

Abstract
The Nε-amino group of lysine residues can be transiently modified by the addition of an acetyl group. Recognized functions of Nε-lysine acetylation include regulation of activity, molecular stabilization and conformational assembly of a protein. For more than forty years lysine acetylation was thought to occur only in the cytosol and nucleus. Targets included cytoskeletal-associated proteins as well as transcription factors, histone proteins and proteins involved in DNA recombination and repair. However, in 2007 we reported that a type I membrane protein involved in the pathogenesis of Alzheimer’s disease was transiently acetylated on the ε amino group of seven lysine residues while transiting along the secretory pathway. Surprisingly, the acetylation occurred in the lumen of the endoplasmic reticulum (ER) forcing us to reconsider old paradigms. Indeed, if lysine acetylation can occur in the lumen of the ER, then all the essential biochemical elements of the reaction must be available in the lumen of the organelle. Follow-up studies revealed the existence of ER-based acetyl-CoA:lysine acetyltransferases as well as a membrane transporter that translocates acetyl-CoA from the cytosol into the ER lumen. Large-scale proteomics showed that the list of substrates of the ER-based acetylation machinery includes both transiting and resident proteins. Finally, genetic studies revealed that this machinery is tightly linked to human diseases. Here, we describe these exciting findings as well as recent biochemical and cellular advances, and discuss possible impact on both human physiology and pathology.
Keywords: Endoplasmic reticulum, Lysine acetylation, ATases, AT-1, PCSK9/NARC-1, Autophagy/ERAD(II)
1. Introduction
The ε amino group of lysine residues can undergo different modifications, including acetylation and other forms of acylation, sumoylation, biotinylation, ubiquitylation and methylation. Although extensively studied, the precise function(s) of the above modifications are still largely unknown. Lysine acetylation was originally identified more than forty years ago on the N-terminal tail of histone proteins, where its transient status regulates the interaction with chromatin and, as a result, DNA transcription [1]. Soon after, a large number of cytosolic and nuclear proteins were reported to be transiently acetylated in one or more lysine residues. In addition to histones, they include proteins involved in DNA recombination and repair, transcription factors, cytoskeletal proteins, chaperones, signaling proteins, and several metabolic enzymes [2, 3]. Recognized functions of Nε-lysine acetylation include regulation of activity, molecular stabilization and conformational assembly of a protein [4]. The acetylation of the ε amino group of a lysine residue (more typically referred to as lysine acetylation) differs from Nα-acetylation and O-acetylation. Nα-acetylation (also called N-terminal acetylation) occurs on the α-amino group at the N-terminus of nascent cytosolic proteins and is thought to be irreversible. It is mainly a co-translational event and predominantly affects alanine, serine and methionine residues; additionally it can also occur-albeit less often-on glycine, threonine, valine and cysteine residues [5]. O-acetylation was described on serine and threonine residues by two independent groups while studying the virulence of bacteria of the genus Yersinia [6, 7]. Although the acetyltransferase activity of the bacterial effector Yersinia outer protein J (YopJ) can target host proteins, an endogenous serine/threonine O-acetyltransferase activity has not been identified in mammalian cells. Both Nα- and O-acetylation are still poorly understood and will not be discussed by this review.
From the biochemical perspective, lysine acetylation requires three essential components: (1) an acceptor of the acetyl group (a protein that has the appropriate lysine residues); (2) a donor of the acetyl group (acetyl-CoA); and (3) an enzyme able to transfer the acetyl group from the donor to the acceptor (an acetyl-CoA:lysine acetyltransferase or simply called acetyltransferase). The above three components were initially identified only in the cytoplasm and nucleus; as a result, it was assumed that lysine acetylation could only occur in the cytosol or in the nucleus [4, 8]. However, in 2006 Schwer et al. reported the transient lysine acetylation of the mitochondrial matrix protein acetyl-CoA synthetase [9], whereas in 2007 Costantini et al. reported the transient lysine acetylation of the nascent endoplasmic reticulum (ER)-based form of the membrane protein β-site APP cleaving enzyme 1 (BACE1) [10]. After these initial findings, large-scale proteomic approaches reported that several proteins localized in the mitochondrial matrix undergo Nε-lysine acetylation [11, 12]; their acetylation status regulates the metabolism of the cell in response to nutrient availability [13]. Similarly, many membrane and secreted proteins were reported to undergo transient lysine acetylation in the ER lumen [14–16]. Finally, high-scale proteomics also identified ER-resident chaperones and enzymes to be Nε-lysine acetylated in their luminal portion [12, 16]. Therefore, what was once a cytosolic and nuclear event now appears to be an essential component of mitochondria and ER functions as well.
2. A Novel Form of Post-Translational Regulation in the ER
That Nε-lysine acetylation can occur in the lumen of the ER became evident in 2007 when we discovered that the ceramide-mediated regulation of BACE1 metabolism required transient acetylation of the nascent protein in the ER [10]. BACE1 is a type I membrane protein; it is synthesized in the ER and then transported to the plasma membrane along the secretory pathway. During biosynthesis, the N-terminal ectodomain faces the lumen of the ER while the short C-terminal tail faces the cytosol. Since the short C-tail has one single lysine residue we initially thought that, although it was the nascent ER-based form of the protein to be modified, the acetylation was still a cytoplasmic event. However, biochemical assessment as well as mass spectrometry revealed that the modified lysine residues were all in the ectodomain of the protein. This finding posed an immediate biochemical challenge: in order for the reaction to occur, both the donor (acetyl-CoA) and the enzyme (acetyltransferase) of the reaction must be available in the lumen of the organelle when BACE1 is synthesized. Subsequent efforts resulted in the identification of an ER membrane acetyl-CoA transporter [15] and two ER-based acetyltransferases [17].
2.1 The Transporter
Acetyl-CoA serves as the common donor of the acetyl group for the reaction of lysine acetylation (Box 1). Coenzyme A (CoA) is the carrier of the acetyl group, which is linked to the β-mercaptoethylamine end of CoA by a high-energy thioester bond. Acetyl-CoA originates from the break-down of carbohydrates, fatty acids and amino acids. Mammalian cells have three main and distinct pools of acetyl-CoA: a cytosolic/nuclear pool, a mitochondrial pool, and a peroxisomal pool. The cytosolic and nuclear pools are here considered as one because acetyl-CoA can freely pass through the nuclear pore complex [18]. A fourth and smaller pool of acetyl-CoA, which depends on active import from the cytosol, also exists in the ER and is essential for ER-based acetylation (see below). Because of its highly charged structure, acetyl-CoA cannot cross a lipid bilayer and is completely impermeable to cell membranes. This obstacle is resolved by the existence of specific membrane transporters that ensure continuous supply from both the mitochondria and the peroxisomes.
BOX 1. Acetyl-CoA biosynthesis in the cytosol.
The generation of Acetyl-CoA in the cytosol requires conversion of panthothenate (Vit B5) into Coenzyme A (CoA), a process that involves five enzymatic steps [117]. The first reaction, phosphorylation of pantothenate to 4-phospho-pantothenate, appears to act as the rate-limiting step and is catalyzed by pantothenate kinase (Pank) (see Figure 1). Mammals have four different genes encoding different versions of Pank. PANK1 encodes Pank1α and Pank1β, both found exclusively in the cytosol; PANK2 encodes a mitochondrial as well as at least one shorter cytosolic isoform (the catalytic activity of two additional shorter isoforms has not been demonstrated); PANK3 and PANK4 encode one cytosolic protein each (the catalytic signature of Pank4 has not been proven). Defects in Pank2 are associated with Hallervorden-Spatz syndrome (HSS), also known as pantothenate kinase-associated neurodegeneration (PKAN), an autosomal recessive neurodegenerative disorder that affects the central nervous system [118]. Genetic disruption of the common PANK gene, fumble (fbl) , in D. Melanogaster results in increased lethality as well as infertility in the surviving animals [119], while disruption of mouse Pank2 results in retinal degeneration and azoospermia [120]. The possible involvement of the other Pank proteins in human diseases is currently unknown.
The two main enzymes responsible for the biosynthesis of acetyl-CoA in the cytosol are the ATP-citrate lyase (Acly) and the acetyl-CoA synthetase (AceCS) (see Figure 1). Homozygous disruption of Acly in the mouse is lethal while heterozygous disruption is viable [121]. No information on the physiological as well as pathological impact of AceCS is currently available; however, genetic disruption of AceCS2 (also known as ACSS1) which is exclusively found in the mitochondria, leads to defects in thermoregulation and energy production as well as high degree of mortality [122]. Mitochondria and peroxisome pathways that contribute to cytosolic acetyl-CoA are described in the text.
Mitochondrial acetyl-CoA is transported to the cytosol as citrate by the tricarboxylate membrane transport system. Once in the cytosol, citrate is rapidly converted into oxaloacetate by the ATP-citrate lyase (Acly) releasing acetyl-CoA (Figure 1). The reaction requires ATP and free CoA. The oxaloacetate is then converted into pyruvate by the sequential action of the malate dehydrogenase and malic enzyme and transported back into the mitochondria by the pyruvate membrane transporter. Here, pyruvate re-enters into the “pyruvate-citrate cycle” ensuring continuous transfer of acetyl-CoA from the mitochondria to the cytosol [19]. A carnitine acetyltransferase-carnitine acetyl translocase (CAT) system also appears to contribute to the active transport of mitochondrial acetyl-CoA into the cytosol [20].
FIGURE 1. Biochemical pathways that contribute to cytosolic Acetyl-CoA.

Pathways as well as specific key enzymatic steps are described in the text and in Box 1.
Peroxisomal acetyl-CoA is transported to the cytosol as part of different pathways, depending on the organism and cell type [21, 22] (Figure 1). The first involves conversion of acetyl-CoA into acetyl-carnitine by the peroxisomal enzyme carnitine acetyltransferase. Following export from the peroxisome, acetyl-carnitine can enter the mitochondria for further metabolism or feed into the cytosolic/nuclear pool of acetyl-CoA [21, 23–25]. Peroxisomal acetyl-CoA can also enter the glyoxylate cycle to produce succinate, which is subsequently transported to the cytosol and mitochondria for further metabolism [21]. Finally, acetyl-CoA can be hydrolyzed within the peroxisome to acetate and CoA; it has been suggested that this free acetate can be exported to the cytosol where it is used to generate acetyl-CoA [22, 26]. The mechanisms of acetyl-CoA transport from the peroxisomes to the cytosol are still poorly understood and the relative contribution of the above pathways in mammals is currently uncertain.
In addition to the conversion of citrate by Acly, cytoplasmic acetyl-CoA also originates from the condensation of free acetate and CoA by acetyl-CoA synthetase (AceCS) (Figure 1). Both Acly and AceCS are also present in the nucleus, suggesting that acetyl-CoA production can also occur in the nuclear compartment (independently from the cytosolic pool), where it regulates histone acetylation and, therefore, gene expression [27].
The cytosolic pool of acetyl-CoA serves as the donor of the acetyl group for the reaction of lysine acetylation occurring in the cytosol, nucleus and ER lumen. As discussed above, the highly charged nature of acetyl-CoA impedes free passage across the lipid bilayer. Therefore, if lysine acetylation occurs in the lumen of the ER, a specific transporter must exist in the ER membrane. Alternatively, enzymes responsible for acetyl-CoA synthesis could grant local production of the metabolite, assuming that its precursors are also available in the lumen of the ER. However, no evidence for a luminal synthesis of acetyl-CoA in the ER exists. To address this apparent biochemical obstacle, we initially assessed whether highly purified ER vesicles were able to import free acetyl-CoA [10]. The experiments revealed that ER vesicles were able to import acetyl-CoA in a concentration- and temperature-dependent fashion, suggesting a carrier-mediated process. Importantly, acetyl-CoA transport was found to be saturable with an apparent Km of 14 μM [10], which is in the range of cytosolic acetyl-CoA availability and Km of other acetyl-CoA utilizing enzymes [28–30].
Interestingly, in 1997, ten years before we discovered that lysine acetylation occurs in the lumen of the ER [10], Kanamori et al. reported the identification of a putative acetyl-CoA transporter in the ER membrane [31]. The gene product, solute carrier family 33 member 1 (SLC33A1)/Acetyl-CoA Transporter-1 (AT-1), was identified as part of a screen for the Golgi-resident acetyltransferase responsible for the O-acetylation of complex gangliosides. Surprisingly, SLC33A1/AT-1 (simply called AT-1 later on) displayed features that were consistent with a putative ER membrane transporter and appeared to influence the acetylation of non-lipid constituents rather than gangliosides. As a result, the group led by Hirabayashi proposed a possible role of AT-1 as an ER membrane acetyl-CoA transporter [31, 32]. Further biochemical studies from our laboratory confirmed that AT-1 was indeed the ER membrane acetyl-CoA transporter (Figure 2). Importantly, the functional identification of the transporter was achieved by biochemical reconstitution of the transport activity into artificial liposomes [15].
FIGURE 2. Nε-lysine acetylation in the lumen of the ER.

A, The ER membrane transporter AT-1 ensures a continuous supply of acetyl-CoA into the ER lumen while ER-membrane anchored acetyltransferases (ATase1 and ATase2) catalyze the transfer of the acetyl group to seven lysine residues of BACE1.
B, Covalent modification of the Nε-amino group of a lysine residue by acetylation.
AT-1 is 549 amino acids long, migrates as a ~60–62-kDa band on reducing electrophoresis, and is predicted to have nine-to-twelve transmembrane domains. Finally, it displays an exclusive ER localization, which is consistent with its biochemical function [15]. AT-1-mediated translocation of acetyl-CoA is inhibited with high specificity by CoA, suggesting that the CoA moiety serves as recognition signal [15].
2.2 The Acetyl-CoA:Lysine Acetyltransferases
As with the transporter, the existence of an acetyl-CoA:lysine acetyltransferase activity in the lumen of the ER was first proven with classical biochemistry. In fact, affinity purified BACE1 could be acetylated in vitro when radiolabeled acetyl-CoA was added together with highly purified ER vesicles. Importantly, acetylation was only observed in the presence of permeabilized ER vesicles, indicating that the enzymatic activity resided in the lumen of the organelle [10]. The human genome was then searched for genes encoding proteins that displayed structural similarities to the catalytic domain of existing acetyltransferases. The analysis resulted in the identification of two different ER-based acetyltransferases, which were named ATase1 (also known as camello-like 2 and N-acetyltransferase 8B) and ATase2 (also known as camello-like 1 and N-acetyltransferase 8) [17]. The two transferases are 86% identical at the protein level. Both have one short cytosolic tail, a single transmembrane segment and a larger endolumenal domain with the catalytic activity (Figure 2) [17]. When separated on gel electrophoresis, they have an apparent molecular mass of ~28-kDa and ~25-kDa, respectively.
ATase1 and ATase2 are members of the camello family, which belongs to the GNAT (Gcn5-related N-acetyltransferase) superfamily of N-acetyltransferases. The GNAT superfamily includes over 10,000 members grouped in more than 12 protein families with diverse cellular functions and substrates [33, 34]. Members of this superfamily share the GNAT (acetyltransferase) domain, which contains four conserved sequence motifs, known as Motif C, D, A and B (ordered from N-terminus to C-terminus) [33–35]. Although the homology at the level of primary sequence, between different members of the superfamily, might not be outstanding (3–23% pairwise sequence identity), the structure of the GNAT/acetyltransferase domain is highly conserved [34, 35]. The GNAT conserved fold contains an N-terminal beta strand (B1) followed by two alpha helices (H1 and H2), three antiparallel beta strands (B2–B4), a central alpha helix (H3), another beta strand (B5), a fourth alpha helix (H4) and a final beta strand (B6) [34]. The lowest degree of conservation is found in the N-terminal motif C (B1-H1) while the strongest is found in motifs D (B2–B3), A (B4-H3) and B (B5-H4). Together they make the HAT (histone aceytltransferase) core (Figure 3) [33, 35, 36]. Motif A is the most conserved motif from prokaryotes to eukaryotes and is also present in p300/CBP and MYST family (named after its founding members: MOZ, Ybf2/Sas3, Sas2 and Tip60) [37]. It contains the sequence Arg/Gln-X-X-Gly-X-Gly/Ala that is important for acetyl-CoA recognition and binding, establishing most of the direct contacts to bound CoA. Remarkably, in spite of the high degree of sequence identity, the alignment of ATase1 and ATase2 sequences revealed three non-conservative amino acid substitutions inside the HAT core. Further studies are required to determine any functional consequences of these substitutions. Interestingly, two of these substitutions are located inside Motif A and one of them is in the sequence that makes up direct contact to bound CoA (Figure 3). These differences may have important consequences for the kinetics of the enzymatic activity and, perhaps, could explain the high degree of selectivity as well as the lack of off-site effects displayed by ATase1/ATase2 inhibitors [38].
FIGURE 3. Alignment of the primary sequence of the ATases HAT core with prominent members of the GNAT superfamily.
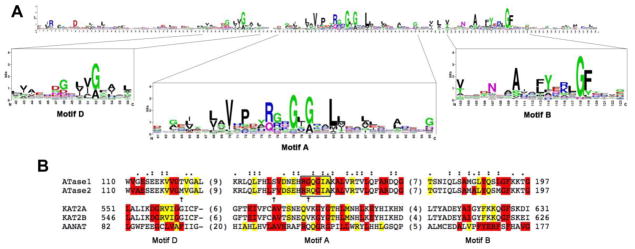
A, Sequence logo of the GNAT domain profile available at PROSITE (Database of protein domains, families and functional sites (http://prosite.expasy.org); PS51186). The sequence logo was generated after multiple sequence alignment of 660 UniProtKB/Swiss-Prot true positive hits. The consensus sequence of the GNAT domain can be read from the top of the stacks. The height of each letter is proportional to the frequency of the corresponding amino acid at the indicated position in the alignment.
B, Primary sequence alignment of the HAT core highly conserved motifs of ATase1 (NAT8B_HUMAN, Q9UHF3), ATase2 (NAT8_HUMAN, Q9UHE5) and other members of the GNAT superfamily, including Lysine acetyltransferase 2A also known as hGCN5 (KAT2A_HUMAN, Q92830), Lysine acetyltransferase 2B also known as PCAF (KAT2B_HUMAN, Q92831) and aralkylamine N-acetyltransferase (AANAT) also known as SNAT (SNAT_HUMAN, Q16613). The first and last columns indicate the amino acid position from the corresponding full sequence. The numbers in parenthesis between the conserved motifs indicate the length of the omitted sequence. The alignment was performed using the Clustal Omega multiple sequence alignment program (http://www.ebi.ac.uk/Tools/msa/clustalo). In red are highlighted fully conserved residues from the consensus sequence indicated in A. The presence of the second most frequently occurring consensus amino acid is indicated in yellow. The “:” (colon) indicates conservation between groups of strongly similar properties (scoring > 0.5 in the Gonnet PAM 250 matrix) after alignment of the ATase1 and ATase2 sequences with the GNAT consensus sequence. The “.” (period) indicates conservation between groups of weakly similar properties (scoring =< 0.5 in the Gonnet PAM 250 matrix) after alignment of the ATase1 and ATase2 sequences with the GNAT consensus sequence. The alignment of ATase1 and ATase2 sequences revealed the presence of non-conservative amino acid substitutions in motifs A and D (arrows). The box in Motif A encloses the residues that have been shown to be important for acetyl-CoA recognition and binding in other members of the GNAT superfamily.
ATase1 and ATase2 are differentially expressed in a variety of immortalized as well as primary cells [38]. Both in the mouse and human brain, they appear to be preferentially -if not exclusively-expressed in neurons [38]. Finally, they are up-regulated in patients affected by Alzheimer’s disease [38], further supporting a possible disease-association.
CML2 (the gene encoding ATase1) and CML1 (the gene encoding ATase2) appear to be the result of a gene duplication event that occurred after the ape-Old World monkey split [39]. Although no disease association has been reported, several single nucleotide polymorphisms (SNPs) have been identified on both genes in the human population. Particularly, one SNP (accession no. rs4852974) on ATase1 generates a stop codon in the middle of the acetyl-CoA:lysine acetyltransferase catalytic domain and is predicted to inactivate the enzyme. The SNP seems to be present in less than 2% of the general population. Whether this results in decreased ATase activity is unknown. Also unknown is the biological relevance of this apparent inactivation.
It is likely that ATase1 and ATase2 are not the only ER-based acetyltransferases. Interestingly, calreticulin, an ER luminal protein that is mostly known as a Ca+2-binding chaperone, appears to have acetyl-CoA:lysine acetyltransferase activity in vitro [40]. Whether calreticulin retains acetyltransferase activity in vivo remains to be determined. Also to be determined are its potential substrates. However, considering the number of ER-resident and -transiting proteins that interact with calreticulin in the ER lumen, this potential acetyltransferase activity could have large impact.
In vitro assessment revealed that ATase1, but not ATase2, undergoes autoacetylation in one or more lysine residues [17]. The biological significance of this different behavior is still unclear. In the case of other acetyltransferases, the autoacetylation event has been described either as part of an allosteric mechanism that regulates the activity of the acetyltransferase or as part of the catalytic signature of the transferase (see below in the Conclusive Remarks). The allosteric switch is meant to regulate the activity of the acetyltransferase without involving transcription/translation of the enzyme. In fact, when “hypo-acetylated” the enzyme displays reduced activity, while when “hyper/auto-acetylated” the enzyme displays increased activity [41, 42]. This scenario would be consistent with the fact that ATase1 appears to act as a constitutive form of acetyltrasferase while ATase2 appears to be tightly controlled at the transcriptional level [38].
2.3 The Disposing Machinery
To further study the role of lysine acetylation in the metabolism of nascent BACE1, we mutated the seven lysine residues that are normally acetylated in the native protein into alanine, arginine or glutamine. The Lys-to-Ala and Lys-to-Arg substitution generates “loss-of-acetylation” mutants whereas the Lys-to-Gln substitution generates a “gain-of-acetylation” mutant form of BACE1. The use of the above mutants revealed that the acetylated intermediates of BACE1 are able to reach the Golgi apparatus and complete maturation while the non-acetylated intermediates are retained in the ER Golgi Intermediate Compartment (ERGIC) [10]. Although efficiently synthesized in the ER and not able to reach the Golgi apparatus, the “loss-of-acetylation” mutants did not appear to accumulate in the ERGIC or to be disposed of by the proteasome machinery [10]. This observation suggested that an alternative pathway was responsible for the disposal of non-acetylated BACE1 intermediates. Subsequent studies revealed that proprotein convertase subtilisin kexin type 9 (PSCK9)/neural-apoptosis-regulated convertase 1 (NARC-1) was an essential component of the disposing machinery [14] (Figure 4).
FIGURE 4. Schematic view of BACE1 acetylation.

Nascent BACE1 is transiently acetylated in the lumen of the ER by ATase1 and ATase2. If acetylated, nascent BACE1 can reach the Golgi apparatus and complete maturation. In contrast, non-acetylated BACE1 is retained and degraded in the ERGIC. The ER-based acetylation also requires translocation of acetylCoA from the cytosol into the lumen of the ER by the ER membrane transporter AT-1. The acetyl group is shown as a red circle.
PCSK9/NARC-1 is a member of the subtilisin (S8) family of serine proteases [43, 44]. More specifically, PCSK9/NARC-1 is part of the S8A subfamily, which so far includes only two members, PCSK9/NARC-1 itself and site-1 protease (S1P), known to regulate the processing and consequent activation of several ER-based transcription factors [45]. PCSK9/NARC-1 is a ~75-kDa proprotein that undergoes apparent autocatalysis to generate a shorter form of ~65-kDa [46, 47]. PCSK9/NARC-1 has received great attention since the discovery that mutations in the Pcsk9/Narc1 gene are associated with hypercholesterolemia. The fact that PCSK9/NARC-1 can regulate the degradation of the low-density lipoprotein (LDL) receptor (LDLR) has caused additional excitement as possible down-regulation or biochemical inhibition of PCSK9/NARC-1 might serve as an alternative approach for reducing hypercholesterolemia. In fact, low levels or activity of PCSK9/NARC-1 would lead to increased expression of LDLR in the liver and increased uptake/degradation of circulating LDL [43, 44].
Although the exact role of PCSK9/NARC-1 in LDLR metabolism is still uncertain, early work showed that one of its essential functions is to regulate the disposal of LDLR in a “post-ER compartment” while transiting along the secretory pathway [48, 49]. The “post-ER compartment” mechanism was briefly put aside by evidence that PCSK9/NARC-1 could also degrade cell-surface LDLR following secretion in the conditioned media [50–52]. However, recent work has spurred new interest in the “post-ER compartment” mechanism [53]. The fact that PCSK9/NARC-1 could affect the levels of the nascent form of LDLR in a post-ER compartment while trafficking along the secretory pathway [48, 49] stimulated our own interest. The possible involvement of PCSK9/NARC-1 in the disposal of non-acetylated intermediate forms of BACE1 in the ERGIC was addressed by a combination of biochemical and molecular biological approaches. In particular, over-expression of PCSK9/NARC-1 in cultured cells decreased the levels of BACE1 whereas siRNA-mediated down-regulation of PCSK9/NARC-1 achieved the opposite results [14]. Finally, the levels of BACE1 were found to be increased in the brain of PCSK9/NARC-1 knockout animals [14]. Importantly, down-regulation of PCSK9/NARC-1 increased the levels of the “loss-of-acetylation” mutant forms of BACE1 but not those of the “gain-of-acetylation” mutants, suggesting that PCSK9/NARC-1 might work in concert with the ER-based acetylation machinery to regulate the disposal of BACE1 non-acetylated intermediates [14]. This conclusion is further supported by the observation that, similarly to BACE1, the nascent ER-form of LDLR is acetylated [14], suggesting a common function for the serine protease.
It is also worth mentioning that the promoter region of Pcsk9/Narc1 contains a sterol regulatory element (SRE). As a result, the mRNA levels of PCSK9/NARC-1 are up-regulated by SRE-binding proteins (SREBPs), ER membrane-based transcription factors that control both biosynthesis and uptake of cholesterol [54, 55]. As a consequence, the expression of PCSK9/NARC-1 is down-regulated by increased cellular cholesterol content [56], and upregulated by HMG-CoA inhibitors (also known as “statins”), which reduce cholesterol content [57]. These results are particularly important considering that several aspects of cholesterol metabolism have been associated with Alzheimer’s disease [58], including the fact that increased cellular levels of cholesterol (which are predicted to decrease PCSK9/NARC-1-mediated disposal of BACE1) increase Aβ generation, whereas reduced levels of cholesterol (which are predicted to increase PCSK9/NARC-1-mediated disposal of BACE1) reduce Aβ generation.
Although initially studied for its implication in LDLR metabolism, it is now evident that PCSK9/NARC-1 “does” more than just regulating LDLR levels. In fact, in addition to BACE1 [14] and APP [15], it also regulates the disposal of other cell-surface membrane proteins [59, 60] and perhaps secreted proteins as well [61], suggesting a more global function. Indeed, a recent proteomic study identified more than 300 proteins that are influenced by PCSK9/NARC-1 activity [62]. The list includes cell-surface proteins as well as intracellular proteins, proteins involved in folding and post-translational modification in the early secretory pathway, proteins involved in vesicle and organelle transport, cytoskeleton as well as cell-adhesion proteins. This global impact of PCSK9/NARC-1 was also demonstrated by two microarrays studies, which implicated this serine protease in many biological pathways [63, 64].
We initially reported that inhibition of the proteasome machinery did not appear to affect the disposal of the “loss-of-acetylation” mutant forms of BACE1 [10]. However, recently Gong et al. have proposed the possible requirement of the E3-ligase activity of the Fbx2-SCF ubiquitin ligase complex, suggesting that the ubiquitin-proteasome system might still be involved with the clearance of non-acetylated species of BACE1 [65]. The ubiquitylation of BACE1 is somewhat specific and seems to occur on some of the seven lysine residues that are acetylated [65]; this could potentially explain why acetylated BACE1 intermediates are resistant to proteasome degradation [10]. Whether there is cross-talk between PCSK9/NARC-1 and SCFFbx2-E3 ligase remains to be assessed. In fact, PCSK9/NARC-1 is a serine protease as well as a chaperone-like protein, while Fbx2 is typically involved in the recognition and binding of high-mannose oligosaccharides on misfolded/unfolded ER luminal proteins, which are subsequently targeted for proteasome-dependent disposal (see also later). Another issue that will also need further investigation is how the Fbx2-SCF ubiquitin ligase complex can have access to the ectodomain of BACE1; in fact, the former is in the cytosol while the latter is in the lumen of the ER. It is likely that nascent non-acetylated BACE1 might need to be “preprocessed” and retro-translocated to the cytosol before becoming a substrate of the Fbx2-SCF ubiquitin ligase complex, as described for other Fbx2-SCF substrates [66].
3. A Novel Mechanism for Quality Control and/or ERAD?
The essential information for the correct assembly of membrane and secreted proteins synthesized in the ER is already present in their primary amino acid sequence. However, the efficiency of folding itself is ensured and enhanced by ER-resident proteins that act as chaperones and/or modifying enzymes. Chaperones assist in the folding by ensuring safe passage of the nascent protein chain through the ER membrane, by retrieving and preventing aggregation of incorrectly folded protein intermediates, and -in some cases- by protecting the nascent polypeptide from interacting with potential ligands in the ER [67, 68]. Chaperones can be solely restricted to the ER, can travel through the secretory pathway together with their substrates or, in some cases, be secreted to the extracellular milieu with their immediate substrates [67–69]. The large family of ER chaperones includes heat-shock proteins such as BiP, calreticulin, calnexin and the receptor associated protein (RAP). In contrast to chaperones, modifying enzymes typically ensure correct folding by completing the information already contained in the primary amino acid sequence. Some examples are the protein disulfide isomerase (PDI), which catalyzes the oxidation of free SH groups of paired cysteine residues to form disulfide bonds (-CH2-S-S-CH2-), and the oligosaccharyltransferase (OST), which transfers a preformed “core” oligosaccharide from a glycolipid donor, the dolichol-P-P-(GlcNAc)2(Man)5(Glc)3, to an asparagine residue in the consensus sequence Asn-X-Ser/Thr of the nascent glycoprotein. Although the above are examples of permanent covalent modifications, there are also transient modifications that are only meant to recognize and salvage incorrectly folded protein intermediates. This is the case of the UDP-glucose:glycoprotein glucosyltransferase (UGGT), which attaches one glucose residue to improperly folded nascent glycoproteins and regulates its interaction with the lectin chaperone calnexin [70, 71].
The above chaperones and modifying enzymes not only ensure fidelity of the “protein code” by assisting nascent polypeptides in assuming the right conformation, which is necessary to their function, but also sequester incorrectly folded intermediates that have failed quality control (QC). These are generally retained in the ER and directed toward the ER associated degradation (ERAD). Incorrectly folded polypeptides that are disposed of by the ERAD are typically retro-translocated to the cytosol and sent to the proteasome for disposal but can also aggregate (in the cytosol or in the ER itself) to form protein inclusions. The fate of the aggregates is uncertain; they can serve as a “sink” for misfolded proteins that failed QC, thus preventing their otherwise toxic functions (this seems to be the case for the cytoplasmic aggregates called “aggresomes”) or can be digested through autophagic-like mechanisms (see below) [68, 72–76]. However, under certain conditions, protein aggregates can be overwhelming for the cell and become toxic themselves. Misfolded protein intermediates can also bypass the above more classical ERAD and be directed to the Golgi apparatus where they are retained or directed to the lysosomes for ultimate degradation [68].
3.1 To protect the good ones
The use of the acetylation mutants revealed that the acetylated intermediates of BACE1 are able to reach the Golgi apparatus and complete maturation while the non-acetylated intermediates are retained in the ERGIC [10]. In addition, the down-regulation of PCSK9/NARC-1 only increased the levels of the “loss-of-acetylation” mutant, suggesting that some sort of selection takes place [14]. These features are reminiscent of a QC event where certain biosynthetic intermediates are selected for “free passage” across the secretory pathway while others (presumably unfolded/misfolded intermediates) are not (see Figure 4). Although the possible connection between lysine acetylation in the ER and QC still needs to be carefully evaluated, this possibility is supported by the “chaperone-like” activity of the acetyltransferases [17]. Perhaps, the ATases can only recognize and acetylate correctly folded intermediates. Perhaps, the Nε-lysine acetylation provides a way to “lock” the acetylated (correctly folded) intermediates to the transferases protecting them from PCSK9/NARC-1 mediated disposal. The locking mechanisms would explain why all seven acetylated lysine residues in BACE1 are located on the same face of the protein [10]. Perhaps, the release of the ATases [17] together with the deacetylation of BACE1 that occurs in the Golgi apparatus [10] serves to “restore” the original “protein code” allowing final maturation and transport to the cell surface. Perhaps, the ATases do not simply serve to recognize already folded protein intermediates but actively participate in the folding itself. In the case of BACE1, the acetylated lysine residues are clustered in areas that are characterized by low-electron density (and intrinsic conformational flexibility). This structural flexibility appears to be essential for the catalytic activity [77–79] as well as for the natural “shedding” [80] of the enzyme. It is possible that the Nε-lysine acetylation -by neutralizing the positive charges of the lysine residues- serves to stabilize and facilitate the folding of kinetically unfavorable areas of the nascent protein [10]. Perhaps, the “stabilization” and “locking” mechanisms delineated above are intimately connected to achieve greater efficiency while processing large quantities of nascent membrane and secreted proteins in the ER. Although we recognize that the above “perhaps” still need to be biochemically proven, our current information appears to preclude other possibilities. This view is also supported by the fact that BACE1 is not alone; in fact, other membrane proteins undergo the same transient post-translational modification [14–16].
3.2 What if it fails?
If the ER-based Nε-lysine acetylation machinery is an essential form of post-translational regulation of nascent proteins, we would expect the global failure of the ER-based acetylation to lead to cellular disaster. The down-regulation of the two transferases, ATase1 and ATase2, did not result in significant cell toxicity [17]. However, it is likely that the ER has more than just two acetyltransferases (discussed above); it is also likely that the transferases share (at least in part) their substrates allowing for one enzyme to compensate for the lack of the other(s). In contrast to the ATases, the down-regulation of AT-1 resulted in massive and widespread cell death, indicating that the translocation of acetyl-CoA into the ER lumen is an essential cellular function [15, 81]. This conclusion is strongly supported by the fact that AT-1 is mutated in patients affected by autosomal dominant spastic paraplegia-42 (SPG42) [82].
SPG42 is part of the highly heterogeneous family of hereditary spastic paraplegias (here collectively called SPGs). The clinical heterogeneity probably reflects the genetic complexity of the disease. A recent list of genes and loci implicated with SPGs has been reported by Salinas et al. [83]. SPGs are generally divided into two groups: uncomplicated and complicated. Uncomplicated SPGs are typically characterized by progressive bilateral spasticity and peripheral neuropathy; urinary symptoms and mild cognitive decline are also present in a large number of patients. The onset of the disease varies greatly from early childhood to as late as 70 years of age. The main pathological feature of uncomplicated SPGs is a progressive degeneration of motor axons of the corticospinal tract in the absence of evident muscle pathology. Loss of volume of the corpus callosum and memory-forming areas of the brain, as well as disseminated lesions of cerebral white matter, have also been reported [83, 84]. Complicated SPGs are characterized by the fact that the lower limb spasticity is accompanied by other features such as severe mental retardation, dementia, epilepsy, cerebellar ataxia, amyotrophy, retinopathy, glaucoma, deafness and icthyosis [83, 84].
Enlarged ER sheets and tubules as well as generalized defects of the ER network constitute a common morphological feature of SPG neurons (reviewed in [85]). Importantly, the vast majority of mutations found in SPGs are concentrated in genes encoding proteins that are localized in the ER and ERGIC, function at the ER-to-Golgi interface or are overall involved in ER network organization, membrane trafficking, and axonal transport of macromolecules and other cargoes (reviewed in [83, 85]). This finding should be viewed together with the fact that the earliest and most striking changes observed in SPG patients are always concentrated in the long axons of descending and ascending spinal neurons. In humans, these processes can measure up to one meter in length. As a result of their morphology, they are heavily dependent on membrane trafficking, rapid axonal transport of cargo molecules from the ER to the plasma membrane and active cytoskeletal organization. A defect in the above events is likely to disrupt their function and lead to degeneration. Interestingly, haploinsufficiency or leaky mutations associated with SPGs only cause defects/degeneration of neurons, further stressing the strong dependence of neuronal cells to the above events [86, 87]. Although ER network organization, membrane trafficking, and transport of macromolecules are essential and common features of every cell, neurons are simply more dependent on them. However, less dependence does not mean no dependence! In fact, a ~95% decrease in AT-1 levels caused massive death of cultured cells under ex vivo conditions [15]. Furthermore, down-regulation of AT-1 in the zebrafish increased embryonic lethality and caused a curved-shaped tail phenotype in the surviving animals, as well as defective axonal growth of spinal cord neurons [82]. The striking phenotype caused by the down-regulation of AT-1 under both ex vivo [15] and in vivo [82] conditions indicates that a constant influx of acetyl-CoA into the lumen of the ER is an essential cellular function (discussed later).
Mutations in AT-1 have also been identified in five patients affected by a complex autosomal-recessive syndrome characterized by psychomotor retardation and severe developmental delay. The patients, who displayed brain atrophy, cerebellar hypoplasia, hypomyelination, congenital chataracts, hearing loss, and multi-organ failure, died prematurely within the first 5 years of their life [88]. In contrast to SPG42 patients, who were heterozygous for the mutation [82], the above were all homozygous [88]. The above patients also displayed low serum copper and ceruloplasmin [88]. However, reassessment of one of the above five patients identified an additional mutation in the copper chaperone for superoxide dismutase (CCS) [89], suggesting that the phenotype might be complicated by the co-presence of more than one mutation.
3.3 Regulation of Autophagy/ERAD(II) by the ER-based acetylation machinery
A close assessment of the modalities of the cell death associated with the down-regulation of AT-1 in culture revealed that cells initially respond to the reduced import of acetyl-CoA into the ER lumen by increasing the size of the ER tubules [15]. This is then followed by the formation of autophagosomes in close proximity of the ER membrane, suggesting that the ER membrane itself participates in the formation of the autophagic elements. If the down-regulation of AT-1 persists, the entire ER breaks down into autophagosomes and ultimately the cell dies by autophagic cell death [15]. Importantly, before widespread digestion of intracellular organelles (final autophagic cell death) the ER is the only organelle that appears to be affected. The above observation clearly indicates that the continuous supply of acetyl-CoA into the ER lumen is an essential feature of this organelle; they also indicate that autophagic digestion of ER tubules is the preferential strategy to deal with the failure of the ER-based acetylation machinery.
Autophagy is a cellular process that allows turnover and/or degradation of subcellular components, which are initially engulfed in double-membrane structures and then diverted toward active lysosomes for final degradation. This process can be part of typical “house-keeping” activities that dispose of unwanted proteins or excessive membranes; it can also serve to ensure supply of nutrients during starvation or to clear the organism of unwanted cells during development [76, 90–93]. As described above, the ER has mechanisms in place that ensure correct folding of secreted/membrane proteins. When these fail, unwanted (unfolded/misfolded) proteins are typically degraded by the proteasome as part of ERAD. However, the proteasome preferentially degrades monomeric proteins that are unfolded prior to retro-translocation across the ER membrane [68]. Large protein aggregates are mostly dealt with by expanding the ER and activating autophagy. The autophagic process allows the cell to counterbalance the ER expansion and dispose of the protein aggregates at the same time [73–75, 94]. This process is still part of ERAD and the terms ERAD(I) and ERAD(II) have also been used to differentiate the ubiquitin/proteasome process from the autophagic process, respectively [95]. Therefore, autophagy must be seen as an essential cellular function that ensures disposal of unwanted material. This is particularly important for neurons where a high threshold for ER stress (discussed above) and inherent cellular features require a continuous and efficient way to dispose of protein aggregates [92, 96–99]. Obviously, if unchecked, autophagy can become terminal (autophagic cell death; also referred to as type II cell death to differentiate it from apoptosis, which is referred to as type I cell death). Both ERAD(I) and ERAD(II) are under the control of the unfolded protein response (UPR), a complex ER-based signaling machinery that corrects possible imbalances that result in ER stress and adjusts the ability of the organelle to cope with the accumulation of unfolded/misfolded protein intermediates [68, 100–102].
That the ER-based acetylation machinery is intimately linked to the UPR and ERAD(II) in the ER is supported by four main recent findings. First, a large number of ER-resident proteins involved with folding and disposal of unfolded/misfolded protein intermediates is acetylated in the lumen of the ER [16]. Second, already known and well characterized ER stress inducers increase the influx of acetyl-CoA into the ER lumen by up-regulating the expression of AT-1 [81]. Third, the up-regulation of AT-1 during ER stress was linked to one of the three main branches of the UPR, specifically to the inositol-requiring protein-1 (IRE1)-dependent signaling pathway [81]. IRE1 is an ER-membrane bound protein that when activated, as a result of ER stress, removes a small fragment (intron) from the mRNA of X-box binding-1 (XBP1). The process requires the endoribonuclease activity of IRE1 and the ligase activity of an unknown ligase [102, 103]. The product of the spliced version of XBP1 (XBP1s) is a potent transcriptional activator of UPR target genes that promote ERAD of unfolded/misfolded protein intermediates and ER biogenesis [104]. Fourth, AT-1 was shown to regulate the activation of autophagy down-stream of XBP1 itself [81]. Therefore, the influx of acetyl-CoA into the ER lumen is a novel and essential feature of the UPR and appears to control the ability of the ER to recognize and select correctly folded protein intermediates (as observed with BACE1), and dispose of unfolded/misfolded protein aggregates through the regulation of autophagy/ERAD(II). At the mechanistic level, the regulation of ERAD(II) involves intraluminal acetylation of the autophagy protein Atg9A [81], which might act as an ER-based “sensor” for the acetylation status of the ER (Figure 5). Indeed, acetylation of Atg9A prevents the induction of autophagy whereas the deacetylation (or lack of acetylation) has the opposite effect [81]. A similar regulatory function of the Nε-lysine acetylation has also been observed with the cytosolic members of the autophagic machinery, specifically Atg5, Atg7, Atg8 and Atg12 [105, 106].
FIGURE 5. Regulation of the influx of acetyl-CoA into the ER lumen as part of the UPR.

The constant influx of acetyl-CoA into the ER lumen is required to control the induction of autophagy/ERAD(II) as part of the UPR. Specifically, AT-1 acts downstream of IRE1/XBP1 signaling. If the influx of acetyl-CoA is maintained, ERAD(II) helps to eliminate unwanted unfolded/misfolded aggregates without inducing cell death (A). However, if the influx is not maintained, programmed Type II (autophagic) cell death is activated (B). The autophagy protein Atg9A seems to act as a “sensor” for the acetylation status of the ER.
It must be noted that the activation of the “ER stress signaling” is both a physiologic and pathologic event. Cells can experience transient periods of ER stress as part of their activity [107–110]. As discussed above, due to their specific functions, neurons may have to cope with a substantial physiological burden and, as a result, have higher basal levels of ER stress. This could potentially explain why a mutation in AT-1 is primarily associated with a disease of the nervous system. However, B cells also experience reorganization and expansion of the ER and activation of the UPR when they differentiate into immunoglobulin secreting plasma cells [111]. The ultimate purpose is to ensure the biosynthesis and secretion of a large quantity of correctly folded immunoglobulins [111, 112]. Interestingly, the differentiation process of B cells into plasma cells is also accompanied by activation of XBP1 signaling and up-regulation of AT-1 [113]. Therefore, AT-1 does not only respond to ER stress-inducing experimental drugs, which are largely non-physiologic, but also to “naturally” occurring conditions that require induction of the ER stress machinery.
4. Conclusive Remarks and Unresolved Questions
Although “young”, the ER-based acetylation machinery is rapidly emerging as a major component of normal ER functions and is poised to grow in importance as we further delineate its impact on physiologic and pathologic events. As we learn more, several currently unresolved questions will need to be addressed. For example:
What is the purpose of the ER-based acetylation?
Studies performed with BACE1 indicate that the Nε-lysine acetylation of the nascent protein might serve to distinguish between correctly folded and unfolded/misfolded protein intermediates. Although it needs to be proven, a similar mechanism might be in place for other ER-transiting proteins, which need to move out of the ER along the secretory pathway. However, why acetylate ER-resident proteins, which are not supposed to leave the organelle? Is there a similar mechanism in place for ER-resident proteins? Perhaps, in the case of ER-resident proteins the acetylation influences activity/function of the proteins rather than their ability to leave the organelle. It is also possible that lysine acetylation is necessary for the assembly of kinetically unfavorable (disordered?) regions of the nascent polypeptide. This scenario could be tested by analyzing the structural signature -when available- of recently identified ER-resident and -transiting proteins that undergo acetylation [16]. Finally, it is highly possible that the cell might have more than one reason to acetylate nascent proteins in the ER lumen.
How many ATases are in the ER?
At this point, it is evident that the ER has more than two acetyltransferases. Although some overlap is possible, it is likely that they have different substrates. Identification of the other ATases as well as their immediate substrates will help us target the acetylation machinery for therapeutic purposes. ATase1 and ATase2 are attractive targets for Alzheimer’s disease therapeutics [17, 38]. It is likely that other ATases might represent viable targets for other diseases.
What is the catalytic signature of the ATases?
Most of the known acetyltransferases employ two different catalytic mechanisms: the ternary complex and the ping-pong mechanism [114] (Figure 6). The ternary complex mechanism involves direct transfer of the acetyl group from the donor (acetyl-CoA) to the acceptor lysine residue. This process requires the enzyme to interact and bind both the donor and the acceptor at the same time (hence, ternary complex). In contrast, the ping-pong mechanism involves initial transfer of the acetyl group from the donor (acetyl-CoA) to the enzyme, which becomes acetylated. The intermediate acetylated enzyme will then transfer the acetyl group to the acceptor lysine residue. Therefore, this process requires the acetyltransferase to interact separately with the donor and the acceptor and to act as an acetylated intermediate species (hence, ping-pong). These mechanistic differences are very important because different mechanisms imply different classes of inhibitors and different forms of regulation [114].
FIGURE 6. Schematic view of the two main catalytic mechanisms employed by acetyl-CoA:lysine acetyltransferases.

Description is in the text.
If there are acetyltransferases, are there deacetylases as well?
Our initial studies with BACE1 indicated that the nascent protein is normally acetylated in the lumen of the ER and then deacetylated in the lumen of the Golgi apparatus [10]. Indeed, in vitro biochemical assessment revealed that the Golgi apparatus possess deacetylase activity [10]. Importantly, the activity was found to reside in the lumen of the organelle [10]. However, computer-based searches for possible candidates have -so far- failed to yield solid candidates, suggesting that “more traditional” protein purification approaches will be required to ensure the identification of another piece of this puzzle. Similar strategies might also be necessary to determine how transient acetylation/deacetylation is achieved for ER-resident proteins.
Can AT-1 be targeted for therapeutic purposes?
Initial studies indicate that AT-1 is essential for cell viability [15, 82]. Indeed, down-regulation of AT-1 in mammalian cells triggers the activation of the autophagic machinery and results in widespread type I (autophagic) cell death [15, 81]. As such, we should predict AT-1 not to be a viable target. However, what is toxic for normal cells might not be as toxic for sick cells. Indeed, mice expressing a mutant form of the Cu,Zn superoxide dismutase (SOD1) linked to amyotrophic lateral sclerosis (ALS) benefit from an excessive activation of the autophagic machinery [115, 116]. This appears to be due to a more efficient removal of toxic protein aggregates. Perhaps, a balance between the rate of protein aggregates formation and the rate of autophagy-mediated removal of the aggregates must be ensured. As such, excessive autophagy would be deleterious in a normal cell with normal levels of protein aggregates, but beneficial in a “sick” cell with excessive formation/accumulation of protein aggregates. Importantly, the protective effect of a “hyper-active” autophagic machinery in the above ALS-like model was achieved by down-regulating XBP1 [115, 116], which appears to act through AT-1 itself [81]. Therefore, AT-1 might be a possible target for therapeutics, albeit only in a selected group of patients.
How is the cross-talk between AT-1 and the autophagic machinery ensured?
Recent results indicate that the influx of acetyl-CoA into the ER lumen is “sensed” by Atg9A, the only membrane bound autophagy protein [81]. However, the molecular details are still unknown. Perhaps the acetylation status of Atg9A regulates its trafficking to the Golgi apparatus and the consequent formation of the “autophagic membrane”. Or, perhaps, the acetylation status of Atg9A affects its ability to interact with the other elements of the autophagic machinery.
Is the proteasome machinery involved?
How can we reconcile the fact that the disposal of non-acetylated BACE1 mutants is not affected by proteasome inhibitors [10] with the fact that the lysine residues that are acetylated appear to be necessary for the in vitro ubiquitylation of BACE1 by the Fbx2-SCF ubiquitin ligase complex [65]? Are these two mechanisms related? Or are they meant to recognize different intermediate species of the nascent protein?
Although many questions remain to be answered, one thing is certain: what was once a cytosolic and nuclear event is now an essential component of ER functions. Dissection of the biochemistry that governs the ER-based acetylation machinery will certain lead us to a better understanding of fundamental biological events that influence human physiology and pathology.
Acknowledgments
The Authors wish to thank Dr. Rozalyn Anderson for critical reading of an early version of this manuscript. Luigi Puglielli is supported by grants from the NIH/NIA (AG028569 and AG033514) and the Department of Veterans Affairs (Merit Award). Mariana Pehar is partially supported by NIH (8P20GM103542).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yang XJ, Gregoire S. Metabolism, cytoskeleton and cellular signalling in the grip of protein Nepsilon - and O-acetylation. EMBO Rep. 2007;8:556–562. doi: 10.1038/sj.embor.7400977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 3.Xiong Y, Guan KL. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J Cell Biol. 2012;198:155–164. doi: 10.1083/jcb.201202056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? Embo J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Polevoda B, Sherman F. N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J Mol Biol. 2003;325:595–622. doi: 10.1016/s0022-2836(02)01269-x. [DOI] [PubMed] [Google Scholar]
- 6.Mittal R, Peak-Chew SY, McMahon HT. Acetylation of MEK2 and I kappa B kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc Natl Acad Sci U S A. 2006;103:18574–18579. doi: 10.1073/pnas.0608995103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukherjee S, Keitany G, Li Y, Wang Y, Ball HL, Goldsmith EJ, Orth K. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science. 2006;312:1211–1214. doi: 10.1126/science.1126867. [DOI] [PubMed] [Google Scholar]
- 8.Wade PA, Pruss D, Wolffe AP. Histone acetylation: chromatin in action. Trends Biochem Sci. 1997;22:128–132. doi: 10.1016/s0968-0004(97)01016-5. [DOI] [PubMed] [Google Scholar]
- 9.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costantini C, Ko MH, Jonas MC, Puglielli L. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem J. 2007;407:383–395. doi: 10.1042/BJ20070040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 12.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 13.Guan KL, Xiong Y. Regulation of intermediary metabolism by protein acetylation. Trends Biochem Sci. 2011;36:108–116. doi: 10.1016/j.tibs.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jonas MC, Costantini C, Puglielli L. PCSK9 is required for the disposal of non-acetylated intermediates of the nascent membrane protein BACE1. EMBO Rep. 2008;9:916–922. doi: 10.1038/embor.2008.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jonas MC, Pehar M, Puglielli L. AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability. J Cell Sci. 2010;123:3378–3388. doi: 10.1242/jcs.068841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pehar M, Lehnus M, Karst A, Puglielli L. Proteomic Assessment Shows That Many Endoplasmic Reticulum (ER)-resident Proteins Are Targeted by N{epsilon}-Lysine Acetylation in the Lumen of the Organelle and Predicts Broad Biological Impact. J Biol Chem. 2012;287:22436–22440. doi: 10.1074/jbc.C112.362871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ko MH, Puglielli L. Two Endoplasmic Reticulum (ER)/ER Golgi Intermediate Compartment-based Lysine Acetyltransferases Post-translationally Regulate BACE1 Levels. J Biol Chem. 2009;284:2482–2492. doi: 10.1074/jbc.M804901200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell. 2006;23:207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 19.Voet D, Voet JG, editors. Biochemistry. 2. John Wiley & Sons, Inc; 1995. p. 687. [Google Scholar]
- 20.Lysiak W, Lilly K, DiLisa F, Toth PP, Bieber LL. Quantitation of the effect of L-carnitine on the levels of acid-soluble short-chain acyl-CoA and CoASH in rat heart and liver mitochondria. J Biol Chem. 1988;263:1151–1156. [PubMed] [Google Scholar]
- 21.van Roermund CW, Hettema EH, van den Berg M, Tabak HF, Wanders RJ. Molecular characterization of carnitine-dependent transport of acetyl-CoA from peroxisomes to mitochondria in Saccharomyces cerevisiae and identification of a plasma membrane carnitine transporter, Agp2p. Embo J. 1999;18:5843–5852. doi: 10.1093/emboj/18.21.5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wanders RJ, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006;75:295–332. doi: 10.1146/annurev.biochem.74.082803.133329. [DOI] [PubMed] [Google Scholar]
- 23.Jakobs BS, Wanders RJ. Fatty acid beta-oxidation in peroxisomes and mitochondria: the first, unequivocal evidence for the involvement of carnitine in shuttling propionyl-CoA from peroxisomes to mitochondria. Biochem Biophys Res Commun. 1995;213:1035–1041. doi: 10.1006/bbrc.1995.2232. [DOI] [PubMed] [Google Scholar]
- 24.Palmieri L, Lasorsa FM, Iacobazzi V, Runswick MJ, Palmieri F, Walker JE. Identification of the mitochondrial carnitine carrier in Saccharomyces cerevisiae. FEBS Lett. 1999;462:472–476. doi: 10.1016/s0014-5793(99)01555-0. [DOI] [PubMed] [Google Scholar]
- 25.Madiraju P, Pande SV, Prentki M, Madiraju SR. Mitochondrial acetylcarnitine provides acetyl groups for nuclear histone acetylation. Epigenetics: official journal of the DNA Methylation Society. 2009;4:399–403. doi: 10.4161/epi.4.6.9767. [DOI] [PubMed] [Google Scholar]
- 26.Leighton F, Bergseth S, Rortveit T, Christiansen EN, Bremer J. Free acetate production by rat hepatocytes during peroxisomal fatty acid and dicarboxylic acid oxidation. J Biol Chem. 1989;264:10347–10350. [PubMed] [Google Scholar]
- 27.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Snoswell AM, Koundakjian PP. Relationships between carnitine and coenzyme A esters in tissues of normal and alloxan-diabetic sheep. Biochem J. 1972;127:133–141. doi: 10.1042/bj1270133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knowles SE, Jarrett IG, Filsell OH, Ballard FJ. Production and utilization of acetate in mammals. Biochem J. 1974;142:401–411. doi: 10.1042/bj1420401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mackall JC, Lane MD. Changes in mammary-gland acetyl-coenzyme A carboxylase associated with lactogenic differentiation. Biochem J. 1977;162:635–642. doi: 10.1042/bj1620635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanamori A, Nakayama J, Fukuda MN, Stallcup WB, Sasaki K, Fukuda M, Hirabayashi Y. Expression cloning and characterization of a cDNA encoding a novel membrane protein required for the formation of O-acetylated ganglioside: a putative acetyl-CoA transporter. Proc Natl Acad Sci U S A. 1997;94:2897–2902. doi: 10.1073/pnas.94.7.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirabayashi Y, Kanamori A, Nomura KH, Nomura K. The acetyl-CoA transporter family SLC33. Pflugers Arch. 2004;447:760–762. doi: 10.1007/s00424-003-1071-6. [DOI] [PubMed] [Google Scholar]
- 33.Neuwald AF, Landsman D. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem Sci. 1997;22:154–155. doi: 10.1016/s0968-0004(97)01034-7. [DOI] [PubMed] [Google Scholar]
- 34.Vetting MW, SdC LP, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Structure and functions of the GNAT superfamily of acetyltransferases. Archives of biochemistry and biophysics. 2005;433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Dyda F, Klein DC, Hickman AB. GCN5-related N-acetyltransferases: a structural overview. Annual review of biophysics and biomolecular structure. 2000;29:81–103. doi: 10.1146/annurev.biophys.29.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He H, Ding Y, Bartlam M, Sun F, Le Y, Qin X, Tang H, Zhang R, Joachimiak A, Liu J, Zhao N, Rao Z. Crystal structure of tabtoxin resistance protein complexed with acetyl coenzyme A reveals the mechanism for beta-lactam acetylation. J Mol Biol. 2003;325:1019–1030. doi: 10.1016/s0022-2836(02)01284-6. [DOI] [PubMed] [Google Scholar]
- 37.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 38.Ding Y, Ko MH, Pehar M, Kotch F, Peters NR, Luo Y, Salamat SM, Puglielli L. Biochemical inhibition of the acetyltransferases ATase1 and ATase2 reduces beta-secretase (BACE1) levels and Abeta generation. J Biol Chem. 2012;287:8424–8433. doi: 10.1074/jbc.M111.310136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hahn Y, Lee B. Human-specific nonsense mutations identified by genome sequence comparisons. Hum Genet. 2006;119:169–178. doi: 10.1007/s00439-005-0125-6. [DOI] [PubMed] [Google Scholar]
- 40.Singh P, Ponnan P, Priya N, Tyagi TK, Gaspari M, Krishnan S, Cuda G, Joshi P, Gambhir JK, Sharma SK, Prasad AK, Saso L, Rastogi RC, Parmar VS, Raj HG. Protein acyltransferase function of purified calreticulin: the exclusive role of P-domain in mediating protein acylation utilizing acyloxycoumarins and acetyl CoA as the acyl group donors. Protein and peptide letters. 2011;18:507–517. doi: 10.2174/092986611794927938. [DOI] [PubMed] [Google Scholar]
- 41.Thompson PR, Wang D, Wang L, Fulco M, Pediconi N, Zhang D, An W, Ge Q, Roeder RG, Wong J, Levrero M, Sartorelli V, Cotter RJ, Cole PA. Regulation of the p300 HAT domain via a novel activation loop. Nat Struct Mol Biol. 2004;11:308–315. doi: 10.1038/nsmb740. [DOI] [PubMed] [Google Scholar]
- 42.Karanam B, Jiang L, Wang L, Kelleher NL, Cole PA. Kinetic and mass spectrometric analysis of p300 histone acetyltransferase domain autoacetylation. J Biol Chem. 2006;281:40292–40301. doi: 10.1074/jbc.M608813200. [DOI] [PubMed] [Google Scholar]
- 43.Attie AD. The mystery of PCSK9. Arterioscler Thromb Vasc Biol. 2004;24:1337–1339. doi: 10.1161/01.ATV.0000137288.82390.04. [DOI] [PubMed] [Google Scholar]
- 44.Maxwell KN, Breslow JL. Proprotein convertase subtilisin kexin 9: the third locus implicated in autosomal dominant hypercholesterolemia. Curr Opin Lipidol. 2005;16:167–172. doi: 10.1097/01.mol.0000162321.31925.a3. [DOI] [PubMed] [Google Scholar]
- 45.Yoshida H. ER stress and diseases. Febs J. 2007;274:630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 46.Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, Basak A, Prat A, Chretien M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A. 2003;100:928–933. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, Asselin MC, Hamelin J, Varret M, Allard D, Trillard M, Abifadel M, Tebon A, Attie AD, Rader DJ, Boileau C, Brissette L, Chretien M, Prat A, Seidah NG. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. 2004;279:48865–48875. doi: 10.1074/jbc.M409699200. [DOI] [PubMed] [Google Scholar]
- 48.Maxwell KN, Breslow JL. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci U S A. 2004;101:7100–7105. doi: 10.1073/pnas.0402133101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maxwell KN, Fisher EA, Breslow JL. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci U S A. 2005;102:2069–2074. doi: 10.1073/pnas.0409736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park SW, Moon YA, Horton JD. Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J Biol Chem. 2004;279:50630–50638. doi: 10.1074/jbc.M410077200. [DOI] [PubMed] [Google Scholar]
- 51.Benjannet S, Rhainds D, Hamelin J, Nassoury N, Seidah NG. The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: functional consequences of natural mutations and post-translational modifications. J Biol Chem. 2006;281:30561–30572. doi: 10.1074/jbc.M606495200. [DOI] [PubMed] [Google Scholar]
- 52.Qian YW, Schmidt RJ, Zhang Y, Chu S, Lin A, Wang H, Wang X, Beyer TP, Bensch WR, Li W, Ehsani ME, Lu D, Konrad RJ, Eacho PI, Moller DE, Karathanasis SK, Cao G. Secreted PCSK9 downregulates low density lipoprotein receptor through receptor-mediated endocytosis. J Lipid Res. 2007;48:1488–1498. doi: 10.1194/jlr.M700071-JLR200. [DOI] [PubMed] [Google Scholar]
- 53.Poirier S, Mayer G, Poupon V, McPherson PS, Desjardins R, Ly K, Asselin MC, Day R, Duclos FJ, Witmer M, Parker R, Prat A, Seidah NG. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J Biol Chem. 2009;284:28856–28864. doi: 10.1074/jbc.M109.037085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA, Horton JD. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci U S A. 2005;102:5374–5379. doi: 10.1073/pnas.0501652102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maxwell KN, Soccio RE, Duncan EM, Sehayek E, Breslow JL. Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J Lipid Res. 2003;44:2109–2119. doi: 10.1194/jlr.M300203-JLR200. [DOI] [PubMed] [Google Scholar]
- 57.Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah NG, Bernier L, Prat A. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24:1454–1459. doi: 10.1161/01.ATV.0000134621.14315.43. [DOI] [PubMed] [Google Scholar]
- 58.Puglielli L, Tanzi RE, Kovacs DM. Alzheimer’s disease: the cholesterol connection. Nat Neurosci. 2003;6:345–351. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- 59.Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, Mayer H, Nimpf J, Prat A, Seidah NG. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem. 2008;283:2363–2372. doi: 10.1074/jbc.M708098200. [DOI] [PubMed] [Google Scholar]
- 60.Labonte P, Begley S, Guevin C, Asselin MC, Nassoury N, Mayer G, Prat A, Seidah NG. PCSK9 impedes hepatitis C virus infection in vitro and modulates liver CD81 expression. Hepatology. 2009;50:17–24. doi: 10.1002/hep.22911. [DOI] [PubMed] [Google Scholar]
- 61.Mbikay M, Sirois F, Mayne J, Wang GS, Chen A, Dewpura T, Prat A, Seidah NG, Chretien M, Scott FW. PCSK9-deficient mice exhibit impaired glucose tolerance and pancreatic islet abnormalities. FEBS Lett. 2010;584:701–706. doi: 10.1016/j.febslet.2009.12.018. [DOI] [PubMed] [Google Scholar]
- 62.Denis N, Palmer-Smith H, Elisma F, Busuttil A, Wright TG, Bou Khalil M, Prat A, Seidah NG, Chretien M, Mayne J, Figeys D. Quantitative proteomic analysis of PCSK9 gain of function in human hepatic HuH7 cells. J Proteome Res. 2011;10:2011–2026. doi: 10.1021/pr2000072. [DOI] [PubMed] [Google Scholar]
- 63.Ranheim T, Mattingsdal M, Lindvall JM, Holla OL, Berge KE, Kulseth MA, Leren TP. Genome-wide expression analysis of cells expressing gain of function mutant D374Y-PCSK9. J Cell Physiol. 2008;217:459–467. doi: 10.1002/jcp.21519. [DOI] [PubMed] [Google Scholar]
- 64.Lan H, Pang L, Smith MM, Levitan D, Ding W, Liu L, Shan L, Shah VV, Laverty M, Arreaza G, Zhang Q, Murgolo NJ, Hernandez M, Greene JR, Gustafson EL, Bayne ML, Davis HR, Hedrick JA. Proprotein convertase subtilisin/kexin type 9 (PCSK9) affects gene expression pathways beyond cholesterol metabolism in liver cells. J Cell Physiol. 2010;224:273–281. doi: 10.1002/jcp.22130. [DOI] [PubMed] [Google Scholar]
- 65.Gong B, Chen F, Pan Y, Arrieta-Cruz I, Yoshida Y, Haroutunian V, Pasinetti GM. SCFFbx2-E3-ligase-mediated degradation of BACE1 attenuates Alzheimer’s disease amyloidosis and improves synaptic function. Aging cell. 2010;9:1018–1031. doi: 10.1111/j.1474-9726.2010.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoshida Y, Tanaka K. Lectin-like ERAD players in ER and cytosol. Biochimica et biophysica acta. 2010;1800:172–180. doi: 10.1016/j.bbagen.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 67.Bu G, Schwartz AL. RAP, a novel type of ER chaperone. Trends Cell Biol. 1998;8:272–276. doi: 10.1016/s0962-8924(98)01283-5. [DOI] [PubMed] [Google Scholar]
- 68.Trombetta ES, Parodi AJ. Quality control and protein folding in the secretory pathway. Annu Rev Cell Dev Biol. 2003;19:649–676. doi: 10.1146/annurev.cellbio.19.110701.153949. [DOI] [PubMed] [Google Scholar]
- 69.Henderson B, Pockley AG. Molecular chaperones and protein-folding catalysts as intercellular signaling regulators in immunity and inflammation. J Leukoc Biol. 2010;88:445–462. doi: 10.1189/jlb.1209779. [DOI] [PubMed] [Google Scholar]
- 70.Dempski RE, Jr, Imperiali B. Oligosaccharyl transferase: gatekeeper to the secretory pathway. Curr Opin Chem Biol. 2002;6:844–850. doi: 10.1016/s1367-5931(02)00390-3. [DOI] [PubMed] [Google Scholar]
- 71.Kleizen B, Braakman I. Protein folding and quality control in the endoplasmic reticulum. Curr Opin Cell Biol. 2004;16:343–349. doi: 10.1016/j.ceb.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 72.Hoseki J, Ushioda R, Nagata K. Mechanism and components of endoplasmic reticulum-associated degradation. J Biochem. 2010;147:19–25. doi: 10.1093/jb/mvp194. [DOI] [PubMed] [Google Scholar]
- 73.Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702–4710. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- 76.He C, Klionsky DJ. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Patel S, Vuillard L, Cleasby A, Murray CW, Yon J. Apo and inhibitor complex structures of BACE (beta-secretase) J Mol Biol. 2004;343:407–416. doi: 10.1016/j.jmb.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 78.Xiong B, Huang XQ, Shen LL, Shen JH, Luo XM, Shen X, Jiang HL, Chen KX. Conformational flexibility of beta-secretase: molecular dynamics simulation and essential dynamics analysis. Acta Pharmacol Sin. 2004;25:705–713. [PubMed] [Google Scholar]
- 79.Hong L, Tang J. Flap position of free memapsin 2 (beta-secretase), a model for flap opening in aspartic protease catalysis. Biochemistry. 2004;43:4689–4695. doi: 10.1021/bi0498252. [DOI] [PubMed] [Google Scholar]
- 80.Zhou L, Chavez-Gutierrez L, Bockstael K, Sannerud R, Annaert W, May PC, Karran E, De Strooper B. Inhibition of beta-secretase in vivo via antibody binding to unique loops (D and F) of BACE1. J Biol Chem. 2011;286:8677–8687. doi: 10.1074/jbc.M110.194860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pehar M, Jonas MC, Hare TM, Puglielli L. SLC33A1/AT-1 Protein Regulates the Induction of Autophagy Downstream of IRE1/XBP1 Pathway. J Biol Chem. 2012;287:29921–29930. doi: 10.1074/jbc.M112.363911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lin P, Li J, Liu Q, Mao F, Qiu R, Hu H, Song Y, Yang Y, Gao G, Yan C, Yang W, Shao C, Gong Y. A missense mutation in SLC33A1, which encodes the acetyl-CoA transporter, causes autosomal-dominant spastic paraplegia (SPG42) Am J Hum Genet. 2008;83:752–759. doi: 10.1016/j.ajhg.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. doi: 10.1016/S1474-4422(08)70258-8. [DOI] [PubMed] [Google Scholar]
- 84.Depienne C, Stevanin G, Brice A, Durr A. Hereditary spastic paraplegias: an update. Curr Opin Neurol. 2007;20:674–680. doi: 10.1097/WCO.0b013e3282f190ba. [DOI] [PubMed] [Google Scholar]
- 85.Park SH, Blackstone C. Further assembly required: construction and dynamics of the endoplasmic reticulum network. EMBO Rep. 2010;11:515–521. doi: 10.1038/embor.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Svenson IK, Ashley-Koch AE, Pericak-Vance MA, Marchuk DA. A second leaky splice-site mutation in the spastin gene. Am J Hum Genet. 2001;69:1407–1409. doi: 10.1086/324593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Svenson IK, Ashley-Koch AE, Gaskell PC, Riney TJ, Cumming WJ, Kingston HM, Hogan EL, Boustany RM, Vance JM, Nance MA, Pericak-Vance MA, Marchuk DA. Identification and expression analysis of spastin gene mutations in hereditary spastic paraplegia. Am J Hum Genet. 2001;68:1077–1085. doi: 10.1086/320111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huppke P, Brendel C, Kalscheuer V, Korenke GC, Marquardt I, Freisinger P, Christodoulou J, Hillebrand M, Pitelet G, Wilson C, Gruber-Sedlmayr U, Ullmann R, Haas S, Elpeleg O, Nurnberg G, Nurnberg P, Dad S, Moller LB, Kaler SG, Gartner J. Mutations in SLC33A1 cause a lethal autosomal-recessive disorder with congenital cataracts, hearing loss, and low serum copper and ceruloplasmin. Am J Hum Genet. 2012;90:61–68. doi: 10.1016/j.ajhg.2011.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huppke P, Brendel C, Korenke GC, Marquardt I, Donsante A, Yi L, Hicks JD, Steinbach PJ, Wilson C, Elpeleg O, Moller LB, Christodoulou J, Kaler SG, Gartner J. Molecular and biochemical characterization of a unique mutation in CCS, the human copper chaperone to superoxide dismutase. Human mutation. 2012;33:1207–1215. doi: 10.1002/humu.22099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Juhasz G, Neufeld TP. Autophagy: a forty-year search for a missing membrane source. PLoS Biol. 2006;4:e36. doi: 10.1371/journal.pbio.0040036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281:30299–30304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Klionsky DJ. Neurodegeneration: good riddance to bad rubbish. Nature. 2006;441:819–820. doi: 10.1038/441819a. [DOI] [PubMed] [Google Scholar]
- 93.Bergmann A. Autophagy and cell death: no longer at odds. Cell. 2007;131:1032–1034. doi: 10.1016/j.cell.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, Hayashi YK, Momoi T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II) Hum Mol Genet. 2007;16:618–629. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- 96.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 98.Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee JA. Autophagy in neurodegeneration: two sides of the same coin. BMB Rep. 2009;42:324–330. doi: 10.5483/bmbrep.2009.42.6.324. [DOI] [PubMed] [Google Scholar]
- 100.Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–454. doi: 10.1016/s0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- 101.Patil C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol. 2001;13:349–355. doi: 10.1016/s0955-0674(00)00219-2. [DOI] [PubMed] [Google Scholar]
- 102.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 103.Iwawaki T, Tokuda M. Function of yeast and amphioxus tRNA ligase in IRE1alpha-dependent XBP1 mRNA splicing. Biochem Biophys Res Commun. 2011;413:527–531. doi: 10.1016/j.bbrc.2011.08.129. [DOI] [PubMed] [Google Scholar]
- 104.Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, Mori K. A time-dependent phase shift in the mammalian unfolded protein response. Dev Cell. 2003;4:265–271. doi: 10.1016/s1534-5807(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 105.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee IH, Finkel T. Regulation of autophagy by the p300 acetyltransferase. J Biol Chem. 2009;284:6322–6328. doi: 10.1074/jbc.M807135200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 108.Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. Embo J. 2005;24:4368–4380. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, Blumberg RS. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wiest DL, Burkhardt JK, Hester S, Hortsch M, Meyer DI, Argon Y. Membrane biogenesis during B cell differentiation: most endoplasmic reticulum proteins are expressed coordinately. J Cell Biol. 1990;110:1501–1511. doi: 10.1083/jcb.110.5.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Calame KL, Lin KI, Tunyaplin C. Regulatory mechanisms that determine the development and function of plasma cells. Annu Rev Immunol. 2003;21:205–230. doi: 10.1146/annurev.immunol.21.120601.141138. [DOI] [PubMed] [Google Scholar]
- 113.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 114.Hodawadekar SC, Marmorstein R. Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene. 2007;26:5528–5540. doi: 10.1038/sj.onc.1210619. [DOI] [PubMed] [Google Scholar]
- 115.Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23:2294–2306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Madeo F, Eisenberg T, Kroemer G. Autophagy for the avoidance of neurodegeneration. Genes Dev. 2009;23:2253–2259. doi: 10.1101/gad.1858009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leonardi R, Zhang YM, Rock CO, Jackowski S. Coenzyme A: back in action. Progress in lipid research. 2005;44:125–153. doi: 10.1016/j.plipres.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 118.Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nature genetics. 2001;28:345–349. doi: 10.1038/ng572. [DOI] [PubMed] [Google Scholar]
- 119.Afshar K, Gonczy P, DiNardo S, Wasserman SA. fumble encodes a pantothenate kinase homolog required for proper mitosis and meiosis in Drosophila melanogaster. Genetics. 2001;157:1267–1276. doi: 10.1093/genetics/157.3.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kuo YM, Duncan JL, Westaway SK, Yang H, Nune G, Xu EY, Hayflick SJ, Gitschier J. Deficiency of pantothenate kinase 2 (Pank2) in mice leads to retinal degeneration and azoospermia. Hum Mol Genet. 2005;14:49–57. doi: 10.1093/hmg/ddi005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Beigneux AP, Kosinski C, Gavino B, Horton JD, Skarnes WC, Young SG. ATP-citrate lyase deficiency in the mouse. J Biol Chem. 2004;279:9557–9564. doi: 10.1074/jbc.M310512200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sakakibara I, Fujino T, Ishii M, Tanaka T, Shimosawa T, Miura S, Zhang W, Tokutake Y, Yamamoto J, Awano M, Iwasaki S, Motoike T, Okamura M, Inagaki T, Kita K, Ezaki O, Naito M, Kuwaki T, Chohnan S, Yamamoto TT, Hammer RE, Kodama T, Yanagisawa M, Sakai J. Fasting-induced hypothermia and reduced energy production in mice lacking acetyl-CoA synthetase 2. Cell metabolism. 2009;9:191–202. doi: 10.1016/j.cmet.2008.12.008. [DOI] [PubMed] [Google Scholar]