p62 was introduced, mitochondrial morphology did revert to a ribbon-like tubulo-reticular network indicative of normal mitochondria. (Fig. 2B). These results suggest that morphological changes in mitochondria may be directly correlated with mitochondrial localization of p62. Fragmentation is directly associated with mitochondrial depolarization and energetics defects [42]. Since p62 localizes to mitochondria, restores normal mitochondrial morphology and possibly spans the mitochondrial membranes [37], we reasoned p62 could be in a position to affect mitochondrial depolarization and energetics.
3.3 p62 is required for normal mitochondrial function
Growing lines of evidence show a positive correlation between mitochondria fragmentation and loss of membrane potential, decreased mitochondrial energetics, and mtDNA damage response [19,24,26,29]. Based on initial observations of atypical mitochondrial morphology in p62−/− cells, we anticipated elevated mitochondrial dysfunction and genomic instability. In order to evaluate if p62−/− mitochondrial morphology correlated with mitochondrial energetics, we examined Δψm in WT and p62−/− cells by staining with JC-1. JC-1 selectively enters potentiometric mitochondria and reversibly changes color from red to green as Δψm decreases. p62−/− cells showed an increase in green fluorescence when examined by flow cytometry (Fig. 3A) indicating a significant portion of the mitochondria displayed decreased membrane potential when compared to WT cells. Defects in Δψm have been shown to negatively impact mitochondrial dynamics and energy production [43], therefore, we sought to examine if p62 might play a role in maintaining functional mitochondrial energetics.
Fig. 3. Abnormal mitochondrial metabolism and increased oxidative stress in p62−/− cells.

(A) Membrane potential was analyzed using JC-1 staining in WT or p62−/− MEF cells. Cells were counted by flow cytometry. (B) Mitochondrial ATP production in WT, p62−/− MEF cells minus/plus exogenous myc-p62. Cells were grown in DMEM (D), Galacotose media (G) or Galactose media plus oligomycin (G/O). (C) WT or p62−/− MEF cells minus/plus exogenous myc-p62 were analyzed for ROS production by detection with H2DCFDA at 485nm at times indicated. (D) Comparison of the oxidative damage of mitochondrial DNA fragments in WT and p62−/− mice. Data for all graphs are mean +/− s.e.m., P< 0.05.
To address p62 effects on energy production, we measured the capacity of mitochondria to synthesize ATP in MEF cells plus or minus p62. Significant impairment in ATP production was observed in p62−/− cells cultured in DMEM media with glucose as the major energy substrate suggesting p62 is required for normal cellular energetics through glycolysis (Fig. 3B). When cells were cultured in glucose free media containing galactose as the energy source to force mitochondrial respiration, ATP levels were likewise depressed in the absence of p62. We did not observe an increase in ATP production when ATP synthase was inhibited with oligomycin seemingly indicating the lack of p62 does not cause reversal of ATP synthase [44, 45]. Moreover, the decrease in ATP production was abrogated when p62 was introduced into a null background showing ATP production can be rescued by p62 restoration. Glucose tolerance tests in aged p62−/− mice have also shown them to be defective in glucose uptake and insulin tolerance [4] indicative of glycolytic defects. Interestingly, overexpression of p62 not only recovered ATP production to WT levels, but also appeared to increase overall ATP levels above control (Fig. 4B). While decreased Δψm of p62−/− MEFs no doubt affects ATP levels in mitochondria, restoration of ATP above WT levels seems to be a clear indication that presence of p62 is integral to mitochondrial energetics in a manner that extends beyond simply impacting membrane potential.
Fig. 4. p62 protects mitochondrial genome integrity.

(A) Quantitative analysis of mtDNA copy in WT and p62−/− mouse brain was analyzed across ages. (B) mtDNA copy number in tissue from WT and p62−/− mice. (C) mtDNA copy number quantitated in WT and p62−/− MEF cells with overexpression of myc-p62. (D) Mitochondrial TFAM import was analyzed by Western blot. Mitochondria were isolated from WT and p62−/− MEFs transfected with myc-p62 by Percol gradient centrifugation and levels of TFAM in the mitochondria analyzed. Resulting blots were analyzed by densitometry and graphed for relative TFAM intensity. Blots shown are representative of 4 different experiments. Data for all graphs are mean +/− s.e.m., P< 0.05.
Changes in Δψm are initiating events during activation of NADPH oxidase and ROS production [46]. We reasoned that decreased Δψm of p62−/− MEFs along with deregulation of OXPHOS would result in an increase in ROS production [47]. Indeed, analysis of ROS from culture media of WT and p62−/− MEFs showed increased levels in p62−/− MEFs which was again abrogated with the addition of p62 (Fig. 3C). mtDNA damage is a hallmark response to ROS-induced oxidative stress [48]. p62 has been shown to protect nuclear DNA from oxidative damage [31]. We therefore hypothesized that p62’s association with mitochondria could also confer protection to mtDNA. To evaluate this possibility, we examined damage to mtDNA in the hippocampus of WT and p62−/− mice. We observed an increase in in vivo oxidative damage from mtDNA fragments from p62−/− mice (Fig. 3D). Similar results have been seen in p62’s role in the Keap1-Nrf2-Nqo1 pathway and mitochondrial aging [49]. Consequently, the observed correlation between elevated ROS levels and increased mtDNA damage in the absence of p62 as well as the fact that p62 can abrogate the effects of mtDNA damage in a null background was consistent with our hypothesis that p62 plays a general role in mediating oxidative stress. Furthermore, ROS acts as a key modulator of mtDNA copy number [50] while oxidative stress promotes mitochondria fission in neurons [13]. Overall, our results indicate a deficiency in p62 could be an underlying cause of increased oxidative stress related damage to mtDNA.
3.4 p62 is integral to maintaining mitochondrial stability and mtDNA biogenesis
Normal mitochondrial dynamics maintain mtDNA copy number while fragmentation results in mtDNA mutations and loss of mtDNA [13,22]. In addition to mtDNA damage, total quantity of mtDNA is reported to regulate energy metabolism [24]. We investigated if loss of p62 affected the physical volume of the mitochondrial genome pool in p62−/− mice. We examined total DNA copy number in mitochondria from brain of age matched WT and p62−/− mice (Fig. 4A). mtDNA copy number accumulated with age, peaking at 12-months in both genotypes.. Increased levels of mtDNA have been observed in both human and mouse brain tissue previously [51; 52] possibly due to a p62 dependent role in mitophagy [16]. However, p62−/− consistently measured below WT levels after 3 months of age. In order to investigate if mtDNA variation was tissue specific, we also examined mtDNA copy number in mouse liver and adipose tissues at 9 months (Fig. 4B). Decreases in mtDNA levels were seen in both tissues indicating loss of mtDNA copy number in p62−/− mice could be universal. mtDNA copy number of p62−/− mice decreased compared to age-matched WT mice which correlates with the appearance of early AD phenotypes [32]. This result is consistent with investigations that propose mtDNA decrease occurs as an early event preceding loss of mitochondria function [27,53,54].
Loss of copy number was further seen in p62−/− MEF cells where mtDNA levels decreased compared with WT cells (Fig. 4C). Of note, when p62 was introduced to p62−/− MEF cells, mtDNA levels returned to a point consistent with WT whereas overexpression of p62 in WT cells resulted in a 3-fold increase in mtDNA. This is significant in that restoration of p62 not only rescued the damaged mitochondrial phenotype of p62−/− MEFS, but overexpression also increased the levels of mtDNA in normal cells. mtDNA levels are correlated with functional energetics [24] and our data demonstrate that the presence of p62 is required for normal mitochondrial energy production (Fig. 3B). Collectively, our results lead to the unequivocal conclusion that p62 plays a central role in mitochondrial biogenesis and mitochondrial energetics. The full magnitude of this observed p62/mitochondria association remains largely unexplored and presents a fertile area for subsequent research.
One aspect of the p62/mitochondria relationship we investigated was related to the observation of a physical association between p62 and mitochondrial membranes. p62 has been shown to be a component of the nuclear pore complex, directly implicating it in protein import [55]. Recently, parkin was reported to protect the mitochondrial genome by association with mitochondrial transcription factor A (TFAM) prior to its import into mitochondria [56,57]. TFAM promotes mtDNA replication/transcription [58], protects mtDNA from damage [59,60] and affects repair of oxidatively damaged mtDNA [59,61]. Moreover, replication of mtDNA is dependent on TFAM import and activated transcription [62]. As levels of mtDNA were restored in p62−/− cells and increased in WT cells overexpressing p62, we reasoned that p62 might play a role in TFAM/mitochondria association and import. Such a relationship might represent one avenue by which p62 contributes to stabilized energetics and protection of the mitochondrial genome. We isolated mitochondria from WT and p62−/− MEF cells and analyzed levels of TFAM protein (Fig. 4D). WT cells showed significantly more mature TFAM localized into the mitochondria than did p62−/− MEFs while mitochondrial protein TOM40 remained relatively unchanged.. Interestingly, this correlates well with the levels of mtDNA observed in Fig. 4C indicating TFAM transcriptional activity is not affected. When myc-p62 constructs were expressed to increase p62 levels, a noticeable increase in TFAM protein was detected in WT mitochondria while restoration of p62 in p62−/− MEFs increased TFAM in the mitochondria to levels approaching WT basal expression. Post-translational modification (maturing) of TFAM occurs following import into mitochondria [63], thus increases in mature TFAM protein in transfected p62−/− MEFs provide clear evidence that mitochondrial import was restored.
A direct correlation between TFAM and mtDNA copy number has been seen in multiple systems [58,59,64]. Moreover, decreased mtDNA levels and respiratory chain function were found in TFAM−/− adipose cells [65]. We demonstrated a decrease in mitochondrial TFAM protein levels along with associated energetic defects and decreased mtDNA copy number in p62−/− MEFs. Expression of p62 in a null background system effectively restores mitochondrial energetics, in many instances approaching WT levels. Our data also indicated that while restoring p62 expression resulted in normal levels of mitochondrial import, overexpression of p62 increased TFAM import into mitochondria resulting in higher levels of total mtDNA. This indicates p62 is integral to maintaining normal mitochondrial function and biogenesis. The exact in vivo import mechanism is yet unknown but the evidence indicates it is multifactorial. Lack of p62 obviously results in decreased Δψm (Fig 3A). Variation in membrane potential is well known to directly affect import dynamics [66] and therefore no doubt accounts for some proportion of the observed p62/TFAM import relationship. But importantly, based on increased levels of TFAM in mitochondria as well as the increase in total mtDNA from p62 overexpressed cells, p62 plays an important, independent role in the functionality of normal mitochondrial dynamics which extends beyond solely altering Δψm.
5. Conclusion
In conclusion, our study not only confirmed that p62 localizes to the mitochondria under non-stressed, physiological conditions but also further defines a critical role for p62 in the normal functioning of mitochondria. We show that p62 is integral to normal mitochondrial dynamics and, by regulation of the mitochondrial transcription factor TFAM, maintains mitochondrial genome stability. These functions appear to be the results of direct interactions of p62 with mitochondria that extend beyond generalized physiological responses.
Highlights.
p62 is localized to the mitochondria where it plays a physiological role.
p62 affects both mitochondrial morphology and function.
p62 is required to maintaininvolved in maintenance of mitochondrial genome stability.
p62 regulates the import of TFAM to mitochondria.
mtDNA biogenesis is affected by p62 beyond regulation provided by Δψm.
Acknowledgments
We thank Paul Cobine for critical reading. This manuscript is dedicated to M.W.W. This work was supported by NIH-2RO1NS033661 (MWW).
Footnotes
We have no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vadlamudi RK, Joung I, Strominger JL, Shin J. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem. 1996;271:20235–20237. doi: 10.1074/jbc.271.34.20235. [DOI] [PubMed] [Google Scholar]
- 2.Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, Kleinert R, Prinz M, Aguzzi A, Denk H. p62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002;160:255–263. doi: 10.1016/S0002-9440(10)64369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moscat J, Diaz-Meco MT, Wooten MW. Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci. 2007;32:95–100. doi: 10.1016/j.tibs.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez A, Durán A, Selloum M, Champy MF, Diez-Guerra FJ, Flores JM, Serrano M, Auwerx J, Diaz-Meco MT, Moscat J. Mature-onset obesity and insulin resistance in in mice deficient in the signaling adapter p62. Cell Metab. 2006;3:211–222. doi: 10.1016/j.cmet.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 5.Wooten MW, Hu X, Babu JR, Seibenhener ML, Geetha T, Paine MG, Wooten MC. Signaling, polyubiquitination, trafficking, and inclusions: sequestosome 1/p62’s role in neurodegenerative disease. J Biomed Biotechnol. 2006;2006:1–12. doi: 10.1155/JBB/2006/62079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 9.Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 10.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 11.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, Tanaka K, Matsuda N. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15:887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta. 2008;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol. 2010;189:671–679. doi: 10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 17.Narendra DP, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1–17. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campello S, Scorrano L. Mitochondrial shape changes: orchestrating cell pathophysiology. EMBO Rep. 2010;11:678–684. doi: 10.1038/embor.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 20.Frank S. Dysregulation of mitochondrial fusion and fission: an emerging concept in neurodegeneration. Acta Neuropathol. 2006;111:93–100. doi: 10.1007/s00401-005-0002-3. [DOI] [PubMed] [Google Scholar]
- 21.Guillery O, Malka F, Frachon P, Milea D, Rojo M, Lombès A. Modulation of mitochondrial morphology by bioenergetics defects in primary human fibroblasts. Neuromuscul Disord. 2008;18:319–330. doi: 10.1016/j.nmd.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 22.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 23.Chen H, Chan DC. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–R176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rocher C, Taanman JW, Pierron D, Faustin B, Benard G, Rossignol R, Malgat M, Pedespan L, Letellier T. Influence of mitochondrial DNA level on cellular energy metabolism: implications for mitochondrial diseases. J Bioenerg Biomembr. 2008;40:59–67. doi: 10.1007/s10863-008-9130-5. [DOI] [PubMed] [Google Scholar]
- 25.Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. 1999;1410:103–123. doi: 10.1016/s0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- 26.Santos JH, Hunakova L, Chen Y, Bortner C, Van Houten B. Cell sorting experiments link persistent mitochondrial DNA damage with loss of mitochondrial membrane potential and apoptotic cell death. J Biol Chem. 2003;278:1728–1734. doi: 10.1074/jbc.M208752200. [DOI] [PubMed] [Google Scholar]
- 27.Acevedo-Torres K, Berríos L, Rosario N, Dufault V, Skatchkov S, Eaton MJ, Torres-Ramos CA, Ayala-Torres S. Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington’s disease. DNA Repair (Amst) 2009;8:126–136. doi: 10.1016/j.dnarep.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hauptmann S, Scherping I, Dröse S, Brandt U, Schulz KL, Jendrach M, Leuner K, Eckert A, Müller WE. Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging. 2009;30:1574–1586. doi: 10.1016/j.neurobiolaging.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 29.Gilkerson RW, Margineantu DH, Capaldi RA, Selker JM. Mitochondrial DNA depletion causes morphological changes in the mitochondrial reticulum of cultured human cells. FEBS Lett. 2000;474:1–4. doi: 10.1016/s0014-5793(00)01527-1. [DOI] [PubMed] [Google Scholar]
- 30.Du Y, Wooten MC, Gearing M, Wooten MW. Age-associated oxidative damage to the p62 promoter: implications for Alzheimer disease. Free Radic Biol Med. 2009;46:492–501. doi: 10.1016/j.freeradbiomed.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du Y, Wooten MC, Wooten MW. Oxidative damage to the promoter region of SQSTM1/p62 is common to neurodegenerative disease. Neurobiol Dis. 2009;35:302–310. doi: 10.1016/j.nbd.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Babu JR, Seibenhener ML, Peng J, Strom AL, Kemppainen R, Cox N, Zhu H, Wooten MC, Diaz-Meco MT, Moscat J, Wooten MW. Genetic inactivation of p62 leads to accumulation of hyperphosphrylated tau and neurodegeneration. J Neurochem. 2008;106:107–120. doi: 10.1111/j.1471-4159.2008.05340.x. [DOI] [PubMed] [Google Scholar]
- 33.Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nature Prot. 2009;4:1582–1590. doi: 10.1038/nprot.2009.151. [DOI] [PubMed] [Google Scholar]
- 34.Liu J, Lillo C, Jonsson PA, Velde CV, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brannstrom T, Gredal O, wong PC, Williams DS, Cleveland DW. Toxicity of Familial ALS-Linked SOD1 Mutants from Selective Recruitment to Spinal Mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 35.Mortiboys H, Johansen KK, Aasly JO, Bandmann O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology. 2010;75:2017–2020. doi: 10.1212/WNL.0b013e3181ff9685. [DOI] [PubMed] [Google Scholar]
- 36.Navarro A, Boveris A. Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Front Aging Neurosci. 2010;2:1–11. doi: 10.3389/fnagi.2010.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee M, Shin J. Triage of oxidation-prone proteins by Sqstm1/p62 within the mitochondria. Biochem Biophys Res Commun. 2011;413:122–127. doi: 10.1016/j.bbrc.2011.08.067. [DOI] [PubMed] [Google Scholar]
- 38.Goetz JG, Nabi IR. Interaction of the smooth endoplasmic reticulum and mitochondria. Biochem Soc Trans. 2006;34:370–373. doi: 10.1042/BST0340370. [DOI] [PubMed] [Google Scholar]
- 39.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zinchuk V, Zinchuk O, Okada T. Quantitative colocalization analysis of multicolor confocal immunofluorescence microscopy images: Pushing pixels to explore biological phenomena. Acta Histochem Cytochem. 2007;40:101–111. doi: 10.1267/ahc.07002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilquin B, Taillebourg E, Cherradi N, Hubstenberger A, Gay O, Merle N, Assard N, Fauvarque MO, Tomohiro S, Kuge O, Baudier J. The AAA+ ATPase ATAD3A controls mitochondrial dynamics at the interface of the inner and outer membranes. Mol Cell Biol. 2010;30:1984–1996. doi: 10.1128/MCB.00007-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, Chang CR, Tsai YS. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol. 2012;32:309–319. doi: 10.1128/MCB.05603-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ward MW, Concannon CG, Whyte J, Walsh CM, Corley B, Prehn JH. The amyloid precursor protein intracellular domain (AICD) disrupts actin dynamics and mitochondrial bioenergetics. J Neurochem. 2010;113:275–284. doi: 10.1111/j.1471-4159.2010.06615.x. [DOI] [PubMed] [Google Scholar]
- 44.Porcelli AM, Angelin A, Ghelli A, Marlani E, Martinuzzi A, Carelli V, Petronelli V, Bernardi P, Rugolo M. Respiratory complex I dysfunction due to mitochondrial DNA mutations shifts the voltage threshold for opening of the permeability transition pore toward resting levels. J Biol Chem. 2009;284:2045–2052. doi: 10.1074/jbc.M807321200. [DOI] [PubMed] [Google Scholar]
- 45.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nature Cell Biol. 2010;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsuzaki I, Chatterjee S, Debolt K, Manevich Y, Zhang Q, Fisher AB. Membrane depolarization and NADPH oxidase activation in aortic endothelium during ischemia reflect altered mechanotransduction. Am J Physiol Heart Circ Physiol. 2005;288:H336–343. doi: 10.1152/ajpheart.00025.2004. [DOI] [PubMed] [Google Scholar]
- 47.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Salazar JJ, Van Houten B. Preferential mitochondrial DNA injury caused by glucose oxidase as a steady generator for hydrogen peroxide in human fibroblasts. Mutat Res. 1997;385:139–149. doi: 10.1016/s0921-8777(97)00047-5. [DOI] [PubMed] [Google Scholar]
- 49.Kwon J, Han E, Bui C-B, Shin W, Lee J, Choi Y-B, Lee A-H, Lee K-H, Park C, Obin MS, Park SK, Seo YJ, Oh GT, Lee H-W, Shin J. Assurance of mitochondrial integrity and mammalian longevity by the p62-Keap1-Nrf2-Nqo1 cascade. EMBO Reports. 2112;13:150–156. doi: 10.1038/embor.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hori AA, Yoshida M, Shibata T, Ling F. Reactive oxygen species regulate DNA copy number in isolated yeast mitochondria by triggering recombination-mediated replication. Nucleic Acids Res. 2009;37:749–761. doi: 10.1093/nar/gkn993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barrientos AJ, Casademont, Cardellach F, Estivell X, Urbano-Marquez A, Nunes V. Reduced steady-state levels of mitochondrial RNA and increased mitochondrial DNA amount in human brain with aging. Mol Brain Res. 1997;52:284–289. doi: 10.1016/s0169-328x(97)00278-7. [DOI] [PubMed] [Google Scholar]
- 52.Masuyama M, Iida R, Takatsuka H, Yasuda T, Matsuki T. Quantitative change in mitochondrial DNA content in various mouse tissues during aging. Biochim Biophys Acta. 2005;1723:302–308. doi: 10.1016/j.bbagen.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 53.Gegg ME, Cooper JM, Schapira AH, Taanman JW. Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS One. 2009;4:e4756. doi: 10.1371/journal.pone.0004756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Graziewicz MA, Day BJ, Copeland WC. The mitochondrial DNA polymerase as a target of oxidative damage. Nucleic Acids Res. 2002;30:2817–2824. doi: 10.1093/nar/gkf392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu T, Guan T, Gerace L. Molecular and functional characterization of the p62 complex, an assembly of nuclear pore complex glycoproteins. J Cell Biol. 1996;134:589–601. doi: 10.1083/jcb.134.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M, Azuma H, Matsumoto T. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet. 2006;15:883–895. doi: 10.1093/hmg/ddl006. [DOI] [PubMed] [Google Scholar]
- 57.Rothfuss O, Fischer H, Hasegawa T, Maisel M, Leitner P, Miesel F, Sharma M, Bornemann A, Berg D, Gasser T, Patenge N. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum Mol Genet. 2009;18:3832–3850. doi: 10.1093/hmg/ddp327. [DOI] [PubMed] [Google Scholar]
- 58.Gensler S, Weber K, Schmitt WE, Pérez-Martos A, Enriquez JA, Montoya J, Wiesner RJ. Mechanism of mammalian mitochondrial DNA replication: import of mitochondrial transcription factor A into isolated mitochondria stimulates 7S DNA synthesis. Nucleic Acids Res. 2001;29:3657–3663. doi: 10.1093/nar/29.17.3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kang D, Kim SH, Hamasaki N. Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion. 2007;7:39–44. doi: 10.1016/j.mito.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 60.Kaufman BA, Durisic N, Mativetsky JM, Costantino S, Hancock MA, Grutter P, Shoubridge EA. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol Biol Cell. 2007;18:3225–3236. doi: 10.1091/mbc.E07-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoshida Y, Izumi H, Ise T, Uramoto H, Torigoe T, Ishiguchi H, Murakami T, Tanabe M, Nakayama Y, Itoh H, Kasai H, Kohno K. Human mitochondrial transcription factor A binds preferentially to oxidatively damaged DNA. Biochem Biophys Res Commun. 2002;295:945–951. doi: 10.1016/s0006-291x(02)00757-x. [DOI] [PubMed] [Google Scholar]
- 62.Gauthier BR, Wiederkehr A, Baquié M, Dai C, Powers AC, Kerr-Conte J, Pattou F, MacDonald RJ, Ferrer J, Wollheim CB. PDX1 deficiency causes mitochondrial dysfunction and defective insulin secretion through TFAM suppression. Cell Metab. 2009;10:110–118. doi: 10.1016/j.cmet.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Larsson N-G, Garman JD, Oldfors A, Barsh GS, Clayton DA. A single mouse gene encodes the mitochondrial transcription factor A and a testis-specific nuclear HMG-box protein. Nature Genetics. 1996;13:296–302. doi: 10.1038/ng0796-296. [DOI] [PubMed] [Google Scholar]
- 64.Kang D, Hamasaki N. Mitochondrial transcription factor A in the maintenance of mitochondrial DNA: overview of its multiple roles. Ann N Y Acad Sci. 2005;1042:101–108. doi: 10.1196/annals.1338.010. [DOI] [PubMed] [Google Scholar]
- 65.Shi X, Burkart A, Nicoloro SM, Czech MP, Straubhaar J, Corvera S. Paradoxical effect of mitochondrial respiratory chain impairment on insulin signaling and glucose transport in adipose cells. J Biol Chem. 2008;283:30658–30667. doi: 10.1074/jbc.M800510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mokranjac D, Neupert W. Protein import into mitochondria. Biochem Soc Trans. 2005;33:1019–1023. doi: 10.1042/BST20051019. [DOI] [PubMed] [Google Scholar]