

Abstract
Voltage gated calcium channels (Ca2+ channels) are key mediators of depolarization induced calcium influx into excitable cells, and thereby play pivotal roles in a wide array of physiological responses. This review focuses on the inhibition of CaV2 (N- and P/Q-type) Ca2+-channels by G protein coupled receptors (GPCRs), which exerts important autocrine/paracrine control over synaptic transmission and neuroendocrine secretion. Voltage-dependent inhibition is the most widespread mechanism, and involves direct binding of the G protein βγ dimer (Gβγ) to the α1 subunit of CaV2 channels. GPCRs can also recruit several other distinct mechanisms including phosphorylation, lipid signaling pathways, and channel trafficking that result in voltage-independent inhibition. Current knowledge of Gβγ-mediated inhibition is reviewed, including the molecular interactions involved, determinants of voltage-dependence, and crosstalk with other cell signaling pathways. A summary of recent developments in understanding the voltage-independent mechanisms prominent in sympathetic and sensory neurons is also included.
Keywords: Calcium channel, G protein coupled receptor, inhibition, Gβγ, PKC, tyrosine kinase, PiP2, arachidonic acid, splice variant, SNARE
1. Voltage gated calcium channels
Voltage gated calcium channels (Ca2+ channels) are key mediators of depolarization induced calcium influx into excitable cells, which in turn mediates a wide array of physiological responses including the activation of calcium dependent enzymes, smooth muscle contraction, pacemaker activity and neurotransmitter release [1-8]. Ca2+ channels are also associated with a wide range of pathologies, including pain, epilepsy, migraine, cardiac arrhythmias and autism [9-14]. It is widely known that there are subtypes of Ca2+ channels with different pharmacological and biophysical properties, and distinct cellular and physiological functions [15-17]. In neurons, certain L-type Ca2+ channel isoforms are expressed at cell bodies and dendrites, and one of their key functions is the initiation of calcium dependent gene transcription events [18-22]. Other L-type channel subtypes are expressed in cochlear hair cells and photoreceptor nerve terminals where they regulate neurotransmitter release at ribbon synapses [23, 24]. T-type calcium channels are expressed in cell bodies as well as dendrites and one of their key functions is to regulate cellular excitability and neuronal firing properties [25-27], in addition to participating in secretion [28-30]. N-type and P/Q-type calcium channels are expressed at synaptic nerve terminals where their opening results in the release of neurotransmitters [1, 19, 31-34].
All Ca2+ channels are comprised of a pore forming Cavα1 subunit that contains the major structural features required for permeation, activation, and inactivation. The mammalian genome encodes ten different Cavα1 subunits that fall into three major families - Cav1 (L-type channels), Cav2 (N, P/Q- and R-types), and Cav3 (T-types) [17, 35]. The CaV1 and CaV2 families are high voltage activated (HVA) channels, and are heteromers comprised of a pore forming Cavα1 subunit as well as Cavα2-δ and Cavβ subunits [36-38] (Fig 1). In addition, these channels associate with calmodulin which is now considered part of the HVA channel macromolecular complex [39-44]. The Cavα1 subunit determines the Ca2+ channel subtype and is a large (~175-225 kDa) protein with four homologous transmembrane domains that are connected by cytoplasmic loops and bracketed by cytoplasmic N- and C-termini [37] (Fig 1). These cytoplasmic regions are key targets for second messenger regulation including protein kinases and G proteins, as we discuss here in detail. The Cavβ subunits are cytoplasmic proteins that associate with HVA α1 subunits at a highly conserved region within the domain I-II linker (termed the Alpha Interaction Domain – AID) [45-47]. These subunits are encoded by four different genes (for review see [48, 49]). The Cavα2-δ subunits are transcribed from one of four different Cavα2-δ genes, proteolytically cleaved and then reconnected via a disulfide bond (for review, see [50]). The α2 portion is located at the extracellular side of the channel, whereas the δ portion either spans the membrane or may be linked to the extracellular leaflet of the plasma membrane through a glycosylphosphatidylinositol (GPI) anchor [51]. The function of these ancillary subunits is to regulate channel properties and promote Cavα1 subunit trafficking to and stabilization at the plasma membrane [52-54] (for reviews see [48, 49, 55-57]. As we will outline below, Cavβ subunits also alter second messenger regulation the channel complex [58-61]. Finally, it should be noted that most Ca2+ channel subunits are subject to alternate mRNA splicing, thus greatly increasing the functional diversity of calcium channels [62-65]. Recently described RNA editing events that alter channel function add further complexity [66]. This then makes it challenging to precisely reconstitute all specific features of native calcium currents in transient expression systems. In this review, we focus on the CaV2 family of channels, and in particular their regulation by G protein coupled receptors.
Figure 1.
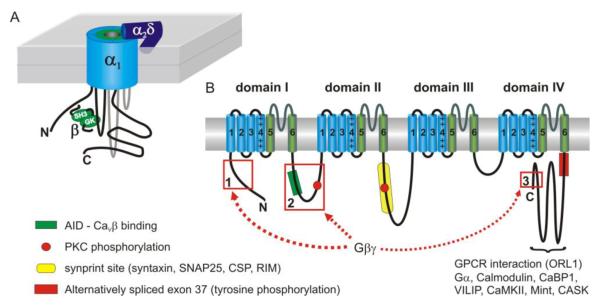
Schematic depiction of the topology and subunit composition of CaV2 voltage-gated Ca2+ channels. (A) Cartoon showing the 3D topology along with channel auxiliary subunits. The intracellular β subunit interacts through its guanylate kinase-like domain (GK) with the I-II linker of the α1 subunit (at the α-interaction domain or AID). The α2δ subunit is largely extracellular and likely GPI-anchored to the plasma membrane. (B) Topology of the pore forming α1 subunit. Four homologous repeats (domain I through domain IV) each consist of six transmembrane spanning α-helices (S1-S6) (blue or green cylinders) and a ‘P-loop’ between S5 and S6. The S5-S6 helices and P-loop comprise the pore domain of the channel (colored green), while S1-S4 (in particular S4 that has multiple charged residues) comprises the voltage sensor (colored blue). The intracellular N- and C-termini and the cytoplasmic loops connecting domains I-IV are important for interaction with other proteins including the auxiliary β subunit, synaptic proteins, Gβγ, GPCRs, calmodulin and other Ca2+ binding proteins (CaBP1, VILIP). These cytoplasmic domains are also targeted by second messenger pathways including phosphorylation by PKC, CaMKII, and tyrosine kinases. Alternative splicing greatly increases the functional diversity of the channels. For example, alternative splicing of exon37 on the proximal C-terminus controls inhibition of CaV2.2 channels by GPCRs in sensory neurons (see section 12 for more details).
2. G protein coupled receptors and heterotrimeric G proteins
G protein coupled receptors (GPCRs) are a large family of membrane proteins encoded by almost 800 human genes, and represent an important class of therapeutic targets [67, 68]. GPCRs are characterized by an extracellular N-terminus, seven transmembrane spanning alpha helices, and an intracellular C-terminus which couples to heterotrimeric G proteins. Extracellular ligand binding to the receptor leads to activation of the G proteins and a myriad of downstream intracellular signaling cascades. In human, sixteen genes encode G protein α subunits (Gα), and these are classified into four major families: Gs, Gi, Gq, and G12, in addition to transducin (Gαt) which is found in the retina. Five genes encode Gβ subunits, and twelve genes encode Gγ subunits (for reviews see[69-72]. Binding of agonist to the GPCR catalyzes the exchange of GDP to GTP on Gα causing conformational changes/dissociation of the Gα and Gβγ heterodimer [71, 73]. The liberated Gα and Gβγ are both capable of signaling to multiple downstream effectors, including voltage-gated Ca2+-channels as discussed in this article. Signaling is terminated by intrinsic GTPase activity of Gα and subsequent reassociation of the Gα-GDP subunit with the Gβγ heterodimer. This GTPase activity can be accelerated by a family of RGS proteins (regulator of G protein signaling) which thus influence the extent and duration of downstream events [74]. Receptor desensitization in the continued presence of agonist can also terminate signaling. Desensitization is complex, involving phosphorylation by PKA, PKC, or G protein coupled receptor kinases (GRKs) and uncoupling of the receptor from the downstream G proteins. Endocytic removal of the GPCR from the plasma membrane can also occur. GRKs recruited by Gβγ phosphorylate the C-terminus of the GPCR leading to recruitment of arrestins and the endocytic machinery [75, 76]. As discussed below (section 10), direct interaction of GPCRs and Ca2+ channels might result in co-internalization adding another dimension to channel modulation.
4. Inhibition of CaV2 channels by G protein coupled receptors
Neurotransmitter mediated inhibition of Ca2+ channels was first demonstrated ~30 years ago by Dunlap and Fischbach who reported that norepinephrine reduced the duration of action potentials [77] and the amplitude of ICa [78] in chick sensory neurons. It is now apparent that a variety of different neurotransmitters/neuromodulators acting on their cognate GPCRs inhibit ICa, and that this is important for controlling neurosecretion (for reviews see [79-85]). It is also known that GPCRs can recruit several distinct signaling pathways that converge on Ca2+ channels. The most widespread and intensively studied of these involves direct binding of Gβγ to the α1 subunit of CaV2 channels. As detailed below (section 5), Gβγ-mediated inhibition shifts the voltage-dependence of channel activation, is less prominent at depolarized membrane potentials, and is transiently relieved by large depolarizing voltage steps. Consequently, this mechanism is often termed voltage-dependent inhibition. GPCRs can also elicit voltage-independent inhibition of ICa which is mediated by several other distinct and generally less well characterized pathways including phosphorylation, lipid signaling pathways, and channel trafficking (see sections 10-12). While voltage-dependent inhibition is widespread throughout the nervous system, voltage-independent inhibition is more variable in extent and mechanism but seems particularly prominent in sensory and sympathetic neurons. In this review we first consider Gβγ-mediated inhibition, including recent developments and crosstalk with other cell signaling pathways. Then we outline some of the voltage-independent mechanisms prominent in sympathetic and sensory neurons.
5. Voltage-dependent inhibition mediated by Gβγ
Voltage-dependent inhibition primarily targets CaV2.1 (P/Q-type) and CaV2.2 (N-type) channels, although CaV2.3 channels are also inhibited by similar mechanisms (see section 5.4 below). The voltage-dependent nature of the inhibition was first demonstrated by Bean [86], who showed that the decrease in current amplitude was not due to a loss of channels per se, but rather a shift in the gating properties that could be overcome by strong depolarization. Several hallmarks are characteristic of this voltage-dependent mechanism: In whole cell recordings, the inhibition of peak ICa amplitude is diminished at depolarized membrane potentials; activation kinetics are slowed; the voltage-dependence of activation is shifted to more depolarized potentials; a conditioning prepulse to depolarized potentials relieves most of the inhibition and normalizes channel kinetics (termed prepulse relief or prepulse facilitation). Figure 2 shows an example of voltage-dependent inhibition of ICa. Prominent slowing of activation kinetics and prepulse relief of the inhibition is clearly seen. Voltage-dependent relief of the inhibition can also occur at least to some extent during more physiologically relevant stimuli such as high frequency trains of action potential-like waveforms [87-92]. In turn, this might contribute to short term synaptic plasticity at some synapses [93].
Figure 2.
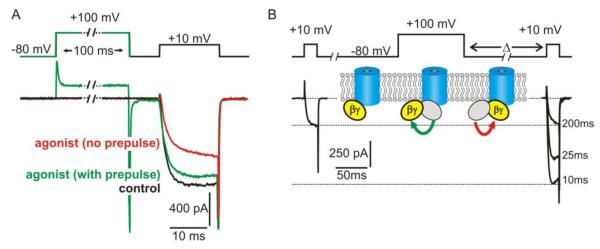
Functional effects of voltage-dependent inhibition on CaV2 channels. (A) “Whole cell” patch clamp recording of ICa from an adrenal chromaffin cell which express purinergic P2Y autoreceptors. Application of a P2Y receptor agonist (red trace) inhibited ICa compared to control conditions (black trace) with the hallmark features of voltage-dependent inhibition. Peak amplitude was reduced with prominent slowing of the activation kinetics, and both of these effects were reversed by a conditioning prepulse to +100mV (green trace). (B) Voltage-dependent relief of inhibition reflects transient dissociation of Gβγ from the channel. Shown is an example of “whole cell” ICa recorded from recombinant CaV2.2 channels expressed with β1b, α2δ and Gβγ in HEK293 cells. Gβγ produced tonic inhibition of ICa that was reversed by a conditioning prepulse to +100 mV. The magnitude of this reversal (prepulse facilitation) diminished as the interval between prepulse and test pulse (Δ) was increased (examples shown are with Δ = 10 ms, 25 ms, and 200 ms). As illustrated by the inset cartoon, prepulse facilitation is thought to reflect dissociation of Gβγ from an inhibitory binding site on the channel at the depolarized membrane potential. Upon return to the hyperpolarized membrane potential, Gβγ rebinds to (and re-inhibits) the channel. This re-inhibition of ICa is monoexponential, and the rate depends on the local concentration of Gβγ.
Bean also introduced the “willing and reluctant” model to explain these functional effects [86], a framework that persists to this day [94-97]. The channels exhibit two functional gating states, “willing” and “reluctant”. In the absence of Gβγ, the “willing” state predominates, whilst binding of Gβγ favors the “reluctant” state which displays the shifts in channel gating noted above. Voltage-dependent relief of the inhibition is thought to reflect a shift of the channels from “reluctant” to “willing” due to transient dissociation of Gβγ (Fig 2B). This was supported by kinetic analyses of prepulse relief as a function of agonist or Gβγ concentration. Increasing the concentration of Gβγ did not alter the rate of relief during the prepulse, but did accelerate the rate of reinhibition following the prepulse [98-101], as expected for voltage-dependent dissociation and rebinding of Gβγ. Further investigations revealed that the kinetics of reinhibition were consistent with binding and unbinding of a single Gβγ dimer with the channel [101].
5.1. Single channel investigations
Single channel studies provided early evidence that the inhibition did not involve a diffusible second messenger. In the “cell-attached” (“on-cell”) recording configuration, bath application of agonist did not inhibit the channels whereas agonist in the patch pipette did [99, 102, 103]. This led to the conclusion that the inhibition was “direct” or “membrane delimited”. Single channel recording also directly revealed “reluctant” gating of inhibited channels. Upon membrane depolarization, the latency (delay) to first channel opening was increased during inhibition whereas there was little impact on other single channel parameters [95, 104]. As a result, the inhibited (“reluctant”) channels appeared essentially silenced, unable to open until Gβγ dissociated and the channels shifted to the “willing” state. Subsequently it has been reported that CaV2.2 (N-type) but not CaV2.1 (P/Q-type) channels can display very brief channel openings from the “reluctant” state (i.e. without Gβγ unbinding), although the probability of such events was low [96, 97].
Overall, the dominant effects of inhibition observed in all studies are the shift in activation and prolonged latency to first channel opening. The slow activation kinetics seen in whole cell recording (Fig 2) and longer latency in single channel recordings reflect the conformational changes and subsequent dissociation of Gβγ from the channel upon membrane depolarization. This diminished binding of Gβγ at depolarized potentials also results in little inhibition of whole cell ICa when neurotransmitter agonists are rapidly applied during a depolarizing voltage-step [105].
5.2. Alteration of gating currents by Gβγ
Further evidence for altered activation comes from recording of channel “gating currents”. Gating currents are not due to ionic flux through the channel pore, but rather reflect movement of the charged voltage-sensor domain of the channels in response to membrane potential changes. Expression of recombinant CaV2.2 in HEK293 cells enables recording of these gating currents in isolation as the cells lack other endogenous voltage-gated channels. G proteins were found to reduce the amplitude, and shift the voltage-dependence of gating currents to more depolarized potentials [106]. G proteins also produced a significant separation in the voltage-dependent activation of gating current and ionic current [106]. These data suggest that Gβγ binding slows movement of the voltage-sensor and uncouples this movement from opening of the activation gate. Modulation of gating currents by G proteins has also been reported in rat sympathetic neurons [107, 108].
5.3. Gβγ and channel inactivation
In addition to these dominant effects on channel activation, evidence supports the idea that Gβγ can also modulate inactivation of CaV2.2 channels [109, 110]. Inactivation of Ca2+ channels is complex and mediated by several voltage-dependent and Ca2+-dependent mechanisms [111-113]. The precise molecular correlates remain somewhat unclear, but fast voltage-dependent inactivation might involve a “hinged lid” type mechanism in which the intracellular loop connecting domains I and II of the α1-subunit serves as the “inactivation gate” [112, 114] (but see [115]). The I-II loop is also an important binding site for Gβγ on the channel [116-119] (Fig 1) (see section 7 for more discussion). Therefore, it is possible that binding of Gβγ disrupts movement of this putative inactivation gate, or its interaction with other channel domains. Inactivation of CaV2.2 can also occur from intermediate closed state(s) of the channel favored during trains of brief repetitive stimuli [104]. If Gβγ were to reduce the probability that the channels populate this state (from which inactivation is preferred) it might reduce the cumulative inactivation throughout a stimulus train. Further investigations are needed to determine quite how G protein modulation and channel inactivation interact.
Ca2+-dependent inactivation is mediated by calmodulin bound to the C-terminus of the channel α1 subunit [42, 120-123]. Strong intracellular Ca2+ buffering (EGTA or BAPTA in the patch pipette solution) blocks Ca2+-dependent inactivation of CaV2 channels indicating that it is mediated by a “global” elevation rather than a “local” microdomain of Ca2+. The reduction of Ca2+-dependent inactivation by Gβγ [109] might therefore result from fewer channels opening and a diminished “global” Ca2+ signal, although more complex interactions are also possible, and direct in vitro binding of Ca2+-calmodulin to Gβγ has been reported [124].
5.4. Differential inhibition of CaV2 channels by Gβγ
Originally demonstrated for N-type channels (CaV2.2) in sensory and sympathetic neurons (for example, [78, 125-127]), it subsequently became clear that CaV2.1 (P/Q-type) channels are also modulated by Gβγ in a similar manner [128]. Initially it was thought that CaV2.3 channels were insensitive to G proteins [129-131], although other studies did find some degree of inhibition [132-135]. Chimeric approaches suggested that the lack of (or poor) responsiveness of CaV2.3 resided in several regions within the N-terminus, domain I, and I-II linker of the channels [136-138]. Subsequently it was discovered that alternative splicing of the N-terminus conferred G protein sensitivity to the channels [139]. Truncation of 50 amino acids abolished inhibition, whereas a splice variant with full length N-terminus did display inhibition, albeit to a lesser extent than CaV2.2 channels [139]. While inhibition of CaV2.3 channels can occur (depending on splice variation), it is generally to a lesser extent and remains less well understood than for CaV2.1 and CaV2.2 channels. In part this might be due to difficulty in isolating these channels in neuronal cell types. The focus on CaV2.1 and CaV2.2 channels is also driven by their prominence in triggering neurotransmitter release, and most of the following discussion revolves around those two channels.
Although the basic mechanism of inhibition is similar for CaV2.1 and CaV2.2 (direct binding of Gβγ to the channel), subtle differences have emerged. As noted above, single channel recording showed that CaV2.2 but not CaV2.1 channels display very brief duration, low probability “reluctant openings” [96, 97]. Differences are also apparent with macroscopic (whole-cell) recordings: activation of GPCRs or expression of Gβγ reduces the peak amplitude of ICa to a significantly greater extent for CaV2.2 than CaV2.1 [129, 140, 141]. Moreover, trains of action potential-like stimuli reverse a greater proportion of CaV2.1 inhibition than CaV2.2 inhibition [92]. These effects can be explained by differences in the affinity of Gβγ binding to the channels. The apparent affinity of Gβγ for the channel can be inferred from prepulse relief and re-inhibition experiments, and is quite similar for the two channels at hyperpolarized or very depolarized potentials. However, at moderately depolarized potentials (< +30mV), within the physiologically relevant range of action potentials, there is a significant divergence in the affinity of Gβγ binding to the two channel types [96]. Subtle differences in binding of Gβγ to the channels is also suggested when comparing the inhibition produced by different Gβ subunits (Gβ1-5), all paired with the same Gγ2 subunit. Such experiments revealed a different rank order of inhibition for CaV2.1 and CaV2.2 channels [142]. Point mutations on the Gβ1 subunit also have distinct effects on the inhibition of CaV2.1 and CaV2.2 channels [143].
It would appear that subtle differences in the binding affinity of Gβγ to the CaV2.1 and CaV2.2 channels results in differential inhibition: CaV2.2 ICa is inhibited to greater extent and this inhibition is more resistant to reversal by high frequency bursts of action potentials. The relative expression level of the two channel types varies between neurons, and even between neighboring synapses arising from the same neuron. Therefore, differential inhibition of CaV2.1 and CaV2.2 could lead to cell and/or synapse specific neuromodulation by GPCRs. Functional differences might also arise from variable interactions or crosstalk with other signaling pathways such as PKC (see section 9).
6. Structural determinants on Gβγ that govern modulation of CaV2 channels
Gβγ is thought to be an obligate heterodimer and there are several high resolution crystal structures in isolation or bound to interacting proteins including Gα, GRK2, and phosducin [144-148]. Figure 3 shows a rendering of the heterotrimer (Gαiβ1γ2) (Fig 3A), and heterodimer (the Gβ1γ2 ) (Fig 3B) based on the structure reported by Wall et al [144] (PDB ID: 1GP2). Gβ adopts a seven blade β-propeller structure with an α-helical N-terminal domain that binds to the α-helical N-terminus of the Gγ subunit (Fig 3B). In the heterotrimeric complex, Gα interacts with multiple residues on the top face of Gβ and the side aspect of propeller blade 1 (Fig 3A). Gβγ interacts with multiple downstream effectors and mutagenesis approaches have been used to map the interaction sites important for binding to these targets. Many effectors bind to a protein interaction “hot spot” on the surface of Gβ that interacts with Gα, with overlapping subsets of residues involved in binding to different effectors [149]. A number of residues identified in mutagenesis studies to contribute to inhibition of Ca2+ channels are highlighted in figure 4. Most of these are on the Gα interacting surface (Fig 3C) and are masked when Gα is present [85, 109, 150-152], although residues on the reverse face of Gβ1 have also been implicated [152-154] (Fig 3D). Also of note, Asn35 and Asn36 on Gβ1 mediate the ability of PKC to antagonize inhibition of CaV2.2 [153]. Thr422 on the rat CaV2.2 I-II linker has been identified as the phosphorylation site for PKC that mediates this effect [155], so it is tempting to speculate that this region of the channel and Gβγ come into close proximity with one another (see section 7).
Figure 3.

Structural determinants on Gβγ that govern modulation of CaV2 channels. (A, B) Ribbon diagram renderings of the heterotrimeric G protein structure in panel A, and the Gβγ dimer in panel B (Gαi - green; Gβ1 - red; and Gγ2 blue). Gβ adopts a seven blade β-propeller structure with an α-helical N-terminal domain that binds to the α-helical N-terminus of Gγ. Gα interacts with multiple residues on the top face of Gβ and the side aspect of propeller blade 1. Many effectors bind to a protein interaction “hot spot” on the surface of Gβ that is masked by Gα in the heterotrimer. (C, D) Molecular surface rendering of the Gβγ dimer (Gβ - red; Gγ - blue). Panel C shows the Gα interacting face of Gβγ, and panel D is rotated ~180° to show opposite face of Gβγ. Residues marked in yellow have been reported to disrupt inhibition of CaV2 channels. Residues marked in green are involved in crosstalk between Gβ1 and PKC phosphorylation of CaV2.2. Molecular graphics images based on data reported by Wall et al [144] (PDB ID: 1GP2) were produced using the UCSF Chimera package [256, 257] from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.
Figure 4.

Model depicting the molecular interactions that underlie Gβγ-mediated inhibition of CaV2 channels. Panels A and B (upper three images) depict a channel with a CaVβ subunit, while panel C (lower images) depicts the situation in which the CaVβ subunit is absent. Currently, data suggest the binding site for Gβγ is comprised from multiple sites on the N-terminus, I-II linker, and probably C-terminus of the channel. Binding of Gβγ causes a conformational shift that promotes interaction of the N-terminus “inhibitory module” with the initial one-third of the I-II-linker (panel Aii). This (and perhaps other interactions) shifts gating charge movement to more depolarized potentials and uncouples voltage-sensor movement from channel activation. Strong membrane depolarization (panel B) leads to conformational changes that result in unbinding of Gβγ and loss of interaction between the N-terminus and I-II linker. This depends upon binding of a CaVβ subunit to the AID on the I-II linker that induces a rigid α-helical connection to the upstream IS6 region of the pore and voltage-sensor. In the absence of CaVβ subunit binding, inhibition still occurs (panel Ci) but cannot be reversed by strong depolarization (panel Cii).
Another study reported that a peptide mimicking the N-terminal 25 amino acids of Gβ2 reduced inhibition of CaV2.1 [156]. The Gβ N-terminal peptide disrupted FRET interaction between the Gβ2 and Gγ3 subunits suggesting a conformational shift or reorientation of the heterodimer that could disrupt interaction with the channels. A few studies have also shown that the subtype of Gγ within the Gβγ heterodimer can influence the extent of inhibition, with Gγ2 generally eliciting greater inhibition than Gγ1, Gγ3 or Gγ13 [157, 158]. The molecular basis for why the Gγ subtype influences inhibition of ICa is not clear, but it is interesting to note that the II-III linker (of the channel α1 subunit) contains a G-gamma-like (GGL) domain [159].
7. Structural determinants on the channel α1 subunit that govern modulation by Gβγ
Although there is currently no crystal structure for voltage-gated calcium channels that could be used to visualize their interactions with G proteins, site directed mutagenesis, chimeric, and biochemical approaches have been used to elucidate channel structural determinants involved in modulation. The first investigations involved chimeras between Cav2.1 and Cav2.2 channels [140]. These chimeras were expressed in Xenopus oocytes and their sensitivities to G proteins assessed via two electrode voltage clamp. These experiments identified domain I as a key determinant of G protein inhibition, along with the C-terminus of the channel. Subsequent biochemical studies using in vitro translated Gβγ subunits revealed two spatially distinct regions on the I-II linker of CaV2.1 as possible Gβγ targets [117]. The existence of two separate Gβγ binding domains in the domain I-II linker was also observed in functional assays. Zamponi et al. [160] showed that intracellular dialysis of tsA-201 cells with ~20 amino acid peptides directed against different regions of the I-II linker of both Cav2.1 and Cav2.2 channels prevented the ability of exogenously delivered Gβγ subunits to mediate voltage dependent inhibition of the channels. The first site contains a QXXER consensus sequence (QQIER in all three CaV2 family members) found in other Gβγ binding partners. This site also overlapped partially with the putative Cavβ subunit binding domain on the channel (the AID). Subsequent co-crystal structures of the Cavβ subunit bound to its interaction site on the isolated domain I-II linker revealed that only part of the 20 amino acid stretch forming the putative Gβγ interaction site is likely to be accessible in the presence of a bound Cavβ subunit [46]. This may suggest two possibilities: Either the Cavβ subunit partially dissociates from regions involved in Gβγ binding, or alternatively Gβγ interacts with those residues that remain exposed after Cavβ docking.
Further support for the involvement of the I-II linker came from scanning mutagenesis of the amino acids in each of the two binding regions in rat CaV2.2 channels [161]. Mutation of two residues (Arg376 and Val416 to alanine) out of thirty tested significantly reduced the magnitude of voltage-dependent inhibition while mutation of Arg376 to phenylalanine increased inhibition. Irrespective of the precise nature of the Gβγ interaction on the domain I-II linker, this general region has been implicated as being important for functional channel inhibition by a number of other groups [116, 131, 136]. These studies contrast with work from Qin and colleagues [134] whose data implicated the C-terminus rather than the domain I-II linker as the critical element for G protein modulation. While likely playing an auxiliary role, the C-terminus region does not appear to be essential for N-type channel inhibition as large parts can be deleted with only small consequences on the extent of receptor mediated voltage-dependent modulation [155, 162, 163]
Several other groups attributed an important role to the N-terminus of the channel based on site directed mutagenesis work [136, 138, 151, 164]. The Dolphin lab identified the N-terminal 55 amino acids of CaV2.2, and in particular an eleven amino acid stretch (45-55) that is predicted to form an α-helix [165], to be critical for Gβγ-mediated inhibition of the channels. The Yue group demonstrated direct interaction of the N-terminus with Gβγ [151] and that the N-terminus (residues 56-95) also binds directly to the I-II linker. Thus, the N-terminus contributes both to binding of Gβγ, and as an “inhibitory module” which binds the I-II linker to perhaps mediate the shift from willing to reluctant gating states. Finally, a recent study revealed that a point mutation (S218L) in the domain I S4-S5 linker of Cav2.1 that is found in patients with familial hemiplegic migraine (FHM) facilitates recovery of the channels from Gβγ inhibition, perhaps by facilitating the dissociation of the G protein dimer [166]. Two other FHM mutations (R192Q, Y1245C) have also been reported to diminish Gβγ-mediated inhibition [167, 168].
Taken together, several sites on both the CaV2 α1 subunit and the Gβγ heterodimer have been implicated in voltage-dependent inhibition. On the Cav2 α1 subunit, the domain I-II linker and N-terminus are essential structural elements (Fig 4). Ultimately crystal structure data will be needed to precisely determine how G protein subunits interact with these channel loci in the presence and the absence of the Cavβ subunit.
8. Contribution of the CaVβ subunit to voltage-dependent inhibition
The subtype of CaVβ can influence the extent and kinetics of Gβγ-mediated inhibition and this depends on the subtype of Gβ involved [169, 170]. However, the precise role of CaVβ subunits in voltage-dependent inhibition of ICa has been unclear (for reviews see [49, 79]). Overlapping binding sites for the two proteins have been identified on the I-II linker, and one fundamental question that arose was whether CaVβ and Gβγ can bind to the channel at the same time, or whether they compete in a mutually exclusive manner. Seemingly contradictory data including FRET analyses suggested either competition [171] or synergistic binding [172]. Some of this confusion might stem from endogenous CaVβ subunits found in some heterologous expression systems (including Xenopus oocytes), or confounding shifts in the voltage-dependence of activation by some CaVβ subunits (see [79]). Evidence from the Dolphin and Yang labs outlined below now suggest that both proteins can interact with the channel simultaneously, and that binding of the CaVβ subunit is required to confer voltage-dependent reversal to Gβγ-mediated inhibition (Fig 4) [173-176].
The Dolphin lab introduced a mutation (W391A) into the AID on the I-II linker of CaV2.2 channels which reduces CaVβ subunit binding affinity by ~1000 fold. [174]. While the extent of Gβγ-mediated inhibition was similar for mutant (W391A) and wild-type channels, prepulse reversal of the inhibition was almost abolished in the mutant. Expression of wild type CaV2.2 along with α2δ but without CaVβ resulted in similar findings, and the voltage-independent inhibition in the absence of the CaVβ was blocked by overexpression of transducin which acts to scavenge free Gβγ subunits [175]. Thus, in the absence of CaVβ binding to the I-II linker, Gβγ-mediated inhibition of the channels was still present but could no longer be reversed in a voltage-dependent manner. The experiments outlined above used the β1b subunit, but when β2a was expressed with the W391A channels voltage-dependent relief of the Gβγ mediated inhibition was restored. Unlike β1b, the β2a subunit is palmitoylated at two N-terminal cysteine residues, and mutation of these residues led to loss of voltage-dependent relief (i.e. the data resembled β1b). The authors proposed that palmitoylation increased the local plasma membrane concentration of β2a such that low affinity interaction with α1 could still take place and permit voltage-dependent relief of the inhibition.
The Yang lab came to similar conclusions for CaV2.1 channels [176]. In this case the authors mutated CaVβ to reduce the affinity for the AID. The channels were expressed in Xenopus oocytes and macroscopic currents were recorded from giant inside-out patches that contained many channels. Washing the cytoplasmic face of the patches resulted in dissociation of CaVβ (due to the reduced binding affinity of the mutant), and this was confirmed by the expected shifts in channel kinetics compared to wild type. In these channels lacking CaVβ, purified Gβγ still inhibited the currents but prepulse reversal was abolished.
The CaVβ subunit consists of SH3 and GK domains separated by a variable HOOK region [48, 49]. Binding to the AID on the I-II linker of α1 is mediated by the GK domain, although interaction between the SH3 and HOOK domains elsewhere on the α1 subunit might also modulate functional properties. In terms of Gβγ effects, voltage-dependent reversal was restored even by binding of the isolated GK domain of CaVβ to the AID [175, 176]. In the absence of such binding the AID adopts a random coil, but the presence of CaVβ induces an α-helical conformation that extends back to the interface with IS6 [46, 47, 177, 178]. The Yang lab introduced seven glycines between the AID and IS6 to disrupt this α-helical structure and found that this prevented the ability of CaVβ to confer voltage-dependence to the inhibition [176]. Conversely, introducing seven alanines (not expected to disrupt the α-helix) maintained the ability of CaVβ to confer voltage-dependence to Gβγ-mediated inhibition. It is possible that binding of CaVβ to the AID induces a rigid α-helical link with domain IS6, and this transmits movement of the voltage-sensor and activation gate (including IS6) to the I-II linker to alter the Gβγ binding pocket at depolarized potentials. It is also worth noting that Gβγ-mediated inhibition was still present in both channel types lacking CaVβ, and in the CaV2.1 channels containing the seven glycine insert [175, 176]. Apparently the rigid α-helical link to the upstream activation gate and voltage-sensor is not required per se to transduce binding of Gβγ into functional inhibition. CaVβ might also influence Gβγ-mediated inhibition in other ways. For example, deletion of the HOOK domain promoted tonic inhibition of CaV2.2 channels, perhaps due to increased affinity for the basal level of free Gβγ in the cells [175].
9. Crosstalk between N-type channels, Gβγ, kinases and synaptic proteins
9.1. Protein kinase C
Most cell signaling events do not occur in isolation, but instead in an integrated fashion. G protein regulation of voltage-gated calcium channels is no exception. This is exemplified by the modulation of voltage dependent Gβγ inhibition of N-type channels by protein kinase C ( PKC). In peripheral neurons, activation of PKC was shown to reduce the extent of subsequent G protein modulation by a number of different receptor pathways, including GABA-B, adenosine and muscarinic receptors [179-181]. Such an interference with G protein inhibition could be due to PKC dependent phosphorylation of the G protein interaction site on the channel, the G protein coupled receptor, or the G protein itself. The first hint supporting the first mechanism came from experiments showing that in vitro phosphorylated domain I-II linker peptides could no longer effectively interact with Gβγ peptides [160]. Subsequent work showed that a threonine residue within the putative I-II linker Gβγ interaction site was responsible for this effect. When phosphorylated, or substituted for glutamic acid, this residue destabilizes the interaction of the channel with Gβγ, and its substitution for alanine precludes the antagonistic effects of PKC [155]. Interestingly, only Gβ1 mediated signaling (not other Gβ isoforms) was subject to this type of PKC crosstalk [182], and this was attributed to a single locus unique to Gβ1 [153] (Fig 3C). This observation suggests that activation of Gq-coupled receptors can modulate signaling of certain types of Gβ1 linked receptors to N-type calcium channels. It should also be noted that PKC activation not only results in antagonistic effects on Gβγ-mediated inhibition but, depending on N-type channel splice variant, can also promote direct enhancement of current activity [155]. This is mediated by phosphorylation of both the above noted threonine residue and an adjacent serine and adds further complexity to the PKC-G protein signaling crosstalk.
9.2. Synaptic proteins
The two types of calcium channels that are most susceptible to the effects of Gβγ also control neurotransmitter release at CNS synapses [1]. Both Cav2.1 and Cav2.2 channels physically associate with proteins that are involved in synaptic vesicle release, such as syntaxin 1A and SNAP25. These SNARE proteins bind directly to a synaptic protein interaction (synprint) site on the II-III linker (Fig 1) which serves to bring the channels into close proximity of the synaptic vesicle release sites [183-189]. RIMs (rab3 interacting molecules) have also emerged as important organizers of the presynaptic active zone [190], and bind CaV2 channels both directly and through RIM binding proteins to control their density and localization at release sites [191-195]. Binding of syntaxin 1A to both Cav2.1 and Cav2.2 also results in a hyperpolarizing shift in the voltage dependence of channel inactivation [185, 196-198] (for review see [199]). In addition to this effect on channel gating, syntaxin 1A modulates G protein regulation of the channels. Coexpression of syntaxin 1A with N-type channels in tsA-201 cells induces tonic inhibition mediated by Gβγ [200]. Syntaxin 1A physically associates with Gβγ at a site distinct from that involved in binding to the N-type channel [198, 201], suggesting the possibility that syntaxin 1A serves to colocalize the channels and Gβγ to ultimately promote a form of tonic inhibition. In contrast, syntaxin 1B does not mediate such an effect, even though it is capable of binding to both the channel and Gβγ [202]. This may suggest that the spatial orientation of the syntaxin/G protein complex relative to the N-type channel complex is critical for functional modulation. Gβγ interaction with SNARE proteins might also serve to directly regulate neurotransmitter release in both lamprey and mammalian neurons and neuroendocrine cells [203-206].
Several other types of synaptic proteins have been shown to alter G protein regulation of N-and P/Q-type channels. Cysteine string protein (CSP) interacts with G proteins and the synprint site and mediates an effect similar to that seen with syntaxin 1A [207]. In addition, CSP appears to stimulate Gα subunit activity by promoting the exchange of GTP for GDP in a receptor independent manner [208]. In contrast to these enhancing effects of syntaxin 1A and CSP, coexpression of Rim1 with Cav2.2 in HEK293 cells promotes “deinhibition” (recovery from Gβγ-mediated inhibition during depolarization) in addition to substantially slowing channel inactivation [209].
9.3. Calcium channel γ subunits
Another protein of note is stargazin, a member of the calcium channel γ subunit family. Skeletal muscle CaV1.1 channels have been shown to associate with a γ1 subunit in addition to β and α2δ subunits. Several neuronal γ subunits have been identified although it remains uncertain that these constitute bona fide channel subunits. Indeed the γ2 isoform (also called stargazin) and related proteins (γ3-7) associate with and modulate glutamatergic AMPA receptors [210]. However, it has been shown that stargazin can bind Gβγ in vitro, and acts to scavenge Gβγ and reduce inhibition of CaV2.2 channels in Xenopus oocytes [211]. Altogether, these findings highlight the notion that Gβγ modulation of calcium channels does not occur in isolation, but is tightly controlled by a wide range of cellular processes and signalling pathways.
10. Direct GPCR/N-type calcium channel interactions
Efficient signaling necessitates close proximity between GPCRs and effectors such as ion channels. This can be accomplished through the formation of large macromolecular signaling complexes between receptors, channels, G proteins, and kinase anchoring proteins [212-215]. In addition, a physical association between receptors and channels provides for a possible mechanism by which receptors can control channel function in an agonist independent manner. This was first shown by Kitano and colleagues [216] who identified a physical association of Cav2.1 channels with metabotropic glutamate receptors that results in altered P/Q-type channel function. For N-type channels, the formation of a signaling complex between Cav2.2 and the NOP (a.k.a. nociceptin) receptor was demonstrated in dorsal root ganglion neurons, and shown to promote tonic voltage-dependent modulation in the absence of receptor ligand, presumably reflecting constitutive receptor activity [217]. Similar observations have been reported for δ- and μ-opioid receptors coexpressed with N-type channels in tsA-201 cells [218, 219].
Association of channels with receptors also provides for an additional level of control through regulation of channel density in the plasma membrane. NOP receptors coexpressed with N-type channels not only promote the cell surface expression of the channels (Fig 5A), but also trigger an agonist mediated co-internalization of the channel/receptor complex into lysosomes [219, 220], thus giving rise to a new form of voltage-independent inhibition (Fig 5B). The extent to which this occurs in neurons is up to some debate. While imaging studies show a clear NOP receptor mediated internalization of channels in cultured DRG neurons, and reduced calcium entry in response to prolonged activation of receptors [220], there do not appear to be clear effects on whole cell current densities in nociceptin treated neurons [221]. It is possible that receptor mediated internalization is offset by kinase pathways that augment the activities of channels remaining in the plasma membrane. D1 and D2 dopamine receptors also associate with N-type calcium channels [222, 223], but while NOP receptors and Cav2.2 channels interact via their C-termini, D1 and D2 receptors also interact with other regions of the Cav2.2 channel α1 subunit such as the domain II-III linker. As with the NOP receptor, D1 or D2 receptor coxpression facilitates trafficking of the channels to the plasma membrane, and allows for receptor-channel co-internalization. In prefrontal cortex neurons, the D1 receptor appears to target N-type channels to dendritric sites [222]. It is likely that other types of GPCR may form complexes with N-type and perhaps P/Q-type calcium channels, however this will need to be confirmed experimentally. Altogether, the formation of macromolecular signaling complexes between receptors and channels provides for previously unrecognized means for controlling channel activity/density.
Figure 5.
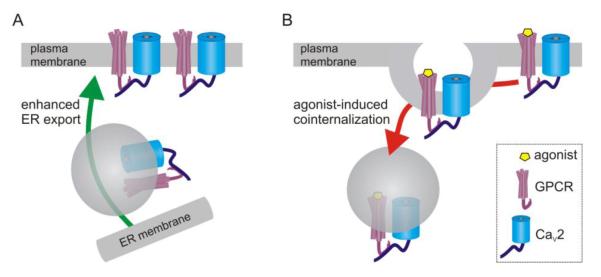
Trafficking-mediated modulation of CaV2.2 channels due to direct interaction with GPCRs. (A) Nociceptin receptors (NOP) interact directly with the Cav2.2 α1 subunit via their C-termini. D1 and D2 receptors also interact with additional regions of the α1 subunit such as the domain II-III linker. Coexpression of these GPCRs with CaV2.2 facilitates trafficking of the channels to the plasma membrane, and the D1 receptor appears to target N-type channels to dendritric sites in prefrontal cortex [222]. (B) Prolonged NOP agonist application has been reported to promote co-internalization of the receptor/channel complex into lysosomes in cultured sensory neurons, thus giving rise to a new form of voltage-independent inhibition [220] (but see [221]).
11. Voltage-independent inhibition of CaV2 channels by Gq-coupled GPCRs
Sympathetic neurons have been used extensively to investigate modulation of CaV2.2 (N-type) channels, and at least two distinct pathways have been identified: the “fast” pathway, mediated by pertussis toxin sensitive Gi/o-coupled GPCRs, is due to direct, voltage-dependent inhibition by Gβγ; a “slow”, voltage-independent pathway takes tens of seconds to develop, is mediated by Gq-coupled GPCRs, involves a diffusible second messenger, and is sensitive to intracellular [BAPTA] (for reviews see [224, 225]). Typical Gq-coupled signaling pathways downstream of phospholipase Cβ including IP3/Ca2+, diacylglycerol, and PKC were shown not to mediate the inhibition, and the pathway remained elusive for quite some time. In the past decade evidence has mounted implicating depletion of plasma membrane PIP2 and/or generation of arachidonic acid as possible mediators of this inhibition [226-229]. Here we outline the proposed mechanisms along with some recent developments.
PIP2 is required for a variety of ion channels to function (for reviews see [229-232]). The first evidence that this included Ca2+ channels was the demonstration that time-dependent “rundown” (loss) of CaV2.1 channel activity in excised membrane patches was slowed by application of PIP2 and accelerated by depleting or sequestering PIP2[233]. Similar effects were subsequently reported for N-type (CaV2.2) channels, along with evidence that the “slow” inhibition by Gq-coupled GPCRs in sympathetic neurons was due to phospholipase C mediated PIP2 hydrolysis [234]. For example, inhibition by muscarinic receptors was blunted by including PIP2 in the patch pipette, whereas recovery from inhibition was slowed by blocking PI-4 kinases which replenish the depleted PIP2. The overall picture that has emerged is that PIP2 is required for channels to open in response to membrane potential changes. This may involve dynamic low affinity interaction of PIP2 with the channels and perhaps additional higher affinity binding to a distinct channel domain. It has been postulated that such interactions might “crosslink” hydrophobic and hydrophilic domains and favor protein conformations conducive to active channel states. Similar to Gq-mediated modulation of M-type potassium channels [226, 230], this model proposes that depletion of local PIP2 by phospholipase C mediated hydrolysis removes this permissive interaction and is both necessary and sufficient to inhibit channel activity.
An alternative, although related, lipid signaling pathway has been proposed by the Rittenhouse lab [227] who reported that arachidonic acid elicits bidirectional modulation of N-type channels; ICa was enhanced at relatively hyperpolarized test potentials and inhibited at more depolarized potentials [235, 236]. The enhancement seems to involve extracellular actions of arachidonic acid [237], whereas the inhibition is mediated at the cytoplasmic face of the membrane. Arachidonic acid can be produced either by the action of phospholipase A2 on PIP2 and other membrane phospholipids, or by the action of diacylglycerol-lipase on diacylglycerol [227]. It is postulated that activation of these lipases by muscarinic receptors cleaves PIP2 and generates arachidonic acid which binds to the channel. This binding has the opposite effect to PIP2 such that it stabilizes closed/inactivated states of the channel and thus leads to inhibition. The involvement of arachidonic acid is a matter of some debate, in part due to conflicting reports on the ability of DAG-lipase inhibitors to block channel inhibition [226, 227, 234, 236].
A recent study from the Hille lab has provided evidence in support of the PIP2 depletion model [238]. To avoid downstream and parallel signaling pathways subsequent to Gq-coupled receptor activation, the authors used controlled activation of exogenous polyphosphoinositide 5-phosphatases which convert PIP2 into PI(4)P [239, 240]. In one approach, cells were transfected with a voltage-sensitive phosphatase (VSP) that enabled rapid (~1s) and reversible depletion of PIP2. Another approach involved chemical dimerization to translocate transfected yeast INP54p 5-phosphatase to the membrane and irreversibly deplete PIP2. In both cases ICa was inhibited by PIP2 depletion, and the rate/extent of recovery from this inhibition tracked the rate/extent of PIP2 resynthesis [238]. These data support the idea that depletion of PIP2 in of itself is sufficient to inhibit ICa. However, it was noted that the magnitude of ICa inhibition was less than that produced by muscarinic receptors, even though the predicted depletion of PIP2 is comparable. Therefore, it is possible that another signal, perhaps arachidonic acid, also contributes to the Gq-mediated inhibition, perhaps through synergistic actions with PIP2 depletion [226, 227].
11.1. CaVβ and intracellular Ca2+ modulate Gq-mediated inhibition
Notably, it has been reported that the CaV β2a subunit opposes inhibition of ICa by arachidonic acid or PIP2 depletion. The β2a subunit is palmitoylated at two N-terminal residues and it is this lipidation that diminishes the inhibition. Indeed, N-type channels containing β2a are enhanced rather than inhibited by Gq-coupled GPCRs and arachidonic acid [60, 227, 241]. Rittenhouse and colleagues postulated that the palmitoyl groups interact directly with the α1 subunit of the channel and thereby mask an inhibitory binding site for arachidonic acid. Inhibition of ICa by PIP2 depletion (using a voltage-sensitive phosphatase) is also diminished in channels containing a palmitoylated β2a subunit [61]. It is speculated that the palmitoyl groups of CaVβ2a, the lipid tail of PIP2, and arachidonic acid compete for binding to a site(s) on the α1 subunit of the channel. Binding of the palmitoyl groups or lipid tails of PIP2 favors active channel conformations whereas loss of this interaction and/or binding of arachidonic acid favors closed/inactive channel conformations.
Highlighting the complexity of neuronal Ca2+ channel regulation, it is noteworthy that even in the same cell, not all Gq-coupled GPCRs elicit inhibition of ICa [234]. This correlates with the ability of the different receptors to elicit significant IP3 mediated release of intracellular Ca2+ stores which likely relates to the proximity of the GPCR, IP3 receptors, and other components of a macromolecular signaling complex [226, 242]. In turn, such Ca2+ elevations are postulated to promote phosphatidylinositol 4-kinase activity thereby preventing local PIP2 depletion and inhibition of ICa [226, 234, 243, 244]. Thus, the extent of this voltage-independent inhibition depends on the subunit composition of the channels (CaVβ isoform), and colocalization of GPCRs and various phosphoinositide and Ca2+ signaling components in macromolecular signaling complexes. Adding further to this complexity, another type of “fast”, voltage-independent inhibition of N-type ICa mediated by a distinct pathway(s) perhaps involving both Gα and Gβγ signaling has also been described in these same sympathetic neurons [245, 246].
12. Kinase-mediated, voltage-independent inhibition of CaV2 channels in sensory neurons
Voltage-independent inhibition that appears to involve channel phosphorylation has also been described. For example, PKC has been implicated in the inhibition of N-type ICa in chick sensory neurons [247, 248] In frog and mammalian neurons PKC can also potentiate ICa, might target multiple phosphorylation sites with opposing actions, antagonize Gβγ mediated inhibition, or modulate channel trafficking (see sections 9, 10) [155, 180, 181, 249, 250]. It was also reported that rapid activation of a tyrosine kinase by GABAB receptors resulted in voltage-independent inhibition of N-type ICa in chick sensory neurons [251]. More recently, the Lipscombe lab demonstrated that manifestation of this tyrosine kinase mediated inhibition in mammalian neurons is controlled by alternative splicing of the CaV2.2 C-terminus (Fig 6) [252, 253]. There are two mutually exclusive forms of exon 37 (e37a and e37b). Gβγ-mediated, voltage-dependent inhibition is identical in recombinant channels containing either e37a or e37b. However, channels containing exon 37a are also inhibited by another voltage-independent pathway. This second pathway involves rapid activation of pp60c-src tyrosine kinase and requires a tyrosine residue (Y1747) present in exon 37a that is replaced by phenylalanine in exon 37b. Of particular note, expression of exon 37a is restricted to dorsal root ganglia, and preferentially expressed in capsaicin sensitive, nociceptive neurons [254]. The gene encoding chicken CaV2.2 only has one exon 37 which is similar to e37a and includes a tyrosine residue [252]. This then explains the restriction of this pathway to nociceptive neurons in mammals and its prevalence in chick neurons. This expression pattern also suggests that the e37a splice variant might be tailored to play a role in pain transmission [255]. Recently, using an exon replacement strategy in mice, it was shown that basal nociceptive transmission was unaltered by loss of e37a, but the analgesic effects of intrathecal morphine were diminished [253].
Figure 6.
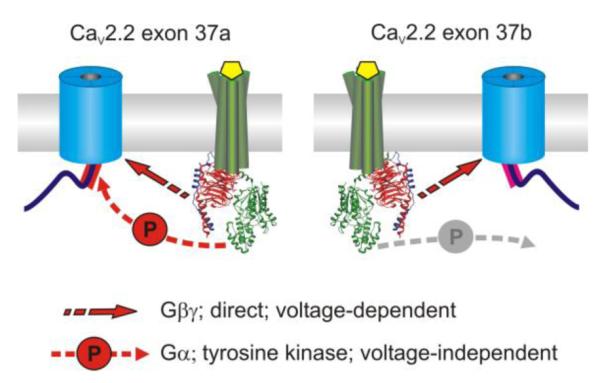
Alternative splicing of CaV2.2 controls voltage-independent inhibition of N-type ICa in sensory neurons. Two mutually exclusive forms of exon 37 encode the proximal C-terminus of CaV2.2. Expression of exon 37a is restricted to dorsal root ganglia, preferentially in nociceptive neurons, while exon 37b is widely expressed throughout the nervous system [254]. Gβγ-mediated, voltage-dependent inhibition of ICa is identical in channels containing either isoform of exon 37. An additional Gα-mediated, voltage-independent pathway involving pp60c-src tyrosine kinase inhibits channels containing exon 37a but not exon 37b. Thus alternative splicing of CaV2.2 results in cell-type specific alteration in the magnitude and mechanisms of GPCR-mediated inhibition.
13. Concluding remarks
In this review we have highlighted the complex inhibition of CaV2 channels by G protein coupled receptors. Voltage-dependent inhibition, mediated by direct binding of Gβγ to the Ca2+ channel α1 subunit, is the most common and best understood mechanism. Membrane potential, firing patterns, channel subunit composition/splice variants, and Gβγ heterodimer composition all modulate the extent and/or kinetics of voltage-dependent inhibition. Although less well understood and perhaps less widespread, there are also several mechanisms leading to voltage-independent inhibition of CaV2 channels. These include direct interaction with GPCRs, inhibition through lipid signaling pathways, and channel phosphorylation. CaV2 channels are also subject to a variety of other regulatory mechanisms, notably Ca2+-dependent feedback (both inactivation and facilitation). Thus, GPCRs in combination with Ca2+ channels sense and integrate a complex array of inputs in order to fine tune the spatiotemporal aspects of Ca2+ entry that play such pivotal roles in cellular physiology and synaptic transmission.
Highlights.
CaV2 channels play pivotal roles in neurotransmitter and hormone release
G protein coupled receptors orchestrate precise control of CaV2 channels
Voltage-dependent inhibition is mediated by direct binding of Gβγ to the channels
Voltage-independent inhibition is mediated by several other distinct pathways
Current understanding of these important mechanisms is provided in this review
Acknowledgements
Work in the Currie lab is supported by the National Institutes of Health, National Institute Of Neurological Disorders And Stroke [Grant R01-NS052446], and by the American Heart Association. GWZ is supported by the Canadian Institutes of Health Research, is an AI-HS Scientist and a Canada Research Chair. The molecular graphics images in figure 3 were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR001081)[256, 257].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- [1].Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- [2].Turner TJ, Dunlap K. Pharmacological characterization of presynaptic calcium channels using subsecond biochemical measurements of synaptosomal neurosecretion. Neuropharmacology. 1995;34:1469–1478. doi: 10.1016/0028-3908(95)00133-q. [DOI] [PubMed] [Google Scholar]
- [3].Goonasekera SA, Chen SR, Dirksen RT. Reconstitution of local Ca2+ signaling between cardiac L-type Ca2+ channels and ryanodine receptors: insights into regulation by FKBP12.6. American journal of physiology. 2005;289:C1476–1484. doi: 10.1152/ajpcell.00250.2005. [DOI] [PubMed] [Google Scholar]
- [4].Cooper PJ, Soeller C, Cannell MB. Excitation-contraction coupling in human heart failure examined by action potential clamp in rat cardiac myocytes. J Mol Cell Cardiol. 2010;49:911–917. doi: 10.1016/j.yjmcc.2010.04.012. [DOI] [PubMed] [Google Scholar]
- [5].Zhao X, Yamazaki D, Kakizawa S, Pan Z, Takeshima H, Ma J. Molecular architecture of Ca2+ signaling control in muscle and heart cells. Channels (Austin, Tex. 2011;5:391–396. doi: 10.4161/chan.5.5.16467. [DOI] [PubMed] [Google Scholar]
- [6].Braun AP. Multi-tasking at the protein level: L-type calcium channels function as ionotropic and metabotropic activators of smooth muscle contraction. Channels (Austin, Tex. 2011;5:459–460. doi: 10.4161/chan.5.6.17996. [DOI] [PubMed] [Google Scholar]
- [7].Marger L, Mesirca P, Alig J, Torrente A, Dubel S, Engeland B, Kanani S, Fontanaud P, Striessnig J, Shin HS, Isbrandt D, Ehmke H, Nargeot J, Mangoni ME. Functional roles of Ca(v)1.3, Ca(v)3.1 and HCN channels in automaticity of mouse atrioventricular cells: insights into the atrioventricular pacemaker mechanism. Channels (Austin, Tex. 2011;5:251–261. doi: 10.4161/chan.5.3.15266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Marger L, Mesirca P, Alig J, Torrente A, Dubel S, Engeland B, Kanani S, Fontanaud P, Striessnig J, Shin HS, Isbrandt D, Ehmke H, Nargeot J, Mangoni ME. Pacemaker activity and ionic currents in mouse atrioventricular node cells. Channels (Austin, Tex. 2011;5:241–250. doi: 10.4161/chan.5.3.15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- [10].Tottene A, Urbani A, Pietrobon D. Role of different voltage-gated Ca2+ channels in cortical spreading depression: specific requirement of P/Q-type Ca2+ channels. Channels (Austin, Tex. 2011;5:110–114. doi: 10.4161/chan.5.2.14149. [DOI] [PubMed] [Google Scholar]
- [11].Pietrobon D, Striessnig J. Neurological diseases: Neurobiology of migraine. Nat Rev Neurosci. 2003;4:386–398. doi: 10.1038/nrn1102. [DOI] [PubMed] [Google Scholar]
- [12].Iftinca MC, Zamponi GW. Regulation of neuronal T-type calcium channels. Trends Pharmacol Sci. 2009;30:32–40. doi: 10.1016/j.tips.2008.10.004. [DOI] [PubMed] [Google Scholar]
- [13].Khosravani H, Zamponi GW. Voltage-gated calcium channels and idiopathic generalized epilepsies. Physiol Rev. 2006;86:941–966. doi: 10.1152/physrev.00002.2006. [DOI] [PubMed] [Google Scholar]
- [14].Liao P, Soong TW. CaV1.2 channelopathies: from arrhythmias to autism, bipolar disorder, and immunodeficiency. Pflugers Arch. 2010;460:353–359. doi: 10.1007/s00424-009-0753-0. [DOI] [PubMed] [Google Scholar]
- [15].Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- [16].Bean BP. Classes of calcium channels in vertebrate cells. Annu Rev Physiol. 1989;51:367–384. doi: 10.1146/annurev.ph.51.030189.002055. [DOI] [PubMed] [Google Scholar]
- [17].Yokoyama CT, Myers SJ, Fu J, Mockus SM, Scheuer T, Catterall WA. Mechanism of SNARE protein binding and regulation of Cav2 channels by phosphorylation of the synaptic protein interaction site. Mol Cell Neurosci. 2005;28:1–17. doi: 10.1016/j.mcn.2004.08.019. [DOI] [PubMed] [Google Scholar]
- [18].Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Westenbroek RE, Hoskins L, Catterall WA. Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J Neurosci. 1998;18:6319–6330. doi: 10.1523/JNEUROSCI.18-16-06319.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wheeler DG, Barrett CF, Groth RD, Safa P, Tsien RW. CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J Cell Biol. 2008;183:849–863. doi: 10.1083/jcb.200805048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- [22].Brittain JM, Wang Y, Wilson SM, Khanna R. Regulation of CREB signaling through L-type Ca ( 2+) channels by Nipsnap-2. Channels (Austin, Tex. 2012;6:94–102. doi: 10.4161/chan.19415. [DOI] [PubMed] [Google Scholar]
- [23].Baig SM, Koschak A, Lieb A, Gebhart M, Dafinger C, Nurnberg G, Ali A, Ahmad I, Sinnegger-Brauns MJ, Brandt N, Engel J, Mangoni ME, Farooq M, Khan HU, Nurnberg P, Striessnig J, Bolz HJ. Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci. 2011;14:77–84. doi: 10.1038/nn.2694. [DOI] [PubMed] [Google Scholar]
- [24].Doering CJ, Rehak R, Bonfield S, Peloquin JB, Stell WK, Mema SC, Sauve Y, McRory JE. Modified Ca(v)1.4 expression in the Cacna1f(nob2) mouse due to alternative splicing of an ETn inserted in exon 2. PLoS One. 2008;3:e2538. doi: 10.1371/journal.pone.0002538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].McKay BE, McRory JE, Molineux ML, Hamid J, Snutch TP, Zamponi GW, Turner RW. Ca(V)3 T-type calcium channel isoforms differentially distribute to somatic and dendritic compartments in rat central neurons. Eur J Neurosci. 2006;24:2581–2594. doi: 10.1111/j.1460-9568.2006.05136.x. [DOI] [PubMed] [Google Scholar]
- [26].Molineux ML, McRory JE, McKay BE, Hamid J, Mehaffey WH, Rehak R, Snutch TP, Zamponi GW, Turner RW. Specific T-type calcium channel isoforms are associated with distinct burst phenotypes in deep cerebellar nuclear neurons. Proc Natl Acad Sci U S A. 2006;103:5555–5560. doi: 10.1073/pnas.0601261103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- [28].Giancippoli A, Novara M, de Luca A, Baldelli P, Marcantoni A, Carbone E, Carabelli V. Low-threshold exocytosis induced by cAMP-recruited CaV3.2 (alpha1H) channels in rat chromaffin cells. Biophys J. 2006;90:1830–1841. doi: 10.1529/biophysj.105.071647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Weiss N, Hameed S, Fernandez-Fernandez JM, Fablet K, Karmazinova M, Poillot C, Proft J, Chen L, Bidaud I, Monteil A, Huc-Brandt S, Lacinova L, Lory P, Zamponi GW, De Waard M. A Ca(v)3.2/syntaxin-1A signaling complex controls T-type channel activity and low-threshold exocytosis. J Biol Chem. 2012;287:2810–2818. doi: 10.1074/jbc.M111.290882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Carbone E, Giancippoli A, Marcantoni A, Guido D, Carabelli V. A new role for T-type channels in fast “low-threshold” exocytosis. Cell Calcium. 2006;40:147–154. doi: 10.1016/j.ceca.2006.04.019. [DOI] [PubMed] [Google Scholar]
- [31].Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron. 1992;9:1099–1115. doi: 10.1016/0896-6273(92)90069-p. [DOI] [PubMed] [Google Scholar]
- [32].Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP, Catterall WA. Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J Neurosci. 1995;15:6403–6418. doi: 10.1523/JNEUROSCI.15-10-06403.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Khanna R, Li Q, Bewersdorf J, Stanley EF. The presynaptic CaV2.2 channel-transmitter release site core complex. Eur J Neurosci. 2007;26:547–559. doi: 10.1111/j.1460-9568.2007.05680.x. [DOI] [PubMed] [Google Scholar]
- [34].Reid CA, Bekkers JM, Clements JD. Presynaptic Ca2+ channels: a functional patchwork. Trends Neurosci. 2003;26:683–687. doi: 10.1016/j.tins.2003.10.003. [DOI] [PubMed] [Google Scholar]
- [35].Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall WA. Nomenclature of voltage-gated calcium channels. Neuron. 2000;25:533–535. doi: 10.1016/s0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- [36].Hofmann F, Lacinova L, Klugbauer N. Voltage-dependent calcium channels: from structure to function. Rev Physiol Biochem Pharmacol. 1999;139:33–87. doi: 10.1007/BFb0033648. [DOI] [PubMed] [Google Scholar]
- [37].Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- [38].Stotz SC, Jarvis SE, Zamponi GW. Functional roles of cytoplasmic loops and pore lining transmembrane helices in the voltage-dependent inactivation of HVA calcium channels. J Physiol. 2004;554:263–273. doi: 10.1113/jphysiol.2003.047068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang HG, George MS, Kim J, Wang C, Pitt GS. Ca2+/calmodulin regulates trafficking of Ca(V)1.2 Ca2+ channels in cultured hippocampal neurons. J Neurosci. 2007;27:9086–9093. doi: 10.1523/JNEUROSCI.1720-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Minor DL, Jr., Findeisen F. Progress in the structural understanding of voltage-gated calcium channel (CaV) function and modulation. Channels (Austin, Tex. 2010;4:459–474. doi: 10.4161/chan.4.6.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Findeisen F, Tolia A, Arant R, Kim EY, Isacoff E, Minor DL., Jr. Calmodulin overexpression does not alter Cav1.2 function or oligomerization state. Channels (Austin, Tex. 2011;5:320–324. doi: 10.4161/chan.5.4.16821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- [43].Pitt GS, Zuhlke RD, Hudmon A, Schulman H, Reuter H, Tsien RW. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem. 2001;276:30794–30802. doi: 10.1074/jbc.M104959200. [DOI] [PubMed] [Google Scholar]
- [44].Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron. 2001;31:973–985. doi: 10.1016/s0896-6273(01)00438-x. [DOI] [PubMed] [Google Scholar]
- [45].Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel beta-subunit binds to a conserved motif in the I-II cytoplasmic linker of the alpha 1-subunit. Nature. 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- [46].Van Petegem F, Clark KA, Chatelain FC, Minor DL., Jr. Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature. 2004;429:671–675. doi: 10.1038/nature02588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Opatowsky Y, Chen CC, Campbell KP, Hirsch JA. Structural analysis of the voltage-dependent calcium channel beta subunit functional core and its complex with the alpha 1 interaction domain. Neuron. 2004;42:387–399. doi: 10.1016/s0896-6273(04)00250-8. [DOI] [PubMed] [Google Scholar]
- [48].Dolphin AC. Beta subunits of voltage-gated calcium channels. J Bioenerg Biomembr. 2003;35:599–620. doi: 10.1023/b:jobb.0000008026.37790.5a. [DOI] [PubMed] [Google Scholar]
- [49].Buraei Z, Yang J. The beta subunit of voltage-gated Ca2+ channels. Physiol Rev. 2010;90:1461–1506. doi: 10.1152/physrev.00057.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Klugbauer N, Marais E, Hofmann F. Calcium channel alpha2delta subunits: differential expression, function, and drug binding. J Bioenerg Biomembr. 2003;35:639–647. doi: 10.1023/b:jobb.0000008028.41056.58. [DOI] [PubMed] [Google Scholar]
- [51].Davies A, Kadurin I, Alvarez-Laviada A, Douglas L, Nieto-Rostro M, Bauer CS, Pratt WS, Dolphin AC. The alpha2delta subunits of voltage-gated calcium channels form GPI-anchored proteins, a posttranslational modification essential for function. Proc Natl Acad Sci U S A. 2010;107:1654–1659. doi: 10.1073/pnas.0908735107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fang K, Colecraft HM. Mechanism of auxiliary beta-subunit-mediated membrane targeting of L-type (Ca(V)1.2) channels. J Physiol. 2011;589:4437–4455. doi: 10.1113/jphysiol.2011.214247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Waithe D, Ferron L, Page KM, Chaggar K, Dolphin AC. Beta-subunits promote the expression of Ca(V)2.2 channels by reducing their proteasomal degradation. J Biol Chem. 2011;286:9598–9611. doi: 10.1074/jbc.M110.195909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Altier C, Garcia-Caballero A, Simms B, You H, Chen L, Walcher J, Tedford HW, Hermosilla T, Zamponi GW. The Cavbeta subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat Neurosci. 2011;14:173–180. doi: 10.1038/nn.2712. [DOI] [PubMed] [Google Scholar]
- [55].Bauer CS, Tran-Van-Minh A, Kadurin I, Dolphin AC. A new look at calcium channel alpha2delta subunits. Curr Opin Neurobiol. 2010;20:563–571. doi: 10.1016/j.conb.2010.05.007. [DOI] [PubMed] [Google Scholar]
- [56].Arikkath J, Campbell KP. Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr Opin Neurobiol. 2003;13:298–307. doi: 10.1016/s0959-4388(03)00066-7. [DOI] [PubMed] [Google Scholar]
- [57].Simms BA, Zamponi GW. Trafficking and stability of voltage-gated calcium channels. Cell Mol Life Sci. 2012;69:843–856. doi: 10.1007/s00018-011-0843-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Abiria SA, Colbran RJ. CaMKII associates with CaV1.2 L-type calcium channels via selected beta subunits to enhance regulatory phosphorylation. J Neurochem. 2010;112:150–161. doi: 10.1111/j.1471-4159.2009.06436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hermosilla T, Moreno C, Itfinca M, Altier C, Armisen R, Stutzin A, Zamponi GW, Varela D. L-type calcium channel beta subunit modulates angiotensin II responses in cardiomyocytes. Channels (Austin, Tex. 2011;5:280–286. doi: 10.4161/chan.5.3.15833. [DOI] [PubMed] [Google Scholar]
- [60].Heneghan JF, Mitra-Ganguli T, Stanish LF, Liu L, Zhao R, Rittenhouse AR. The Ca2+ channel beta subunit determines whether stimulation of Gq-coupled receptors enhances or inhibits N current. J Gen Physiol. 2009;134:369–384. doi: 10.1085/jgp.200910203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Suh BC, Kim DI, Falkenburger BH, Hille B. Membrane-localized beta-subunits alter the PIP2 regulation of high-voltage activated Ca2+ channels. Proc Natl Acad Sci U S A. 2012;109:3161–3166. doi: 10.1073/pnas.1121434109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Flucher BE, Tuluc P. A new L-type calcium channel isoform required for normal patterning of the developing neuromuscular junction. Channels (Austin, Tex. 2011;5:518–524. doi: 10.4161/chan.5.6.17951. [DOI] [PubMed] [Google Scholar]
- [63].Gray AC, Raingo J, Lipscombe D. Neuronal calcium channels: splicing for optimal performance. Cell Calcium. 2007;42:409–417. doi: 10.1016/j.ceca.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lieb A, Scharinger A, Sartori S, Sinnegger-Brauns MJ, Striessnig J. Structural determinants of CaV 1.3 L-type calcium channel gating. Channels (Austin, Tex. 2012;6 doi: 10.4161/chan.21002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Liao P, Yong TF, Liang MC, Yue DT, Soong TW. Splicing for alternative structures of Cav1.2 Ca2+ channels in cardiac and smooth muscles. Cardiovascular research. 2005;68:197–203. doi: 10.1016/j.cardiores.2005.06.024. [DOI] [PubMed] [Google Scholar]
- [66].Huang H, Tan BZ, Shen Y, Tao J, Jiang F, Sung YY, Ng CK, Raida M, Kohr G, Higuchi M, Fatemi-Shariatpanahi H, Harden B, Yue DT, Soong TW. RNA editing of the IQ domain in Ca(v)1.3 channels modulates their Ca(2)(+)-dependent inactivation. Neuron. 2012;73:304–316. doi: 10.1016/j.neuron.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bjarnadottir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, Schioth HB. Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics. 2006;88:263–273. doi: 10.1016/j.ygeno.2006.04.001. [DOI] [PubMed] [Google Scholar]
- [68].Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- [69].Kristiansen K. Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol Ther. 2004;103:21–80. doi: 10.1016/j.pharmthera.2004.05.002. [DOI] [PubMed] [Google Scholar]
- [70].Downes GB, Gautam N. The G protein subunit gene families. Genomics. 1999;62:544–552. doi: 10.1006/geno.1999.5992. [DOI] [PubMed] [Google Scholar]
- [71].Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- [72].Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].McIntire WE. Structural determinants involved in the formation and activation of G protein betagamma dimers. Neurosignals. 2009;17:82–99. doi: 10.1159/000186692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hollinger S, Hepler JR. Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev. 2002;54:527–559. doi: 10.1124/pr.54.3.527. [DOI] [PubMed] [Google Scholar]
- [75].Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- [76].Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- [77].Dunlap K, Fischbach GD. Neurotransmitters decrease the calcium ocmponent of sensory neurone action potentials. Nature. 1978;276:837–839. doi: 10.1038/276837a0. [DOI] [PubMed] [Google Scholar]
- [78].Dunlap K, Fischbach GD. Neurotransmitters decrease the calcium conductance activated by depolarization of embryonic chick sensory neurones. J Physiol. 1981;317:519–535. doi: 10.1113/jphysiol.1981.sp013841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Dolphin AC. G protein modulation of voltage-gated calcium channels. Pharmacol Rev. 2003;55:607–627. doi: 10.1124/pr.55.4.3. [DOI] [PubMed] [Google Scholar]
- [80].Tedford HW, Zamponi GW. Direct G protein modulation of Cav2 calcium channels. Pharmacol Rev. 2006;58:837–862. doi: 10.1124/pr.58.4.11. [DOI] [PubMed] [Google Scholar]
- [81].Ikeda SR, Dunlap K. Voltage-dependent modulation of N-type calcium channels: role of G protein subunits. Adv Second Messenger Phosphoprotein Res. 1999;33:131–151. doi: 10.1016/s1040-7952(99)80008-1. [DOI] [PubMed] [Google Scholar]
- [82].Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- [83].Stephens GJ. G-protein-coupled-receptor-mediated presynaptic inhibition in the cerebellum. Trends Pharmacol Sci. 2009;30:421–430. doi: 10.1016/j.tips.2009.05.008. [DOI] [PubMed] [Google Scholar]
- [84].Currie KP. Inhibition of Ca2+ channels and adrenal catecholamine release by G protein coupled receptors. Cell Mol Neurobiol. 2010;30:1201–1208. doi: 10.1007/s10571-010-9596-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Currie KP. G protein inhibition of CaV2 calcium channels. Channels (Austin, Tex. 2010;4:497–509. doi: 10.4161/chan.4.6.12871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- [87].Womack MD, McCleskey EW. Interaction of opioids and membrane potential to modulate Ca2+ channels in rat dorsal root ganglion neurons. J Neurophysiol. 1995;73:1793–1798. doi: 10.1152/jn.1995.73.5.1793. [DOI] [PubMed] [Google Scholar]
- [88].Williams S, Serafin M, Muhlethaler M, Bernheim L. Facilitation of N-type calcium current is dependent on the frequency of action potential-like depolarizations in dissociated cholinergic basal forebrain neurons of the guinea pig. J Neurosci. 1997;17:1625–1632. doi: 10.1523/JNEUROSCI.17-05-01625.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Park D, Dunlap K. Dynamic regulation of calcium influx by G-proteins, action potential waveform, and neuronal firing frequency. J Neurosci. 1998;18:6757–6766. doi: 10.1523/JNEUROSCI.18-17-06757.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Tosetti P, Taglietti V, Toselli M. Action-potential-like depolarizations relieve opioid inhibition of N-type Ca2+ channels in NG108-15 cells. Pflugers Arch. 1999;437:441–448. doi: 10.1007/s004240050799. [DOI] [PubMed] [Google Scholar]
- [91].Brody DL, Patil PG, Mulle JG, Snutch TP, Yue DT. Bursts of action potential waveforms relieve G-protein inhibition of recombinant P/Q-type Ca2+ channels in HEK 293 cells. J Physiol. 1997;499(Pt 3):637–644. doi: 10.1113/jphysiol.1997.sp021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Currie KPM, Fox AP. Differential facilitation of N- and P/Q-type calcium channels during trains of action potential-like waveforms. J Physiol. 2002;539:419–431. doi: 10.1113/jphysiol.2001.013206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Brody DL, Yue DT. Relief of G-protein inhibition of calcium channels and short-term synaptic facilitation in cultured hippocampal neurons. J Neurosci. 2000;20:889–898. doi: 10.1523/JNEUROSCI.20-03-00889.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Elmslie KS, Zhou W, Jones SW. LHRH and GTP-gamma-S modify calcium current activation in bullfrog sympathetic neurons. Neuron. 1990;5:75–80. doi: 10.1016/0896-6273(90)90035-e. [DOI] [PubMed] [Google Scholar]
- [95].Carabelli V, Lovallo M, Magnelli V, Zucker H, Carbone E. Voltage-dependent modulation of single N-Type Ca2+ channel kinetics by receptor agonists in IMR32 cells. Biophys J. 1996;70:2144–2154. doi: 10.1016/S0006-3495(96)79780-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Colecraft HM, Patil PG, Yue DT. Differential occurrence of reluctant openings in G-protein-inhibited N- and P/Q-type calcium channels. J Gen Physiol. 2000;115:175–192. doi: 10.1085/jgp.115.2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Lee HK, Elmslie KS. Reluctant gating of single N-type calcium channels during neurotransmitter-induced inhibition in bullfrog sympathetic neurons. J Neurosci. 2000;20:3115–3128. doi: 10.1523/JNEUROSCI.20-09-03115.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Golard A, Siegelbaum SA. Kinetic basis for the voltage-dependent inhibition of N-type calcium current by somatostatin and norepinephrine in chick sympathetic neurons. J Neurosci. 1993;13:3884–3894. doi: 10.1523/JNEUROSCI.13-09-03884.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Elmslie KS, Jones SW. Concentration dependence of neurotransmitter effects on calcium current kinetics in frog sympathetic neurones. J Physiol. 1994;481(Pt 1):35–46. doi: 10.1113/jphysiol.1994.sp020417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Delmas P, Brown DA, Dayrell M, Abogadie FC, Caulfield MP, Buckley NJ. On the role of endogenous G-protein beta gamma subunits in N-type Ca2+ current inhibition by neurotransmitters in rat sympathetic neurones. J Physiol. 1998;506(Pt 2):319–329. doi: 10.1111/j.1469-7793.1998.319bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Zamponi GW, Snutch TP. Modulation of voltage-dependent calcium channels by G proteins. Curr Opin Neurobiol. 1998;8:351–356. doi: 10.1016/s0959-4388(98)80060-3. [DOI] [PubMed] [Google Scholar]
- [102].Forscher P, Oxford GS, Schulz D. Noradrenaline modulates calcium channels in avian dorsal root ganglion cells through tight receptor-channel coupling. J Physiol. 1986;379:131–144. doi: 10.1113/jphysiol.1986.sp016244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Bernheim L, Beech DJ, Hille B. A diffusible second messenger mediates one of the pathways coupling receptors to calcium channels in rat sympathetic neurons. Neuron. 1991;6:859–867. doi: 10.1016/0896-6273(91)90226-p. [DOI] [PubMed] [Google Scholar]
- [104].Patil PG, de Leon M, Reed RR, Dubel S, Snutch TP, Yue DT. Elementary events underlying voltage-dependent G-protein inhibition of N-type calcium channels. Biophys J. 1996;71:2509–2521. doi: 10.1016/S0006-3495(96)79444-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Boland LM, Bean BP. Modulation of N-type calcium channels in bullfrog sympathetic neurons by luteinizing hormone-releasing hormone: kinetics and voltage dependence. J Neurosci. 1993;13:516–533. doi: 10.1523/JNEUROSCI.13-02-00516.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Jones SW, Elmslie KS. Transmitter modulation of neuronal calcium channels. J Membr Biol. 1997;155:1–10. doi: 10.1007/s002329900153. [DOI] [PubMed] [Google Scholar]
- [107].Rebolledo-Antunez S, Farias JM, Arenas I, Garcia DE. Gating charges per channel of Ca(V)2.2 channels are modified by G protein activation in rat sympathetic neurons. Arch Biochem Biophys. 2009;486:51–57. doi: 10.1016/j.abb.2009.04.002. [DOI] [PubMed] [Google Scholar]
- [108].Hernandez-Ochoa EO, Garcia-Ferreiro RE, Garcia DE. G protein activation inhibits gating charge movement in rat sympathetic neurons. American journal of physiology. 2007;292:C2226–2238. doi: 10.1152/ajpcell.00540.2006. [DOI] [PubMed] [Google Scholar]
- [109].McDavid S, Currie KP. G-proteins modulate cumulative inactivation of N-type (Cav2.2) calcium channels. J Neurosci. 2006;26:13373–13383. doi: 10.1523/JNEUROSCI.3332-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Weiss N, Tadmouri A, Mikati M, Ronjat M, De Waard M. Importance of voltage-dependent inactivation in N-type calcium channel regulation by G-proteins. Pflugers Arch. 2007;454:115–129. doi: 10.1007/s00424-006-0184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Cens T, Rousset M, Leyris JP, Fesquet P, Charnet P. Voltage- and calcium-dependent inactivation in high voltage-gated Ca(2+) channels. Prog Biophys Mol Biol. 2006;90:104–117. doi: 10.1016/j.pbiomolbio.2005.05.013. [DOI] [PubMed] [Google Scholar]
- [112].Stotz SC, Zamponi GW. Structural determinants of fast inactivation of high voltage-activated Ca(2+) channels. Trends Neurosci. 2001;24:176–181. doi: 10.1016/s0166-2236(00)01738-0. [DOI] [PubMed] [Google Scholar]
- [113].Hering S, Berjukow S, Sokolov S, Marksteiner R, Weiss RG, Kraus R, Timin EN. Molecular determinants of inactivation in voltage-gated Ca2+ channels. J Physiol. 2000;528(Pt 2):237–249. doi: 10.1111/j.1469-7793.2000.t01-1-00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Tadross MR, Ben Johny M, Yue DT. Molecular endpoints of Ca2+/calmodulin- and voltage-dependent inactivation of Ca(v)1.3 channels. J Gen Physiol. 2010;135:197–215. doi: 10.1085/jgp.200910308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Findeisen F, Minor DL., Jr. Disruption of the IS6-AID linker affects voltage-gated calcium channel inactivation and facilitation. J Gen Physiol. 2009;133:327–343. doi: 10.1085/jgp.200810143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Herlitze S, Hockerman GH, Scheuer T, Catterall WA. Molecular determinants of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel alpha1A subunit. Proc Natl Acad Sci U S A. 1997;94:1512–1516. doi: 10.1073/pnas.94.4.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].De Waard M, Liu H, Walker D, Scott VE, Gurnett CA, Campbell KP. Direct binding of G-protein betagamma complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- [118].De Waard M, Hering J, Weiss N, Feltz A. How do G proteins directly control neuronal Ca2+ channel function? Trends Pharmacol Sci. 2005;26:427–436. doi: 10.1016/j.tips.2005.06.008. [DOI] [PubMed] [Google Scholar]
- [119].Schiff ML, Siderovski DP, Jordan JD, Brothers G, Snow B, De Vries L, Ortiz DF, Diverse-Pierluissi M. Tyrosine-kinase-dependent recruitment of RGS12 to the N-type calcium channel. Nature. 2000;408:723–727. doi: 10.1038/35047093. [DOI] [PubMed] [Google Scholar]
- [120].Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- [121].Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- [122].Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, Yue DT. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- [123].Lee A, Zhou H, Scheuer T, Catterall WA. Molecular determinants of Ca(2+)/calmodulin-dependent regulation of Ca(v)2.1 channels. Proc Natl Acad Sci U S A. 2003;100:16059–16064. doi: 10.1073/pnas.2237000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Liu M, Yu B, Nakanishi O, Wieland T, Simon M. The Ca2+-dependent binding of calmodulin to an N-terminal motif of the heterotrimeric G protein beta subunit. J Biol Chem. 1997;272:18801–18807. doi: 10.1074/jbc.272.30.18801. [DOI] [PubMed] [Google Scholar]
- [125].Dolphin AC, Scott RH. Calcium channel currents and their inhibition by (-)-baclofen in rat sensory neurones: modulation by guanine nucleotides. J Physiol. 1987;386:1–17. doi: 10.1113/jphysiol.1987.sp016518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Marchetti C, Carbone E, Lux HD. Effects of dopamine and noradrenaline on Ca channels of cultured sensory and sympathetic neurons of chick. Pflugers Arch. 1986;406:104–111. doi: 10.1007/BF00586670. [DOI] [PubMed] [Google Scholar]
- [127].Lipscombe D, Kongsamut S, Tsien RW. Alpha-adrenergic inhibition of sympathetic neurotransmitter release mediated by modulation of N-type calcium-channel gating. Nature. 1989;340:639–642. doi: 10.1038/340639a0. [DOI] [PubMed] [Google Scholar]
- [128].Mintz IM, Bean BP. GABAB receptor inhibition of P-type Ca2+ channels in central neurons. Neuron. 1993;10:889–898. doi: 10.1016/0896-6273(93)90204-5. [DOI] [PubMed] [Google Scholar]
- [129].Bourinet E, Soong TW, Stea A, Snutch TP. Determinants of the G protein-dependent opioid modulation of neuronal calcium channels. Proc Natl Acad Sci U S A. 1996;93:1486–1491. doi: 10.1073/pnas.93.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Toth PT, Shekter LR, Ma GH, Philipson LH, Miller RJ. Selective G-protein regulation of neuronal calcium channels. J Neurosci. 1996;16:4617–4624. doi: 10.1523/JNEUROSCI.16-15-04617.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Page KM, Stephens GJ, Berrow NS, Dolphin AC. The intracellular loop between domains I and II of the B-type calcium channel confers aspects of G-protein sensitivity to the E-type calcium channel. J Neurosci. 1997;17:1330–1338. doi: 10.1523/JNEUROSCI.17-04-01330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Mehrke G, Pereverzev A, Grabsch H, Hescheler J, Schneider T. Receptor-mediated modulation of recombinant neuronal class E calcium channels. FEBS Lett. 1997;408:261–270. doi: 10.1016/s0014-5793(97)00437-7. [DOI] [PubMed] [Google Scholar]
- [133].Shekter LR, Taussig R, Gillard SE, Miller RJ. Regulation of human neuronal calcium channels by G protein betagamma subunits expressed in human embryonic kidney 293 cells. Mol Pharmacol. 1997;52:282–291. doi: 10.1124/mol.52.2.282. [DOI] [PubMed] [Google Scholar]
- [134].Qin N, Platano D, Olcese R, Stefani E, Birnbaumer L. Direct interaction of gbetagamma with a C-terminal gbetagamma-binding domain of the Ca2+ channel alpha1 subunit is responsible for channel inhibition by G protein-coupled receptors. Proc Natl Acad Sci U S A. 1997;94:8866–8871. doi: 10.1073/pnas.94.16.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Ottolia M, Platano D, Qin N, Noceti F, Birnbaumer M, Toro L, Birnbaumer L, Stefani E, Olcese R. Functional coupling between human E-type Ca2+ channels and mu opioid receptors expressed in Xenopus oocytes. FEBS Lett. 1998;427:96–102. doi: 10.1016/s0014-5793(98)00401-3. [DOI] [PubMed] [Google Scholar]
- [136].Simen AA, Miller RJ. Structural features determining differential receptor regulation of neuronal Ca channels. J Neurosci. 1998;18:3689–3698. doi: 10.1523/JNEUROSCI.18-10-03689.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Simen AA, Miller RJ. Involvement of regions in domain I in the opioid receptor sensitivity of alpha1B Ca(2+) channels. Mol Pharmacol. 2000;57:1064–1074. [PubMed] [Google Scholar]
- [138].Stephens GJ, Canti C, Page KM, Dolphin AC. Role of domain I of neuronal Ca2+ channel alpha1 subunits in G protein modulation. J Physiol. 1998;509(Pt 1):163–169. doi: 10.1111/j.1469-7793.1998.163bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Page KM, Canti C, Stephens GJ, Berrow NS, Dolphin AC. Identification of the amino terminus of neuronal Ca2+ channel alpha1 subunits alpha1B and alpha1E as an essential determinant of G-protein modulation. J Neurosci. 1998;18:4815–4824. doi: 10.1523/JNEUROSCI.18-13-04815.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Zhang JF, Ellinor PT, Aldrich RW, Tsien RW. Multiple structural elements in voltage-dependent Ca2+ channels support their inhibition by G proteins. Neuron. 1996;17:991–1003. doi: 10.1016/s0896-6273(00)80229-9. [DOI] [PubMed] [Google Scholar]
- [141].Currie KPM, Fox AP. Comparison of N- and P/Q-type voltage-gated calcium channel current inhibition. J Neurosci. 1997;17:4570–4579. doi: 10.1523/JNEUROSCI.17-12-04570.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Arnot MI, Stotz SC, Jarvis SE, Zamponi GW. Differential modulation of N-type 1B and P/Q-type 1A calcium channels by different G protein subunit isoforms. J Physiol. 2000;527(Pt 2):203–212. doi: 10.1111/j.1469-7793.2000.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Agler HL, Evans J, Colecraft HM, Yue DT. Custom distinctions in the interaction of G-protein beta subunits with N-type (CaV2.2) versus P/Q-type (CaV2.1) calcium channels. J Gen Physiol. 2003;121:495–510. doi: 10.1085/jgp.200208770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, Sprang SR. The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- [145].Sondek J, Bohm A, Lambright DG, Hamm HE, Sigler PB. Crystal structure of a G-protein beta gamma dimer at 2.1A resolution. Nature. 1996;379:369–374. doi: 10.1038/379369a0. [DOI] [PubMed] [Google Scholar]
- [146].Lambright DG, Sondek J, Bohm A, Skiba NP, Hamm HE, Sigler PB. The 2.0 A crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- [147].Gaudet R, Bohm A, Sigler PB. Crystal structure at 2.4 angstroms resolution of the complex of transducin betagamma and its regulator, phosducin. Cell. 1996;87:577–588. doi: 10.1016/s0092-8674(00)81376-8. [DOI] [PubMed] [Google Scholar]
- [148].Lodowski DT, Pitcher JA, Capel WD, Lefkowitz RJ, Tesmer JJ. Keeping G proteins at bay: a complex between G protein-coupled receptor kinase 2 and Gbetagamma. Science. 2003;300:1256–1262. doi: 10.1126/science.1082348. [DOI] [PubMed] [Google Scholar]
- [149].Smrcka AV. G protein betagamma subunits: central mediators of G protein-coupled receptor signaling. Cell Mol Life Sci. 2008;65:2191–2214. doi: 10.1007/s00018-008-8006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Ford CE, Skiba NP, Bae H, Daaka Y, Reuveny E, Shekter LR, Rosal R, Weng G, Yang CS, Iyengar R, Miller RJ, Jan LY, Lefkowitz RJ, Hamm HE. Molecular basis for interactions of G protein betagamma subunits with effectors. Science. 1998;280:1271–1274. doi: 10.1126/science.280.5367.1271. [DOI] [PubMed] [Google Scholar]
- [151].Agler HL, Evans J, Tay LH, Anderson MJ, Colecraft HM, Yue DT. G protein-gated inhibitory module of N-type (ca(v)2.2) ca2+ channels. Neuron. 2005;46:891–904. doi: 10.1016/j.neuron.2005.05.011. [DOI] [PubMed] [Google Scholar]
- [152].Tedford HW, Kisilevsky AE, Peloquin JB, Zamponi GW. Scanning mutagenesis reveals a role for serine 189 of the heterotrimeric G-protein beta 1 subunit in the inhibition of N-type calcium channels. J Neurophysiol. 2006;96:465–470. doi: 10.1152/jn.00216.2006. [DOI] [PubMed] [Google Scholar]
- [153].Doering CJ, Kisilevsky AE, Feng ZP, Arnot MI, Peloquin J, Hamid J, Barr W, Nirdosh A, Simms B, Winkfein RJ, Zamponi GW. A single Gbeta subunit locus controls crosstalk between PKC and G protein regulation of N-type calcium channels. J Biol Chem. 2004 doi: 10.1074/jbc.M308693200. [DOI] [PubMed] [Google Scholar]
- [154].Mirshahi T, Mittal V, Zhang H, Linder ME, Logothetis DE. Distinct sites on G protein beta gamma subunits regulate different effector functions. J Biol Chem. 2002;277:36345–36350. doi: 10.1074/jbc.M205359200. [DOI] [PubMed] [Google Scholar]
- [155].Hamid J, Nelson D, Spaetgens R, Dubel SJ, Snutch TP, Zamponi GW. Identification of an integration center for cross-talk between protein kinase C and G protein modulation of N-type calcium channels. J Biol Chem. 1999;274:6195–6202. doi: 10.1074/jbc.274.10.6195. [DOI] [PubMed] [Google Scholar]
- [156].Li X, Hummer A, Han J, Xie M, Melnik-Martinez K, Moreno RL, Buck M, Mark MD, Herlitze S. G protein beta2 subunit-derived peptides for inhibition and induction of G protein pathways. Examination of voltage-gated Ca2+ and G protein inwardly rectifying K+ channels. J Biol Chem. 2005;280:23945–23959. doi: 10.1074/jbc.M414078200. [DOI] [PubMed] [Google Scholar]
- [157].Zhou JY, Siderovski DP, Miller RJ. Selective regulation of N-type Ca channels by different combinations of G-protein beta/gamma subunits and RGS proteins. J Neurosci. 2000;20:7143–7148. doi: 10.1523/JNEUROSCI.20-19-07143.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Blake BL, Wing MR, Zhou JY, Lei Q, Hillmann JR, Behe CI, Morris RA, Harden TK, Bayliss DA, Miller RJ, Siderovski DP. G beta association and effector interaction selectivities of the divergent G gamma subunit G gamma(13) J Biol Chem. 2001;276:49267–49274. doi: 10.1074/jbc.M106565200. [DOI] [PubMed] [Google Scholar]
- [159].Fathallah M, Sandoz G, Mabrouk K, Geib S, Urbani J, Villaz M, Ronjat M, Sabatier JM, De Waard M. Modelling of the III-IV loop, a domain involved in calcium channel Ca(v)2.1 inactivation, highlights a structural homology with the gamma subunit of G proteins. Eur J Neurosci. 2002;16:219–228. doi: 10.1046/j.1460-9568.2002.02074.x. [DOI] [PubMed] [Google Scholar]
- [160].Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- [161].Tedford HW, Kisilevsky AE, Vieira LB, Varela D, Chen L, Zamponi GW. Scanning mutagenesis of the I-II loop of the Cav2.2 calcium channel identifies residues Arginine 376 and Valine 416 as molecular determinants of voltage dependent G protein inhibition. Mol Brain. 2010;3:6. doi: 10.1186/1756-6606-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Li B, Zhong H, Scheuer T, Catterall WA. Functional role of a C-terminal Gbetagamma-binding domain of Ca(v)2.2 channels. Mol Pharmacol. 2004;66:761–769. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- [163].Furukawa T, Nukada T, Mori Y, Wakamori M, Fujita Y, Ishida H, Fukuda K, Kato S, Yoshii M. Differential interactions of the C terminus and the cytoplasmic I-II loop of neuronal Ca2+ channels with G-protein alpha and beta gamma subunits. I. Molecular determination. J Biol Chem. 1998;273:17585–17594. doi: 10.1074/jbc.273.28.17585. [DOI] [PubMed] [Google Scholar]
- [164].Canti C, Page KM, Stephens GJ, Dolphin AC. Identification of residues in the N terminus of alpha1B critical for inhibition of the voltage-dependent calcium channel by Gbeta gamma. J Neurosci. 1999;19:6855–6864. doi: 10.1523/JNEUROSCI.19-16-06855.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Page KM, Heblich F, Margas W, Pratt WS, Nieto-Rostro M, Chaggar K, Sandhu K, Davies A, Dolphin AC. N terminus is key to the dominant negative suppression of Ca(V)2 calcium channels: implications for episodic ataxia type 2. J Biol Chem. 2010;285:835–844. doi: 10.1074/jbc.M109.065045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Weiss N, Sandoval A, Felix R, Van den Maagdenberg A, De Waard M. The S218L familial hemiplegic migraine mutation promotes deinhibition of Ca(v)2.1 calcium channels during direct G-protein regulation. Pflugers Arch. 2008;457:315–326. doi: 10.1007/s00424-008-0541-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Melliti K, Grabner M, Seabrook GR. The familial hemiplegic migraine mutation R192Q reduces G-protein-mediated inhibition of P/Q-type (Ca(V)2.1) calcium channels expressed in human embryonic kidney cells. J Physiol. 2003;546:337–347. doi: 10.1113/jphysiol.2002.026716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Serra SA, Fernandez-Castillo N, Macaya A, Cormand B, Valverde MA, Fernandez-Fernandez JM. The hemiplegic migraine-associated Y1245C mutation in CACNA1A results in a gain of channel function due to its effect on the voltage sensor and G-protein-mediated inhibition. Pflugers Arch. 2009;458:489–502. doi: 10.1007/s00424-009-0637-3. [DOI] [PubMed] [Google Scholar]
- [169].Canti C, Bogdanov Y, Dolphin AC. Interaction between G proteins and accessory subunits in the regulation of 1B calcium channels in Xenopus oocytes. J Physiol. 2000;527(Pt 3):419–432. doi: 10.1111/j.1469-7793.2000.t01-1-00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Feng ZP, Arnot MI, Doering CJ, Zamponi GW. Calcium channel beta subunits differentially regulate the inhibition of N-type channels by individual Gbeta isoforms. J Biol Chem. 2001;276:45051–45058. doi: 10.1074/jbc.M107784200. [DOI] [PubMed] [Google Scholar]
- [171].Sandoz G, Lopez-Gonzalez I, Stamboulian S, Weiss N, Arnoult C, De Waard M. Repositioning of charged I-II loop amino acid residues within the electric field by beta subunit as a novel working hypothesis for the control of fast P/Q calcium channel inactivation. Eur J Neurosci. 2004;19:1759–1772. doi: 10.1111/j.1460-9568.2004.03216.x. [DOI] [PubMed] [Google Scholar]
- [172].Hummer A, Delzeith O, Gomez SR, Moreno RL, Mark MD, Herlitze S. Competitive and synergistic interactions of G protein beta(2) and Ca(2+) channel beta(1b) subunits with Ca(v)2.1 channels, revealed by mammalian two-hybrid and fluorescence resonance energy transfer measurements. J Biol Chem. 2003;278:49386–49400. doi: 10.1074/jbc.M306645200. [DOI] [PubMed] [Google Scholar]
- [173].Meir A, Bell DC, Stephens GJ, Page KM, Dolphin AC. Calcium channel beta subunit promotes voltage-dependent modulation of alpha 1 B by G beta gamma. Biophys J. 2000;79:731–746. doi: 10.1016/S0006-3495(00)76331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Leroy J, Richards MW, Butcher AJ, Nieto-Rostro M, Pratt WS, Davies A, Dolphin AC. Interaction via a key tryptophan in the I-II linker of N-type calcium channels is required for beta1 but not for palmitoylated beta2, implicating an additional binding site in the regulation of channel voltage-dependent properties. J Neurosci. 2005;25:6984–6996. doi: 10.1523/JNEUROSCI.1137-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Dresviannikov AV, Page KM, Leroy J, Pratt WS, Dolphin AC. Determinants of the voltage dependence of G protein modulation within calcium channel beta subunits. Pflugers Arch. 2009;457:743–756. doi: 10.1007/s00424-008-0549-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Zhang Y, Chen YH, Bangaru SD, He L, Abele K, Tanabe S, Kozasa T, Yang J. Origin of the voltage dependence of G-protein regulation of P/Q-type Ca2+ channels. J Neurosci. 2008;28:14176–14188. doi: 10.1523/JNEUROSCI.1350-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L, Yang J. Structural basis of the alpha1-beta subunit interaction of voltage-gated Ca2+ channels. Nature. 2004;429:675–680. doi: 10.1038/nature02641. [DOI] [PubMed] [Google Scholar]
- [178].Arias JM, Murbartian J, Vitko I, Lee JH, Perez-Reyes E. Transfer of beta subunit regulation from high to low voltage-gated Ca2+ channels. FEBS Lett. 2005;579:3907–3912. doi: 10.1016/j.febslet.2005.06.008. [DOI] [PubMed] [Google Scholar]
- [179].Swartz KJ. Modulation of Ca2+ channels by protein kinase C in rat central and peripheral neurons: disruption of G protein-mediated inhibition. Neuron. 1993;11:305–320. doi: 10.1016/0896-6273(93)90186-u. [DOI] [PubMed] [Google Scholar]
- [180].Zhu Y, Ikeda SR. VIP inhibits N-type Ca2+ channels of sympathetic neurons via a pertussis toxin-insensitive but cholera toxin-sensitive pathway. Neuron. 1994;13:657–669. doi: 10.1016/0896-6273(94)90033-7. [DOI] [PubMed] [Google Scholar]
- [181].Barrett CF, Rittenhouse AR. Modulation of N-type calcium channel activity by G-proteins and protein kinase C. J Gen Physiol. 2000;115:277–286. doi: 10.1085/jgp.115.3.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Cooper CB, Arnot MI, Feng ZP, Jarvis SE, Hamid J, Zamponi GW. Cross-talk between G-protein and protein kinase C modulation of N-type calcium channels is dependent on the G-protein beta subunit isoform. J Biol Chem. 2000;275:40777–40781. doi: 10.1074/jbc.C000673200. [DOI] [PubMed] [Google Scholar]
- [183].Sheng ZH, Rettig J, Takahashi M, Catterall WA. Identification of a syntaxin-binding site on N-type calcium channels. Neuron. 1994;13:1303–1313. doi: 10.1016/0896-6273(94)90417-0. [DOI] [PubMed] [Google Scholar]
- [184].Sheng ZH, Yokoyama CT, Catterall WA. Interaction of the synprint site of N-type Ca2+ channels with the C2B domain of synaptotagmin I. Proc Natl Acad Sci U S A. 1997;94:5405–5410. doi: 10.1073/pnas.94.10.5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Bezprozvanny I, Scheller RH, Tsien RW. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- [186].Rettig J, Sheng ZH, Kim DK, Hodson CD, Snutch TP, Catterall WA. Isoform-specific interaction of the alpha1A subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proc Natl Acad Sci U S A. 1996;93:7363–7368. doi: 10.1073/pnas.93.14.7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Wiser O, Tobi D, Trus M, Atlas D. Synaptotagmin restores kinetic properties of a syntaxin-associated N-type voltage sensitive calcium channel. FEBS Lett. 1997;404:203–207. doi: 10.1016/s0014-5793(97)00130-0. [DOI] [PubMed] [Google Scholar]
- [188].Zhong H, Yokoyama CT, Scheuer T, Catterall WA. Reciprocal regulation of P/Q-type Ca2+ channels by SNAP-25, syntaxin and synaptotagmin. Nat Neurosci. 1999;2:939–941. doi: 10.1038/14721. [DOI] [PubMed] [Google Scholar]
- [189].Mochida S, Sheng ZH, Baker C, Kobayashi H, Catterall WA. Inhibition of neurotransmission by peptides containing the synaptic protein interaction site of N-type Ca2+ channels. Neuron. 1996;17:781–788. doi: 10.1016/s0896-6273(00)80209-3. [DOI] [PubMed] [Google Scholar]
- [190].Sudhof TC. The presynaptic active zone. Neuron. 2012;75:11–25. doi: 10.1016/j.neuron.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Han Y, Kaeser PS, Sudhof TC, Schneggenburger R. RIM determines Ca(2)+ channel density and vesicle docking at the presynaptic active zone. Neuron. 2011;69:304–316. doi: 10.1016/j.neuron.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, Sudhof TC. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell. 2011;144:282–295. doi: 10.1016/j.cell.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Hibino H, Pironkova R, Onwumere O, Vologodskaia M, Hudspeth AJ, Lesage F. RIM binding proteins (RBPs) couple Rab3-interacting molecules (RIMs) to voltage-gated Ca(2+) channels. Neuron. 2002;34:411–423. doi: 10.1016/s0896-6273(02)00667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Kiyonaka S, Wakamori M, Miki T, Uriu Y, Nonaka M, Bito H, Beedle AM, Mori E, Hara Y, De Waard M, Kanagawa M, Itakura M, Takahashi M, Campbell KP, Mori Y. RIM1 confers sustained activity and neurotransmitter vesicle anchoring to presynaptic Ca2+ channels. Nat Neurosci. 2007;10:691–701. doi: 10.1038/nn1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Gandini MA, Felix R. Functional interactions between voltage-gated Ca(2+) channels and Rab3-interacting molecules (RIMs): new insights into stimulus-secretion coupling. Biochim Biophys Acta. 2012;1818:551–558. doi: 10.1016/j.bbamem.2011.12.011. [DOI] [PubMed] [Google Scholar]
- [196].Bergsman JB, Tsien RW. Syntaxin modulation of calcium channels in cortical synaptosomes as revealed by botulinum toxin C1. J Neurosci. 2000;20:4368–4378. doi: 10.1523/JNEUROSCI.20-12-04368.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Jarvis SE, Zamponi GW. Interactions between presynaptic Ca2+ channels, cytoplasmic messengers and proteins of the synaptic vesicle release complex. Trends Pharmacol Sci. 2001;22:519–525. doi: 10.1016/s0165-6147(00)01800-9. [DOI] [PubMed] [Google Scholar]
- [198].Davies JN, Jarvis SE, Zamponi GW. Bipartite syntaxin 1A interactions mediate CaV2.2 calcium channel regulation. Biochem Biophys Res Commun. 2011;411:562–568. doi: 10.1016/j.bbrc.2011.06.185. [DOI] [PubMed] [Google Scholar]
- [199].Davies JN, Zamponi GW. Old proteins, developing roles: The regulation of calcium channels by synaptic proteins. Channels (Austin, Tex. 2008;2:130–138. doi: 10.4161/chan.2.2.6214. [DOI] [PubMed] [Google Scholar]
- [200].Jarvis SE, Magga JM, Beedle AM, Braun JE, Zamponi GW. G protein modulation of N-type calcium channels is facilitated by physical interactions between syntaxin 1A and G betagamma. Journal of Biological Chemistry. 2000;275:6388–6394. doi: 10.1074/jbc.275.9.6388. [DOI] [PubMed] [Google Scholar]
- [201].Jarvis SE, Barr W, Feng ZP, Hamid J, Zamponi GW. Molecular determinants of syntaxin 1 modulation of N-type calcium channels. J Biol Chem. 2002;277:44399–44407. doi: 10.1074/jbc.M206902200. [DOI] [PubMed] [Google Scholar]
- [202].Lu Q, AtKisson MS, Jarvis SE, Feng ZP, Zamponi GW, Dunlap K. Syntaxin 1A supports voltage-dependent inhibition of alpha1B Ca2+ channels by Gbetagamma in chick sensory neurons. J Neurosci. 2001;21:2949–2957. doi: 10.1523/JNEUROSCI.21-09-02949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Blackmer T, Larsen EC, Takahashi M, Martin TF, Alford S, Hamm HE. G protein betagamma subunit-mediated presynaptic inhibition: regulation of exocytotic fusion downstream of Ca2+ entry. Science. 2001;292:293–297. doi: 10.1126/science.1058803. [DOI] [PubMed] [Google Scholar]
- [204].Gerachshenko T, Blackmer T, Yoon EJ, Bartleson C, Hamm HE, Alford S. Gbetagamma acts at the C terminus of SNAP-25 to mediate presynaptic inhibition. Nat Neurosci. 2005;8:597–605. doi: 10.1038/nn1439. [DOI] [PubMed] [Google Scholar]
- [205].Yoon EJ, Hamm HE, Currie KP. G protein betagamma subunits modulate the number and nature of exocytotic fusion events in adrenal chromaffin cells independent of calcium entry. J Neurophysiol. 2008;100:2929–2939. doi: 10.1152/jn.90839.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Yoon EJ, Gerachshenko T, Spiegelberg BD, Alford S, Hamm HE. Gbetagamma interferes with Ca2+-dependent binding of synaptotagmin to the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex. Mol Pharmacol. 2007;72:1210–1219. doi: 10.1124/mol.107.039446. [DOI] [PubMed] [Google Scholar]
- [207].Magga JM, Jarvis SE, Arnot MI, Zamponi GW, Braun JE. Cysteine string protein regulates G protein modulation of N-type calcium channels. Neuron. 2000;28:195–204. doi: 10.1016/s0896-6273(00)00096-9. [DOI] [PubMed] [Google Scholar]
- [208].Natochin M, Campbell TN, Barren B, Miller LC, Hameed S, Artemyev NO, Braun JE. Characterization of the G alpha(s) regulator cysteine string protein. J Biol Chem. 2005;280:30236–30241. doi: 10.1074/jbc.M500722200. [DOI] [PubMed] [Google Scholar]
- [209].Weiss N, Sandoval A, Kyonaka S, Felix R, Mori Y, De Waard M. Rim1 modulates direct G-protein regulation of Ca(v)2.2 channels. Pflugers Arch. 2011;461:447–459. doi: 10.1007/s00424-011-0926-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Diaz E. Regulation of AMPA receptors by transmembrane accessory proteins. Eur J Neurosci. 2010;32:261–268. doi: 10.1111/j.1460-9568.2010.07357.x. [DOI] [PubMed] [Google Scholar]
- [211].Tselnicker I, Tsemakhovich VA, Dessauer CW, Dascal N. Stargazin modulates neuronal voltage-dependent Ca(2+) channel Ca(v)2.2 by a Gbetagamma-dependent mechanism. J Biol Chem. 2010;285:20462–20471. doi: 10.1074/jbc.M110.121277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Davare MA, Dong F, Rubin CS, Hell JW. The A-kinase anchor protein MAP2B and cAMP-dependent protein kinase are associated with class C L-type calcium channels in neurons. J Biol Chem. 1999;274:30280–30287. doi: 10.1074/jbc.274.42.30280. [DOI] [PubMed] [Google Scholar]
- [213].Turner RW, Anderson D, Zamponi GW. Signaling complexes of voltage-gated calcium channels. Channels (Austin, Tex. 2011;5:440–448. doi: 10.4161/chan.5.5.16473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Altier C, Dubel SJ, Barrere C, Jarvis SE, Stotz SC, Scott JD, Nargeot J, Zamponi GW, Bourinet E. AKAP79 modulation of L-type channels involves disruption of intramolecular interactions in the CaV 1.2 subunit. Channels (Austin, Tex. 2012;6 doi: 10.4161/chan.20865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Beene DL, Scott JD. A-kinase anchoring proteins take shape. Curr Opin Cell Biol. 2007;19:192–198. doi: 10.1016/j.ceb.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Kitano J, Nishida M, Itsukaichi Y, Minami I, Ogawa M, Hirano T, Mori Y, Nakanishi S. Direct interaction and functional coupling between metabotropic glutamate receptor subtype 1 and voltage-sensitive Cav2.1 Ca2+ channel. J Biol Chem. 2003;278:25101–25108. doi: 10.1074/jbc.M303266200. [DOI] [PubMed] [Google Scholar]
- [217].Beedle AM, McRory JE, Poirot O, Doering CJ, Altier C, Barrere C, Hamid J, Nargeot J, Bourinet E, Zamponi GW. Agonist-independent modulation of N-type calcium channels by ORL1 receptors. Nat Neurosci. 2004;7:118–125. doi: 10.1038/nn1180. [DOI] [PubMed] [Google Scholar]
- [218].Chee MJ, Morl K, Lindner D, Merten N, Zamponi GW, Light PE, Beck-Sickinger AG, Colmers WF. The third intracellular loop stabilizes the inactive state of the neuropeptide Y1 receptor. J Biol Chem. 2008;283:33337–33346. doi: 10.1074/jbc.M804671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Evans RM, You H, Hameed S, Altier C, Mezghrani A, Bourinet E, Zamponi GW. Heterodimerization of ORL1 and opioid receptors and its consequences for N-type calcium channel regulation. J Biol Chem. 2010;285:1032–1040. doi: 10.1074/jbc.M109.040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Altier C, Khosravani H, Evans RM, Hameed S, Peloquin JB, Vartian BA, Chen L, Beedle AM, Ferguson SS, Mezghrani A, Dubel SJ, Bourinet E, McRory JE, Zamponi GW. ORL1 receptor-mediated internalization of N-type calcium channels. Nat Neurosci. 2006;9:31–40. doi: 10.1038/nn1605. [DOI] [PubMed] [Google Scholar]
- [221].Murali SS, Napier IA, Rycroft BK, Christie MJ. Opioid-related (ORL1) receptors are enriched in a subpopulation of sensory neurons and prolonged activation produces no functional loss of surface N-type calcium channels. J Physiol. 2012;590:1655–1667. doi: 10.1113/jphysiol.2012.228429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Kisilevsky AE, Mulligan SJ, Altier C, Iftinca MC, Varela D, Tai C, Chen L, Hameed S, Hamid J, Macvicar BA, Zamponi GW. D1 receptors physically interact with N-type calcium channels to regulate channel distribution and dendritic calcium entry. Neuron. 2008;58:557–570. doi: 10.1016/j.neuron.2008.03.002. [DOI] [PubMed] [Google Scholar]
- [223].Kisilevsky AE, Zamponi GW. D2 dopamine receptors interact directly with N-type calcium channels and regulate channel surface expression levels. Channels (Austin, Tex. 2008;2:269–277. doi: 10.4161/chan.2.4.6402. [DOI] [PubMed] [Google Scholar]
- [224].Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- [225].Elmslie KS. Neurotransmitter modulation of neuronal calcium channels. J Bioenerg Biomembr. 2003;35:477–489. doi: 10.1023/b:jobb.0000008021.55853.18. [DOI] [PubMed] [Google Scholar]
- [226].Delmas P, Coste B, Gamper N, Shapiro MS. Phosphoinositide lipid second messengers: new paradigms for calcium channel modulation. Neuron. 2005;47:179–182. doi: 10.1016/j.neuron.2005.07.001. [DOI] [PubMed] [Google Scholar]
- [227].Roberts-Crowley ML, Mitra-Ganguli T, Liu L, Rittenhouse AR. Regulation of voltage-gated Ca2+ channels by lipids. Cell Calcium. 2009;45:589–601. doi: 10.1016/j.ceca.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Michailidis IE, Zhang Y, Yang J. The lipid connection-regulation of voltage-gated Ca(2+) channels by phosphoinositides. Pflugers Arch. 2007;455:147–155. doi: 10.1007/s00424-007-0272-9. [DOI] [PubMed] [Google Scholar]
- [229].Suh BC, Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys. 2008;37:175–195. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Hernandez CC, Zaika O, Tolstykh GP, Shapiro MS. Regulation of neural KCNQ channels: signalling pathways, structural motifs and functional implications. J Physiol. 2008;586:1811–1821. doi: 10.1113/jphysiol.2007.148304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Logothetis DE, Jin T, Lupyan D, Rosenhouse-Dantsker A. Phosphoinositide-mediated gating of inwardly rectifying K(+) channels. Pflugers Arch. 2007;455:83–95. doi: 10.1007/s00424-007-0276-5. [DOI] [PubMed] [Google Scholar]
- [232].Rohacs T. Regulation of TRP channels by PIP(2) Pflugers Arch. 2007;453:753–762. doi: 10.1007/s00424-006-0153-7. [DOI] [PubMed] [Google Scholar]
- [233].Wu X, Kushwaha N, Albert PR, Penington NJ. A critical protein kinase C phosphorylation site on the 5-HT(1A) receptor controlling coupling to N-type calcium channels. J Physiol. 2002;538:41–51. doi: 10.1113/jphysiol.2001.012668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Gamper N, Reznikov V, Yamada Y, Yang J, Shapiro MS. Phosphatidylinositol [correction] 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J Neurosci. 2004;24:10980–10992. doi: 10.1523/JNEUROSCI.3869-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Liu L, Barrett CF, Rittenhouse AR. Arachidonic acid both inhibits and enhances whole cell calcium currents in rat sympathetic neurons. American journal of physiology. 2001;280:C1293–1305. doi: 10.1152/ajpcell.2001.280.5.C1293. [DOI] [PubMed] [Google Scholar]
- [236].Liu L, Rittenhouse AR. Arachidonic acid mediates muscarinic inhibition and enhancement of N-type Ca2+ current in sympathetic neurons. Proc Natl Acad Sci U S A. 2003;100:295–300. doi: 10.1073/pnas.0136826100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Barrett CF, Liu L, Rittenhouse AR. Arachidonic acid reversibly enhances N-type calcium current at an extracellular site. American journal of physiology. 2001;280:C1306–1318. doi: 10.1152/ajpcell.2001.280.5.C1306. [DOI] [PubMed] [Google Scholar]
- [238].Suh BC, Leal K, Hille B. Modulation of high-voltage activated Ca(2+) channels by membrane phosphatidylinositol 4,5-bisphosphate. Neuron. 2010;67:224–238. doi: 10.1016/j.neuron.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Okamura Y, Murata Y, Iwasaki H. Voltage-sensing phosphatase: actions and potentials. J Physiol. 2009;587:513–520. doi: 10.1113/jphysiol.2008.163097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Suh BC, Inoue T, Meyer T, Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science. 2006;314:1454–1457. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Mitra-Ganguli T, Vitko I, Perez-Reyes E, Rittenhouse AR. Orientation of palmitoylated CaVbeta2a relative to CaV2.2 is critical for slow pathway modulation of N-type Ca2+ current by tachykinin receptor activation. J Gen Physiol. 2009;134:385–396. doi: 10.1085/jgp.200910204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Delmas P, Wanaverbecq N, Abogadie FC, Mistry M, Brown DA. Signaling microdomains define the specificity of receptor-mediated InsP(3) pathways in neurons. Neuron. 2002;34:209–220. doi: 10.1016/s0896-6273(02)00641-4. [DOI] [PubMed] [Google Scholar]
- [243].Zaika O, Zhang J, Shapiro MS. Combined phosphoinositide and Ca2+ signals mediating receptor specificity toward neuronal Ca2+ channels. J Biol Chem. 2011;286:830–841. doi: 10.1074/jbc.M110.166033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Winks JS, Hughes S, Filippov AK, Tatulian L, Abogadie FC, Brown DA, Marsh SJ. Relationship between membrane phosphatidylinositol-4,5-bisphosphate and receptor-mediated inhibition of native neuronal M channels. J Neurosci. 2005;25:3400–3413. doi: 10.1523/JNEUROSCI.3231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Beech DJ, Bernheim L, Hille B. Pertussis toxin and voltage dependence distinguish multiple pathways modulating calcium channels of rat sympathetic neurons. Neuron. 1992;8:97–106. doi: 10.1016/0896-6273(92)90111-p. [DOI] [PubMed] [Google Scholar]
- [246].Kammermeier PJ, Ruiz-Velasco V, Ikeda SR. A voltage-independent calcium current inhibitory pathway activated by muscarinic agonists in rat sympathetic neurons requires both Galpha q/11 and Gbeta gamma. J Neurosci. 2000;20:5623–5629. doi: 10.1523/JNEUROSCI.20-15-05623.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Rane SG, Dunlap K. Kinase C activator 1,2-oleoylacetylglycerol attenuates voltage-dependent calcium current in sensory neurons. Proc Natl Acad Sci U S A. 1986;83:184–188. doi: 10.1073/pnas.83.1.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Luebke JI, Dunlap K. Sensory neuron N-type calcium currents are inhibited by both voltage-dependent and -independent mechanisms. Pflugers Arch. 1994;428:499–507. doi: 10.1007/BF00374571. [DOI] [PubMed] [Google Scholar]
- [249].Yang J, Tsien RW. Enhancement of N- and L-type calcium channel currents by protein kinase C in frog sympathetic neurons. Neuron. 1993;10:127–136. doi: 10.1016/0896-6273(93)90305-b. [DOI] [PubMed] [Google Scholar]
- [250].Rajagopal S, Fang H, Oronce CI, Jhaveri S, Taneja S, Dehlin EM, Snyder SL, Sando JJ, Kamatchi GL. Site-specific regulation of CA(V)2.2 channels by protein kinase C isozymes betaII and epsilon. Neuroscience. 2009;159:618–628. doi: 10.1016/j.neuroscience.2008.12.047. [DOI] [PubMed] [Google Scholar]
- [251].Diverse-Pierluissi M, Remmers AE, Neubig RR, Dunlap K. Novel form of crosstalk between G protein and tyrosine kinase pathways. Proc Natl Acad Sci U S A. 1997;94:5417–5421. doi: 10.1073/pnas.94.10.5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Raingo J, Castiglioni AJ, Lipscombe D. Alternative splicing controls G protein-dependent inhibition of N-type calcium channels in nociceptors. Nat Neurosci. 2007;10:285–292. doi: 10.1038/nn1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Andrade A, Denome S, Jiang YQ, Marangoudakis S, Lipscombe D. Opioid inhibition of N-type Ca2+ channels and spinal analgesia couple to alternative splicing. Nat Neurosci. 2010;13:1249–1256. doi: 10.1038/nn.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Bell TJ, Thaler C, Castiglioni AJ, Helton TD, Lipscombe D. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron. 2004;41:127–138. doi: 10.1016/s0896-6273(03)00801-8. [DOI] [PubMed] [Google Scholar]
- [255].Altier C, Dale CS, Kisilevsky AE, Chapman K, Castiglioni AJ, Matthews EA, Evans RM, Dickenson AH, Lipscombe D, Vergnolle N, Zamponi GW. Differential role of N-type calcium channel splice isoforms in pain. J Neurosci. 2007;27:6363–6373. doi: 10.1523/JNEUROSCI.0307-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- [257].Sanner MF, Olson AJ, Spehner JC. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]