

Abstract
Hermansky-Pudlak syndrome (HPS) is a group of rare autosomal recessive disorders characterized by oculocutaneous albinism, a bleeding tendency, and sporadic pulmonary fibrosis, granulomatous colitis or infections. Nine HPS-causing genes have been identified in humans. HPS-1 is the most severe subtype with a prevalence of ~1/1800 in northwest Puerto Rico due to a founder mutation in the HPS1 gene. Mutations in HPS genes affect the biogenesis of lysosome-related organelles such as melanosomes in melanocytes and platelet dense granules. Two of these genes (HPS1 and HPS4) encode the HPS1 and HPS4 proteins, which assemble to form a complex known as Biogenesis of Lysosome-related Organelle Complex 3 (BLOC-3). We report the identification of the interacting regions in HPS1 and HPS4 required for the formation of this complex. Two regions in HPS1, spanning amino acids 1–249 and 506–700 are required for binding to HPS4; the middle portion of HPS1 (residues 250–505) is not required for this interaction. Further interaction studies showed that the N-termini of HPS1 and HPS4 interact with each other and that a discrete region of HPS4 (residues 340–528) interacts with both the N- and C-termini of the HPS1 protein. Several missense mutations found in HPS-1 patients did not affect interaction with HPS4, but some mutations involving regions interacting with HPS4 caused instability of HPS1. These observations extend our understanding of BLOC-3 assembly and represent an important first step in the identification of domains responsible for the biogenesis of lysosome-related organelles.
Keywords: HPS, Hermansky-Pudlak Syndrome, oculocutaneous albinism, bleeding, BLOC-3, lysosome-related organelles
1.1 introduction
Hermansky-Pudlak syndrome (HPS) [MIM#203300] is a group of autosomal recessive conditions characterized by hypopigmentation and a bleeding tendency caused by defective biogenesis of lysosome-related organelles such as melanosomes and platelet dense granules [1–4]. Some HPS subtypes develop other complications such as pulmonary fibrosis and/or granulomatous colitis [3]. In humans, nine different genes have been found to cause HPS but in mouse, mutations in at least 16 genes cause HPS-like phenotypes [5–12]. The majority of these genes encode proteins of unknown structure and function. Others encode subunits of well-characterized complexes involved in vesicular trafficking such as AP-3 and HOPS [6]. HPS gene products have been identified as subunits of at least three multi-protein complexes named BLOC (Biogenesis of Lysosome-related Organelles Complex) -1 to 3. The precise functions of BLOC complexes have not been well established, although physical interactions between components of these complexes have been suggested and defined [13]. In particular, BLOC-3 consists of two cytosolic proteins, HPS1 and HPS4 [14–17].
Patients with mutations in either subunit of the heterodimer generally develop fatal lung fibrosis in their fourth to fifth decade of life or earlier in some cases [3]. A frameshift mutation consisting of duplication of a 16 base pairs sequence in exon 15 of the HPS1 gene is the most common mutation in the northwest region of the island of Puerto Rico [5]. The HPS1 gene encodes a 700 amino acids-long protein with a molecular weight of 79.3 kDa [5], whereas HPS4 codes for a 76.9-kDa polypeptide of 708 amino acids. Both proteins exhibit a cytosolic distribution with approximately 10% associated to membranes, but association to a specific cellular organelle has not been established [14–16]. Co-immunoprecipitation of epitope-tagged and endogenous proteins demonstrated a tight interaction between these two proteins. However, this interaction was not evident on yeast two hybrid analysis, which suggested that additional components of BLOC-3 may exist [14–16]. Rab9, a small GTPase that localizes in late endosomes, was recently shown to interact with BLOC-3 [17]. Bioinformatic analyses have suggested that HPS proteins exhibit some homology to yeast proteins, including some that participate in intracellular vesicular trafficking [18, 19]. In particular, HPS4 contains a conserved N-terminal domain of approximately 200 amino acids, which has been termed CHiPS (for CCZ1-HPS4) [18]. In addition, a central region of approximately 200 residues of HPS1 has homology to the C-terminal domain of members of the PAT family (for Perilipin, Adipophilin, and TIP47 (tail-interacting protein of 47 kDa) [18]. In order to elucidate and understand the molecular function of the BLOC-3 complex, it is important to assess the organization and interacting regions critical for complex formation.
In this study, we identified the regions in HPS1 and HPS4 required for the formation of BLOC-3. Point mutations in the interacting regions destabilize the proteins, suggesting that they are important for complex stability. These observations extend our understanding of BLOC-3 assembly and represent an important first step in identifying novel protein domains responsible for the biogenesis of lysosome-related organelles.
1.2 Materials And Methods
1.2.1. Antibodies
The following mouse monoclonal antibodies were used: anti-Myc (9E10), anti-GFP (B-2) and anti-V5 (Santa Cruz Biotechnology), hemagglutinin (HA) tag (HA.11; Covance, Princeton, NJ), anti-Akt, anti-Phospho Akt (Cell Signaling Technology, Beverly, MA), anti-HPS1 (hHPS5, a gift of Dr. Richard Spritz, University of Colorado) and the rabbit polyclonal antibody anti-LAMP2 (Abcam), anti-HPS4 (a gift of Dr. Juan Bonifacino). Donkey alexa-488 anti-mouse IgG and alexa-555 anti rabbit IgG were purchased from Molecular probes (Eugene, OR). Horseradish peroxidase-conjugated anti-mouse and anti-rabbit IgGs were purchased from Amersham Biosciences, Piscataway, NJ.
1.2.2. Cell culture and Transfections
Normal (GM00037H) and Puerto Rican HPS1 16bp duplication mutant fibroblasts (GM14609) were obtained from the Coriell Institute for Medical Research and maintained in MEM Eagle-Earle supplemented with 15% fetal bovine serum (FBS). HeLa and M1 cells (a gift of Dr. Juan Bonifacino, NICHD) were maintained in Dulbecco’s Modified Eagle’s medium (Invitrogen, Carlsbad, CA) with 10% (v/v) heat-inactivated fetal bovine serum (FBS; Invitrogen). For the stably transfected M1 clone 26, expressing Myc3-HPS4, the medium was supplemented with 600μg/ml G-418. All cell cultures were supplemented with 2mM L-glutamine, 100U/ml penicillin, and 100μg/ml streptomycin and grown in a humidified incubator with 5% CO2 at 37°C. Transfections were carried out using the Lipofectamine™ 2000 reagent (Invitrogen) according to the manufacturer’s instructions.
1.2.3. cDNA tissue panel PCR
For tissue-specific expression studies of alternatively spliced RNA isoforms, human multiple tissue cDNA panels (human MTC panels I and II) were purchased from Clontech/BD Biosciences. We designed a primer set for PCR amplification of the regions where the splice variant(s) differed and generated products of different lengths. The forward primer 5′-GGCAACTTCCTGTATGTCCTTCACCTG -3′ was located before exon 5 and the reverse primer 5′-CTGCTATCTGAAGGGCATCC-3′ hybridizes after exon 9.
1.2.4. DNA constructs and Mutations
The pCI-HA3-HPS1 and pCI-Myc3-HPS4 vectors were generated as described [14]. The full length coding sequence of HPS1 was obtained by PCR amplification of cDNA and cloned in frame into the Xho1 and EcoR1 sites of the pEGFP1-C1 vector (BD Biosciences Clontech, Palo Alto, CA). Mutagenesis of expression constructs was performed using the QuikChange Site-Directed Mutagenesis kit (Stratagene) as recommended by the manufacturer. To generate the pCI-Myc3-HPS4340–708 and pCI-Myc3-HPS4528–708 constructs, the desired portions of the HPS4 coding region were amplified by PCR using the pCI-Myc3-HPS4 plasmid as template. PCR products were sub-cloned into the EcoRI and XbaI sites of the pCI-Myc3 vector. All the constructs were sequenced on both strands to confirm their identity and the presence of the desired mutations, if applicable. Sequencing was performed using ABI BigDye Terminator chemistry (Applied Biosystems, Foster City, CA) with detection on an ABI 310 or a 3130xl Genetic Analyzer (Applied Biosystems). Electrophoregrams were analyzed using Sequencher v4.9 software (Gene Codes Corporation, Ann Arbor, MI).
1.2.5. Co-immunoprecipitations and Western Blotting
Transfected cells were rinsed twice with ice-cold phosphate-buffered saline (PBS) and incubated in 400 μL of lysis buffer [50 mM Tris-HCl pH 7.4, 300 mM NaCl, 0.5% w/v Triton X-100, 5mM EDTA, 0.1% BSA,1X protease inhibitor cocktail (Roche, Indianapolis, IN)]. After 30 min of incubation at 4°C, lysates were centrifuged at 16,000 × g for 15 min. The supernatants were then pre-cleared by incubation for 60 min at 4°C with G-Sepharose beads (Amersham Pharmacia Biotech, Piscataway, NJ). The pre-cleared lysates were subsequently incubated overnight at 4°C with G-Sepharose beads and mouse monoclonal antibody against the specific tag. The beads were washed three times with 1 ml ice-cold lysis buffer and once with ice-cold PBS. Bound proteins were eluted by boiling in 30μL Laemmli buffer at 95°C for 5 min. Samples were analyzed by SDS-PAGE and immunoblotting. SDS–PAGE analysis and electroblotting onto nitrocellulose membranes was performed using the NuPAGE® Bis-Tris Gel system (Invitrogen), according to the manufacturer’s instructions. Nitrocellulose membranes were incubated with primary and horseradish peroxidase-labeled secondary antibodies and reactive proteins were detected using ECL Western Blotting Substrate from Pierce (Rockford, IL).
1.2.6. Cell fractionation
To prepare cytosolic and membrane fractions, transfected cells were washed twice in phosphate-buffered saline (PBS), detached by scraping, suspended in buffer A (25mM HEPES pH 7.4, 0.25M sucrose, 1mM EGTA, 0.5 EGTA, 1 mM dithiothreitol, and protease inhibitor cocktail). Cells were mechanically disrupted by successive passages through a 25-gauge needle. Extracts were centrifuged at 800 × g for 10 min, and the resulting post-nuclear supernatants were then centrifuged at 120,000 × g for 45 min at 4°C to yield cytosolic and membrane fractions. Membranes were resuspended in equal volume of buffer A containing 0.1% Triton-X100. Both fractions were analyzed by immunoblotting.
1.2.7. Pulse-chase Assay
The pEGFP-C1-HPS1 and HPS1 mutant constructs were transfected in M1 cells using 1μg of each plasmid. One day after transfection, cells were washed with PBS and translation was inhibited using 100μg/mL cycloheximide and 40μg/mL chloramphenicol in 1mL of media. Samples were then collected at 0, 1, 3, and 6 hours after translation inhibition. Collected cells were lysed using 200μL of lysis buffer. Equal volumes of each sample were analyzed by SDS-PAGE and immunoblotting.
1.2.8. Immunofluorescence Analysis
Transfected M1 cells were washed twice with PBS containing Ca+2/Mg+2, fixed in 4% PFA in PBS, and permeabilized for 10 min with 0.2% (wt/vol) Triton X-100 in PBS. After permeabilization, cells were blocked for 30 min with 0.2% (wt/vol) porcine skin gelatin in PBS and incubated in a humid chamber for 1hr at 37°C with the primary antibody, washed with PBS Ca+2/Mg+2 for 5 min at room temperature, and incubated for 30min at 37°C with Alexa 488-conjugated anti-rabbit IgG secondary antibody. Stained samples were washed with PBS with Ca+2/Mg+2 and mounted on glass slides using VECTASHIELD® (Vector Laboratories, Burlingame, CA). Images were acquired on a Zeiss LSM510 META confocal laser-scanning microscope (Carl Zeiss, Microimaging Inc., Thornwood, NY).
1.2.9. Treatment with PI3 Kinase inhibitor
For each treatment, human fibroblasts were grown in 24-well tissue culture plates until approximately 95% confluency and starved with reduced serum Opti-MEM overnight. After overnight starvation, cells were treated with PI3-Kinase inhibitors (wortmannin, LY294002) for 8 hours. After treatments, cells were either washed with PBS Ca+2/Mg+2 and fixed with 4% PFA to perform immunofluorescence for HPS1 and HPS4 or lysed for immunoblotting purposes.
1.3. Results
1.3.1. An HPS1 isoform encoded by a transcript lacking exon 9 interacts with HPS4
Sequence analysis of the HPS1 cDNA products generated by RT-PCR revealed a previously reported HPS1 transcript variant [20], that lacks exon 9 (HPS1Δex9). We tested the distribution of HPS1Δex9 in different human tissues. PCR amplification using a primer pair spanning the two HPS1 transcripts revealed that the major HPS1 mRNA isoform amplified as a 766-bp fragment, which was highly expressed in all tissues, as expected [21]. The HPS1Δex9 (667-bp fragment) isoform was expressed in tissues in which the major HPS1 isoform was expressed (Figure 1A). Additional bands seen in the RT-PCR analysis reflect amplification of minor splice variants, which are considered to be NMD substrates and were not considered in this study.
Figure 1. Expression and Co-immunoprecipitation of HPS1Δex9.
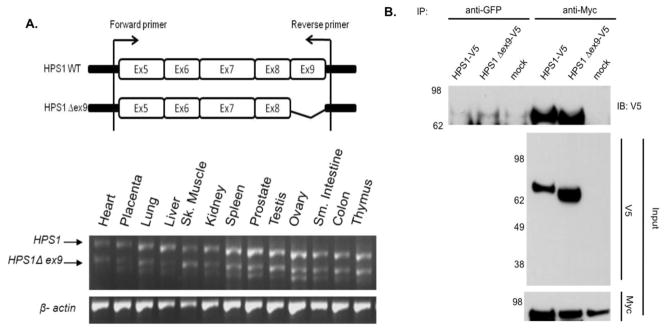
(A) Top. Schematic representation of the HPS1 gene and location of the primers used for PCR analysis to detect HPS1 splice variants. Bottom. A panel of first-strand cDNAs derived from several human tissues (Clontech) was PCR amplified using a primer pair spanning exons 5–9. The two major transcripts were amplified as 766- bp (HPS1) and 667-bp (HPS1Δex9) fragments. β-Actin was utilized as loading control. Additional lower molecular weight bands reflect amplification of minor splice variants. (B) Stably transfected M1 cells expressing Myc3-HPS4 (clone number 26) were transiently transfected with constructs containing either HPS1 full-length or HPS1Δex9. An aliquot of these crude extracts corresponding to 1% of the material available for IP, was analyzed by immunoblotting (IB) using a mouse antibody against the V5 epitope and a mouse monoclonal antibody against the Myc tag. The crude extracts were used in immunoprecipitation (IP) reactions using a mouse monoclonal antibody against the Myc epitope. The immunoprecipitates were analyzed by 4–12% gradient SDS-PAGE followed by immunoblotting using a monoclonal antibody against the V5 epitope. The positions of molecular weight markers are indicated on the left.
To determine whether the HPS1Δex9 protein product interacted with HPS4, constructs coding for V5-tagged HPS1 and HPS1Δex9 proteins were transiently transfected in M1 clone 26 cells expressing Myc3-tagged HPS4. Tagged protein interactions were detected by co-immunoprecipitation assays. HPS4 interacted with the HPS1 full length as well as with HPS1Δex9 (Figure 1B), suggesting that the region encoded by exon 9 (residues 256–289) is not required for the interaction with full-length HPS4. This result sparked our interest in determining which regions in each protein are important for the interaction between HPS1 and HPS4 in BLOC-3.
1.3.2. Mapping of the HPS regions recognized by HPS4
To further define the region in HPS1 required for the interaction with HPS4, we analyzed the predicted secondary structure of the protein (Network Protein Sequence Analysis of the Pôle Bio-informatique Lyonnais). The candidate regions for interacting domains were selected based on their predicted structural content. We generated five HPS1 deletion constructs (Figure 2A), that were GFP tagged and transiently transfected in M1 clone 26 cells expressing Myc3-tagged HPS4. Co-immunoprecipitation analysis revealed an interaction of HPS4 with the N-terminus of HPS1 (residues 1–249) (Figure 2B), suggesting that this region of high α-helical content is critical for interaction with full-length HPS4. Interestingly, the C-terminus of HPS1 (residues 506–700) also showed interaction with HPS4 (Figure 2B). These results are consistent with the presence of two regions in HPS1, located at the N- and C-termini that are independently recognized by HPS4. As far as we have determined, no internal region of HPS1 is required to interact with HPS4. Surprisingly, HPS1250–700 did not show any interaction with HPS4, possibly due to autoinhibition of intermolecular binding by an intramolecular interaction of the C- terminal region of HPS1 with a region towards the N-terminal, as has been shown to occur with GGA1/3 [22] and the kinesin-2 motor KIF17 [23].
Figure 2. Divalent interaction of HPS1 with HPS4.

(A) Top. The consensus secondary structure prediction for HPS1 as determined by the Network Protein Sequence Analysis of the Pôle Bio-informatique Lyonnais. Blue areas represent α-helices, while red areas indicate predicted β-extended strand, and solid purple lines denote random coils. Bottom. GFP-tagged HPS1 constructs used to map the HPS4 binding sites on HPS1 are represented by solid bars with residue numbers indicated on the left. (B) GFP-tagged HPS1 constructs were transiently transfected into M1 clone 26 cells expressing Myc3-tagged HPS4. Coimmunoprecipitation analysis revealed an interaction of HPS4 with the first 249 amino acids of HPS1, suggesting that the N-terminus of HPS1 contains a region required for interaction with full-length HPS4. Interestingly, a construct containing the HPS1 C-terminus (HPS1506-700) also displayed an interaction with HPS4. These results are consistent with HPS4 recognizing two different regions, located at the N-terminus (first 249 amino acids) and the C-terminus (506– 700 amino acids) of HPS1. Co-immunoprecipitation with an irrelevant anti-HA antibody was used as control.
1.3.3. Two separate binding sites in HPS4 are recognized by HPS1
A similar approach was used to identify the regions in HPS4 responsible for the interaction with HPS1. After analyzing the predicted secondary structure of HPS4 (Figure 3A), we generated a truncated HPS4 protein that contained amino acids 1–230, and two additional constructs spanning residues 340–708 and 528–708 (Figure 3A). Myc3-tagged HPS4 constructs were transiently transfected in M1 clone 5 cells expressing HA3-tagged HPS1 (Figure 3B). Co-immunoprecipitation of lysates from transfected cells revealed an interaction of HPS1 with the first 230 amino acids of HPS4, suggesting that the N-terminus of HPS4 plays a role in the interaction with full-length HPS1 (Figure 3B). Interestingly, although the N-terminus of the HPS4 is absent from the HPS4340–708 construct, an interaction with HPS1 was nonetheless evident. No interaction was observed when the HPS4528–708construct was used (Figure 3B). When the partial overlap of constructs HPS4340–708 and HPS4528–708 is considered, we can delineate an area between residues 340 and 528 of HPS4 with relatively low α-helix content as a second region recognized by HPS1. This result suggests that the N-terminal and a central region of HPS4 are important to interact with HPS1.
Figure 3. HPS4 shows a dual interaction with HPS1.

(A) Top. Consensus secondary structure prediction for HPS4 as determined using the Network Protein Sequence Analysis of the Pôle Bio-informatique Lyonnais. Bottom. Three Myc3-tagged truncations of HPS4 are represented by solid bars with residue numbers indicated on the left. (B) Stably transfected M1 clone expressing HA3-PS1 (clone 5) were transiently transfected with constructs coding for either full length Myc3-HPS4 or truncated proteins. Aliquots of the crude extracts corresponding to 1% of the material available for immunoprecipitation (IP) were analyzed by immunoblotting (IB) using an anti-Myc monoclonal antibody. The extracts were then subjected to an IP with mouse monoclonal anti-Myc. The immunoprecipitates were analyzed by 4–12% gradient SDS-PAGE gel followed by immunoblotting using an antibody against HA-coupled to horseradish peroxidase. IP with irrelevant mouse monoclonal antibodies against GFP was performed as control. The positions of molecular weight markers are indicated on the left.
1.3.4. The N-terminus of the HPS1 protein interacts with the N-terminus of HPS4
Bioinformatic analyses suggested the presence of possible domains at the N-terminus of HPS1 and HPS4 that could be important for the stability of their interaction [18, 19]. To assess this possibility, HeLa cells were co-transfected with vectors encoding Myc3-tagged HPS4 truncations, i.e. HPS41–230 in combination with either GFP-tagged HPS11–249 (N-terminus), HPS1250–505 (middle) or HPS1506–700 (C-terminus). Consistent with the results shown in figures 2 and 3, Myc3-tagged HPS41–230 interacted only with the N-terminal region of HPS1, but not with the middle or C- terminal regions of HPS1 (Figure 4A).
Figure 4. Mutual interaction of the N-terminal fragments of HPS1 and HPS4, while the central region of HPS4 shows divalent interaction with the N- and C- termini of HPS1.

To delineate the structural determinants of the HPS1 and HPS4 interactions, HeLa cells were co-transfected with vectors encoding Myc3- tagged HPS41–230(A), HPS4340–708 (B) or HPS4528–708 (C) truncations in combination with GFP-tagged HPS1 truncations representing N-terminal, middle and C-terminal portions of the protein. The Myc3-HPS4-N terminal region interacts with the N-terminus of HPS1, but not with the C-terminus or the middle region of this protein. Myc3-tagged HPS4340–708 interacts with both the N- and the C- termini of HPS1. The C- terminus of HPS4506–708 did not show any interaction with portions of the HPS1 protein.
1.3.5. The central region of HPS4 protein interacts dually with either the N or C- terminus of HPS1
We then analyzed the interactions of the HPS4 central region with HPS1. To this end, we performed double transfections using vectors encoding the Myc3-tagged HPS4 truncations HPS4340–708 and HPS4528–708 in combination with either GFP-tagged HPS11–249, HPS1250–505 or HPS1506–700. Myc3-tagged HPS4340–708 terminal interacted with either the N- or C- terminus of HPS1 but not with its central fragment (Figure 4B). In contrast, no interaction was detected with HPS4528–708, indicating that the C-terminal region of HPS4 does not play a role in complex assembly (Figure 4C). The results in Figure 4C corroborate the results shown in Figure 3B, where the HPS4528–708 protein failed to interact with the HPS1 fragments tested, indicating that the C-terminal of HPS4 does not participate in the interaction with HPS1, although we cannot exclude the possibility of this region participating in complexes with other, unidentified proteins. Taken together, these experiments show that the central region of HPS4 (residues 340–528) interacts dually with either the N- or C- termini of HPS1.
1.3.6. Interacting regions of HPS1 and HPS4 exist as soluble and membrane-associated forms
Full-length HPS1 and HPS4 exhibit a cytosolic distribution with approximately 10% associated to membranes [14]. To test whether this was also the case for the binding regions identified in the HPS1 and HPS4 proteins, M1 cells were transfected with vectors encoding either GFP-tagged HPS11–249, GFP-tagged HPS1506–700, Myc3-tagged HPS41–230 or Myc3-tagged HPS4340–708. Cells were lysed in the absence of detergents and soluble and membrane fractions were isolated by differential centrifugation.
Immunoblot analysis using anti-GFP showed that HPS11–249 localized mostly in the cytosol, while HPS1506–700 was mainly membrane-associated (Figure 5A). In the case of HPS4 fragments, Myc3-tagged HPS41–230 and HPS4340–708 were recovered from both cytosolic and membrane-associated (Figure 5B) fractions. Consistent with the results shown in Figures 5A and B, immunofluorescence microscopic analysis agreed with the distribution found by immunoblot for each HPS1 and HPS4 region analyzed (Figure 5C-F), where the HPS11–249 localized mostly in the cytosol, while HPS1506–700 was mainly membrane-associated. In the case of Myc3-tagged HPS41–230 and HPS4340–708, these two protein fragments share distribution between cytosol and membrane-like structures. These observations suggest that the reported membrane association of full-length HPS1 and HPS4 could be mediated through the HPS1506–700, HPS41–230, and HPS4340–708 binding regions.
Figure 5. HPS1 and HPS4 binding regions exist in soluble and membrane associated forms.

Transfected M1 cells were mechanically disrupted by passing them through a 25-gauge needle in buffer A. The crude lysates were centrifuged at 800 x g and the resulting post-nuclear supernatants were centrifuged further to yield cytosolic (cyt) and membrane (memb) fractions as described (Materials and Methods). (A) The presence of GFP-HPS11–249, HPS1506–700 (upper panel) and (B) Myc3-HPS41–230, HPS4340–708 (upper panel) or Lamp-2 (lower panel, integral membrane control) was assessed by immunoblotting. The positions of molecular weight markers are indicated on the left. Transfected M1 cells were fixed, and GFP tagged proteins were visualized directly (C and D) to detect GFP-HPS11–249, HPS1506–700. Transfected M1 cells were stained with mouse anti-Myc antibody (E and F) to detect Myc3-HPS41–230 and HPS4340–708, followed by Alexa 488- conjugated anti-mouse IgG. Bar, 10μm.
1.3.7. The N-terminus of the HPS1-Puerto Rican mutant protein can interact with HPS4
Following mapping of the regions required for BLOC-3 assembly, we analyzed whether clinically relevant HPS1 truncated proteins exhibit altered interaction with HPS4. To this end, we mimicked the most common HPS1 mutation in the Puerto Rican population, i.e. an insertion of 16 base pairs (c.1472_1487dup16), which causes disruption of the predicted secondary structure of the C-terminal domain of the protein (p.H497QfsX90) and addition of 90 new amino acids due to this frameshift mutation (Figure 6A), while the first 497 amino acids remain intact. We inserted the 16-bp region into the cDNA region corresponding to exon 15 of the HA3-tagged HPS1 full-length construct by site-directed mutagenesis. To test for physical association between the HA-tagged truncated HPS1 (HA3-HPS116bp) and full-length HPS4, epitope-tagged proteins were co-immunoprecipitated under mild, nondenaturing conditions from lysates of transfected M1 clone 26 cells. As shown in Figure 6, Myc3-HPS4 was detected in immunoprecipitates of both HA3-HPS1 and HA3-HPS116bp transfected cells (Figure 6B). Hence, the HPS1 N-terminus is sufficient for the interaction with HPS4. These results suggest that the interactions of the N- and C- termini of HPS1 may be independent from one another. The interaction of the HA3-HPS116bp with HPS4 suggests that truncated forms of HPS1 may retain partial function, which may be of clinical relevance. Since the 16-bp duplication mutation is subject to nonsense-mediated RNA decay (NMD) [24,25], we decided to inhibit NMD by affecting the hSMG-1 kinase activity, a member of the PI3K family, using wortmannin or LY294002. Human fibroblasts from a Puerto Rican HPS-1 patient homozygous for the 16-bp HPS1 duplication were treated with different concentrations of wortmannin for 8h (Figure 6C). Western Blot analysis against endogenous HPS1 showed a faint lower molecular weight band after treatment with the hSMG-1 inhibitor, wortmannin; suggesting the presence of truncated form of HPS1 (Figure 6C). Treatment with wortmannin (data not shown) or LY294002 resulted in detection of HPS1 by immunofluorescence in HPS-1 mutant cells (Figure 6D). Interestingly, the levels of HPS4 were restored in those cells that showed positive staining for the truncated form of HPS1 (similar results were obtained after treatment with wortmannin, data not shown). These results suggest that truncated form of HPS1 can stabilize endogenous HPS4, supporting the notion that the intact N-terminal of the truncated form is able to complex with HPS4 endogenously.
Figure 6. Co-immunoprecipitation of HA-tagged HPS1 and HPS1–16bp.

(A) The consensus secondary structure prediction for the wild type and the predicted mutant protein HPS116bp–ins was determined using the Network Protein Sequence Analysis of the Pôle Bio-informatique Lyonnais. Note that the predicted secondary structures of the N- terminus of wild type and mutant HPS1 proteins are identical up to amino acid 500. The frameshift mutation in the HPS1 gene causes the loss of the last 200 amino acids compared to wild type HPS1 and the addition of 89 new residues due to the resulting frameshift. Blue areas denote α-helices, red areas indicate predicted β-extended strand, and solid purple lines represent random coils. (B) Stably transfected M1 cells expressing Myc3-HPS4 were transiently transfected with either HA3-HPS1 or HA3-HPS116bp-ins. Aliquots of the extracts corresponding to 1% of the material available for IP were analyzed by immunoblotting (IB) using an anti-HA mAb. The extracts were then subjected to an immunoprecipitation (IP) with mouse monoclonal anti-HA as indicated. Irrelevant mouse monoclonal antibodies (GFP) were included as controls. The immunoprecipitates were analyzed by 4–12% SDS-PAGE gradient gels followed by immunoblotting using an antibody against the Myc-tag. The positions of molecular weight markers are indicated on the left. Serum-starved HPS-1 skin fibroblasts were treated with or without 20 μM wortmannin or LY294002 for 8 h in reduced serum medium. (C) Western blot analysis of HPS1 from cell extracts after wortmannin treatment. Proteins (100 μg per lane) extracted with cell lysis buffer were separated by SDS-PAGE (4–12%) in the presence of β-mercaptoethanol and transferred to nitrocellulose membranes. Treatment with different concentrations of wortmannin suppressed phosphorylation of Akt (control of treatment). A faint band of the expected molecular weight of 64 KDa was observed after treatment with 20 μM wortmannin, corresponding to the truncated form of HPS1 (D) Fibroblasts were fixed with 4% PFA, non specific sites were blocked and a dual incubation with mouse monoclonal anti-HPS1 and rabbit polyclonal anti-HPS4 was performed for 30 min in a humid chamber. Staining with secondary Alexa 488-conjugated goat anti- mouse and Alexa 555- conjugated goat anti-rabbit for 30 min. Treatment with LY294002 for 8 hours showed an intracellular expression of truncated form of HPS1 and a restoration of HPS4 protein as compared with untreated defective cells. Bar, 10μm.
1.3.8. Proposed model of interaction between HPS1 and HPS4
Taken together, these co-immunoprecipitation results show that the interactions between HPS1 and HPS4 are mainly stabilized by their N-termini. The N-terminal residues of HPS1 are predicted to form a structured reason composed of β-sheets accompanied by α-helices that resemble a DUF254 domain (a distant homolog of the mu-adaptin longin domain found in clathrin adapter protein (AP) complexes). The DUF254-like domain of HPS1 interacts with the 230 amino acids of the HPS4 N-terminal CHiPS-like domain (Figure 7). In addition, our data indicate that the unstructured middle region of HPS4 (residues 340–528) interacts dually with both the N-terminal (1–249) and C-terminal (506–700) region of HPS1.
Figure 7. Proposed model for the interaction of HPS1 and HPS4.
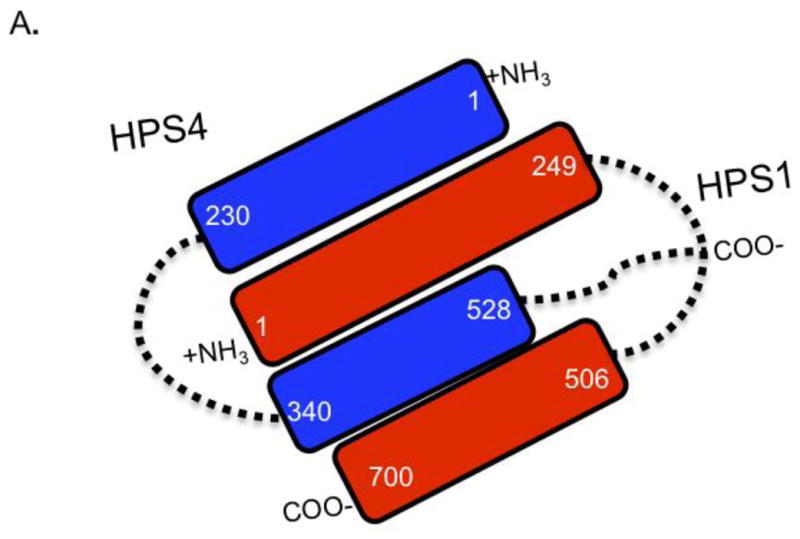
The results of the co-immunoprecipitation analyses (Figures 1–4 and 6) are consistent with a model where the N- and C- termini of HPS1and the N-terminal and middle region of HPS4 are involved in the interactions, leading to BLOC-3 formation. Specifically, N-termini of HPS4 and HPS1 interact with each other, and the central portion of HPS4 interacts with both the N- and the C- termini of HPS1.
1.3.9. Missense mutations within interacting regions of HPS1 alter its stability
The proposed model of interaction between HPS1 and HPS4 was tested by correlating the binding regions mapped in HPS1 with the pathogenicity of HPS. We analyzed a series of missense mutations found in HPS-1 patients, to determine whether these residues play an important role in the assembly with HPS4 and stabilization of the BLOC-3 complex. The substitutions introduced in HPS1 included L239P, G313S, V547L, Y646C and E666K. The GFP-tagged HPS1 constructs containing these missense mutations were transiently transfected in M1 cells stably expressing full length Myc3-tagged HPS4. Coimmunoprecipitation experiments indicate that all mutants interacted with Myc3-HPS4 in a manner similar to that of the HPS1 wild type construct (Figure 8A). Since these missense substitutions did not affect the association with HPS4, we examined the stability of the HPS1 mutants by pulse-chase analysis. Transiently transfected M1 cells were treated with chloroamphenicol and cycloheximide to inhibit translation, and proteins were chased for different times (0, 1, 3, 6 h) at 37°C. Equal amounts of total protein were resolved by SDS-PAGE followed by Western blot analysis. Importantly, most of the HPS1 mutants showed decreased stability compared to the wild type protein (Figure 8B–C), suggesting that alterations within regions of interaction with HPS4 can affect the stability of the protein and the proper formation of BLOC-3. These results support the notion that these missense mutations are pathogenic, with the exception of E666K, which did not show stability changes (Figure 8B–C).
Figure 8. Missense mutations in HPS1 within putative interacting regions cause protein instability.

(A) M1 cells expressing Myc3-HPS4 were transiently transfected with GFP-HPS1 or GFP-HPS1 constructs containing mutations identified in HPS patients. Aliquots of the extracts representing 1% of the material available for IP were analyzed by immunoblotting (IB) using anti-GFP. The extracts were then subjected to an immunoprecipitation (IP) with mouse monoclonal anti-Myc as indicated. Irrelevant IgG were included as controls. The immunoprecipitates were analyzed by 4–12% SDS-PAGE gradients followed by immunoblotting using an antibody against GFP. The positions of molecular weight markers are indicated on the left. (B) Transiently transfected M1 cells were treated with chloroamphenicol and cycloheximide. Proteins were chased for different periods (0, 1, 3, and 6 h) at 37°C. Equivalent amounts of homogenate were resolved by SDS-PAGE followed by western blotting. (C) Bands were quantified using ImageJ 1.44 software and the percentage of each mutant at each chase time was calculated relative to time zero (T0).
1.4. Discussion
1.4.1. Discussion of results
The present work was undertaken to determine the domains involved in the assembly and stabilization of BLOC-3. Our data demonstrated that the product of the HPS1Δex9 transcript variant interacted with HPS4. This interaction indicates that the exon 9 nucleotides encoding residues 256–289 are not required for interaction with HPS4. This isoform is expressed in all tissues examined and its interaction with the major form of HPS4 suggests that interaction between this HPS1 isoform and HPS4 may be physiologically significant. Western blot analysis of endogenous HPS1 yields typical doublet bands, likely corresponding to the major and minor HPS1 isoforms (79.3 KDa and 75.9KDa) that result from alternative splicing of HPS mRNA [21]. HPS1 doublet bands have been detected in HPS4 immunoprecipitates [16] suggesting that both isoforms are able to interact with HPS4. Although additional experiments are needed, we propose that HPS1Δex9 is the HPS1 isoform that corresponds to the minor band that normally appears in Western blot analyses of endogenous HPS1 [20], since the shorter transcript variant of HPS1 has a much lower predicted molecular weight.
To define the binding regions in HPS1 and HPS4 required for the formation of BLOC-3, we first examined the predicted secondary structures of both proteins. They each have well-structured N- and C-termini mainly composed of α-helices and β-strands connected by disordered segments of different lengths. The N-termini of both proteins were predicted to contain potential CHiPS or longin domains, respectively [18,19]. Co-immunoprecipitation analysis showed assembly of the N- terminus of each protein with its full-length partner. Moreover, expression of protein fragments in melanoma cells showed that the N-terminus of HPS4 interacted with the N-terminus of HPS1, indicating that both possible domains are related and required for the formation of BLOC-3. Interestingly, these proteins showed a second region of interaction. The HPS1 C- terminus assembled with HPS4340–708, but not with HPS4528–708. Excluding the overlapping amino acid residues between these two HPS4 constructs, we proposed that the interaction of the HPS1-C-terminus is mediated by the middle region of HPS4 comprised by residues 340–528. Despite recent evidence suggesting that the central region of HPS4 is not involved in the interaction with HPS1 [17], we showed that this region in HPS4, predicted to be relatively unstructured, plays an important role in BLOC-3 formation. Also, the middle region in HPS4 appears to interact with the N-terminus of HPS1. Hence, the poorly conserved and unstructured region in HPS4 appeared to be responsible for dual interactions with both the N- and C- termini of HPS1. This result indicates that both proteins are involved in a series of interdomain interactions, where the HPS1 N- and C-terminal and the middle portions of HPS4 are expected to assume a compact structure.
Our results are generally consistent with previous studies on HPS1•HPS4 interactions [17], except that we did not find any participation of the C-terminus of HPS4 in the interactions required to form BLOC-3, even though the C-terminus of HPS4 is well-organized and a previous study suggested its participation [17]. The discrepancy between our results and other published studies [17] may be due to the different approaches (in vitro solubility/aggregation and proteolysis studies of purified full length proteins expressed in insect cells or truncated proteins expressed in bacteria, versus, co-immunoprecipitations of tagged proteins expressed in human cell lines) used to determine the regions that participate in the interactions. We chose to evaluate the HPS1•HPS4 interactions by co-immunoprecipitation because this approach was successfully used with epitope-tagged proteins to assemble and form BLOC-3 [14,15]. Overexpression of proteins can trigger aggregation and thus raise concerns of false interactions being detected. Our experiments were performed using the soluble portion of the whole cell lysates to avoid artifacts caused by aggregated proteins, which are usually found in the insoluble fraction. In addition, our truncated protein forms demonstrate cytosolic and/or membrane-associated patterns with low degree of aggregation. The endogenous HPS1 and HPS4 protein have been described to be mainly cytosolic with approximately 10% associated to membranes [14–16]. To this end, we are confident that the interactions we are detecting are of physiologic relevance and not artifacts of aggregation events. Other approaches, that should include structural studies, have to be examined in order to confirm our findings.
Our data support the hypothesis that membrane association of HPS1 and HPS4 depends upon specific regions of these proteins. Specifically, the N-terminal portion of HPS1 is mostly cytosolic, with a small fraction associated with membranes; the C-terminus was found mainly in the membrane fraction. The HPS4 fragments at the N- and C-termini (HPS41–230 and HPS4340–708) were found in both the cytosolic and membrane fractions. Previous studies have shown that the HPS1•HPS4 complex is mainly cytosolic, with a small membrane-bound fraction [14], and that endogenous HPS1 associates with tubulovesicular and pre-melanosome structures [21]. The role of HPS1 membrane association and its implication in the biogenesis of lysosome-related organelles remains to be determined.
In vitro assays with the truncated HPS116bp, mimicking the common mutation found in Puerto Rican HPS-1 patients, showed interaction with HPS4, suggesting the possibility to generate BLOC-3 in these patients after creating a truncated form of HPS1. To corroborate the in vitro finding, we used small molecules such as wortmannin and LY24002 to impair the NMD pathway and generate a truncated HPS1 protein in skin fibroblasts derived from an HPS-1 patient homozygous for the 16-bp duplication. We found that after inhibiting NMD, a lower molecular weight band was generated and interestingly, the levels of HPS4 were restored. Interactions of HPS4 with truncated form of HPS1 may be mediated through the N-terminus region of HPS1, since HPS116bp retains an intact N-terminus compared to the wild type. This supports the notion that lacking one of the two binding region in HPS can be compensated partially by the other binding region. Although the BLOC-3-like complex may have decreased stability and we did not test its functionality or other biochemical properties, these results warrant examination of the functionality of truncated forms of HPS1 in patient cells using small molecules or other genetic approaches.
Other missense mutations of HPS-1 patients, located within the regions that interact with HPS4, also retained their ability to interact with HPS4 but, in general, had decreased stability. These mutations presumably disrupt the structure of BLOC-3, leading to degradation, which explains the pathogenesis in patients harboring homozygous missense mutations in HPS1. Patients carrying missense mutations in the binding regions (e.g., L668P) manifest a moderate disease phenotype [26]. It is possible that a mutation in one of the two binding regions of HPS1 may be compensated by the other interacting region, allowing BLOC-3 to form but with decreased stability.
1.4.2. Conclusions
Our experiments demonstrate that the molecular determinants of the HPS1•HPS4 interaction reside in the N- and C-termini of HPS1 and in the N-terminal and middle region of HPS4. These novel protein interaction domains may also function in other proteins. In addition, these findings support the possibility of generating a truncated form of BLOC-3 that could potentially retain partial functionality. Structural and functional studies are needed to better understand the nature of BLOCs and their roles in the pathogenesis of HPS.
Clinically, since HPS1 requires two interacting regions to stabilize HPS4 in BLOC-3 formation, the rest of the protein may be favorably manipulated by molecular approaches to generate a truncated form of HPS1 that retains the required interacting regions. Generation of truncated proteins has been successfully applied in patients, where the truncated protein retains function and restores the missing function [27–29]. HPS1 mutations in regions that are not required for the interaction with HPS4, such as the 16-bp duplication mutation, found 1 in 1,800 Puerto Ricans in the northwest region of the island, may be a good target. Hopefully, generation of a truncated form of HPS1 in patients can partially restore the missing function and may impact the development of the pulmonary fibrosis associated with the lack of this protein in HPS-1 patients. Further studies are needed to determine if this approach may be translated to an effective therapy for this life-threatening complication in HPS-1 and HPS-4.
Highlights.
An HPS1 isoform encoded by a transcript lacking exon 9 interacts with HPS4
The central region of HPS4 protein interacts dually, with either the N- or C- terminus of HPS1
Interacting regions of HPS1 and HPS4 exist as soluble and membrane-associated forms
Missense mutations within interacting regions of HPS1 alter its stability
Membrane association of HPS1 and HPS4 depends upon specific regions of these proteins
Acknowledgments
We thank Drs. J. S. Bonifacino, R. Mattera, G. A. Mardones, P. V. Burgos and R.A. Spritz for kind gifts of reagents and/or critical reading of the manuscript. This work was partially supported by the NIH-IRTA Program and grant MBRS RISE R25GM068138. Infrastructure support was provided in part by grants from the National Center for Research Resources (2G12RR003051) and the National Institute on Minority Health and Health Disparities (8G12MD007600). Funds to support this project were also provided by the UPR School of Medicine Associate Deanship of Biomedical Science.
Abbreviations used
- BLOC
biogenesis of lysosome-related organelles
- DUF
domain of unknown function
- DMEM
Dulbecco’s modified Eagle’s medium
- EGFP
enhanced green fluorescent protein
- FBS
fetal bovine serum
- HA
hemagglutinin
- HPS
Hermansky-Pudlak syndrome
- IgG
immunoglobulin G
- LAMP-2
lysosome-associated membrane protein 2
- NaCl
sodium chloride
- PAT
perilipin adipophilin, TIP47
- RT-PCR
reverse transcriptase-polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hermansky F, Pudlak P. Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: report of two cases with histochemical studies. Blood. 1959;14:162–169. [PubMed] [Google Scholar]
- 2.Schinella RA, Greco MA, Cobert BL, Denmark LW, Cox RP. Hermansky-Pudlak Syndrome with granulomatous colitis. Ann Intern Med. 1980;9:220–23. doi: 10.7326/0003-4819-92-1-20. [DOI] [PubMed] [Google Scholar]
- 3.Gahl WA, Brantly M, Kaiser-Kupfer MI, Iwata F, Hazelwood S, Shotelersuk V, Duffy LF, Kuehl EM, Troendle J, Bernardini I. Genetic defect and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky- Pudlak syndrome) N Engl J Med. 1998;338:1258–1264. doi: 10.1056/NEJM199804303381803. [DOI] [PubMed] [Google Scholar]
- 4.Brantly M, Avila NA, Shotelersuk V, Lucero C, Huizing M, Gahl WA. Pulmonary function and high-resolution CT finding in patients with an inherited form of pulmonary fibrosis, Hermansky-Pudlak syndrome, due to mutations in HPS-1. Chest. 2000;117:129–136. doi: 10.1378/chest.117.1.129. [DOI] [PubMed] [Google Scholar]
- 5.Oh J, Bailin T, Fukai K, Feng GH, Ho L, Mao JI, Frenk E, Tamura N, Spritz RA. Positional cloning of a gene for Hermansky-Pudlak syndrome, a disorder of cytoplasmic organelles. Nat Genet. 1996;14:300–306. doi: 10.1038/ng1196-300. [DOI] [PubMed] [Google Scholar]
- 6.Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the b3A subunit of the AP-3 adaptor. Mol Cell. 1999;3:11–21. doi: 10.1016/s1097-2765(00)80170-7. [DOI] [PubMed] [Google Scholar]
- 7.Anikster Y, Huizing M, White J, Shevchenko YO, Fitzpatrick DL, Touchman JW, Compton JG, Bale SJ, Swank RT, Gahl WA, Toro JR. Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat Genet. 2001;28:376–380. doi: 10.1038/ng576. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki T, Li W, Zhang Q, Karim A, Novak EK, Sviderskaya EV, Hill SP, Bennett DC, Levin AV, Nieuwenhuis HK, Fong CT, Castellan C, Miterski B, Swank RT, Spritz RA. Hermansky-Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light-ear gene. Nat Genet. 2002;30:321–324. doi: 10.1038/ng835. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Q, Zhao B, Li W, Oiso N, Novak EK, Rusiniak ME, Gautam R, Chintala S, O’Brien EP, Zhang Y, Roe BA, Elliott RW, Eicher EM, Liang P, Kratz C, Legius E, Spritz RA, O’Sullivan TN, Copeland NG, Jenkins NA. Swank RT. Ru2 and Ru encode mouse orthologs of the genes mutated in human Hermansky-Pudlak syndrome types 5 and 6. Nat Genet. 2003;33:145–153. doi: 10.1038/ng1087. [DOI] [PubMed] [Google Scholar]
- 10.Li W, Zhang Q, Oiso N, Novak EK, Gautam R, O’Brien EP, Tinsley CL, Blake DJ, Spritz RA, Copeland NG, Jenkins NA, Amato D, Roe BA, Starcevic M, Dell’Angelica EC, Elliott RW, Mishra V, Kingsmore SF, Paylor RE, Swank RT. Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1) Nat Genet. 2003;35:84–89. doi: 10.1038/ng1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan NV, Pasha S, Johnson CA, Ainsworth JR, Eady RA, Dawood B, McKeown C, Trembath RC, Wilde J, Watson SP, Maher ER. A germline mutation in BLOC1S3/reduced pigmentation causes a novel variant of Hermansky-Pudlak syndrome (HPS8) Am J Hum Genet. 2006;78:160–166. doi: 10.1086/499338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cullinane AR, Curry JA, Carmona-Rivera C, Summers CG, Ciccone C, Dorward H, Hess RA, White JG, Adams D, Huizing M, Gahl WA. A BLOC-1 Mutation Screen Reveals PLDN (Pallidin) is Mutated in Hermansky- Pudlak Syndrome Type 9 (HPS-9) Am J Hum Genet. 2011;88:778–787. doi: 10.1016/j.ajhg.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Di Pietro SM, Falcón-Pérez JM, Dell’Angelica EC. Characterization of BLOC-2, a complex containing the Hermansky-Pudlak syndrome proteins HPS3, HPS5 and HPS6. Traffic. 2004;5:276–283. doi: 10.1111/j.1600-0854.2004.0171.x. [DOI] [PubMed] [Google Scholar]
- 14.Martina JA, Moriyama K, Bonifacino JS. BLOC-3, a protein complex containing the Hermansky-Pudlak syndrome gene products HPS1 and HPS4. J Biol Chem. 2003;278:29376–29384. doi: 10.1074/jbc.M301294200. [DOI] [PubMed] [Google Scholar]
- 15.Nazarian R, Falcón-Pérez JM, Dell’Angelica EC. Biogenesis of lysosome-related organelles complex 3 (BLOC-3): a complex containing the Hermansky-Pudlak syndrome (HPS) protein HPS1 and HPS4. Proc Natl Acad Sci USA. 2003;100:8770–8775. doi: 10.1073/pnas.1532040100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiang PW, Oiso N, Gautam R, Suzuki T, Swank RT, Spritz RA. The Hermanky-Pudlak syndrome 1 (HPS1) and HPS4 proteins are components of two complexes, BLOC-3 and BLOC-4, involved in the biogenesis of lysosomerelated organelles. J Biol Chem. 2003;278:20332–20337. doi: 10.1074/jbc.M300090200. [DOI] [PubMed] [Google Scholar]
- 17.Kloer DP, Rojas R, Ivan V, Moriyama K, van Vlijmen T, Murthy N, Ghirlando R, van der Sluijs P, Hurley JH, Bonifacino JS. Assembly of the biogenesis of lysosome-related organelles complex-3 (BLOC-3) and its interaction with Rab9. J Biol Chem. 2010;285:7794–7804. doi: 10.1074/jbc.M109.069088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoffman-Sommer M, Grynberg M, Kucharczyk R, Rytka J. The CHiPS Domain-ancient traces for the Hermansky-Pudlak syndrome. Traffic. 2005;6:534–538. doi: 10.1111/j.1600-0854.2005.00301.x. [DOI] [PubMed] [Google Scholar]
- 19.Kinch LN, Grishin NV. Longin-like folds identified in CHiPS and DUF254 proteins: vesicle trafficking complexes conserved in eukaryotic evolution. Protein Sci. 2006;15:2669–2674. doi: 10.1110/ps.062419006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bailin T, Oh J, Feng GH, Fukai K, Spritz RA. Organization and nucleotide sequence of the human Hermansky-Pudlak syndrome (HPS) gene. J Invest Dermatol. 1997;108:923–927. doi: 10.1111/1523-1747.ep12294634. [DOI] [PubMed] [Google Scholar]
- 21.Oh J, Liu ZX, Feng GH, Raposo G, Spritz RA. The Hermansky- Pudlak syndrome (HPS) protein is part of a high molecular weight complex involved in biogenesis of early melanosomes. Hum Mol Genet. 2000;9:375–378. doi: 10.1093/hmg/9.3.375. [DOI] [PubMed] [Google Scholar]
- 22.Doray B, Bruns K, Ghosh P, Kornfeld SA. Autoinhibition of the ligand-binding site of GGA1/3 VHS domains by an internal acidic cluster-dileucine motif. Proc Natl Acad Sci USA. 2002:8072–8077. doi: 10.1073/pnas.082235699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammond JW, Blasius TL, Soppina V, Cai D, Verhey KJ. Autoinhibition of the kinesin-2 motor KIF17 via dual intramolecular mechanisms. J Cell Biol. 2010;189:1013–1025. doi: 10.1083/jcb.201001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hazelwood S, Shotelersuk V, Wildenberg SC, Chen D, Iwata F, Kaiser-Kupfer MI, White JG, King RA, Gahl WA. Evidence for locus heterogeneity in Puerto Ricans with Hermansky–Pudlak syndrome. Am J Hum Genet. 1997;61:1088–1094. doi: 10.1086/301611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shotelersuk V, Hazelwood S, Larson D, Iwata F, Kaiser-Kupfer MI, Kuehl E, Bernardini I, Gahl WA. Three new mutations in a gene causing Hermansky-Pudlak syndrome: clinical correlations. Mol Genet Metab. 1998;2:99–107. doi: 10.1006/mgme.1998.2679. [DOI] [PubMed] [Google Scholar]
- 26.Ito S, Suzuki T, Inagaki K, Suzuki N, Takamori K, Yamada T, Nakazawa M, Hatano M, Takiwaki H, Kakuta Y, Spritz RA, Tomita Y. High frequency of Hermansky-Pudlak syndrome type 1 (HPS1) among Japanese albinism patients and functional analysis of HPS1 mutant protein. J Invest Dermatol. 2005;125:715–720. doi: 10.1111/j.0022-202X.2005.23884.x. [DOI] [PubMed] [Google Scholar]
- 27.van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, den Dunnen JT, Koop K, van der Kooi AJ, Goemans NM, de Kimpe SJ, Ekhart PF, Venneker EH, Platenburg GJ, Verschuuren JJ, van Ommen GJ. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357:2677–2686. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 28.Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C, Guglieri M, Ashton E, Abbs S, Nihoyannopoulos P, Garralda ME, Rutherford M, McCulley C, Popplewell L, Graham IR, Dickson G, Wood MJ, Wells DJ, Wilton SD, Kole R, Straub V, Bushby K, Sewry C, Morgan JE, Muntoni F. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, doseescalation, proof-of-concept study. Lancet Neurol. 2009;8:918–928. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, Dickson G, Wood MJ, Wilton SD, Straub V, Kole R, Shrewsbury SB, Sewry C, Morgan JE, Bushby K, Muntoni F. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]