

Abstract
Background
Human pluripotent stem cells (hPSCs) hold great promise for treating ischemic heart disease. However, current protocols for differentiating hPSCs either result in low yields or require expensive cytokines.
Methods
Here we developed a novel two dimensional (2D) stepwise differentiation system that generates a high yield of cardiomyocytes (CMs) from hPSCs without using special cytokines. Initially, undifferentiated hPSCs were transferred onto Matrigel-coated plates without forming embryoid bodies (EBs) for a few days and were cultured in bFGF-depleted human embryonic stem cells (hESCs) medium. When linear cell aggregation appeared in the margins of the hPSC colonies, the medium was changed to DMEM supplemented with 10% fetal bovine serum (FBS). Thereafter when cell clusters became visible, the medium was changed to DMEM with 20% FBS.
Results and Conclusions
At about two weeks of culture, contracting clusters began to appear and the number of contracting clusters continuously increased, reaching approximately 70% of all clusters. These clusters were dissociated by two-step enzyme treatment to monolayered CMs, of which ~90% showed CM phenotypes confirmed by an α –myosin heavy chain reporter system. Electrophysiologic studies demonstrated that the hPSC-derived CMs showed three major CM action potential types with 61 to 78% having a ventricular-CM phenotype. This differentiation system showed a clear spatiotemporal role of the surrounding endodermal cells for differentiation of mesodermal cell clusters into CMs. In conclusion, this system provides a novel platform to generate CMs from hPSCs at high yield without using cytokines and to study the development of hPSCs into CMs.
Keywords: Human embryonic stem cells, Human induced pluripotent stem cells, Cardiomyocytes, Directed differentiation
Introduction
Heart failure is the leading cause of death worldwide. Current therapies including surgical and pharmacological interventions are only capable of delaying the progression of this devastating disease [1]. Since human CMs have very little or no regenerative capacity, the damage to CMs resulting from various heart diseases is irreversible and frequently leads to heart failure [2]. Recent discovery of pluripotent stem cells (PSCs) such as embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) has allowed the use of PSCs for cell therapy [3]. hESCs proliferate unlimitedly and differentiate into cells of all three germ layers [4]. hESC-derived CMs (hESC-CMs) hold similar structural and functional properties to native CMs and showed a potential to integrate into the recipient heart when transplanted in vivo [5–8]. Thus, hESCs can be an appropriate source for cardiac cell-based therapy. However, use of hESCs for therapy has been limited due to ethical controversy. Recent discovery of hiPSCs opened a new avenue for using similar PSCs while avoiding ethical controversy and immunological mismatches associated with hESCs. hiPSCs have been generated from various human somatic cells by introducing pluripotency factors through viral and non-viral delivery techniques, and possess almost identical cell biologic characteristics to hESCs [9–13]. Several independent groups have successfully generated CMs from human PSCs [14–19]. These differentiated CMs displayed many of the properties of primary CMs including the appearance of spontaneous contraction, expression of CM-specific genes and proteins, and electrophysiological characteristics including the expected responses to known pharmacological agents [20–22]. The most common method to induce CM differentiation is EB-mediated differentiation. EBs are round multi-cellular three dimensional aggregates which are formed via suspension culture and consist of cells representing all three germ layers [23, 24]. Typically, a part of the aggregated EBs mature into spontaneously contracting CM-like cells during culture [16, 18]. While the results of EB-mediated CM differentiation have been fairly successful, the method has several limitations. Conventional methods to form EBs yield significant variations in the size and cell number of EBs, and human EBs (hEBs) have different differentiation capabilities dependent on the size [25]. hEBs do not induce specific distribution patterns of three germ layers, and their descendent lineages are heterogeneous, resulting in variations in cardiomyogenic differentiation [23, 24]. Moreover, EBs are often fused together to form large aggregates causing extensive cell death [23]. Finally, the process for forming EBs is labor-intensive and therefore is considered an obstacle for mass production for therapeutic use.
Another approach for inducing CM differentiation is to co-culture PSCs with a mouse visceral endoderm-like cell line, END-2 [6]. It has been demonstrated that direct cell-cell interactions or secreted factors from the END-2 cells are capable of stimulating CM differentiation in vitro [26]. Although this system is reliable for generating CMs, it results in a relatively heterogeneous and undefined mixture of other cell types, which may require another step for isolating a pure population of CMs [14]. Recently, a high-density monolayer model has been introduced for CM differentiation [15, 17]. In this protocol, PSCs are cultured in a feeder-free system for cell expansion and undergo cardiomyogenic differentiation by the addition of serum-free medium supplemented with several developmental-stage-specific cytokines such as BMP4, Activin A and VEGF [15, 17]. However, this system requires expensive cytokines and yielded ~30% CMs. Common problems for the above methods are the difficulties in enrichment of CMs, as these systems result in heterogeneous cell mixtures. To purify CMs, Percoll density gradient or antibody-based cell isolation for KDR have been developed; however, the yield was variable and KDR+ cell sorting resulted in cardiac progenitor cells but not pure CMs [3, 15, 27].
In the present study, we developed a novel 2D, directed differentiation system which can generate a high yield of differentiated CMs without using cytokines. The identity of differentiated CMs was verified by immunocytochemistry, gene expression studies, electrophysiology, and a CM-specific viral reporter system. Of note, we discovered the significant contribution of endodermal lineage cells to the differentiation and maturation of CMs as seen in developmental cardiomyogenesis. This differentiation system can serve as an important platform to investigate cardiomyogenesis.
1. Materials and methods
The authors of this manuscript have certified that they comply with the Principles of Ethical Publishing in the International Journal of Cardiology [28].
1.1. hPSC culture
hiPSC lines were generated by the lentivirally-mediated four transcription factors (OCT4, SOX2, c-MYC and KLF4) as previously described [29]. In this study, we used a hiPSC line derived from human dermal fibroblasts (BJ1) and a hESC line H1 [4, 29]. Briefly, undifferentiated hPSCs were grown on mitotically inactivated STO cells (ATCC, Manassas, VA) in DMEM/F12 (50:50; Gibco BRL, Gaithersberg, MD) supplemented with 20% (vol/vol) serum replacement (Gibco) and basic hESC medium, including 1 mM L-glutamine (Gibco), 1% nonessential amino acids (Gibco), 100 mM mercaptoethanol (Gibco), and 4 ng/ml basic fibroblast growth factor (bFGF; Sigma Aldrich Inc., St. Louis, MO) [30]. The medium was changed every 24 hours, and hPSCs were transferred to new feeder cells every 5 to 7 days using dissecting pipettes.
1.2. Differentiation of hPSCs into CMs using a 2D system
For hPSC-CMs differentiation by the 2D system, cultured hPSCs were plated at 3 × 105 cells per well on Matrigel (BD Biosciences, Bedford, MA)-coated 6-well plates (Nunc, Roskilde, Denmark) without feeder cells as previously described [31]. Attached hPSCs were expanded in hESC media for 4 days (Stage 1), and then cultured in hESC media without bFGF for three days (Stage 2). Next, the aggregated cells were transferred into DMEM containing 10% FBS (HyClone, Logan, UT) and cultured for 7 days to form clusters (Stage 3). Finally, FBS was increased to 20% and the clusters were more clearly demarked, with some clusters showing contraction within 7 days (Stage 4). All culture medium was changed every 24 hours.
1.3. Isolation of CMs using two step enzyme treatments
To isolate purified CMs from contracting clusters at stage 4, we used a two-step enzyme treatment. First, peripheral cells surrounding contracting clusters were removed by 0.05% trypsin-EDTA treatment (Invitrogen, Grand Island, NY). The remaining contracting clusters were detached by 0.25% trypsin-EDTA (Fig. 1C). The isolated contracting clusters were replated onto 0.1% gelatin-coated plates and cultured with DMEM containing 2% FBS [18]. Under these conditions, the contracting clusters formed a monolayer-like structure (Fig. 1E, left panel). We further digested these compact clusters by incubation with 0.25% trypsin-EDTA and made true monolayered cell sheets by culturing the cells on 0.1% gelatin (Sigma)-coated plates. Single cells and small clusters composed of less than 10 cells continued to beat even after digestion (supplemental online Movie 4).
Fig. 1.

The differentiation and purification of contracting CMs from hPSCs in a 2D-directed differentiation system. (A) Overall strategy to differentiate hPSCs into CMs and representative images in each stage. (B) Contracting clusters at stage 4 before treatment with 0.05% trypsin-EDTA. (C) Cluster morphology after the enzyme treatment. (D) Isolated contracting clusters after 0.25% trypsin-EDTA treatment. Phase contrast (left panel) and fluorescent images (center and right panels) showing expression of cTnT and α-MHC. Bar 50 μm. (E) Monolayered cells dissociated from contracting clusters on 0.1% gelatin-coated plates maintain expression of α-MHC as shown by immunocytochemistry. Bar 100 μm. (F) Cells dissociated from contracting clusters after 0.25% trypsin-EDTA treatment and plated on 0.1% gelatin-coated plates still beat and expressed various mature CM proteins. (G) Percentages of cells expressing CM proteins among monolayered cells after digestion measured by immunocytochemistry.
1.4. Real-time RT-PCR (qRT-PCR)
To analyze gene expression patterns at each stage (Fig. 1A, cell populations indicated by white arrows), we performed real-time RT-PCR (qRT-PCR) as described previously [32]. Total RNA was isolated using TRIzol Reagent (Molecular Research Center, OH) according to the manufacturer’s instructions. Two μg of RNA was used for cDNA synthesis with SuperScript II reverse transcriptase according to the manufacturer’s instructions (Invitrogen). Real time PCR was performed with SYBER Green PCR master mix using 7500 Fast RT-PCR System. GAPDH cDNA was used as an internal control. Primer sequences are listed in Supplemental online Table 1.
1.5. Construction of α-MHC viral reporter system
We utilized lentiviral vectors (courtesy of Dr. Miyoshi) as backbones to construct a human α-myosin heavy chain (MHC) viral reporter system. (http://www.brc.riken.go.jp/lab/cfm/Subteam_for_Manipulation_of_Cell_Fate/Plasmid_List.html). CSII-EF-Venus was incubated with AgeI to delete its EF-1 promoter. The digested vector was self-ligated with T4 DNA ligase. CSII-ΔEF-Venus was double-digested by BamHI and XbaI, then ligated with the fragment of IRES2-BSD containing the same double-digestion sites from CSII-CMV-MCS-IRES2-BSD. To construct CSII-ΔEF-GFP-IRES2-BSD, eGFP was amplified by PCR using the following primers:
5′ ATATATGCGGCCGCAAGGATCCCCGGGTACCGGTCGCCACC, 5′ ATATATAGATCTTTACTTGTACAGCTCGTCCATG,
with pEGFP-N1 as a template, and were double-digested by NotI and BglII. The eGFP fragment was pasted into the sites of NotI and BamHI in CSII-ΔEF- IRES2-BSD. The fragment of human α-MHC promoter (3068 bp) was amplified by PCR using the following primers
5′ ATATATGCGGCCGCCCAGACAGAGCTCCCTCAAACCA, 5′ ATATATGGATCCTCCTCAAAGCTCCAGTTCCTTTAT,
with human genomic DNA as a template. These synthetic DNAs were double-digested by NotI and BamHI and ligated with NotI/BamHI-digested CSII-ΔEF-GFP-IRES2-BSD for the construction of α-MHC-GFP-IRES2-BSD. Lentiviral particles were produced in 293T cells using an α-MHC reporter with packaging plasmids pMDL-gp-RRE and pCMV-VSV-G-RSV-Rev. The sequences of the constructs were verified by DNA sequencing.
1.6. Immunocytochemistry
Immunocytochemistry was performed as described previously [33]. Briefly, cells were fixed with 4% paraformaldehyde (PFA) for 20 minutes and permeabilized with 0.1% Triton X-100 in phosphate buffered saline (PBS, Sigma) for 5 minutes. After treatment with 5% normal goat serum for 30 minutes, the cells were incubated for 12 hours at 4°C with antibodies against the following proteins: a pluripotency marker OCT4 (Santa Cruz Biotechnology Inc., Santa Cruz, CA), mesodermal lineage markers Brachyury (Abcam Inc., Cambridge, MA) and HAND1 (Santa Cruz Biotechnology), cardiac-lineage markers NKX2–5, cardiac troponin T (cTnT), α-MHC (Abcam), connexin 43, GATA4 (BD Bioscience), MEF2C (Abgent Inc, San Diego, CA), β-MHC (Millipore, Billerica, MA), MLC2a, and MLC2v (Synaptic Systems, Goettingen, Germany), and endodermal lineage markers FOXA2 (Abcam) and AFP (Abcam). Cells were washed three times with PBS and then incubated with rhodamine or FITC-conjugated secondary antibodies (Molecular Probes Inc., Eugene, OR) for 1 hour. DAPI counterstaining was performed. All images were analyzed with an LSM 510 META confocal microscope (Carl Zeiss Inc., Oberkochen, Germany).
1.7. Electrophysiologic study
The intracellular AP was recorded as described previously with slight modification [5]. The beating cells isolated from contracting clusters by two step enzyme treatment at stage 4 were transferred and cultured on 0.1% gelatin-coated glass bottom microwell dishes for 5 to 7 days. For intracellular recording, the 35-mm dishes were mounted on an inverted microscope (Olympus IX71, Japan) and heated by a heating/cooling bath temperature controller (DTC-200, Dagan Corporation, Minneapolis, MN). The contracting clusters were perfused with Tyrode’s solution containing (mmol/L) 140 NaCl, 5.4 KC1, 1 MgCl2, 10 HEPES, 10 glucose, 1.8 CaCl2, pH 7.4 with NAOH 37°C. Glass microelectrodes were fabricated from borosilicate glass (PG52151-4, World Precision Instruments, Inc., Sarasota, FL) and pulled on a P-87 Flaming/Brown puller (Sutter Instrument Company, Novato, CA). The tip resistance of the microelectrode was 40–80 M when filled with a 3 mol/L KCl solution. Intracellular recordings of membrane potential were performed using an EPC 7 amplifier (List Medical, Darmstadt, Germany) in current clamp mode at 37 ± 0.5°C. The junction potential between the microelectrode solution and the bath solution was adjusted to zero and the microelectrodes’ capacitance was compensated. Spontaneously contracting cell clusters were impaled with the sharp microelectrodes and the spontaneous APs were filtered at 10 kHz and digitized on a computer at 10 kHz. APs were analyzed using Origin 6.0 software (Microcal Inc, Northampton, MA).
1.8. Induction of myocardial infarction and cell transplantation
All protocols for animal experiments were approved by the Institutional Animal Care and Use Committee of Emory University and conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Myocardial infarction (MI) and cell transplantation was performed in athymic male nude rats (Rh-rnu/rnu, 200–250g, Harlan, IN) as previously described by our group (n = 7) [33]. Briefly, MI was induced by ligation of the left coronary artery. Immediately after ligation, the enriched hPSC-CMs were injected into the border zone surrounding the infarcted region (1×106 cells in 100 μl PBS). Before cell transplantation, the cells were labeled with a red fluorescent dye, CM-DiI (Molecular Probes, NY) for the detection of injected cells in histologic sections.
1. 9. Histological analysis
Rats were euthanized after 7 weeks and the heart tissues were retrieved. Specimens were embedded in OCT compound (TISSUE-TEK® 4583, Sakura Finetek USA Inc., CA), frozen, and cut into 8–12 μ thick sections at −20°C. For examining fibrosis and tumor formation, specimens were stained with Masson’s trichrome. For examining engraftment, the tissue sections were stained with a primary antibody against TnT (Abcam Inc., Cambridge, MA) or Connexin 43 (BD Biosciences, Bedford, MA) followed by FITC-conjugated secondary antibodies (Molecular Probes). All images of were acquired using a confocal microscope (Carl Zeiss Inc., Oberkochen, Germany).
1.10. Statistical analysis
All experiments were performed at least 3 times. Quantitative data are expressed as the mean ± standard derivation (SD). Student’s paired t-test or one-way ANOVA was performed to analyze statistical significance. Prespecified comparisons between groups were made where appropriate with post hoc testing using Tukey’s method (SPSS, version 17). A P value< 0.05 was considered statistically significant.
2. Results
2.1. Cardiomyogenic differentiation of hPSCs by a FBS-induced 2D-directed differentiation system
In this study, we have developed a new FBS-induced 2D-directed CM differentiation system to obtain highly purified CMs from hPSCs. Undifferentiated hPSCs cultured on STO feeder cells (H1 or BJ1, supplemental online Fig. 1) were transferred to Matrigel-coated 6 well plates (Fig. 1A, Day -5) and cultured in hESC medium for 4 days. Colonies of undifferentiated hPSCs were expanded (Fig. 1A, white arrows in Stage 1). The cells were then cultured in bFGF-depleted hESC medium for 3 days and became aggregated at the interfaces of growing hPSC colonies (Fig. 1A, white arrows in Stage 2). The aggregated cells were cultured under DMEM/10%FBS for another 7 days and grew further and formed round clusters (Fig. 1A, white arrows in Stage 3). Next, FBS was increased to 20% to enhance cardiomyogenic differentiation, and the first contracting clusters appeared after ~7 days (Fig. 1A, white arrow in Stage 4). A majority of the clusters contracted over the next 4 to 5 days (supplemental online Movie 1). When 20% FBS was used instead of 10% FBS at stage 3, more floating cells were observed (white arrows in supplemental online Fig. 2A). Most floating cells were apoptotic based on trypan blue staining (supplemental online Fig. 2B). In contrast, continuous use of 10% FBS throughout stage 4 led to a lower number of contracting clusters (data not shown). Based on these results, 10% FBS was used to decrease apoptotic cells at stage 3 and switched to 20% FBS to enhance contracting cluster formation at stage 4.
2.2. Enrichment of CMs from contracting clusters
Cardiomyocytes were enriched from contracting clusters by using a two-step enzyme treatment. First, peripheral cells in contracting clusters (Fig. 1B) were removed by 0.05% trypsin-EDTA treatment (Fig. 1C). Subsequently, the remaining compact cells in the center of the clusters (Fig. 1C) were treated with 0.25% trypsin-EDTA treatment (Fig. 1D, left panel). Such dissociated clusters remained contractile while expressing cTnT and α-MHC (Fig. 1D and supplemental online Movie 2). These clusters were replated onto 0.1% gelatin-coated dishes and cultured under 2% FBS [15]. These dissociated clusters formed a monolayer (Fig. 1E, left panel) while contracting, and expressed α-MHC (Fig. 1E and supplemental online Movie 3). Next, these clusters were digested into finer pieces by incubating with 0.25% trypsin-EDTA, and cultured on 0.1% gelatin-coated plates. Single cells and small clusters composed of less than ten cells were observed (Fig. 1F and supplemental online Movie 4). Most of the contracting cells expressed CM specific markers, such as α-MHC, cTNT, NKX2–5, CONNEXIN 43, β-MHC, GATA4 and MEF2C (Fig. 1F). Next, we determined the purification efficiency measured by the proportion of CMs stained positive for GATA4, NKX2–5, α-MHC or cTNT divided by DAPI-positive cells and found that more than 90% of the monolayered cells were cardiomyogenically differentiated cells (Fig. 1G). This new 2D-directed CM differentiation and enrichment system yields highly purified CMs from hPSCs.
2.3. Confirmation of enriched CMs using a lentiviral α-MHC reporter system
Next, we sought to confirm the efficiency of CM generation using a viral promoter system. To do this, we constructed α-MHC reporter system using a lentiviral vector system (Fig. 2A). Viral particles expressing α-MHC-driven GFP were applied twice during stage 2 and 3 (Fig. 2B). GFP-expressing contractile clusters appeared at stage 4 (Fig. 2C and supplemental online Movie 5). We took advantage of antibiotic resistance for CM selection, because this reporter contains a Blasticidin resistance gene driven by an IRES sequence, whose expression is dependent on GFP expression (Fig. 2A). Treatment with Blasticidin could eliminate non-GFP-expressing cells at stage 4. Three days after treatment with Blasticidin, GFP-expressing clusters remained alive and contracting (supplemental online Movie 6), but GFP-negative cells were removed (Fig. 2D). GFP-positive clusters were collected with mechanical methods and treated with 0.25% trypsin-EDTA, and the resultant single cells were further cultured on 0.1% gelatin coated plates. All contracting cells expressed GFP (Left panel in Fig. 2E and supplemental online Movie 7) and expressed β-MHC (Fig. 2E, right panel). Our α-MHC reporter system worked similarly in other hESCs (H1 and H9) and hiPSCs (BJ1 and E1)(data not shown). Next, we determined the efficiency of CM differentiation using this reporter system. We counted the number of undifferentiated hPSCs at stage 1 (Fig. 1A, Day-1) and GFP-positive cells at stage 4 (Fig. 2D) and calculated the ratio. Fifty to seventy percent of undifferentiated hPSCs were finally committed to CMs at stage 4. These data indicate that our 2D-directed differentiation system can efficiently differentiate hPSCs into CMs.
Fig. 2.

Purification of CMs by an α-MHC reporter system. (A) Structure of α-MHC reporter. (B) Timing of infections of α-MHC reporter containing lentiviruses during CM differentiation. (C) Contracting clusters infected with viral particles exhibiting α-MHC driven GFP. (D) Cell clusters after removing non-GFP-expressing cells by treatment with Blasticidin at stage 4. (E) GFP-expressing and Blasticidin resistant CMs were digested by enzyme treatment (left panel), α-MHC driven GFP-expressing CMs also expressed β-MHC (right panel).
2.4. Three types of AP recorded from hPSC-CMs
To assess the functional activities of hPSC-CMs, we recorded APs from contracting cells. Stable recordings were obtained from eighteen BJ1-derived and eighteen H1-derived individual contracting cells. Three major types of APs were observed: nodal-like, atrial-like and ventricular-like APs (Fig. 3A). Nodal-like APs (Fig. 3A, top) displayed a fast spontaneous contracting rate, a prominent phase-4 depolarization, a slow upstroke and relative small amplitude. Ventricular-like APs (Fig. 3A, bottom) were characterized by a more negative maximum diastolic potential, a fast upstroke and a significant plateau phase resulting in longer duration and slow contracting rate. Compared to ventricular-like cells, atrial-like APs (Fig. 3A, middle) did not display a plateau phase during repolarization. In addition, the maximum diastolic potential of atrial-like APs was less negative than that of ventricular-like, but more negative than that of nodal-like APs.
Fig. 3.
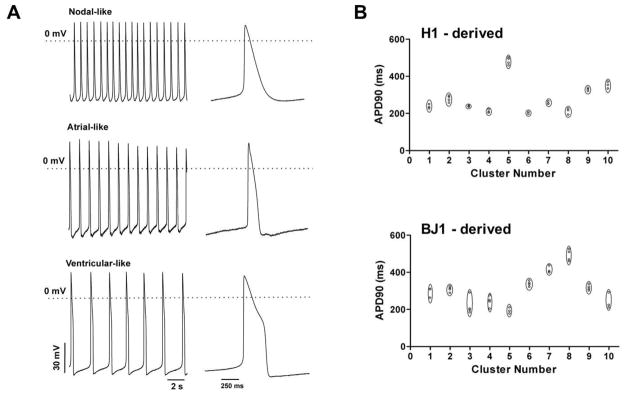
Action potential (AP morphologies of hPSC-CMs. (A) Intracellular recording from 3 different iPS cell-derived CMs demonstrating the 3 major types of AP: nodal-like (top), atrial-like (middle) and ventricular-like (bottom). (B) CMs within a given cluster characterized by similar AP morphology. Plot of APD90 measured for each recording in 10 BJ1-derived (top) or H1-derived (bottom) contracting clusters, from which 3 or more cells were examined.
Table 1 shows the proportions of contracting BJ1 cells displaying the nodal, atrial and ventricular-like APs in comparison to the proportions observed from H1-derived contracting clusters. The proportions of the three cell types were similar between BJ1- and H1-derived contracting cells, suggesting that this differentiation system can generate three types of CMs to a similar extent regardless of the original pluripotent cell types. In order to detect whether the same type of CMs grew together within a given cluster, we recorded APs from three or more cells within one contracting cluster and measured the AP duration from the peak to 90% repolarization (APD90). As shown in Fig. 3B (bottom) the APD90 values were similar in cells obtained from the same contracting clusters, but were different from cells obtained from other clusters. APD90 values of BJ1-derived contracting clusters displayed similar characteristics to those derived from H1 (Fig. 3B, top). The results suggest that one predominant CM type was differentiating within a given cluster.
Table 1.
Type percentage of cardiomyocyte clusters derived from HPSCs
| Total (N) | Nodal-like | Atrial-like | Ventricular-like | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Cluster (N) | Percentage (%) | Cluster (N) | Percentage (%) | Cluster (N) | Percentage (%) | ||
| HI–derived | 18 | 2 | 11 | 5 | 28 | 11 | 61 |
| BJ1 - derived | 18 | 2 | 11 | 2 | 11 | 14 | 78 |
N: the number of contracting clusters examined for each cell line
2.5. Gene expression patterns in the 2D-directed CM differentiation system
We analyzed the gene expression patterns at each stage (Fig. 1A, cell populations indicated by white arrows) by real time RT-PCR (qRT-PCR). Expression levels of mesoderm lineage markers, such as HAND1, Brachyury, KDR, M-CAD, MESP1 and MESP2, were high at stage 2 (cell aggregated region) and 3 (clustered region), but were decreased at stage 4 (Fig. 4A). In contrast, cardiac lineage genes began to appear at stage 3 and increased at stage 4 in both H1 and BJ1 cells (Fig. 4B). Expression of endoderm genes, such as SOX17, FOXA2 and AFP, peaked at stage 3 and decreased at stage 4. TUBB3, an ectoderm and neuronal gene was highly expressed at stage 2, but decreased at stages 3 and 4 (Fig. 4C) [34]. These results imply that using FBS in culture at stage 3 induced a decrease in ectodermal gene expression but increased endodermal gene expression. At stage 4, when contracting clusters were formed, endodermal gene expression was decreased (Fig. 4C).
Fig. 4.

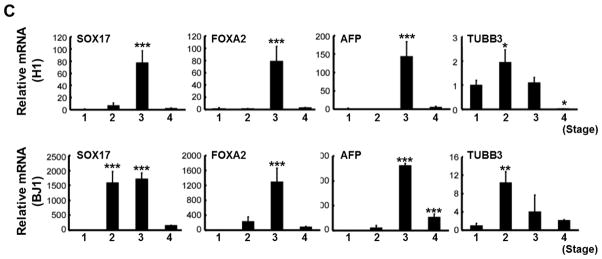
Gene expression patterns of (A) mesodermal, (B) cardiac lineage and (C) endoderm-ectodermal markers measured by real-time RT-PCR (n = 3 per group). *P < 0.05; **, P < 0.01; ***, P < 0.001.
2.6. Spatio-temporal relationship between mesoderm, endoderm and CM
We next examined the interaction between mesoderm, cardiac and endoderm lineage cells during CM differentiation. At stage 1, OCT4 was strongly expressed (Fig. 5A); however, mesoderm and endoderm markers were not yet expressed (data not shown). At stage 2, OCT4 was only expressed in central regions of hPSC colonies (Fig. 5B), but not in the cell aggregated regions (interfaces of two colonies; yellow line within the boxed region). Brachyury expression was restricted to the linear aggregated regions and FOXA2-expressing endodermal cells were observed broadly around aggregated regions (Fig. 5B, right panel). However, cells expressing CM markers were not yet observed (data not shown). These results indicate that differentiating cells begin to appear at the periphery of hPSC colonies, and peripheral cells which are at the interface of original hPSC colonies give rise to mesoderm lineage.
Fig. 5.


Expression patterns of pluripotency and lineage-specific markers in 2D-directed CM differentiation from undifferentiated hPSCs. (A) At stage 1, Oct4 was strongly expressed in hPSCs. (B) At stage 2, Oct4 was expressed in the central region of hPSCs. Mesoderm marker Brachyury and endoderm marker FoxA2 were broadly expressed in outgrowing cells of hPSC colonies. (C) Cell clusters appeared and grew at stage 3. (D) Mesoderm lineage markers were expressed at the peripheral region of the clusters, (E) cardiac lineage markers were expressed in the center region of the clusters and (F) endoderm lineage markers were expressed in the outgrowing regions of clusters in stage 3.
At stage 3, the linearly aggregated cells formed clearly distinguished round to polygonal clusters by addition of 10% FBS. At the center of the clusters (Fig. 5C, white circle), cardiac lineage markers NKX2–5, MEF2C, β-MHC, MLC2v, cTNT, MLC2a, and GATA4 were expressed. At the periphery of the clusters (Fig. 5C, between the white and yellow circles) mesoderm lineage markers, HAND1 and Brachyury were expressed (Fig. 5D and 5E). At the outgrowing region (Fig. 5C, outside of the yellow circle) of the clusters, endodermal lineage markers, FOXA2, and APF were expressed (Fig. 5F). These results suggest that use of 10% FBS drives cardiac lineage differentiation from mesodermal cells in the clustered regions surrounded by endodermal lineage cells at the periphery.
2.7. Contribution of endoderm cells to cardiac lineage commitment
At stage 4, the number of contracting clusters was increased by increasing FBS to 20%, and these cells showed clear CM characteristics. At stage 4, the boundaries of clusters were more clearly defined than the clusters at stage 3 (Fig. 6A), expression of CMs was restricted to the contracting clusters, and mesodermal markers were no longer expressed (Fig. 6A and 6B). Endodermal markers were still present at the periphery of contracting clusters (Fig. 6C). These results suggested that endodermal cells surrounding the clusters could influence the commitment of mesodermal cells into cardiac lineage. To test this hypothesis, peripheral cells were removed by trypsin-EDTA treatment at stage 3 and changes at stage 4 were investigated (Fig. 6D). While the control group induced contracting clusters with decreased expression of Brachyury at the periphery (Fig. 6D, upper panel), in the enzyme treated group, Brachyury expression remained strong in the periphery of the clusters and the number of contracting clusters were reduced more than 10 – fold at stage 4 (Fig. 6D, lower panel, Fig. 6E). These results indicate that endodermal cells play a crucial role in differentiation and commitment of mesodermal cells into cardiac lineage.
Fig. 6.


Formation of contracting clusters by the presence of endoderm cells. (A, B) Cardiac lineage markers were exclusively expressed in contracting clusters. (C) Endoderm lineage markers AFP and FoxA2 were expressed surrounding contracting clusters. (D) Comparison of expression patterns of mesoderm lineage markers Brachyury and β-MHC according to existence of endoderm cells. (E) Comparison of the number of contracting clusters between the control (intact endoderm) and endoderm-depleted groups (n = 7 per group). *P < 0.05.
2. 8. Engraftment of hPSC-derived CMs in the ischemic myocardium
To verify the in vivo behavior of the hPSC-CMs in injured myocardium, we induced myocardial infarction in nude rats and injected DiI-labeled hPSC-CMs which were enriched by the monolayer culture. Seven weeks later, we harvested the rat hearts and conducted histopathologic examination. Masson’s trichrome staining showed islands of patchy myocardium within circumferential fibrosis suggesting engrafted hPSC-CMs (Supplemental online Fig. 4A). Confocal microscopic examination revealed clusters of organized DiI-labeled hPSC-CMs in the infarcted area which expressed cTnT and Connexin 43. These results suggest that implanted hPSC-CMs were well engrafted, showed mature CM phenotypes and survived over 7 weeks post transplantation in the infarcted region (Supplemental online Fig. 4B and 4C). No gross or microscopic tumor was observed in any gross and microscopic specimens (n = 7).
3. Discussion
In the present study, we developed a novel 2D-directed culture system for differentiating human PSCs into functional CMs. This newly devised protocol for CM differentiation produces highly enriched CMs by the simple use of different concentrations of FBS in a timely fashion based on the morphologic changes concordant with developmental stages. Three types of CMs (nodal, atrial and ventricular CMs) were generated, with ventricular type CMs as the major cell type. There were no differences in the yield of CMs between the hESC- and hiPSC lines that we tried. This differentiation system recapitulates cardiac development in vivo, and suggests a critical spatiotemporal role of endodermal cells for differentiation and maturation of CMs from hPSCs.
Various methods have been attempted to differentiate mouse and human PSCs into CMs [6, 14–17]. These include embryoid body (EB)-mediated spontaneous differentiation [15], co-culturing with endodermal cell line END-2 [6], serum-free cytokine induction [17], electrical or mechanical stimulation [35], and pharmacological preconditioning [36]. However, most of these studies have used EBs to induce cardiogenic differentiation as an initial step [24], and these methods can only produce up to 25% differentiated CMs [5, 18]. Our experiments also showed that EB-mediated differentiation was only able to generate about 15% contracting EBs (supplemental online Fig. 3A and supplemental online Movie 8), and heterogeneous populations of cells contained in these contracting EBs (supplemental online Fig. 3B) will require a sorting or purification process before they can be used for cell therapy or drug screening.
The main goal of this study was to develop a system which can generate CMs from hPSCs with high yield and purity. We postulated that a monolayer culture system can enhance the efficiency of CM differentiation by allowing more uniform exposure to growth factors and oxygen tension. A recent study demonstrated that monolayer culture could achieve an 8.5-fold increase in cell numbers compared to EB-mediated culture systems [37]. We initiated culturing hPSCs on growth factor-reduced Matrigel in place of feeder cells to create a more homogeneous cell population and to eliminate the potential hazards from feeder cells. We adopted FBS-induced mesodermal and cardiogenic differentiation as it is simple and widely used for this purpose [34]. Interestingly, in this system we found that morphologic changes correlate with developmental stages and applying higher FBS concentrations at early cardiac commitment stages based on morphologic changes can increase the efficiency of CM differentiation. In this system, 70% of the clusters which appeared in stage 3 (cardiogenic mesoderm) converted to contracting clusters at stage 4. Through continuous culture, these cells expressed all representative CM markers such as α, and β-MHC, cTNT, Connexin 43, MLC2a and MLC2v. When evaluated by a precise α-MHC reporter system, this system produced CMs at 50 to 70% yield compared to the initially seeded hPSC numbers. When two-step enzyme treatment was applied to remove non-CMs, the CM yield increased to 95%.
For embryonic cardiogenesis, physical interactions between cells and substances secreted by different germ layers play vital roles [6, 38]. For instance, BMPs, FGFs and TGF-β secreted by endoderm lineage help commitment to cardiac lineage in mouse development in vivo [38] and mouse ESCs differentiation in vitro [39, 40]. During human ESC differentiation through EBs, endoderm markers SOX17 and FoxA2 were increased concurrently with mesoderm marker Brachyury, suggesting the interplay between the committed cells for mutual differentiation [34]. In another hESC study, CM differentiation was achieved by co-culture with an endoderm lineage cell line (END2) [6]. In addition, endoderm-secreted factors, such as Activin A and BMP4 are known to increase the expression of cardiac-specific genes in undifferentiated hESCs [41]. In our study, when mesodermal cells become aggregated, endodermal cells are surrounding mesodermal cells and appear to interact to induce mesodermal cells into cardiogenic lineages and maturation of CMs. When the peripheral endodermal cells were eliminated at stage 3, the number of contracting clusters was significantly reduced suggesting a crucial role for endoderm in CM maturation. In fact, this is the first study showing the clear spatiotemporal role of endoderm in the cardiomyogenic differentiation of hPSCs in vitro.
This 2-dimentional, morphology based, step-wise differentiation system using FBS is a new system which has many advantages over the previous culture system. First of all, this system allows generation of highly enriched CMs from hPSCs and thereby can enhance the therapeutic efficacy and reduce potential side effects. In fact, when these CMs were implanted into a rat model of myocardial infarction, these cells were engrafted into host myocardium and showed sustained engraftment without forming tumor. By avoiding the use of expensive cytokines, this system has additional economic advantages for clinical application. This system can further function as a useful platform for investigating developmental biologic events as well as drug evaluation. Particularly, CMs selected by α-MHC-reporter expression can provide >95% purity and can be highly useful for pharmacologic testing and genetic studies. As this system recapitulates many of the molecular events occurring during CM differentiation in vivo based purely on morphologic observation [42, 43], it can also enable the investigation of epigenetic changes and discovery of new factors involved in human cardiac differentiation. This novel culture system can be a highly useful platform for generation of hPSC-derived CMs for regenerative cardiac cell therapy, pharmacologic testing and human developmental studies.
Supplementary Material
Fig. 7.

A working model for CM differentiation in our 2D-direct differentiation system. Undifferentiated hPSCs were expanded at stage 1. Meso-endoderm lineage cells were induced from the peripheral region of undifferentiated hPSCs at stage 2. Mesoderm lineage cells were committed to cardiac lineage cells in aggregated clusters and endoderm cells were localized to the periphery of aggregated clusters at stage 3. Contracting clusters were further differentiated into cardiomyocytes at stage 4.
Acknowledgments
This work was supported in part by NIH grant DP3DK094346, HHSN268201000043C, NSF-EBICS grant, CBET-0939511 and ACTSI pilot grant (PHS Grant UL1 RR025008 from the CTSA program, NIH, NCRR).
Abbreviations
- hiPSCs
human induced pluripotent stem cells
- CMs
cardiomyocytes
- hESCs
human embryonic stem cells
- 2D
two dimensional
- hPSCs
human pluripotent stem cells
- EB
embryoid body
- AP
action potential
- APD 90
AP duration from the peak to 90% repolarization
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et al. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 2.Laflamme MA, Murry CE. Regenerating the heart. Nat Biotechnol. 2005;23:845–56. doi: 10.1038/nbt1117. [DOI] [PubMed] [Google Scholar]
- 3.Yi BA, Wernet O, Chien KR. Pregenerative medicine: developmental paradigms in the biology of cardiovascular regeneration. The Journal of clinical investigation. 2010;120:20–8. doi: 10.1172/JCI40820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–7. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 5.He JQ, Ma Y, Lee Y, Thomson JA, Kamp TJ. Human embryonic stem cells develop into multiple types of cardiac myocytes: action potential characterization. Circ Res. 2003;93:32–9. doi: 10.1161/01.RES.0000080317.92718.99. [DOI] [PubMed] [Google Scholar]
- 6.Mummery C, Ward-van Oostwaard D, Doevendans P, Spijker R, van den Brink S, Hassink R, et al. Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation. 2003;107:2733–40. doi: 10.1161/01.CIR.0000068356.38592.68. [DOI] [PubMed] [Google Scholar]
- 7.Kehat I, Khimovich L, Caspi O, Gepstein A, Shofti R, Arbel G, et al. Electromechanical integration of cardiomyocytes derived from human embryonic stem cells. Nat Biotechnol. 2004;22:1282–9. doi: 10.1038/nbt1014. [DOI] [PubMed] [Google Scholar]
- 8.Xue T, Cho HC, Akar FG, Tsang SY, Jones SP, Marban E, et al. Functional integration of electrically active cardiac derivatives from genetically engineered human embryonic stem cells with quiescent recipient ventricular cardiomyocytes: insights into the development of cell-based pacemakers. Circulation. 2005;111:11–20. doi: 10.1161/01.CIR.0000151313.18547.A2. [DOI] [PubMed] [Google Scholar]
- 9.Seki T, Yuasa S, Oda M, Egashira T, Yae K, Kusumoto D, et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell. 2010;7:11–4. doi: 10.1016/j.stem.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Kim D, Kim CH, Moon JI, Chung YG, Chang MY, Han BS, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4:472–6. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 12.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 13.Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–30. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Passier R, Oostwaard DW, Snapper J, Kloots J, Hassink RJ, Kuijk E, et al. Increased cardiomyocyte differentiation from human embryonic stem cells in serum-free cultures. Stem Cells. 2005;23:772–80. doi: 10.1634/stemcells.2004-0184. [DOI] [PubMed] [Google Scholar]
- 15.Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M, et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008;453:524–8. doi: 10.1038/nature06894. [DOI] [PubMed] [Google Scholar]
- 16.Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. The Journal of clinical investigation. 2001;108:407–14. doi: 10.1172/JCI12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015–24. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP, et al. Functional Cardiomyocytes Derived From Human Induced Pluripotent Stem Cells. Circ Res. 2009;104:e30–e41. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, et al. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell. 2011;8:228–40. doi: 10.1016/j.stem.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 20.Zwi L, Caspi O, Arbel G, Huber I, Gepstein A, Park IH, et al. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation. 2009;120:1513–23. doi: 10.1161/CIRCULATIONAHA.109.868885. [DOI] [PubMed] [Google Scholar]
- 21.Narazaki G, Uosaki H, Teranishi M, Okita K, Kim B, Matsuoka S, et al. Directed and systematic differentiation of cardiovascular cells from mouse induced pluripotent stem cells. Circulation. 2008;118:498–506. doi: 10.1161/CIRCULATIONAHA.108.769562. [DOI] [PubMed] [Google Scholar]
- 22.Huber I, Itzhaki I, Caspi O, Arbel G, Tzukerman M, Gepstein A, et al. Identification and selection of cardiomyocytes during human embryonic stem cell differentiation. Faseb J. 2007;21:2551–63. doi: 10.1096/fj.05-5711com. [DOI] [PubMed] [Google Scholar]
- 23.Vidarsson H, Hyllner J, Sartipy P. Differentiation of human embryonic stem cells to cardiomyocytes for in vitro and in vivo applications. Stem cell reviews. 2010;6:108–20. doi: 10.1007/s12015-010-9113-x. [DOI] [PubMed] [Google Scholar]
- 24.Itskovitz-Eldor J, Schuldiner M, Karsenti D, Eden A, Yanuka O, Amit M, et al. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Molecular medicine (Cambridge, Mass. 2000;6:88–95. [PMC free article] [PubMed] [Google Scholar]
- 25.Ungrin MD, Joshi C, Nica A, Bauwens C, Zandstra PW. Reproducible, ultra high-throughput formation of multicellular organization from single cell suspension-derived human embryonic stem cell aggregates. PLoS One. 2008;3:e1565. doi: 10.1371/journal.pone.0001565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graichen R, Xu X, Braam SR, Balakrishnan T, Norfiza S, Sieh S, et al. Enhanced cardiomyogenesis of human embryonic stem cells by a small molecular inhibitor of p38 MAPK. Differentiation; research in biological diversity. 2008;76:357–70. doi: 10.1111/j.1432-0436.2007.00236.x. [DOI] [PubMed] [Google Scholar]
- 27.Xu C, Police S, Rao N, Carpenter MK. Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circ Res. 2002;91:501–8. doi: 10.1161/01.res.0000035254.80718.91. [DOI] [PubMed] [Google Scholar]
- 28.Coats AJ, Shewan LG. Statement on authorship and publishing ethics in the international journal of cardiology. International journal of cardiology. 2011;153:239–40. doi: 10.1016/j.ijcard.2011.10.119. [DOI] [PubMed] [Google Scholar]
- 29.Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–6. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 30.Cho SW, Moon SH, Lee SH, Kang SW, Kim J, Lim JM, et al. Improvement of postnatal neovascularization by human embryonic stem cell derived endothelial-like cell transplantation in a mouse model of hindlimb ischemia. Circulation. 2007;116:2409–19. doi: 10.1161/CIRCULATIONAHA.106.687038. [DOI] [PubMed] [Google Scholar]
- 31.Braam SR, Denning C, Matsa E, Young LE, Passier R, Mummery CL. Feeder-free culture of human embryonic stem cells in conditioned medium for efficient genetic modification. Nature protocols. 2008;3:1435–43. doi: 10.1038/nprot.2008.140. [DOI] [PubMed] [Google Scholar]
- 32.Kim H, Cho HJ, Kim SW, Liu B, Choi YJ, Lee J, et al. CD31+ cells represent highly angiogenic and vasculogenic cells in bone marrow: novel role of nonendothelial CD31+ cells in neovascularization and their therapeutic effects on ischemic vascular disease. Circ Res. 2010;107:602–14. doi: 10.1161/CIRCRESAHA.110.218396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoon YS, Wecker A, Heyd L, Park JS, Tkebuchava T, Kusano K, et al. Clonally expanded novel multipotent stem cells from human bone marrow regenerate myocardium after myocardial infarction. The Journal of clinical investigation. 2005;115:326–38. doi: 10.1172/JCI22326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bettiol E, Sartiani L, Chicha L, Krause KH, Cerbai E, Jaconi ME. Fetal bovine serum enables cardiac differentiation of human embryonic stem cells. Differentiation; research in biological diversity. 2007:669–81. doi: 10.1111/j.1432-0436.2007.00174.x. [DOI] [PubMed] [Google Scholar]
- 35.Heng BC, Haider H, Sim EK, Cao T, Ng SC. Strategies for directing the differentiation of stem cells into the cardiomyogenic lineage in vitro. Cardiovascular research. 2004;62:34–42. doi: 10.1016/j.cardiores.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 36.Rosenblatt-Velin N, Lepore MG, Cartoni C, Beermann F, Pedrazzini T. FGF-2 controls the differentiation of resident cardiac precursors into functional cardiomyocytes. The Journal of clinical investigation. 2005;115:1724–33. doi: 10.1172/JCI23418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oldershaw RA, Baxter MA, Lowe ET, Bates N, Grady LM, Soncin F, et al. Directed differentiation of human embryonic stem cells toward chondrocytes. Nat Biotechnol. 2010;28:1187–94. doi: 10.1038/nbt.1683. [DOI] [PubMed] [Google Scholar]
- 38.Lough J, Sugi Y. Endoderm and heart development. Dev Dyn. 2000;217:327–42. doi: 10.1002/(SICI)1097-0177(200004)217:4<327::AID-DVDY1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 39.Rudy-Reil D, Lough J. Avian precardiac endoderm/mesoderm induces cardiac myocyte differentiation in murine embryonic stem cells. Circ Res. 2004;94:e107–16. doi: 10.1161/01.RES.0000134852.12783.6e. [DOI] [PubMed] [Google Scholar]
- 40.Kubo A, Shinozaki K, Shannon JM, Kouskoff V, Kennedy M, Woo S, et al. Development of definitive endoderm from embryonic stem cells in culture. Development. 2004;131:1651–62. doi: 10.1242/dev.01044. [DOI] [PubMed] [Google Scholar]
- 41.Yao S, Chen S, Clark J, Hao E, Beattie GM, Hayek A, et al. Long-term self-renewal and directed differentiation of human embryonic stem cells in chemically defined conditions. Proc Natl Acad Sci U S A. 2006;103:6907–12. doi: 10.1073/pnas.0602280103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Srivastava D, Olson EN. A genetic blueprint for cardiac development. Nature. 2000;407:221–6. doi: 10.1038/35025190. [DOI] [PubMed] [Google Scholar]
- 43.Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661–80. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.