

Summary
Bacteria utilize multiple regulatory systems to modulate gene expression in response to environmental changes, including two-component signaling systems and partner-switching networks. We recently identified a novel regulatory protein SypE that combines features of both signaling systems. SypE contains a central response regulator receiver domain flanked by putative kinase and phosphatase effector domains with similarity to partner-switching proteins. SypE was previously shown to exert dual control over biofilm formation through the opposing activities of its terminal effector domains. Here, we demonstrate that SypE controls biofilms in Vibrio fischeri by regulating the activity of SypA, a STAS (sulphate transporter and anti-sigma antagonist) domain protein. Using biochemical and genetic approaches, we determined that SypE both phosphorylates and dephosphorylates SypA, and that phosphorylation inhibits SypA’s activity. Furthermore, we found that biofilm formation and symbiotic colonization required active, unphosphorylated SypA, and thus SypA phosphorylation corresponded with a loss of biofilms and impaired host colonization. Finally, expression of a non-phosphorylatable mutant of SypA suppressed both the biofilm and symbiosis defects of a constitutively inhibitory SypE mutant strain. This study demonstrates that regulation of SypA activity by SypE is a critical mechanism by which V. fischeri controls biofilm development and symbiotic colonization.
Introduction
Bacteria continuously monitor their surroundings and coordinate cellular behavior with current environmental conditions. The ability to adaptively respond to perceived changes in the environment is critical for a bacterial cell to colonize and persist within a particular ecological niche. Two-component signaling (TCS) systems are a common mechanism by which bacteria sense and respond to environmental stimuli. The typical two-component system consists of a sensor histidine kinase (SK) and a cognate response regulator (RR) (Stock et al., 2000). Upon stimulus detection, the SK autophosphorylates at a conserved histidine residue and subsequently donates this phosphoryl group to an aspartate residue located in the regulatory receiver (REC) domain of a downstream RR (Bourret et al., 1990; Stock and Guhaniyogi, 2006). REC domain phosphorylation typically regulates the activity of an attached effector domain, often a DNA-binding domain (Bourret, 2010). Alternatively, the RR REC domain may be attached to a variety of other signaling domains, including those involved in enzymatic activity or protein binding (Galperin, 2006; Galperin, 2010).
In addition to TCS systems, bacteria employ a variety of other regulatory devices to coordinate gene expression with environmental cues. Partner-switching systems represent another common mechanism by which bacteria regulate signal transmission and gene expression. The canonical partner switching system consists of several conserved regulatory elements: a serine kinase/anti-sigma factor, a serine phosphatase, and an antagonist protein/anti-sigma factor antagonist (Yang et al., 1996). The partner-switching paradigm was first observed in the Gram-positive bacterium Bacillus subtilis, in which partner-switching proteins were found to regulate sigma factors involved in sporulation (σF) and the general stress response (σB) (Duncan and Losick, 1993; Dufour and Haldenwang, 1994). Partner-switching systems have since been characterized in a variety of bacterial systems, particularly in other Gram-positive organisms. The B. subtilis RsbU-RsbV-RsbW signaling network, which regulates σB of the general stress response, represents one of the most well-characterized examples of a partner-switching system (Fig. 1A). In this regulatory pathway, an anti-sigma/serine kinase RsbW negatively regulates σB activity by binding it and preventing its association with core RNA polymerase (Benson and Haldenwang, 1993a; Benson and Haldenwang, 1993b). σB is released by the action of an antagonist protein, RsbV, which binds RsbW and prevents sequestration of σB (Dufour and Haldenwang, 1994). The ability of RsbV to function as an antagonist is regulated by its phosphorylation state. When phosphorylated by RsbW, RsbV is rendered unable to bind and inhibit RsbW (Dufour and Haldenwang, 1994). Dephosphorylation of RsbV is promoted by a set of PP2C phosphatases, RsbU and RsbP, which are activated in response to cellular stresses (Voelker et al., 1996). Thus, stress detection induces RsbV dephosphorylation, the inhibition of RsbW, and the release of σB and subsequent activation of the σB regulon (Voelker et al., 1996).
Figure 1. Model of V. fischeri biofilm regulation by SypE and SypA.

(A) Model of the Bacillus subtilis partner switching system that regulates the activity of σB in the general stress response pathway (see text for full description). Shown are the relevant domains for partner-switching regulation. (B) SypE contains a central RR REC domain flanked by an N-terminal RsbW-like serine kinase domain and a C-terminal PP2C-like serine phosphatase domain. SypA contains a single STAS-domain conserved in anti-sigma factor antagonists. Under conditions in which SypE is unphosphorylated, its N-terminal kinase domain is active, resulting in the phosphorylation of SypA (on conserved serine residue S56) and inhibition of biofilm formation. When phosphorylated (presumably on conserved residue D192), SypE functions as a phosphatase (Morris et al., 2011). This results in the dephosphorylation of SypA, activating SypA to promote biofilm formation. A D192A mutation “locks” SypE into a constitutive kinase, resulting in SypA phosphorylation and the inhibition of biofilms and colonization. Protein phosphatase 2C (PP2C) domain (black box); Serine/threonine kinase (RsbW) domain (white box); anti-sigma factor antagonist (STAS) domain (grey box); response regulator receiver (REC) domain (light grey box).
TCS and partner-switching systems provide distinct mechanisms by which bacteria transmit cellular signals and regulate target gene expression. Interestingly, regulatory elements of both systems are combined in the biofilm regulator, SypE. SypE is an unusual RR consisting of a central regulatory REC domain flanked by two effector domains that exhibit sequence similarity to partner-switching regulatory elements. SypE’s N-terminal effector domain exhibits sequence similarity to HPK (histidine protein kinase)-like serine kinases, including the serine kinase/anti-sigma RsbW of B. subtilis (Fig. 1)(Morris and Visick, 2010), while its C-terminal effector domain is similar to PP2C-like serine phosphatases, including the B. subtilis phosphatase RsbU (Fig. 1) (Morris and Visick, 2010).
We recently demonstrated that SypE plays a key role in the regulation of host colonization by the bacterium Vibrio fischeri (Morris et al., 2011). V. fischeri is a Gram-negative marine bacterium that forms a symbiosis with the Hawaiian squid Euprymna scolopes. SypE regulates one of the earliest stages of host colonization, the formation of a biofilm-like aggregate on the surface of the squid’s symbiotic light organ (Nyholm et al., 2000). The formation of this biofilm requires the symbiosis polysaccharide (syp) locus, which is controlled at the transcriptional level by the SK RscS and the downstream syp-encoded RR SypG (Yip et al., 2005; Yip et al., 2006; Hussa et al., 2008). In laboratory culture, substantial biofilm formation is observed only when one of these regulators is overexpressed (Yip et al., 2006; Hussa et al., 2008,).
SypE exerts both negative and positive control over RscS-induced biofilms through the opposing activities of its terminal effector domains (Morris et al., 2011). Specifically, SypE’s N-terminal, RsbW-like domain is both necessary and sufficient to inhibit biofilm formation, while the C-terminal, PP2C-like domain alone is sufficient to mediate positive control over biofilm formation (Morris et al., 2011). Furthermore, phosphorylation of SypE’s central REC domain regulates these opposing activities: a SypE mutant disrupted at the predicted site of phosphorylation within the REC domain (SypED192A) inhibited both biofilm formation and colonization in a manner dependent on the N-terminal, putative kinase domain (Morris et al., 2011).
The downstream target of SypE’s regulatory activities and the mechanism by which SypE restricts biofilms and colonization remain unknown. Given the results from our previous study, we hypothesized that SypE likely regulates biofilm formation by controlling the phosphorylation state of a downstream target, possibly another partner switching-like protein. One intriguing possibility for such a target is SypA, which exhibits sequence similarity to the B. subtilis antagonist protein RsbV (Yip et al., 2005; Morris and Visick, 2010). Like RsbV, SypA contains a sulphate transporter and anti-sigma antagonist (STAS) domain indicative of an anti-sigma factor antagonist (Fig. 1B) (Morris and Visick, 2010). A role for SypA in regulating biofilm formation and/or colonization has yet to be identified. However, given the similarity to characterized partner-switching regulators and the genetic proximity of sypA to sypE, we speculated that SypA may represent the target of SypE’s regulatory activities. Here, we investigated the mechanism by which the RR SypE regulates biofilm formation and host colonization. We demonstrate that SypE interacts with SypA and mediates control over biofilm formation and colonization through regulation of SypA phosphorylation and, consequently, SypA activity. Together, our data demonstrate that a key function of SypE is to control the ability of SypA to promote biofilm formation and colonization.
Results
SypE interacts with the putative regulatory protein SypA
The RR SypE controls biofilm formation both negatively and positively through the opposing activities of its putative N-terminal serine kinase and C-terminal serine phosphatase domains (Morris et al., 2011). However, the activity of this protein as a serine kinase/phosphatase has not yet been experimentally established, in large part because the target(s) of these putative activities remained unknown. Due to sequence similarities of SypE and the STAS-domain protein SypA to proteins present in partner-switching systems, we speculated that SypE could function by controlling the phosphorylation state of SypA (Morris and Visick, 2010). To determine if this were the case, we first asked whether SypE interacts with SypA in vivo by performing a co-immunoprecipitation assay. Briefly, we generated FLAG- and HA-epitope tag fusions to the C-termini of SypE and SypA, respectively, and expressed the epitope-tagged alleles in V. fischeri. We then used anti-FLAG or anti-HA antibodies to immunoprecipitate either SypE or SypA, respectively, and detected the immunoprecipitated proteins using western blotting analyses. Upon immunoprecipitation using anti-FLAG antibody, we detected not only immunoprecipitated SypE, but also SypA (Fig. 2A, left panel). In reciprocal experiments, we found that immunoprecipitation of SypA with anti-HA antibody resulted in the co-immunoprecipation of SypE (Fig. 2A, right panel). These results indicate that SypA and SypE are capable of interacting in vivo, a result that is consistent with the model that SypA may serve as a target of SypE’s regulatory activity.
Figure 2. SypE interacts with SypA.

(A) Soluble lysates from V. fischeri ΔsypA ΔsypE cells [KV4716] carrying plasmids expressing FLAG-SypE (pARM80), HA-SypA (pARM36), or untagged SypE (pCLD48) and SypA (pARM13) control plasmids were used in immunoprecipation assays with non-specific anti-rabbit IgG antibody (Lanes 1 and 5), anti-FLAG antibody (Lanes 2–4), or anti-HA antibody (Lanes 6–8). The samples were resolved using SDS-PAGE and subjected to western blot analysis with anti-FLAG (top panel) or anti-HA (bottom panel) antibodies. (+) indicates V. fischeri cells carrying the epitope tagged SypE (pARM80) and/or SypA (pARM36) plasmids. (−) indicates V. fischeri cells carrying the control plasmids expressing untagged SypE (pCLD48) or SypA (pARM13). (B) Soluble lysates from V. fischeri ΔsypA ΔsypE cells [KV4716] carrying plasmids expressing HA-SypA (pARM36) and either FLAG-SypEΔNTD (pARM162) [Lanes 1 and 3] or FLAG-SypENTD (pARM136) [Lanes 2 and 4] were used in immunoprecipation assays with anti-FLAG (or anti-HA antibody. Lanes 5 and 6, lysates from ΔsypA ΔsypE cells [KV4716] carrying both pARM36 and either pARM162 (lane5) or pARM136 (lane 6) immunoprecipitated with non-specific, anti-rabbit IgG. Samples were resolved using SDS-PAGE and subjected to western blot analysis with anti-FLAG (top panel) or anti-HA (bottom panel) antibodies.
We next examined the domains of SypE required for mediating this binding to SypA. Based on similarity to B. subtilis orthologues, in which RsbW directly binds RsbV (Dufour and Haldenwang, 1994), we hypothesized that binding to SypA would depend upon SypE’s N-terminal, RsbW-like domain. We first tested the ability of SypA to interact with an N-terminal deletion mutant of SypE (SypEΔNTD), a protein that lacks the first 135 amino acids (comprising the putative serine kinase domain) but retains positive regulatory activity mediated by the C-terminal domain (Morris et al., 2011). We found that the SypEΔNTD mutant failed to co-immunoprecipitate with SypA (Fig. 2B, lanes 1 and 3). This result suggested that the N-terminus of SypE is required for interaction with SypA. To explore this possibility further, we co-expressed SypA with a SypE mutant (SypENTD) that expressed only the first 140 amino acids of the N-terminus; this mutant retains its inhibitory activity, but lacks its positive activity (Morris et al., 2011). We found that SypENTD co-immunoprecipitated with SypA, indicating that the N-terminal domain alone is sufficient to mediate interaction with SypA (Fig. 2B, lanes 2 and 4). These data support a model in which the N-terminal domain of SypE interacts with SypA in vivo.
The N-terminal domain of SypE phosphorylates SypA in vitro
The identification of SypA as a protein capable of interacting with SypE provided a candidate substrate to test the activities of SypE’s terminal effector domains, e.g., kinase and phosphatase activities. To assess SypE’s kinase activity, we purified glutathione S-transferase (GST)-tagged versions of the proteins (SypE and SypA-FLAG) and incubated purified SypA in kinase buffer either in the absence or presence of purified SypE. We then resolved the samples on SDS-PAGE gels containing Phos-tag™ acrylamide, which permits the separation of phosphorylated and non-phosphorylated forms of proteins by preferentially binding to and retarding the migration of phosphorylated proteins (Kinoshita et al., 2006; Kinoshita-Kikuta et al., 2007). Following electrophoresis, SypA protein was detected by western blotting using an anti-FLAG antibody. As shown in figure 3A, purified SypA incubated in kinase buffer alone (lane 2) migrated as a single species, corresponding to non-phosphorylated SypA. In contrast, upon co-incubation with SypE, two major SypA bands were observed: both a lower, faster migrating SypA band (unphosphorylated SypA) and an upper, slower migrating band corresponding to phosphorylated SypA (SypA~P) (lane 3). This shifted SypA band was not observed upon resolving the reaction samples in parallel on gels lacking Phos-tag™ acrylamide, suggesting that the upper band observed in our Phos-tag™ gels represents phosphorylated SypA (Fig. S1). To confirm that the shifted SypA band indeed represented phosphorylated SypA, we co-incubated SypA with SypE in kinase buffer lacking ATP. As expected, only the lower (unphosphorylated) SypA band was present, indicating that the shifted SypA band represents phosphorylated SypA (Fig. 3A, lane 5). Similar results were observed in kinase assays containing purified SypA-FLAG protein cleaved of its N-terminal GST-tag (Fig. S2 and S3). Together, these data demonstrate that SypE can phosphorylate SypA.
Figure 3. SypE phosphorylates SypA in vitro.

(A) In vitro phosphorylation of SypA by SypE. Purified SypA-FLAG (3 µg) and/or SypE proteins (2 µg) were incubated in kinase buffer in the presence or absence of ATP. Reaction samples were resolved by SDS-PAGE on a 25 µM Phos-tag™ acrylamide gel and the proteins were detected via western blot analysis using an anti-FLAG antibody. Lane 1, SypE incubated in kinase buffer. Lanes 2 and 3, wild-type SypA-FLAG protein incubated in kinase buffer alone (Lane 2) or with SypE (Lane 3). Lane 4, SypAS56A-FLAG protein incubated with SypE in kinase buffer. Lane 5, wild-type SypA-FLAG protein incubated with SypE in kinase buffer lacking ATP. (+) indicates reactions containing wild-type SypA-FLAG or SypE protein. (−) indicates reactions not containing purified protein. (S56A) indicates reactions containing the SypAS56A–FLAG protein. (B) SypE-mediated phosphorylation of SypA in E. coli cells. SypA-FLAG protein was purified from E. coli cells carrying both pARM157 (GST-SypA-FLAG) and either empty vector, pVSV105, (Lane 1) or plasmid pCLD64, which expresses the N-terminal, serine kinase domain of SypE (Lane 2). Samples were resolved using SDS-PAGE on a 25 µM Phos-tag™ acrylamide gel and proteins were detected by anti-FLAG western blot analysis. SypA~P denotes phosphorylated SypA.
Phosphorylation of B. subtilis RsbV, and similar STAS-domain containing orthologs, occurs on a conserved serine residue (Yang et al., 1996). Sequence alignments indicate that this residue corresponds to S56 in SypA (Morris and Visick, 2010). To assess whether this conserved serine was required for SypA phosphorylation, we performed the kinase reactions using a purified SypA mutant in which S56 was substituted with an alanine (SypAS56A). Upon analyzing the samples using Phos-tag™ SDS-PAGE coupled with western blot analysis, we observed only a single (lower) band for the SypAS56A sample, corresponding to unphosphorylated SypA (Fig. 3A, lane 4). Thus, phosphorylation of SypA by SypE depends on the predicted site of phosphorylation (S56) within SypA.
SypE’s serine kinase activity is proposed to reside in the protein’s N-terminal effector domain (Morris et al., 2011). Unfortunately, attempts to assess the kinase activity of purified SypE N-terminal domain alone (SypENTD) in vitro were unsuccessful: the GST-SypENTD protein failed to promote SypA phosphorylation in our in vitro assay (data not shown), perhaps due to misfolding of the fusion protein or interference by the GST tag. As an alternative approach to assess whether the N-terminal domain of SypE possesses kinase activity, we asked whether expression of SypE’s N-terminal domain could promote SypA phosphorylation in E. coli cells. We introduced the GST-SypA plasmid into E. coli cells carrying either an empty vector (pVSV105) or a plasmid (pCLD64) expressing the untagged, N-terminal domain of sypE (SypENTD), which when expressed in V. fischeri is sufficient to inhibit biofilm formation (Morris et al., 2011). We then affinity-purified SypA from these SypENTD-expressing cells and assessed the phosphorylation state of SypA. Whereas SypA purified from vector-containing cells appeared as a single, faster migrating band corresponding to non-phosphorylated SypA (Fig. 3B, lane 1), SypA purified from sypENTD-expressing cells predominately appeared as an upper, slower migrating band corresponding to phosphorylated SypA, although a small amount of unphosphorylated protein could be detected (Fig. 3B, lane 2). We conclude from these data that SypE, and specifically its N-terminal domain, indeed possesses serine kinase activity and can promote phosphorylation of SypA.
The C-terminal domain of SypE dephosphorylates SypA in vitro
Given our finding that the N-terminal domain of SypE can phosphorylate SypA, we next questioned whether SypE’s C-terminal domain possesses phosphatase activity and can dephosphorylate SypA~P. To address this question, we incubated phosphorylated SypA (SypA~P) protein in phosphatase buffer either in the presence or absence of the putative phosphatase domain of SypE (SypECTD) and then assessed the phosphorylation state of SypA. Phosphorylated SypA incubated in phosphatase buffer alone (in the absence of SypE) remained largely phosphorylated as indicated by the presence of the upper (SypA~P) band (Fig. 4, Lanes 3 and 7), relative to the unphosphorylated SypA control (Fig. 4, Lane 2). Upon addition of increasing amounts of SypECTD, we observed a decrease in the intensity of the upper (SypA~P) band and a corresponding increase in the lower, unphosphorylated SypA band (Fig. 4, Lanes 4–6). These data indicate that the C-terminal domain of SypE possesses phosphatase activity and is capable of dephosphorylating SypA in vitro. Similar results were observed in phosphatase assays containing P~SypA protein cleaved of its N-terminal GST-tag (Fig. S2 and S4). Together with the findings from our kinase assays, these results demonstrate that the N- and C-terminal domains of SypE indeed possess enzymatic activity and are sufficient to mediate SypA phosphorylation and dephosphorylation, respectively.
Figure 4. SypE dephosphorylates SypA in vitro.

Western blot analysis of in vitro SypA dephosphorylation samples analyzed on Phos-tag™ acrylamide gels. Purified phosphorylated SypA-FLAG protein (SypA~P; 2 µg) was incubated in Mg2+-containing phosphatase buffer in the presence or absence of increasing concentrations of purified SypE C-terminal phosphatase domain (SypECTD; 2–10 µg). The reactions were terminated at zero or 30 minutes and the samples resolved by SDS-PAGE on a 25 µM Phos-tag™ acrylamide gel. SypA proteins were detected by western blot analysis using an anti-FLAG antibody. Lane 1, SypECTD incubated in buffer alone. Lane 2, non-phosphorylated SypA incubated in buffer alone. Lanes 3 and 7, phosphorylated SypA (SypA~P) incubated in buffer alone for zero (Lane 3) and 30 (Lane 7) minutes. Lanes 4–6, phosphorylated SypA (SypA~P) incubated for 30 minutes in buffer containing 2 µg (Lane 4), 5 µg (Lane 5), or 10 µg (Lane 6) of purified SypECTD. (+) indicates reactions containing SypA-FLAG or SypECTD protein. (−) indicates reactions not containing the indicated purified protein.
SypA is required for RscS-induced biofilm formation
SypE is an established regulator of biofilm formation (Morris et al., 2011), and thus we wondered whether it exerts its effect via control over SypA activity. As a first step towards answering this question, we asked whether SypA itself impacts biofilm formation. We induced biofilm formation by overexpressing the biofilm regulator RscS in either wild-type (WT) or ΔsypA mutant cells. Consistent with previous reports (Yip et al., 2006; Morris et al., 2011), wild-type cells carrying the rscS plasmid exhibited strong biofilm phenotypes, including wrinkled colony morphology and pellicle formation, whereas vector-containing cells failed to form biofilms as indicated by the smooth colony morphology and lack of a pellicle (Fig. 5 A and B and S5, respectively). The ΔsypA mutant consistently failed to form biofilms in response to RscS overexpression, exhibiting smooth colony morphology and little to no pellicle production (Fig. 5C and S5, respectively). To confirm that this loss of biofilm formation was due to the absence of sypA, we introduced a wild-type copy of sypA (sypA+) into the chromosome at the Tn7 site. Single-copy expression of sypA+ fully complemented the ΔsypA mutant, restoring both wild-type wrinkled colony morphology and pellicle formation (Fig. 5D and S5). Together, these results identify SypA as a critical regulator necessary for biofilm formation.
Figure 5. Active SypA is required for RscS-induced biofilm formation.

The RscS plasmid (pARM7) was introduced into wild-type (WT) or indicated sypA strains. Cultures of the following strains were spotted onto LBS medium at 24°C and wrinkled colony formation was assessed at 48 h post spotting: WT cells containing empty Tn7 cassette (EC) [KV4389] and carrying either empty vector pKV282 (A) or pARM7 (B); pARM7-carrying ΔsypA cells containing EC [KV5079] (C), or complemented with wild-type sypA+ [KV5479] (D), sypAS56D [KV5480] (E), or sypAS56A [KV5481] (F). Images are representative of at least three independent experiments. Black bar represents 2 mm.
SypA activity is modulated via its conserved serine residue S56
In other RsbV-like antagonists, phosphorylation of a conserved serine residue (S56 in SypA) inhibits their activity (Dufour and Haldenwang, 1994; Yang et al., 1996). Therefore, we next asked whether phosphorylation impacts SypA’s ability to control biofilm formation by generating sypAS56D and sypAS56A mutant alleles and assessing their ability to complement the ΔsypA mutant for biofilm formation. The aspartate substitution (SypAS56D) was predicted to mimic the phosphorylated, inactive, state (Diederich et al., 1994; Yang et al., 1996), while the alanine substitution (SypAS56A) was predicted to “lock” the protein in the non-phosphorylated, active, state (Yang et al., 1996). Neither mutant produced biofilms in the absence of rscS overexpression (data not shown). When rscS was overexpressed in the ΔsypA mutant containing sypAS56D, biofilm formation occurred, but was severely impaired relative to the sypA+ control (Fig. 5E, S5 and data not shown). This defect could not be attributed to poor expression, as a FLAG-tagged version of this mutant was present at steady-state levels similar to that of FLAG-tagged wild-type SypA protein and the SypAS56A mutant (Fig. S6). In contrast, the alanine substitution mutant (SypAS56A) fully complemented, restoring both wrinkled colony morphology and pellicle formation to the rscS-overexpressing sypA mutant (Fig 5F and S5). Together, these results indicate that S56 of SypA is critical for the regulation of SypA activity, and suggest that phosphorylation at this residue likely inhibits SypA activity.
SypA functions downstream of SypE to control biofilm formation
Our current data indicate that both SypE and SypA contribute to the regulation of syp-dependent biofilm formation, but the relative importance of these regulators in controlling biofilms remains unknown. To further investigate the mechanism by which these regulators control biofilms, we generated a double mutant and assessed its ability to form biofilms relative to that of the two single mutants. The ΔsypE strain exhibited wrinkled colony morphology and pellicle formation similar to wild-type cells (Fig. 6C and S7), albeit with a slight delay in wrinkling (data not shown) as observed previously (Morris et al., 2011). In contrast, the ΔsypA mutant exhibited impaired biofilm phenotypes (Fig. 6D and S7). Similar to the sypA single mutant, the ΔsypA ΔsypE double mutant also failed to produce biofilms, exhibiting both smooth colony morphology (Fig. 6E) and impaired pellicle formation (Fig. S7). Expression of sypA+ at the Tn7 site of the ΔsypA ΔsypE strain fully restored both wrinkled colony development and pellicle formation to a level indistinguishable from the sypE single mutant (Fig. 6F and S7 and data not shown). These data demonstrate that SypA is epistatic to SypE, indicating that SypA functions downstream of SypE in the biofilm regulatory network.
Figure 6. SypA functions downstream of SypE.

Assessment of RscS-induced wrinkled colony formation. Cultures of the following strains were spotted onto LBS medium at 24°C and wrinkled colony formation was assessed at 48 h post-spotting: Wild-type (WT) cells containing empty Tn7 cassette (EC) [KV4389] and carrying empty vector pKV282 (A) or pRscS plasmid pARM7 (B); ΔsypE cells containing EC [KV4390] and carrying pARM7 (C); ΔsypA cells containing EC [KV5079] and carrying pARM7 (D); pARM7-carrying ΔsypA ΔsypE cells containing either EC [KV6392] (E) or wild-type sypA+ [KV6393] (F). Images are representative of at least three independent experiments. Black bar represents 2 mm.
SypE inhibits biofilm formation by inactivating SypA
We previously reported that expression of a mutant sypE allele, sypED192A, constitutively inhibits biofilm formation in laboratory culture and dramatically impairs host colonization in vivo (Morris et al., 2011) (Fig. 1B). The constitutive inhibitory activity of the SypED192A mutant required the protein’s N-terminal kinase domain, suggesting that SypED192A inhibits biofilm formation by constitutively activating the kinase domain to phosphorylate a downstream target protein (Morris et al., 2011). We hypothesized that SypED192A inhibits biofilm formation by promoting the phosphorylation, and therefore the inactivation, of SypA. If so, then the “constitutively active”, non-phosphorylatable SypAS56A mutant should be insensitive to SypE’s inhibitory activity; therefore, expression of sypAS56A should suppress the biofilm defect of the sypED192A mutant. To test this hypothesis, we generated strains expressing combinations of the sypA and sypE alleles: either the sypA+ or sypAS56A allele (expressed from the native sypA locus) was combined with either the wild-type sypE+ or the constitutively inhibitory sypED192A allele (expressed at the Tn7 site of a sypE deletion mutant). As expected, SypE+ strains expressing either sypA+ or sypAS56A exhibited wrinkled colony morphology and pellicle formation (Figs. 7A and B and S8), while a strain that expressed the inhibitory sypED192A allele and wild-type sypA+ failed to produce biofilms (Figs. 7C and S8) (Morris et al., 2011). However, a strain expressing both the inhibitory sypED192A allele and the constitutively active sypAS56A allele was competent to produce wrinkled colony morphology and pellicles (Figs. 7D and S8). These data indicate that expression of sypAS56A suppresses the biofilm defect of the sypED192A mutant and further support our earlier epistasis experiments demonstrating that SypA functions downstream of SypE to control biofilms. We conclude from these studies that SypED192A inhibits biofilm formation through inactivation of SypA (Fig. 1B).
Figure 7. A sypAS56A mutant suppresses the sypED192A biofilm defect.
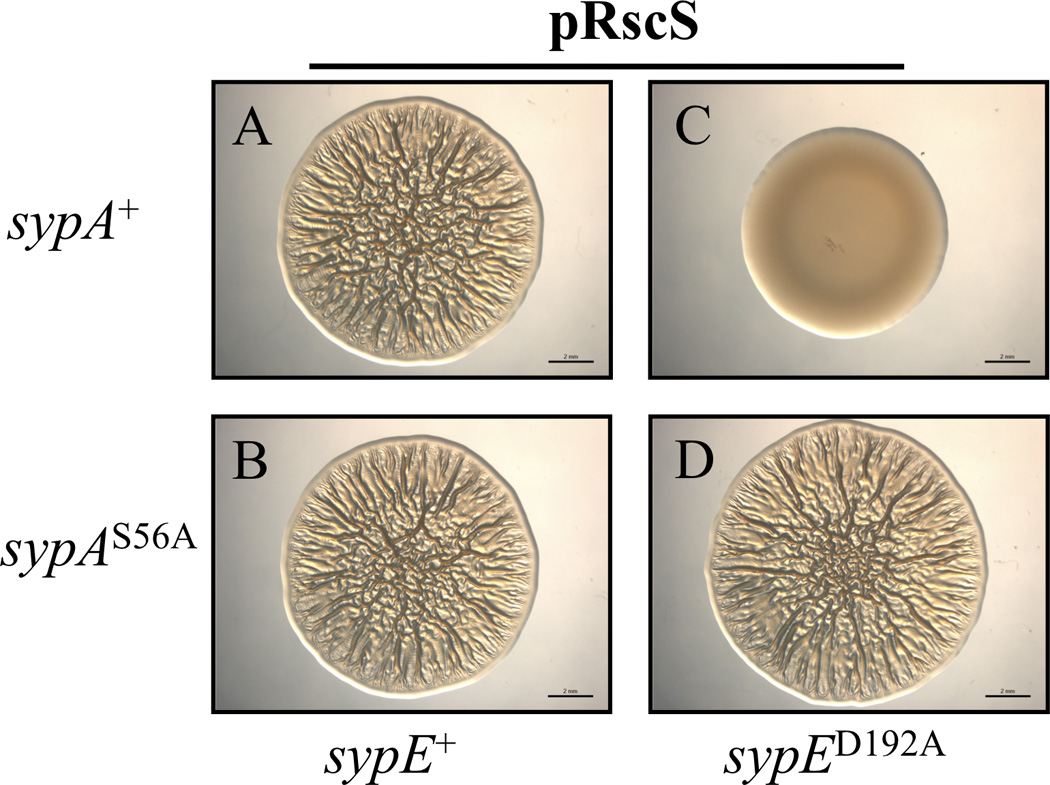
Assessment of RscS-induced wrinkled colony formation. The pRscS plasmid pARM7 was introduced into ΔsypE cells complemented with either wild-type sypE+ or the inhibitory sypED192A allele and expressing either wild-type sypA+ or sypAS56A. Cultures of the following strains were spotted onto LBS medium at 24°C and wrinkled colony formation was assessed at 48 h post spotting: sypE+ cells expressing wild-type sypA [KV6213] (A) or sypAS56A [KV6215] (B); sypED192A cells expressing either wild-type sypA [KV6214] (C) or sypAS56A [KV6216] (D). Images are representative of at least three independent experiments. Black bar represents 2 mm.
SypED192A promotes SypA phosphorylation in vivo
We next questioned whether SypA is indeed phosphorylated in V. fischeri (in vivo), and whether SypA phosphorylation corresponds with a loss of biofilm formation. To determine whether SypE promotes SypA phosphorylation in vivo, we introduced plasmids expressing epitope-tagged SypA or SypAS56A into our V. fischeri strains expressing either sypE+ or the inhibitory sypED192A allele and confirmed their expected biofilm phenotypes (Fig. S9). We next assessed the in vivo phosphorylation state of the tagged SypA proteins using the Phos-tag™ SDS-PAGE assay coupled with western blot analysis. Cells expressing wild-type alleles of sypA and sypE consistently exhibited two bands: a predominant, lower band corresponding to unphosphorylated SypA and a faint, slower migrating band corresponding to phosphorylated SypA (SypA~P) (Fig. 8, Lane 2). In contrast, cells expressing sypA+ and the inhibitory sypED192A allele exhibited only the upper SypA~P band (Fig. 8, Lane 3), indicating that the majority of the SypA protein was phosphorylated under those conditions. The ability of SypED192A to phosphorylate SypA depended upon its kinase domain, as only unphosphorylated SypA was observed in cells expressing SypED192A, N52A, a SypE mutant in which the constitutively inhibitory activity was disrupted by a mutation in the kinase domain (Morris et al., 2011) (Fig. S10). Finally, cells co-expressing the sypAS56A mutant and either sypE+ (Fig. 8, Lane 4) or sypED192A (Fig. 8, Lane 5) exhibited only the lower band corresponding to non-phosphorylated SypA. No shifted SypA-HA bands were observed upon resolving cell lysates on gels lacking Phos-tag™ acrylamide (Fig. S11). These results demonstrate that SypA is indeed phosphorylated in vivo, and that SypA phosphorylation corresponds to inhibition of biofilm formation by SypE (e.g., in cells expressing inhibitory SypED192A). They also corroborate our in vitro analyses indicating that residue S56 is required for SypA phosphorylation. Together, these data support a model in which SypE inhibits biofilms by promoting phosphorylation, and thus inactivation, of SypA (Fig. 1B).
Figure 8. SypE promotes SypA phosphorylation in vivo.

Western blot analysis of V. fischeri cell lysates analyzed on Phos-tag™ acrylamide gels. Soluble lysates from indicated V. fischeri strains were resolved by SDS-PAGE on 25 µM Phos-tag™ acrylamide gels and the proteins were detected by western blot analysis using anti-HA antibody. ΔsypA ΔsypE cells containing wild-type sypE+ [KV6424] and carrying pRscS plasmid (pCLD46) and plasmids expressing either untagged sypA+ (pARM13) (Lane 1), HA-tagged sypA+ (pARM36) (Lane 2), or HA-tagged sypAS56A (pARM78) (Lane 4); ΔsypA ΔsypE cells containing inhibitory sypED192A [KV6425] and carrying pCLD46 and plasmids expressing either HA-tagged sypA+ (pARM36) (Lane 3), or HA-tagged sypAS56A (pARM78) (Lane 5). (+) indicates cells expressing wild-type sypA-HA and/or sypE. (S56A) indicates cells expressing the sypAS56A mutant. (D192A) indicates cells expressing the sypED192A mutant. (−) indicates cells expressing untagged sypA. Images are representative of at least three independent experiments.
SypA functions downstream of SypE to control host colonization
Previous work has shown a correlation between biofilm-forming competence and symbiotic colonization by V. fischeri (Yip et al., 2005; Yip et al., 2006; Morris et al., 2011). Thus, we sought to verify the in vivo relevance of sypA during host colonization using the various sypA mutant strains. Because rscS expressed from the chromosome is sufficient to promote biofilm formation during symbiotic colonization (Yip et al., 2006), we performed these experiments in the absence of rscS overexpression.
First, we competed ΔsypA mutant cells against wild-type cells in mixed inoculation experiments, and found that the sypA mutant exhibited a dramatic competitive colonization defect (mean Log Relative Competitive Index (RCI), 1.38 +/− 0.51) (Fig. 9A). This defect in colonization could be abrogated by complementation with a wild-type allele of sypA+ (mean Log RCI, 0.15 +/− 0.47) (Fig. 9B). These data indicated that sypA is required for efficient colonization of E. scolopes.
Figure 9. Active SypA is required for host colonization.

(A and B) Competitive colonization with wild-type (WT) V. fischeri and select sypA strains. Newly hatched squid were exposed to a mixed inoculum of WT cells and either ΔsypA [KV5079] cells (A) or ΔsypA cells complemented with wild-type sypA+ [KV5479] (B). The Log RCI is plotted on the x-axis. The position of the circles on the y-axis is merely for spacing. Each circle represents a single animal. Closed circles indicate animals containing no sypA mutant cells. The black diamond and errors bars indicate the average Log RCI ± SD for the indicated data set. Data shown are representative of at least three independent experiments. (C) Single-strain colonization by WT and sypA mutant strains. Newly hatched squid were exposed for 18 h to WT cells carrying empty vector (EC) [KV4389] or ΔsypA cells carrying empty vector (EC) [KV5079] or complemented with either wild-type sypA+ [KV5479] or sypAS56D [KV5480]. As a negative control, aposymbiotic (APO) juvenile squid were maintained in bacteria free water. Each circle represents the number of V. fischeri cells recovered from an individual animal. The dashed line indicates the limit of detection (14 CFU/squid). The black bar indicates the average CFU for 10 animals. Data shown are from one experiment and are representative of at least three independent experiments.
Next, we performed single-strain colonization assays. 18 h after exposure to V. fischeri, animals inoculated with wild-type cells contained bacterial levels of about 105 colony-forming units (CFU) per animal (mean= 1.2 ×105 cfu; Fig. 9C). In contrast, animals exposed to ΔsypA cells largely remained uncolonized, while those that were colonized contained significantly fewer (2–3 logs decreased) bacteria (mean= 3.9 × 102 CFU; Fig. 9C). As in the competitive colonization assays, we found that complementation with a wild-type allele of sypA+ restored colonization to levels similar to that of wild-type cells (mean= 8.1 × 104 cfu; Fig. 9C).
Our in vitro biofilm studies suggested that phosphorylation inhibits SypA activity. To explore the dependence of symbiotic colonization on unphosphorylated SypA, we assessed colonization by the ΔsypA mutant expressing the SypAS56D protein, which mimics the phosphorylated state of SypA and fails to promote biofilms. As expected, the SypAS56D strain exhibited a severe colonization defect, similar to that observed for the ΔsypA mutant (mean= 1.1 × 103 cfu; Fig. 9C). The data further support our hypothesis that phosphorylation inhibits or inactivates SypA and thus impairs colonization (Fig. 1B).
Finally, to test our hypothesis that SypE inhibits colonization through phosphorylation of SypA, we examined the ability of the “constitutively active” sypAS56A allele to bypass the inhibition of colonization resulting from the sypED192A (constitutive kinase) allele. As previously observed, animals exposed to wild-type V. fischeri cells contained roughly 105 cfu/animal (mean= 6.8 × 105 cfu; Fig. 10), while sypED192A (sypA+) cells exhibited a severe defect in host colonization: several animals remained un-colonized, while colonized animals exhibited a 2–3 log decrease in bacterial loads (mean = 5.66 × 103 cfu; Fig. 10) (Morris et al., 2011). In contrast, sypED192A cells expressing the sypAS56A allele colonized the animals to levels similar to wild-type V. fischeri cells (mean= 3.68 × 105 cfu; Fig. 10). These data demonstrate that the colonization defect of the sypED192A mutant can be suppressed by a constitutively active, SypE-insensitive derivative of SypA, indicating that the effect of SypED192A during colonization is to inhibit SypA activity. Furthermore, these results support our in vitro biofilm assays, demonstrating that SypE functions through SypA to control both biofilms and colonization (Fig. 1B). Finally, because these experiments utilize V. fischeri strains that do not overexpress rscS, they demonstrate the biological relevance of these regulators in controlling host colonization.
Figure 10. SypE inhibits colonization through inactivation of SypA.

Single-strain colonization by wild-type (WT) V. fischeri and select mutant strains. Newly hatched squid were exposed for 18 h to either WT cells containing empty Tn7 cassette (EC) [KV4389] or sypED192A mutant cells expressing wild-type sypA [KV6214] or sypAS56A [KV6216]. As a negative control, aposymbiotic (APO) juvenile squid were maintained in bacteria free water. Each circle represents the number of V. fischeri cells recovered from an individual animal. The dashed line indicates the limit of detection. The black bar indicates the average CFU for 10 animals. Data shown are from one experiment and are representative of at least three independent experiments.
Discussion
In this study, we sought to determine the mechanism by which the novel RR SypE restricts biofilm formation and host colonization in V. fischeri. SypE was previously shown to both inhibit and promote syp biofilm formation through the opposing activities of its N- and C-terminal effector domains, respectively (Morris et al., 2011). Genetic and bioinformatic analyses suggested that SypE’s inhibitory N-terminal domain might possess serine kinase activity, while its positive acting C-terminal domain might function as a serine phosphatase. We therefore hypothesized that SypE likely regulates biofilms by controlling the phosphorylation state of a downstream target protein(s). In this report, we demonstrated that SypE indeed functions as both a serine kinase and a serine phosphatase, and that SypE exerts its impact on biofilm formation by controlling the phosphorylation state of another syp-encoded regulatory protein, SypA.
We first focused our attention on SypA as a potential target of SypE due to the similarity of these proteins to regulatory proteins found in partner-switching systems. Specifically, the N- and C-terminal effector domains of SypE are similar to the B. subtilis partner-switching proteins RsbW and RsbU, respectively, while SypA is similar to the STAS-domain protein RsbV (Morris and Visick, 2010). In B. subtilis, RsbW and RsbU interact with and regulate the phosphorylation state of RsbV (Yang et al., 1996)(Fig. 1A). The prediction that SypE likewise interacts with SypA was supported by the results of our co-immunoprecipitation studies: antibodies directed against epitope tags present on either SypE or SypA consistently promoted immunoprecipitation of both proteins (Fig. 2A). Interaction with SypA appeared to be mediated by the N-terminus of SypE, as a derivative that included only the N-terminal domain co-immunoprecipitated with SypA, while an N-terminal deletion mutant failed to do so (Fig. 2B). Given our in vitro phosphatase results demonstrating that SypE’s C-terminal domain (SypECTD) is sufficient to promote SypA dephosphorylation (Fig. 4), we propose that the C-terminal domain also interacts with SypA, but that this interaction may be transient and, therefore, not evident in the co-immunoprecipitation assay. The finding that SypE’s RsbW-like domain mediates interaction with SypA is consistent with that observed for characterized B. subtilis orthologs, in which binding of RsbV to RsbW inhibits the anti-sigma activity of RsbW (Dufour and Haldenwang, 1994). Whether the binding between SypE’s N-terminus and SypA plays any role in biofilm regulation remains to be determined. It is possible that binding by SypE may serve to sequester SypA and thereby control SypA activity. This possibility awaits further studies.
In any case, control over SypA’s phosphorylation state is critical for biofilm control: (1) a SypAS56D mutant, predicted to mimic the phosphorylated or “inactive” state of SypA, failed to complement the biofilm defect of the ΔsypA strain, while a non-phosphorylatable SypA mutant (SypAS56A) was fully competent to promote biofilms (Figs. 5 and S5); (2) Phos-tag™ analysis showed that SypA phosphorylation indeed occurs in vivo and its phosphorylation state corresponded with biofilm formation: SypA was largely unphosphorylated under biofilm-promoting conditions (e.g., in cells expressing wild-type sypE), but predominantly phosphorylated under conditions that inhibit biofilm formation (e.g., in cells expressing the inhibitory sypED192A allele) (Figs. 8 and S9); and (3) The non-phosphorylatable SypAS56A mutant promoted biofilm formation even in the presence of the constitutive kinase mutant SypED192A (Fig. 8 and S9). Together, these findings support a model in which SypE regulates biofilm formation by controlling the phosphorylation state, and therefore the activity, of SypA (Fig. 1B).
We also obtained compelling evidence that the phosphorylation state of SypA plays a critical role in controlling host colonization: (1) Similar to the ΔsypA mutant, cells expressing the phosphorylated mimic of sypA (sypAS56D), which failed to support biofilm formation in culture, were dramatically impaired in their ability to colonize (Fig. 9C); and (2) Expression of the non-phosphorylatable version of sypA (sypAS56A) suppressed the severe defect in host colonization (and biofilm formation) caused by the constitutive kinase allele of sypE (sypED192A) (Fig. 10). We conclude from these results that the regulation of SypA activity by SypE is a critical mechanism by which V. fischeri controls biofilm formation and host colonization. Our ability to manipulate and evaluate the in vivo phosphorylation state of SypA and correlate it with biofilm formation and colonization competence provides both substantial insight into the natural processes occurring in V. fischeri and a robust model for probing the control over these processes.
In our analysis of the SypE-SypA signaling network, we confirmed that SypE and SypA indeed function in a single pathway to control biofilms (Fig. 6). Furthermore, we found that SypA functions downstream of SypE in this regulatory pathway. This result stands in contrast to the characterized partner-switching networks described in B. subtilis, in which the serine kinase/anti-sigma (RsbW) functions as the downstream regulatory protein (Yang et al., 1996). In traditional partner-switching systems, the interaction of the switch protein (RsbW) with either of its “partner” proteins (RsbV or σB) dictates the output of the regulatory switch (i.e. sigma factor activity), while the upstream antagonist (RsbV) indirectly controls the output response (sigma factor activity) through antagonism of RsbW (Yang et al., 1996)(Fig. 1A). Our studies, however, demonstrate that the ability of SypE to regulate biofilms and colonization is fully dependent upon SypA and the regulation of SypA activity (Fig. 1B). While we cannot rule out the possibility that SypE has additional regulatory targets, SypA is the clear downstream regulator for the phenotypes investigated here. These results suggest that while SypE and SypA share similarity with partner-switching orthologues, the mechanism by which these regulators control biofilms may deviate from the traditional partner switching paradigm characterized in B. subtilis and other Gram-positives.
Studies are underway to determine the mechanism by which SypA controls biofilm formation and, thus, host colonization. Preliminary evidence suggests that SypA functions to promote biofilm formation at a level below, but dependent on, SypG activation and syp transcription (A.R. Morris and K.L. Visick, unpublished data). Based on the similarity of SypA to RsbV-like antagonist proteins, we hypothesized that SypA may interact with an additional RsbW-like anti-sigma factor, and thus indirectly control the activity of a downstream sigma factor involved in biofilm formation. However, analysis of the V. fischeri genome for RsbW-like orthologues (other than SypE) failed to identify any potential candidate genes. Additionally, preliminary studies have yet to identify a potential sigma factor whose activity might be regulated by SypA.
The possibility also remains that SypA may function in a manner distinct from that described in B. subtilis and the other Gram-positive systems, and thus may not regulate activity of a sigma factor. Partner-switching orthologues have been described in several other Gram-negative systems, including Bordetella bronchiseptica and Chlamydia trachomatis. Interestingly, these reports suggest that while these partner-switching regulators are conserved in these systems, their regulatory mechanisms appear to deviate from the partner-switching paradigm described in B. subtilis. For example, it was reported that the btr partner-switching orthologues of B. bronchiseptica (BtrU-BtrV-BtrW) regulate type III secretion (TTS), but do not impact transcription of the known TTS genes (Mattoo et al., 2004, Kozak et al., 2005). The authors of this study proposed that the Btr partner switching proteins may not regulate a sigma factor, but instead may function post-translationally to control TTS, perhaps by interacting with hypothetical proteins involved in the secretory system (Kozak et al., 2005). Similarly, a study examining partner-switching orthologues in the Gram-negative pathogen Chlamydia trachomatis (RsbU-RsbV1-RsbV2-RsbW) failed to identify an interaction between these proteins and any of the three sigma factors encoded by the C. trachomatis genome (Hua et al., 2006). These studies suggest that while partner-switching orthologues are conserved in Gram-negative species, the regulatory systems in which these proteins function may differ from the traditional partner-switching paradigm observed in B. subtilis and other Gram-positive bacteria. Studies are underway to determine whether SypA promotes biofilms via interaction with a putative regulatory protein (e.g., a sigma factor) or possibly through a post-translational mechanism (e.g., interaction with any of the Syp structural proteins).
Along with previous work, this study demonstrates that biofilm formation in V. fischeri is a tightly controlled process, involving both traditional two-component regulators and partner-switching-like proteins. That regulatory elements from both signaling systems appear to be combined in the RR SypE distinguishes this protein from the traditional RR, and may represent a novel means by which a bacterium can control gene expression. Interestingly, SypE-like RRs also appear to be present in other bacterial systems, including other Vibrios, such as Aliivibrio salmonicida, and non-Vibrio species, including Aeromonas veronii and Pseudomonas aeruginosa (Hsu et al., 2008, Morris and Visick, 2010). What role these SypE-like proteins may play in these other bacterial systems remains largely untested. However, a recent study in the Gram-negative bacterium P. aeruginosa identified a RR, PA3346, that exhibits a domain architecture similar to SypE; PA3346 consists of an N-terminal REC domain, a central PP2C-like domain, and a C-terminal HATP (histidine kinase/ATPase) domain (Hsu et al., 2008). Importantly, the RR PA3346 was shown to regulate swarming motility through a partner-switching mechanism involving a SypA-like protein, PA3347 (Bhuwan et al., 2012). Therefore, SypE-like regulators may be utilized in diverse bacterial species as a means to integrate regulatory elements from distinct signaling systems to provide further control over gene expression.
Experimental Procedures
Bacterial strains, plasmids and media
The bacterial strains and plasmids used in this study are listed in Tables 1 and 2, respectively. V. fischeri strain ES114, an isolate from E. scolopes, was used as the parental strain in this study (Boettcher and Ruby, 1990). All V. fischeri derivatives were generated by conjugation, as previously described (Visick and Skoufos, 2001). E. coli strains Tam1 λ pir (Active Motif, Carlsbad, CA), DH5α (Invitrogen, Carlsbad, CA), and GT115 (Invivogen, San Diego, CA) were used for cloning and conjugative purposes. E. coli strains were grown in Luria Bertani media (LB) (Davis, 1980). V. fischeri strains were grown in complex media [Sea Water Tryptone (SWT) (Yip et al., 2005) or LBS (Graf et al., 1994). Agar was added to a final concentration of 1.5% for solid media. The following antibiotics were added to V. fischeri media, where necessary, at the indicated concentrations: chloramphenicol (Cm) 2.5 µg mL−1, erythromycin at 5 µg mL−1, and tetracycline (Tc) at 5 µg mL−1 in LBS and 30 µg ml−1 in SWT. The following antibiotics were added to E. coli media, where necessary, at the indicated concentrations: Cm at 25 mg/ml−1, kanamycin (Kan) at 50 µg ml−1, Tc at 15 µg/ml−1, or ampicillin (Ap) at 100 µg ml−1.
Table 1.
Strains used in this study.
| Strain | Relevant Genotype | Source or Reference |
|---|---|---|
| E. coli | ||
| DH5α | endA1 hsdR17 (rK− mK+) supE44 thi-1 recA1 relA Δ(lacIZYA-argF)U169 phoA [ϕ80dlac Δ(lacZ)M15] | Invitrogen |
| TAM1 λ pir | mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZ ΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL endA1 nupG attλ::pir+ | Active Motif |
| GT115 | F- mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZ ΔM15 ΔlacX74 recA1 endA1 Δdcm uidA(ΔMluI)::pir-116 ΔsbcC-sbcD | Invivogen |
| V. fischeri | ||
| ES114 | Wild-type V. fischeri | (Boettcher and Ruby, 1990) |
| KV4389 | attTn7:: ermR | (Morris et al., 2011) |
| KV4390 | ΔsypE attTn7:: ermR | (Morris et al., 2011) |
| KV4715 | ΔsypA | This study |
| KV4716 | ΔsypA ΔsypE | This study |
| KV5079 | ΔsypA attTn7:: ermR | This study |
| KV5479 | ΔsypA attTn7:: sypA+ ermR | This study |
| KV5480 | ΔsypA attTn7:: sypAS56D ermR | This study |
| KV5481 | ΔsypA attTn7:: sypAS56A ermR | This study |
| KV6205 | ΔsypE (sypA+) | This study |
| KV6206 | ΔsypE sypAS56D | This study |
| KV6213 | ΔsypE (sypA+) attTn7:: sypE+ ermR | This study |
| KV6214 | ΔsypE (sypA+) attTn7:: sypED192A ermR | This study |
| KV6215 | ΔsypE (sypAS56A) attTn7:: sypE+ ermR | This study |
| KV6216 | ΔsypE (sypAS56A) attTn7:: sypED192A ermR | This study |
| KV6392 | ΔsypA ΔsypE attTn7:: ermR | This study |
| KV6393 | ΔsypA ΔsypE attTn7:: sypA+ ermR | This study |
| KV6424 | ΔsypA ΔsypE attTn7:: sypE+ ermR | This study |
| KV6425 | ΔsypA ΔsypE attTn7:: sypED192A ermR | This study |
| KV6619 | ΔsypA ΔsypE attTn7:: sypEN52A, D192A ermR | This Study |
Table 2.
Plasmids used in this study.
| Name | Description | Derivation of plasmids generated in this study | Source or reference |
|---|---|---|---|
| pARM7 | RscS overexpression construct; tcR | N/A | (Morris et al., 2011) |
| pARM13 | pKV282 + sypA | ~700 bp sypA gene generated with primers 806 and 807 | This study |
| pARM25 | pKV282 + sypAS56D | Generated by site-directed mutagenesis of pARM13 with primers 878 and 849 | This study |
| pARM29 | pKV282 + sypAS56A | Generated by site-directed mutagenesis of pARM13 with primers 877 and 849 | This study |
| pARM35 | pKV282 + sypA-FLAG | ~700 bp sypA-FLAG generated with primers 806 + and 1040 | This study |
| pARM36 | pKV282 + sypA-HA | ~700 bp sypA-HA generated with primers 806 and 1041 | This study |
| pARM37 | pEVS79 + sequences flanking sypA | Sequences flanking sypA were generated with primers 798, 799, 800 and 801 | This study |
| pARM47 | pEVS107 + sypE | N/A | (Morris et al., 2011) |
| pARM54 | pEVS107 + sypED192A | N/A | (Morris et al., 2011) |
| pARM77 | pKV282 + sypAS56D-FLAG | ~ 700 bp sypAS56D-FLAG, generated using pARM25 as a template and primers 806 and 1041 | This study |
| pARM78 | pKV282 + sypAS56A-HA | ~ 700 bp sypAS56A + HA-epitope, generated using pARM29 as a template and primers 806 and 1040 | This study |
| pARM79 | pKV282 + sypAS56A-FLAG | ~ 700 bp sypAS56A + FLAG-epitope, generated using pARM29 as a template and primers 806 and 1041 | This study |
| pARM80 | pVSV105 + sypE-FLAG | N/A | (Morris et al., 2011) |
| pARM101 | pEVS107 + sypEN52A, D192A | N/A | Morris et al., 2011) |
| pARM131 | pEVS107 + sypA | ~0.7 Kb sypA (and upstream region, including native promoter), generated with primers 806 and 807 | This study |
| pARM132 | pEVS107 + sypAS56D | ~ 0.7 Kb sypAS56D (and upstream sequences, including native promoter), generated by site-directed mutagenesis using pARM13 as a template and primers 849 and 878 | This study |
| pARM133 | pEVS107 + sypAS56A | ~ 0.7 Kb sypAS56A (and upstream sequences, including native promoter), generated by site-directed mutagenesis using pARM13 as a template and primers 849 and 877 | This study |
| pARM134 | pJET1.2 + sypA and flanking sequences | sypA and sequences flanking it were generated with primers 423 and 935 | This study |
| pARM135 | pKV363 + sypA and flanking sequences | sypA and flanking sequences were subcloned from pARM134 | This study |
| pARM136 | pVSV105 + sypENTD-FLAG | ~500 bp NTD of sypE generated with primers 462 and 1043 | This study |
| pARM141 | pGEX-5X-1 + sypE | ~1.5 Kb full-length sypE generated with primers 256 and 1475 cloned to construct GST fusion | This study |
| pARM152 | pGEX-5X-1 + sypECTD | ~ 700 bp CTD of sypE, generated with primers 911 and 1475 | This study |
| pARM157 | pGEX-5X-1 + sypA-FLAG | ~350 bp sypA generated with primers 1041 and 1474, cloned to construct GST fusion | This study |
| pARM160 | pKV363 + sypAS56A and sequences flanking native sypA | sypAS56A and sequences flanking native sypA, generated by substituting sypAS56A for sypA+ in pARM134, then subcloned into pKV363 | This study |
| pARM162 | pVSV105 + sypEΔNTD-FLAG | ~1.2 Kb sypEΔNTD-FLAG generated with primers 868 and 921 | This study |
| pCLD46 | pVSV105 + rscS | ~ 2.7 kb rscS1 allele subcloned from pKG11 (Yip et al., 2006) | This study |
| pCLD48 | pVSV105 + sypE+ | N/A | (Hussa et al., 2008) |
| pCLD64 | pVSV105 + sypENTD | ~500 bp sypENTD, generated with primers 462 and 911 | This study |
| pEVS79 | Cloning vector, ColE1 origin, CmR | N/A | {Stabb, 2002 #24 |
| pEVS107 | Mini-Tn7 delivery plasmid; mob; KanR, EmR | N/A | (McCann et al., 2003) |
| pEVS104 | Conjugal helper plasmid (tra trb), KanR | N/A | (Stabb and Ruby, 2002) |
| pGEX-5X-1 | GST tag protein expression vector, ApR | N/A | Amersham Biosciences |
| pJET1.2 | Commercial cloning vector; ApR | N/A | Fermentas |
| pKV282 | Mobilizable vector; TcR | N/A | (Morris et al., 2011) |
| pVSV105 | Mobilizable vector, R6Kori ori(pES213) RP4 oriT cat; cmR | N/A | (Dunn et al., 2006) |
Molecular techniques
The sypA alleles used in this study were generated by PCR amplification using the primers listed in Table 3. PCR products were cloned into the pJET1.2 cloning vector (Fermentas, Glen Burnie, MD), and subsequently subcloned into mobilizable plasmid pKV282 (Morris et al., 2011) using standard molecular techniques. For chromosomal insertion at the Tn7 site, the sypA genes and the upstream PsypA promoter were subcloned into the mini-Tn7 delivery vector pEVS107 (McCann et al., 2003). The alleles were inserted into the chromosomal Tn7 site of V. fischeri using a tetraparental mating as previously described (McCann et al., 2003, Morris et al., 2011). To generate site-directed mutations in sypA, we utilized the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Plasmid pARM13 (Table 2) served as the template for the primer 849 and select sypA mutagenic primers (Table 3). Generation of the desired mutations was confirmed by sequence analysis using the Genomics Core Facility at the Center for Genetic Medicine at Northwestern University (Chicago, IL) and ACGT, inc (Wheeling, IL). FLAG and HA epitope fusions to the C-terminus of SypA and SypE were generated by standard PCR using the primers listed Table 3, and the resulting alleles were cloned into mobilizable vectors, pKV282 (Morris et al., 2011) or pVSV105 (Dunn et al., 2006).
Table 3.
Primers used in this study
| Name | Sequence (5’-3’) |
|---|---|
| 256 | TTTTTCTGCACTTATTGATTCTCAATTAACAGC |
| 393 | GCTACACTTTCACTAGACGC |
| 423 | GGTTGACAGGTTTCTTGGCG |
| 462 | GTCCAAAGAAACCGATTTTTATC |
| 559 | GGTACCTCATTCCGATTCTTCATAG |
| 798 | CTGCAGTTCCATAATAAGCTCCTAGG |
| 799 | CTGCAGCATTAATTAGTGCAAAACACC |
| 800 | TTTTTTGGATCCCCTGATTCTTGAGCATTAC |
| 801 | CAGGAACGAAAATCGCATC |
| 806 | AGCTTCTTCCTTATAGTTATGATG |
| 807 | ATGTGTCATACAGTTAAAATGGTG |
| 849 | CCTGTGTGAAATTGTTATCCG |
| 868 | GTGGTGTAATCATGGAGCGTTCCCCTTCCCAT |
| 877 | GTAGCCTTTTTAGATGCGTCAGGTATTGGCGCT |
| 878 | GTAGCCTTTTTAGATGACTCAGGTATTGGCGCT |
| 911 | CTTAATGGGAAGGGGAACGCTC |
| 921 | ACCCGGGTTATTTATCATCATCATCTTTATAATCTTGATTCTCAATTAACAG |
| 935 | CTGCAGTGTTTTATCCGAAGGTAACCC |
| 1040 | ACCCGGGTTATGCATAATCTGGAACATCATATGGATAATGCGTTGTTTTATTAACAGG |
| 1041 | ACCCGGGTTATTTATCATCATCATCTTTATAATCATGCGTTGTTTTATTAACAGG |
| 1043 | ACCCGGGTTATTTATCATCATCATCTTTATAATCATGGGAAGGGGAACGCTC |
| 1474 | AAAGATCTTGGAACTACATCAATTCGAATCAAATG |
| 1475 | AAAGATCTTGAATTCTACTTTACTTTTTTCAG |
| 1477 | AAAGATCTAAAGATCTTGGCCCATACTCTATTACCACAAG |
To construct the ΔsypA deletion, we used PCR to amplify and clone sequences approximately 2 kb upstream and downstream of sypA into the pJET1.2 cloning vector and subsequently plasmid pEVS79 (Stabb and Ruby, 2002). We then ligated these fragments to form a plasmid, pARM37, which was subsequently used to introduce the ΔsypA deletion into V. fischeri strains as previously described (Hussa et al., 2008). Colony PCR analysis was performed to confirm deletion of sypA.
sypA allelic replacement
To restore wild-type sypA or insert the mutant sypA allele at the native locus, we generated plasmids pARM135 and pARM160, which contained 750 bp upstream and 500 bp downstream of sypA and sypAS56A, respectively. We introduced the plasmids into the recipient strain (KV4716) by conjugation and isolated stably cm resistant (cmR) colonies, indicating integration of the sypA suicide construct into the chromosome. Stable cmR colonies were then cultured in LBS containing 0.2% arabinose to induce expression of the ccdB toxin gene, and plated onto LBS agar plates containing 0.2% arabinose. Arabinose induction selects for those cells that have undergone a second recombination event, resulting in excision of the suicide plasmid and either restoration of the original ΔsypA allele or replacement with the wild-type or sypAS56A allele. Colony PCR was performed to identify those colonies containing the restored sypA allele.
SypA-FLAG western immunoblotting
Indicated V. fischeri strains were cultured in LBS containing Tc overnight at 28 °C with shaking. 1 mL of cell cultures were spun down and cells lysed in 500 µL 2X sample buffer (4% SDS, 40 mM Tris pH 6.3, 10% glycerol). Samples were resolved on 10% SDS-PAGE gels (10% 29:1 acrylamide: N, N’-methylene-bis-acrylamide, 375 mM Tris pH 8.6, 0.1% SDS), and transferred to PVDF membranes. SypA-FLAG proteins were detected by western blot analysis using a rabbit anti-FLAG antibody (Sigma-Aldrich, St. Louis, MO) followed by a secondary, donkey anti-rabbit IgG antibody (Sigma-Aldrich, St. Louis, MO) conjugated to horseradish peroxidase (HRP), and visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fischer Scientific, Rockford, IL).
Co-Immunoprecipitation of SypE and SypA
Plasmids SypA-HA (pARM36) and SypE-FLAG (pARM80), SypENTD-FLAG (pARM136), SypEΔNTD–FLAG (pARM162), or the appropriate empty control vectors, were introduced by conjugation into ΔsypA ΔsypE V. fischeri cells. Bacterial strains were cultured in LBS containing Tc and Cm at 28 °C overnight with shaking and subsequently sub-cultured to an OD600 of 0.5. Cells (~0.10 g) were harvested by centrifugation (13,000 × g for 10 min), and washed in 1 mL of phosphate-buffered saline (PBS). Samples were subsequently prepared and immunoprecipitated using the Dynabeads Co-immunoprecipitation Kit (Invitrogen, Oslo, Norway). Cell samples were re-suspended and lysed in 900 uL of Extraction Buffer (EB) (1X IP buffer, 100 mM NaCl, 1 mM DTT) (Invitrogen, Oslo, Norway). Rabbit anti-FLAG and anti-HA antibodies (25 µg, Sigma-Aldrich, St. Louis, MO) were coupled to magnetic Dynabeads (5 mg, Invitrogen) according the manufacturer’s protocol. As a negative control, dynabeads (1.5 mg) were coupled with non-specific mouse anti-rabbit IgG antibody (5 µg, Promega). For the co-immunoprecipitation, antibody-coupled beads were incubated with 900 µL of whole cell extracts at 4°C with rocking for 1 h. Eluted samples were diluted with 2X sample buffer and resolved using SDS-PAGE. Samples were then transferred to PVDF membrane and proteins were detected using rabbit anti-FLAG and anti-HA antibodies followed by a HRP-conjugated secondary antibody as described earlier.
Purification of SypA and SypE proteins
The sypE (either wild-type SypE or the SypE C-terminal domain alone) and sypA-FLAG (either wild-type SypA or the SypAS56A mutant) alleles were PCR cloned into the GST-fusion vector pGEX-5X-1 (Amersham Biosciences) to generate N-terminal glutathione-S-transferase (GST) fusions (Table 2). The resulting plasmids were transformed into E. coli Tam1 cells. E. coli cultures were grown at 28°C to an OD600 of 0.5 and overexpression of the GST-fusion proteins was induced by the addition of 0.4 mM IPTG followed by further culturing overnight. Cells were harvested by centrifugation (10,000 × g), lysed using BugBuster protein extraction reagent (Novagen, EMD Chemicals Inc, San Diego, CA), and diluted in resuspension buffer (50 mM Na2HPO4, 1 M NaCl, 0.1% Tween-20, pH 8.0). Samples were centrifuged (12,000 × g) and the supernatants applied to a Glutathione Sepharose 4B column (GE Healthcare, Piscataway, NJ), washed with cold 1X PBS, and the bound proteins were eluted by the addition of glutathione elution buffer (10 mM reduced glutathione, 50 mM Tris-HCl, pH 8.0). Eluted proteins were dialyzed using Slide-A-Lyzer dialysis cassettes in storage buffer (50% glycerol, 10 mM Tris-HCl, 100 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol [DTT], 0.1 mM EDTA) (10 K MWCO, Thermo Scientific).
To obtain purified phosphorylated SypA (SypA~P), E. coli GT115 cells were co-transformed with the GST-SypA-FLAG plasmid (pARM157) and either plasmid pCLD64 expressing only the N-terminal kinase domain of SypE, SypENTD, (Morris et al., 2011) or empty vector pVSV105. Overexpression and purification of the protein was performed as described above.
To obtain purified SypA-FLAG protein lacking the N-terminal GST-fusion, the GST-purified SypA-FLAG proteins (either wild-type or phosphorylated SypA; 50 µg) were incubated in cleavage buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM CaCl2) containing Factor XA (50 µg) (New England BioLabs Inc., Ipswich, MA) at 23° C for 16 h. Samples were subsequently applied to a Glutathione Sepharose 4B column to remove any remaining GST-tagged proteins, and the flow-through containing cleaved SypA-FLAG protein were collected. Cleavage of the GST-tag was confirmed by western blotting using an anti-FLAG antibody as described above (see supplemental figure S2).
in vitro kinase assay
Purified GST-SypA containing a C-terminal FLAG epitope tag (either wild-type SypA or SypAS56A; 1 µg) was incubated at 28°C for 10 min in phosphorylation buffer (50 mM Tris-HCl, pH 7.5, 50 mM KCl, 5 mM MgCl2, 1 mM DTT, 0.1 mM EDTA) in the presence or absence of 2 mM ATP. Purified wild-type GST-SypE (2 µg) was added to the reactions and the samples incubated for an additional 30 min. Reactions were terminated by the addition of 2X sample buffer and proteins were resolved on 10% SDS-PAGE gels containing 20 µM Phos-tag™ acrylamide (WAKO chemicals, Richmond, VA) and 40 µM MnCl2. Gels were fixed for 15 min in standard transfer buffer (20% MeOH, 50 mM Tris, 40 mM glycine) containing 1 mM EDTA to remove Mn2+ from the gel. Gels were incubated for an additional 20 min in transfer buffer without EDTA. Proteins were transferred to a PVDF membrane and detected by western blot analysis using an anti-FLAG antibody as described earlier. As a control, the kinase assays were repeated using GST-purified SypA-FLAG protein cleaved of its N-terminal GST-tag (as described above).
in vitro phosphatase assay
Phosphorylated GST-SypA protein containing a C-terminal FLAG epitope tag (SypA~P) was purified from E. coli cells co-expressing the N-terminal kinase domain of SypE (SypENTD) as described earlier. To confirm the phosphorylation state of the purified protein, samples were analyzed using Phos-tag™ acrylamide SDS-PAGE coupled with anti-FLAG western blotting, as described earlier. For the phosphatase assay, purified SypA~P was preincubated alone in phosphatase buffer (50 mM Tris-HCl, 10 mM NaCl, 10 mM MgCl2, 1 mM DTT) at 28°C, followed by the addition of increasing concentrations of GST-purified SypECTD protein. Samples were incubated at 28°C for an additional 30 min and the reactions were terminated by the addition of 2X sample buffer. To assess the phosphorylation state of SypA, samples were analyzed using Phos-tag™ acrylamide SDS-PAGE coupled with anti-FLAG western blot analysis as described for the in vitro kinase assay. As a control, the phosphatase assays were repeated using GST-purified P~SypA protein cleaved of its N-terminal GST-tag (as described above).
Wrinkled colony assay
To observe wrinkled colony formation, strains were streaked onto LBS agar plates containing Tc. Single colonies were then cultured with shaking in LBS broth with Tc overnight at 28°C and then sub-cultured to an OD600 of 0.1 in 5 mL of fresh medium. Cells were spun down, washed twice in 70% artificial seawater (ASW) (0.10 M MgSO4, 19.7 mM CaCl2, 0.6 M NaCl, 20 mM KCl), and re-suspended in 70% ASW and diluted to an OD of 0.1. 10 µL of re-suspended cultures were spotted onto LBS agar plates and grown for 48 h at 22°C. Images of the spotted cultures were acquired at the indicated time points using a Zeiss stemi 2000-C dissecting microscope
Pellicle Assay
Strains were grown with shaking in LBS with Tc at 22°C overnight and then subcultured to an OD of 0.1 in 1.5 mL of fresh medium in 24-well microtiter dishes. Cultures were then grown at room temperature for up to 48 h. To permit visualization, the pellicles were disrupted by probing the air-liquid interface with a sterile pipette tip. A pellicle is observed at a disruption at the culture surface. Cultures with no pellicle were scored as (−); cultures with a weak, easily disrupted pellicle were scored as (+); cultures with an intact pellicle were scored as (++). Images of the spotted cultures were acquired at the indicated time points using a Zeiss stemi 2000-C dissecting microscope.
Analysis of SypA phosphorylation in vivo
Indicated V. fischeri strains were streaked onto LBS agar plates containing Tc and Cm. Single colonies were then cultured overnight in LBS containing Tc and Cm at 24°C with shaking. Aliquots of cells (1 mL) were spun down, washed twice with 1X PBS, and lysed in 2X sample buffer. Samples were resolved on SDS-PAGE gels containing 25 µM Phos-tag™ acrylamide and 50 µM MnCl2. Samples were transferred to PVDF membranes and proteins detected using anti-HA western blot analysis as described for the in vitro kinase assay.
Squid colonization assays
To perform single-strain colonization assays, juvenile squid were placed into artificial seawater (ASW) (Instant Ocean; Aquarium Systems, Mentor, OH) containing roughly 1,000 cells per mL of seawater. Incubation was allowed to proceed for 18–24 h post inoculation, at which time the animals were washed in ASW and homogenized to release the light organ contents. Serial dilutions of the light organs were plated and colony-forming units (CFU) calculated. For competitive colonization assays, juvenile squid were placed into ASW containing approximately 1,000 V. fischeri cells per mL of seawater. Juvenile squid were inoculated with an approximate 1:1 ratio of mutant and wild-type cells, and incubation was allowed to proceed for 18 h. For these assays, sypA+ cells were marked with an erythromycin resistance (EmR) cassette within the chromosome at the Tn7 site. Reciprocal experiments were also performed in which ΔsypA cells contained the EmR marker. The ratio of bacterial strains within the light organs of the animals was assessed through homogenization/plating assays as described previously (Yip et al., 2006). The competitive colonization data are reported as the log-transformed relative competitive index (Log RCI). This index is generated by dividing the ratio of mutant to wild-type in the homogenate by the ratio present in the inoculum and calculating the log10 value of that number.
Supplementary Material
Acknowledgments
We thank Cynthia Darnell and Justin Eddy for their experimental contributions to this work. We also thank Alan Wolfe, Jonathan Visick and members of the Visick lab for their critical reading of the manuscript. This work was supported by NIH grant GM059690 award to K.L.V and a grant from the Conservation Medicine Center of Chicago awarded to A.R.M.
References
- Benson AK, Haldenwang WG. Bacillus subtilis sigma B is regulated by a binding protein (RsbW) that blocks its association with core RNA polymerase. Proc Natl Acad Sci U S A. 1993a;90:2330–2334. doi: 10.1073/pnas.90.6.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson AK, Haldenwang WG. Regulation of sigma B levels and activity in Bacillus subtilis. J Bacteriol. 1993b;175:2347–2356. doi: 10.1128/jb.175.8.2347-2356.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuwan M, Lee HJ, Peng HL, Chang HY. Histidine-containing phosphotransfer protein-B (HptB) regulates swarming motility through partner-switching system in Pseudomonas aeruginosa PAO1 strain. J Biol Chem. 2012;287:1903–1914. doi: 10.1074/jbc.M111.256586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettcher KJ, Ruby EG. Depressed light emission by symbiotic Vibrio fischeri of the sepiolid squid Euprymna scolopes. J Bacteriol. 1990;172:3701–3706. doi: 10.1128/jb.172.7.3701-3706.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourret RB. Receiver domain structure and function in response regulator proteins. Curr Opin Microbiol. 2010;13:142–149. doi: 10.1016/j.mib.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourret RB, Hess JF, Simon MI. Conserved aspartate residues and phosphorylation in signal transduction by the chemotaxis protein CheY. Proc Natl Acad Sci U S A. 1990;87:41–45. doi: 10.1073/pnas.87.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RW, Botstein D, Roth JR. Advanced bacterial genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1980. [Google Scholar]
- Diederich B, Wilkinson JF, Magnin T, Najafi M, Erringston J, Yudkin MD. Role of interactions between SpoIIAA and SpoIIAB in regulating cell-specific transcription factor sigma F of Bacillus subtilis. Genes Dev. 1994;8:2653–2663. doi: 10.1101/gad.8.21.2653. [DOI] [PubMed] [Google Scholar]
- Dufour A, Haldenwang WG. Interactions between a Bacillus subtilis anti-sigma factor (RsbW) and its antagonist (RsbV) J Bacteriol. 1994;176:1813–1820. doi: 10.1128/jb.176.7.1813-1820.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan L, Losick R. SpoIIAB is an anti-sigma factor that binds to and inhibits transcription by regulatory protein sigma F from Bacillus subtilis. Proc Natl Acad Sci U S A. 1993;90:2325–2329. doi: 10.1073/pnas.90.6.2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn AK, Millikan DS, Adin DM, Bose JL, Stabb EV. New rfp- and pES213-derived tools for analyzing symbiotic Vibrio fischeri reveal patterns of infection and lux expression in situ. Appl Environ Microbiol. 2006;72:802–810. doi: 10.1128/AEM.72.1.802-810.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin MY. Structural classification of bacterial response regulators: diversity of output domains and domain combinations. J Bacteriol. 2006;188:4169–4182. doi: 10.1128/JB.01887-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin MY. Diversity of structure and function of response regulator output domains. Curr Opin Microbiol. 2010;13:150–159. doi: 10.1016/j.mib.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf J, Dunlap PV, Ruby EG. Effect of transposon-induced motility mutations on colonization of the host light organ by Vibrio fischeri. J Bacteriol. 1994;176:6986–6991. doi: 10.1128/jb.176.22.6986-6991.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JL, Chen HC, Peng HL, Chang HY. Characterization of the histidine-containing phosphotransfer protein B-mediated multistep phosphorelay system in Pseudomonas aeruginosa PAO1. J Biol Chem. 2008;283:9933–9944. doi: 10.1074/jbc.M708836200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua L, Hefty PS, Lee YJ, Lee YM, Stephens RS, Price CW. Core of the partner switching signalling mechanism is conserved in the obligate intracellular pathogen Chlamydia trachomatis. Mol Microbiol. 2006;59:623–636. doi: 10.1111/j.1365-2958.2005.04962.x. [DOI] [PubMed] [Google Scholar]
- Hussa EA, Darnell CL, Visick KL. RscS functions upstream of SypG to control the syp locus and biofilm formation in Vibrio fischeri. J Bacteriol. 2008;190:4576–4583. doi: 10.1128/JB.00130-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics. 2006;5:749–757. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- Kinoshita-Kikuta E, Aoki Y, Kinoshita E, Koike T. Label-free kinase profiling using phosphate affinity polyacrylamide gel electrophoresis. Mol Cell Proteomics. 2007;6:356–366. doi: 10.1074/mcp.T600044-MCP200. [DOI] [PubMed] [Google Scholar]
- Kozak NA, Mattoo S, Foreman-Wykert AK, Whitelegge JP, Miller JF. Interactions between partner switcher orthologs BtrW and BtrV regulate type III secretion in Bordetella. J Bacteriol. 2005;187:5665–5676. doi: 10.1128/JB.187.16.5665-5676.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattoo S, Yuk MH, Huang LL, Miller JF. Regulation of type III secretion in Bordetella. Mol Microbiol. 2004;52:1201–1214. doi: 10.1111/j.1365-2958.2004.04053.x. [DOI] [PubMed] [Google Scholar]
- McCann J, Stabb EV, Millikan DS, Ruby EG. Population dynamics of Vibrio fischeri during infection of Euprymna scolopes. Appl Environ Microbiol. 2003;69:5928–5934. doi: 10.1128/AEM.69.10.5928-5934.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AR, Darnell CL, Visick KL. Inactivation of a novel response regulator is necessary for biofilm formation and host colonization by Vibrio fischeri. Mol Microbiol. 2011;82:114–130. doi: 10.1111/j.1365-2958.2011.07800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AR, Visick KL. Control of biofilm formation and colonization in Vibrio fischeri: a role for partner switching? Environ Microbiol. 2010;12:2051–2059. doi: 10.1111/j.1462-2920.2010.02269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyholm SV, Stabb EV, Ruby EG, McFall-Ngai MJ. Establishment of an animal-bacterial association: recruiting symbiotic vibrios from the environment. Proc Natl Acad Sci U S A. 2000;97:10231–10235. doi: 10.1073/pnas.97.18.10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabb EV, Ruby EG. RP4-based plasmids for conjugation between Escherichia coli and members of the Vibrionaceae. Methods Enzymol. 2002;358:413–426. doi: 10.1016/s0076-6879(02)58106-4. [DOI] [PubMed] [Google Scholar]
- Stock AM, Guhaniyogi J. A new perspective on response regulator activation. J Bacteriol. 2006;188:7328–7330. doi: 10.1128/JB.01268-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- Visick KL, Skoufos LM. Two-component sensor required for normal symbiotic colonization of Euprymna scolopes by Vibrio fischeri. J Bacteriol. 2001;183:835–842. doi: 10.1128/JB.183.3.835-842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelker U, Voelker A, Haldenwang WG. Reactivation of the Bacillus subtilis anti-sigma B antagonist, RsbV, by stress- or starvation-induced phosphatase activities. J Bacteriol. 1996;178:5456–5463. doi: 10.1128/jb.178.18.5456-5463.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Kang CM, Brody MS, Price CW. Opposing pairs of serine protein kinases and phosphatases transmit signals of environmental stress to activate a bacterial transcription factor. Genes Dev. 1996;10:2265–2275. doi: 10.1101/gad.10.18.2265. [DOI] [PubMed] [Google Scholar]
- Yip ES, Geszvain K, DeLoney-Marino CR, Visick KL. The symbiosis regulator rscS controls the syp gene locus, biofilm formation and symbiotic aggregation by Vibrio fischeri. Mol Microbiol. 2006;62:1586–1600. doi: 10.1111/j.1365-2958.2006.05475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip ES, Grublesky BT, Hussa EA, Visick KL. A novel, conserved cluster of genes promotes symbiotic colonization and σ54-dependent biofilm formation by Vibrio fischeri. Mol Microbiol. 2005;57:1485–1498. doi: 10.1111/j.1365-2958.2005.04784.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.