

Abstract
The bactericidal, cell membrane-targeting lipopeptide antibiotic daptomycin (DAP) is an important agent in treating invasive Staphylococcus aureus infections. However, there have been numerous recent reports of development of daptomycin-resistance (DAP-R) during therapy with this agent. The mechanisms of DAP-R in S. aureus appear to be quite diverse. DAP-R strains often exhibit progressive accumulation of single nucleotide polymorphisms in the multipeptide resistance factor gene (mprF) and the yycFG components of the yycFGHI operon. Both loci are involved in key cell membrane (CM) events, with mprF being responsible for the synthesis and outer CM translocation of the positively-charged phospholipid, lysyl-phosphotidylglycerol (L-PG), while the yyc operon is involved in the generalized response to stressors such as antimicrobials. In addition, other perturbations of the CM have been identified in DAP-R strains including: extremes in CM order; resistance to CM depolarization and permeabilization; and reduced surface binding of DAP. Moreover, modifications of the cell wall (CW) appear to also contribute to DAP-R, including enhanced expression of the dlt operon (involved in D-alanylation of CW teichoic acids) and progressive CW thickening.
Keywords: Staphylococcus aureus, daptomycin, antibiotic resistance, endocarditis
Introduction
The antibiotic management of invasive Staphylococcus aureus infections has become quite complex over the past two decades because of the widespread prevalence of multiple antimicrobial resistances, including methicillin resistance (MRSA), intermediate vancomycin resistance (VISA; MICs of 4–8 ug/ml) and high-level vancomycin resistance (VRSA; MICs ≥ 16 ug/ml).1–6 Moreover, it is becoming abundantly clear that S. aureus strains (especially, but not exclusively MRSA) whose vancomycin MICs fall within the “susceptible breakpoints,” but which are in the 1.5–2 ug/ml range appear to be associated with worse clinical outcomes during vancomycin therapy.7–17 For all these reasons, alternative therapies to vancomycin have been sought. Daptomycin (DAP) was approved by the U.S. Food and Drug Administration in 2003 for treatment of skin and soft tissue infections, and in 2006 for therapy of S. aureus bacteremia and right-sided endocarditis. With the relatively wide-spread use of DAP over the past decade, it has been interesting that there has been little evidence of any overall “creep” in terms of increasing in vitro DAP MICs in the United States among staphylococci.17,a However, an alarming number of clinical reports (~35 patients) have now been published documenting the in vivo development of daptomycin resistance (DAP-R) during treatment with this agent.18–26 Although the official terminology is daptomycin-nonsusceptibility, we will use the term daptomycin-resistance (DAP-R) in this review for ease of presentation. In addition, 34 new cases (2009–2012) of patients with clinical S. aureus infections in which the isolate displayed DAP MICs of ≥ 1 μg/ml were recently presented from Montefiore Hospital (New York City), of which ~40% had DAP MICs > 2 μg/ml.26,b The mechanisms of DAP-R appear to be quite diverse and complex, involving perturbations predominantly in the cell membrane (CM), but also in the cell wall (CW). This review will summarize the current knowledge base as regards the documented mechanisms of DAP-R in S. aureus.
Mechanisms of DAP`cidality against S. aureus
DAP is a complex lipopeptide antibiotic produced by Streptomyces roseosporus. It is a cyclic molecule with a decanoyl fatty acid side chain attached to the exocyclic N-terminal single tryptophan residue (Fig. 1). DAP contains 13 amino acid residues, including several relatively unusual ones such as kynurenine and ornithine.27 The native DAP molecule is anionic in charge; however, its CM targeting in S. aureus absolutely requires the presence of calcium for bactericidal activity.28–30,c Thus, calcium-DAP becomes a de facto “cationic peptide” agent in both charge and mechanism(s) of action. Some studies have suggested that certain amino acid residues are critical targets for initial calcium binding to DAP (e.g., Asp3; Asp7; Trp1; and/or Kyn13).31–33 When calcium is added to DAP in a 1:1 molar ratio, a two-step process appears to be initiated: (1) an initial intramolecular association resulting in a “loosely” oligimerized micellar structure which serves to deliver DAP to the target CM, followed by (2) facilitated insertion of this calcium-DAP complex into the CM to initiate staphylocidal activity.27,29 After CM insertion, presumably involving interaction with the negatively-charged phospholipid head groups of phosphotidylglycerol (PG) and cardiolipin (CL), calcium-DAP induces positive curvature strain on CM lipids. Eventually, these interactions result in CM depolarization and permeabilization, accompanied by leakage of small ions such as potassium, with ultimate cell death, (although the relatively slowly potassium leakage may be also be the result rather the cause of cell death).27,29,32 Recent studies have suggested that staphylocidality of DAP may be a non-lytic event, and can target both exponential and stationary phase cells.34,35 Overall, the complete mechanisms by which DAP causes cell death are not fully understood.
Figure 1.

Chemical structure of calcium-DAP based on NMR analyses. (A) Basic chemical structure of the entire lipo-peptide DAP molecule; (B) and (C) Model of the apostructure and calcium-conjugated structure, respectively. Negatively charged side chains are colored red, while positively charged side chains are colored blue. (E) and (F) Surface representation of the apostructure and calcium-conjugated structure of DAP, respectively, with red representing negative charges, blue representing positive charges, and white representing uncharged regions. Modified from data in Refs. 27–30a.
Because of the CM-targeting properties of DAP it has been assumed that the mechanisms of DAP-R would exclusively involve perturbations in CM structure and/or function. However, several recent studies have suggested that cell wall (CW) modifications may also contribute to DAP-R. These diverse mechanisms are highlighted below, and are in line with early work on the mode-of-action of DAP, which suggested lipoteichoic acid and CW biosynthetic pathways as DAP targets. Since these pathways require the CM for functional organization of the biosynthetic enzymes and a negatively charged phospholipid environment, DAP insertion in such CM areas may well cause pleiotropic effects on CW pathways.
Role of mprF mutations in DAP-R
One rather consistent feature of DAP-R strains has been the progressive accumulation of mutations in a relatively limited cadre of genes in S. aureus. The most frequently identified has been in the mprF gene, generally involving a variety of single nucleotide polymorphisms (SNPs).22,23,36–39 Of note, in studies in which DAP-susceptible (DAP-S) S. aureus strains are passaged serially in vitro in sublethal DAP, such SNPs are usually the first mutation observed as DAP MICs begin to increase, followed by SNPs in yycFG and in rpoB or rpoC.36 The combined accumulation of these latter SNPs during in vitro selection of the DAP-R phenotype has been documented by several laboratories.36,39
MprF is responsible for the lysinylation of PG to generate the positively-charged CM phospholipid, L-PG.40 In addition to this synthetic function, MprF is also involved in the inner-to-outer CM translocation of L-PG.40–42 These two distinct functions of MprF are well correlated with the structure of the mprF locus. The Peschel laboratory in Tübingen, Germany has been instrumental in characterizing the organization, structure and function of the mprF gene in S. aureus. They have shown that the MprF protein is composed of 14 transmembrane domains and a cytosolic C-terminal domain.41 Of the 14 transmembrane segments, the first eight N-terminal domain segment is crucial for “flipping” of L-PG to the outer CM, while the next four transmembrane domains in the center of the protein are “bifunctional”, involved in either L-PG synthesis or flipping.41 The cytosolic C-terminal domain is strictly involved in L-PG synthesis through lysyl-tRNA activity.41 Of interest, depending on which domain they occur in, such mprF SNPs seem to be regularly associated with one or more gain-of-function phenotypes (synthesis and/or translocation). Thus, expression of mprF, which is maximal during exponential phase of growth in DAP-S S. aureus strains, can be observed to still be present during stationary growth phases.37 Although mprF SNPs in association with DAP-R have been scattered throughout the mprF open reading frame in at least 12 loci, there appear to be five-six hotspots within the N-terminal flippase and central bifunctional domains and one hotspot within the C-terminal synthase domain (Fig. 2). Interestingly, there is a compensatory reduction in the proportion of the negatively-charged phospholipid, PG, in the CMs of DAP-R versus DAP-S strains.22
Figure 2.
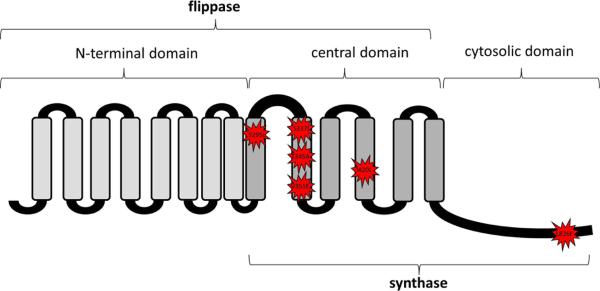
Proposed tri-domain structure-function topology of the MprF molecule by TOPCONS algorithm construction (i.e., C-terminal synthase domain; N-terminal flippase domain; and central bifunctional domains). The sites and amino acid modifications of the five SNPs most commonly observed in association with DAP-R are represented by the star-burst symbols. Modified from Ernst et al.41 (Reproduced with permission of C. Ernst and A. Peschel).
Since mprF gain-of-function can be associated with increased L-PG synthesis and/or flipping, the resultant phenotypic readout is generally an increase in the relative positive surface charge in DAP-R strains.22 This event has been confirmed in several DAP-R strains of S. aureus and has been postulated to render the surface of such S. aureus isolates as a “charge-repulsive milieu” for calcium-complexed DAP. Consistent with this notion, the binding of DAP has been shown to be reduced in DAP-R strains exhibiting gain-of-function mprF SNPs and increased surface positive charge.22 Whether a strictly charge-repulsive mechanism is the sole mechanism of altered cationic peptide interaction with the CM is somewhat questionable. There are several lines of evidence that have somewhat challenged the strict charge–repulsion hypothesis for the DAP-R phenotype. First, our laboratory has recently employed large unilamellar vesicles composed of POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) and varying levels of L-PG, and assessed their interactions with a synthetic fluorescent cationic peptide, tryptophan RP-1 (6WRP-1). These studies showed that kinetics of the initial cationic peptide interactions with the CM were not linearly correlatible with L-PG content, and were not influenced until high L-PG concentrations were incorporated into the vesicles.43 Based on these observations we postulated that negatively-charged POPG represents an initial cationic peptide docking site; “repulsion” of such peptides (including calcium-DAP) would not occur until POPG sites were saturated. In support of this putative mechanism, DAP-R strains exhibit substantially lowered PG content as compared to their isogenic DAP-S parental strains, potentially allowing more rapid saturation of such docking sites and early repulsion of the agent as a secondary event. Furthermore, DAP-R strains of B. subtilis show mutations in their PG synthesis gene (pgs);44,45 in addition, mutations in this same gene have been observed in DAP-R S. aureus strains.46 Importantly, Muraih et al. recently provided in vitro evidence via liposomal and micellar systems that a single molecule of PG is sufficient to trigger DAP oligomerization.47 Thus, reduced PG levels in DAP-R strains may also contribute to this phenotype via reduced DAP-CM interactions. Second, Slavetinsky et al. studied the specificity of MprF for selectively flipping only cationic L-PG.d Of interest, by gene swapping strategies, they showed that: (i) there are two mprF homologs in C. perfringens, and when expressed in a DAP-S S. aureus mprF knockout, the homologs could synthesize both cationic L-PG, as well as zwitterionic alanyl (A)-PG; (ii) by genetic comparisons only the C. perfringens mprF homolog involved in L-PG synthesis included a putative flipping domain (as well as a synthase domain) that could translocate A-PG as well as L-PG; (iii) synthesis and presumed flipping of both A-PG and L-PG (but not synthesis of A-PG alone) in the S. aureus mprF knockout reconstituted parental-level DAP MICs; and (iv) importantly, synthesis of A-PG alone and its translocation by the putative flipping domain alone of the mprF homolog involved in L-PG synthesis/flipping also reconstituted parental-level DAP MICs. These data lend support to the concept that outer CM insertion of either cationic L-PG and/or zwitterionic A-PG can alter CM structure or function by non-charge repulsion mechanisms (for example, by compensatory reductions in PG content as described above).
Whether mprF SNPs are causal in DAP-R remains to be proven. However, recent studies from Rubio et al. lend credence to this hypothesis.48 Using antisense strategies, they showed that blockage of synthesis of mutated forms of MprF reversed the increased DAP MICs associated with mprF gain-in-function mutants. Further, investigations from our laboratory by Yanget al. showed that plasmid complementation of mprF knockout strains with mutated (but not parental) forms of the mprF gene reconstituted elevated DAP MICs.49
Of interest, in recently analyzing the potential mechanism of DAP-R in a clinical methicillin-susceptible strain of S. aureus (MSSA), we demonstrated an mprF SNP in one of the typical “hotspots” mentioned above. However, mprF expression was normal, and there was no evidence of phenotypic gain-of-function.50 In contrast, this strain exhibited increased positive surface charge in association with overexpression of the dlt operon. This latter gene is responsible for encoding a protein that is involved in D-alanylation of wall teichoic acids, and is a known contributor to maintenance of surface charge positivity. Thus, it seems clear that mprF SNPs in DAP-R S. aureus strains are not always causal.
Although the mprF operon has been the signature locus implicated in DAP-R, Kaatz et al. recently identified the absence of an 81 kDa CM protein in a DAP-R MRSA that evolved in a patient with tricuspid valve endocarditis during DAP therapy.38 The loss of this CM protein was associated with reduced surface binding of DAP, and the authors proposed this CM protein to be a chaperone involved in DAP-CM interactions.
Relationship of vancomycin exposures with subsequent DAP-R
There has been a rather common theme between pre-exposures to vancomycin (either in vitro or in vivo) and a laying of the foundation for subsequent DAP-R in S. aureus. This paradigm has taken a number of formats and nuances. For example, several studies have shown a strong positive correlation between reduced susceptibility to vancomycin and DAP among VISA isolates.e In one of these studies (using the 2006 VISA designation51), 70 independent clinical and laboratory VISA isolates with vancomycin MICs between 4–16 ug/ml were tested; > 80% were found to have DAP MICs within the DAP-R range (≥ 2 ug/ml). Also, Mwangi et al. showed that in a patient with recalcitrant MRSA endocarditis treated with long-term vancomycin, the initially vancomycin-susceptible parental isolate evolved a VISA phenotype and a 100-fold increase in DAP MIC without ever being exposed to this latter agent.52 In addition, mutations frequently observed in DAP-R S. aureus (yyc gene cluster and rpoC) also emerged among these strains isolated during vancomycin therapy. Further, Pillai et al. investigated three clinical MRSA strain pairs from vancomycin-treated patients in which all three evolved the VISA phenotype plus elevated DAP MICs in DAP-R range, in the absence of DAP exposures.53 None of the three DAP-R isolates demonstrated mprF mutations. Also, Sakoulas et al. examined the interrelationship between vancomycin and DAP in vitro susceptibilities in: MRSA strains from vancomycin-exposed patients; in VISA strains; and in S. aureus strains passaged in vitro in vancomycin.54 A common theme emerged in terms of a clear correlation between vancomycin and DAP heteroresistance, suggesting that exposures of S. aureus to vancomycin may be a major risk factor for subsequent DAP-R upon subsequent exposure to DAP. In this latter study, the authors suggested that development of the VISA phenotype is likely to be associated with thickened CWs, which may secondarily influence DAP penetration to its CM target. Finally, Cui et al. confirmed the parallel tracking of vancomycin and DAP MICs in VISA strains and strategic variants, and correlated such MICs with CW thickness.55 As in other studies, these authors suggested that the thickened CW phenotype prevented both vancomycin and DAP from reaching each drug's CM site of action (for vancomycin, binding to the CM-bound CW precursor, lipid II; and for DAP, probably nonspecific calcium-DAP micellar binding to the CM).
Thus, in summary, it appears that prior vancomycin exposures may well provide a microbiologic foundation for development of subsequent DAP-R. Such an event may occur with or without (i) DAP therapy; (ii) emergence of heteroresistance to either agent; (iii) a thickened CW phenotype; and/or (iv) evolution of mprF SNPs.
Clinical development of DAP-R in S. aureus
There have been numerous case reports of patients who have developed DAP-R S. aureus strains during treatment with DAP. The vast majority of such cases have been in patients with recalcitrant endocarditis—both right-sided and left-sided infection—in whom vancomycin therapy was utilized prior to switching to DAP. Occasionally, patients with relapsing S. aureus osteomyelitis during DAP treatment have also demonstrated evolution of DAP-R isolates on-therapy. Of interest, in these case reports, the DAP-R phenotype was observed to evolve both in MRSA, as well as MSSA strains. Most of the published reports do not provide enough clinical information concerning DAP dose-regimens to correlate dose-strategies with development of DAP-R on therapy. However, Kaatz et al. reported a patient with right-sided endocarditis who developed a DAP-R MRSA bloodstream isolate after only four days of DAP treatment (which was the primary therapy in this case).38 The authors suggested that a suboptimal dose-regimen may have contributed to the emergence of the DAP-R strain. Sharma et al.26 provided perspective on development of DAP-R on-therapy in a relatively large cohort of S. aureus bacteremic patients. Over a two and a half year period (2004–2006), the authors evaluated all such patients who were treated with DAP for at least two days. Among 18 DAP-treated S. aureus bacteremic patients, 10 had persistent bacteremia, with parental and post-DAP therapy isolate pairs available for study. In 9/10 patients, DAP was given for persistent bacteremia despite other prior therapies (vancomycin in 7/9). In 8/10 patients, an endovascular infection was documented, most commonly intravascular catheter-related or endocarditis-related bacteremia. DAP dose-regimens ranged from 4–6 mg/kg/d. Of the 7 patients in whom both pre-DAP therapy and during or post-therapy isolates were MIC-tested, 4/7 later isolates exhibited an increase from the DAP-S parental MICs into the DAP-R range (2–4 ug/ml).
The largest and most detailed clinical experience in the use of DAP as primary therapy for S. aureus bacteremic syndromes was the seminal study by Fowler et al. in 2006.24 In this multicenter and multinational randomized clinical trial of S. aureus bacteremia or right-sided endocarditis, 120 patients received DAP therapy (6 mg/kg/d) and were compared to 115 patients receiving either vancomycin-based (MRSA) or semisynthetic penicillin-based (MSSA) regimens. Of the 120 DAP-treated patients, 77% had complicated or uncomplicated bacteremia, while 16% had right-sided endocarditis. Of interest, the remaining 7% of patients had unexpected left-sided endocarditis that was not clinically-overt at time of randomization. Of the overall DAP-treated cohort, 6 patients experienced clinical failure coincident with the emergence of DAP-R isolates (MICs ranging from 2–4 ug/ml). All 6 patients with evolving DAP-R organisms had either complicated endocarditis (3 cases), complicated catheter infection (1 case), or undrained localized infections (2 cases).
Therefore, thematically, lessons learned from the development of DAP-R during clinical therapy with DAP include (i) prior vancomycin therapy may be an important risk factor; (ii) high-inoculum infections such as endocarditis may be particularly prone to evolution of DAP-R; (iii) undrained infections (e.g., abscesses) may also provide an optimal scenario for DAP-R emergence; (iv) lower DAP dose-regimens (<6 mg/kg/d) may foster DAP-R in the above infection syndromes; and (v) infectious loci at which DAP penetration may be compromised (for example, infected heart valve vegetations, osteomyelitic bone, and abscesses) may provide an ideal setting for DAP-R isolates to evolve.
CM characteristics contributing to DAP-R
Surface charge
As alluded to above, modification of the staphylococcal surface from a more negative to a relatively more positive charge has been felt to be a major contributor to DAP-R via a charge-repulsion mechanism, leading to reduced surface binding of calcium-DAP micelles. These two characteristics (positive surface charge and reduced DAP binding) have, in fact, been rather consistent phenotypes associated with DAP-R strains.22 However, as pointed out before, charge-repulsion events may follow a more important initial interaction of calcium-DAP with the CM, that is, docking within negatively-charged phospholipid domains. As noted above, one prevailing opinion is that this increase in relative positive surface charge in DAP-R S. aureus strains is principally linked to gain-in-function SNPs within the mprF ORF, resulting in either enhanced synthesis and/or outer CM flipping of the unique positively-charged phospholipid species, L-PG.36,40,41 However, in selected DAP-R strains, an increase in dlt operon expression has been documented, leading to enhanced d-alanylation of CW teichoic acids. Thus, non-L-PG related mechanisms could also contribute to increases in relative surface positive charge. Of interest, when DAP-R strains are selected by serial in vitro passage in sublethal DAP, many of the same phenotypes observed in clinically-derived DAP-R S. aureus strains evolve (e.g., increased L-PG synthesis and flipping);56 however, surface charge became more relatively negative in the fully DAP-R mutants suggesting that (i) a charge repulsion mechanism cannot account for the DAP-R phenotype in all cases, and (ii) host factors likely contribute to the presence of increased positive surface charge in clinically-derived DAP-R isolates. Despite these somewhat paradoxical findings, the importance of relative positive surface charge modulation in DAP-R strains has been emphasized recently. Dhand et al.57 showed that exposures with DAP-nafcillin combinations of a DAP-R MRSA strain from a patient with DAP-unresponsive bacteremia led to enhanced killing of the organism in vitro and clearance of the bacteremia clinically. These events were correlated with the capacity of nafcillin to decrease the surface positive charge in the DAP-R strain to a more electro-negative phenotype, associated with increased DAP CM binding of the drug. The exact mechanism(s) of this nafcillin-induced surface charge modification event remain to be elucidated.
CM order and pigmentation
We have previously shown that relative CM order characteristics (fluidity–rigidity) can have a profound impact on the ability of cationic molecules to interact with and kill S. aureus strains. For example, cationic peptides from mammalian platelets have a suboptimal capacity to kill S. aureus isolates which have evolved a relatively hyper-fluid CM by a variety of diverse mechanisms (e.g., carriage of CM transporters; altered fatty acid or phospholipid content).58–60 This phenomenon has been ascribed to a reduced ability of cationic molecules to bind to and/or penetrate highly fluid CMs. Of note, several recent studies from our laboratory have confirmed that clinically-derived DAP-R S. aureus strains also tend to possess relatively fluid membranes as compared to their isogenic parental strains.22,61 It should be underscored, however, that in vitro-derived DAP-R isolates, in contrast, tend to exhibit more rigid CMs as compared to the wild-type parental strain.56 These seemingly paradoxical observations probably represent the so-called “Goldilocks effect” (i.e., “too much” versus “too little” CM order), and emphasize the notion that there is probably a CM order “sweet spot” for the optimal interaction of a given cationic peptide, like DAP, with the CM. Our recent data also emphasize that individual strains are not universally preprogrammed to adapt their CM order to either a highly fluid or rigid phenotype during every in vitro exposure to DAP.f
S. aureus colonies exhibit their iconic golden color by virtue of their biosynthesis of carotenoid pigments within the CM. Recent data from the Lui laboratory have ascribed an important protective function of staphylococcal carotenoids in the organism's evasion of macrophage-mediated oxidative host defenses.62 Since carotenoids also provide important structural scaffolding to the CM, we investigated whether such pigments could affect the ability of DAP to interact with and target the S. aureus CM. Using a plasmid-based carotenoid-hyperproducing strain, we demonstrated that increased carotenogenesis tracked with both increased DAP MICs as well as enhanced CM rigidity.63 These data further supported the impact of CM order on DAP susceptibility. Of note, Tong and coworkers in the Northern Territories of Australia have recently isolated a community-acquired MRSA strain (clonal complex 75) which commonly causes skin and soft-tissue infections in their aboriginal populations. Of great interest, this clone is naturally deficient for the presence of the carotenoid biosynthetic operon (crtMNOPQ) and produces white colonies on nutrient agar.64 The CMs of this strain are highly fluid, rendering the strain resistant to DAP and other cationic peptides (unpublished data).
Cross-resistance between DAP and host defense cationic peptides
As calcium-DAP is a CM-targeting cationic peptide whose principle mechanisms of action appear to mirror those of many host defense peptides, we investigated whether DAP-R S. aureus strains would also exhibit reduced susceptibility to killing (“cross-resistance”) to these latter molecules. We employed two prototypical mammalian peptides in these analyses including (i) hNP-1 from polymorphonuclear leukocytes (PMNs),61 and (ii) thrombin-induced platelet microbicidal proteins from platelets (tPMPs).65 These two peptides are distinct in terms of size, charge, structure, and mechanisms of action.65 Our initial studies of both clinically-derived, as well as in vitro passage-generated, DAP-R S. aureus strains confirmed that, as compared to their DAP-S isogenic parental strains, the DAP-R phenotype paralleled host defense peptide cross-resistance.22,66 To examine this phenomenon in a larger population of S. aureus strains, we recently analyzed DAP-host defense peptide cross-resistances among 10 well-characterized DAP-S/DAP-R isogenic strain-pairs.61 All DAP-R isolates emerged during failed therapy with this agent. Seven of the 10 DAP-R isolates had SNPs in the mprF locus (with or without yyc operon mutations), while three isolates had neither mutation. Several other phenotypic parameters previously associated with DAP-R were also examined, including CM order and surface charge. As compared to the DAP-S parental strains, their respective DAP-R strains exhibited significantly reduced susceptibility to killing by hNP-1 and tPMPs, as well as increased CM fluidity. Of interest, hNP-1, like the β-defensin, hBD3, has been shown to bind lipid II (see below).67–69 Unexpectedly, DAP-R strains, demonstrated relatively equivalent degrees of cross-resistance and altered CM order in the presence or absence of mprF mutations.61 These compelling data raised a number of provocative questions: (i) Did DAP-R developing in vivo precede the onset of host defense peptide cross-resistance or did they co-evolve?; (ii) Did host defense peptide exposures lay the foundation for subsequent DAP-R during use of this agent?; and (iii) Are the mechanisms of resistance shared between these two peptide genres? Two recent studies have helped shed some light on these queries. First, our laboratory analyzed the relative in vitro susceptibilities to killing by the platelet-derived (tPMPs) and PMN-derived (hNP-1) peptides for 47 initial bloodstream MRSA isolates from DAP treatment-naïve patients.70 Among these DAP-S MRSA from patients who never received DAP, higher DAP MICs (still within the susceptible range) tracked with increased resistance to killing in vitro by the platelet-derived, but not PMN-derived host defense peptides tested. These findings support the concept that endogenous exposures of S. aureus strains within the bloodstream to specific host defense peptides may play an important role in selecting out isolates with an intrinsically higher DAP MIC phenotype. This also underscores the notion that eventual DAP-R may preferentially emerge among S. aureus strains that have been pre-sensitized towards cationic peptide resistance upon subsequent DAP exposures. Moreover, such pre-sensitization may occur in specific body sites where distinct host defense peptides predominate (for example, platelet-derived peptides in the bloodstream versus PMN-derived peptides in abscesses).
Second, the Cremieux laboratory in France recently evaluated the in vivo efficacy of DAP (with or without rifampin) in a model of MRSA prosthetic joint septic arthritis/osteomyelitis in rabbits.71 Despite the overall good efficacy of DAP regimens in this model, several isolates with increased DAP MICs emerged during prolonged DAP therapy. Surprisingly, several isolates with increased DAP MICs also developed during the course of untreated infection (control animals). In comparison with the parental strain, both DAP-treated and DAP-untreated strains with increased DAP MICs exhibited (i) significantly reduced susceptibility to tPMPs and hNP-1 (P < 0.05), (ii) thicker CWs (P < 0.05), (iii) increased synthesis of CM L-PG, (iv) reduced content of CM PG, and (v) SNPs within the mprF locus.72 There were no significant perturbations observed between parental or variant strains in outer CM translocation of L-PG, CM fluidity, CM fatty acid contents, surface charge, or mprF-dltABCD expression profiles. An isolate which underwent the same animal passage, but without evolving, increased DAP MICs, retained exclusively parental phenotypes and genotype. These results suggest that adaptive mechanisms involved in the in vivo emergence of increased MICs to DAP also provide MRSA with enhanced host defense peptide survivability. Moreover, as in the above MRSA bacteremia investigation, increases in DAP MICs may occur in the absence of DAP exposures, and are likely triggered by MRSA-host defense peptide interactions in vivo. These data also emphasize that gain-in-function SNPs within mprF are a likely contributory mechanism in DAP-host defense peptide cross-resistance.
Last, Patel et al. have recently shown that laboratory-derived DAP-R S. aureus strains exhibit cross-resistance to a bacterial produced cationic lantibiotic peptide, nisin.39 Moreover, we recently demonstrated that hBD3-treated S. aureus cells show response patterns similar to treatment with CW antibiotics, and that hBD3 primarily kills through specific binding to lipid II and CW biosynthesis inhibition68,69 underlining the functional similarities between cationic host defense peptides, amino sugar-containing glycopeptide antibiotics, and DAP.
“Natural” resistance to DAP
As a corollary to the putative facilitation of S. aureus strains towards development of DAP-R by their in vivo pre-sensitization via pre-exposures to host defense peptides, recent data by Bhullar et al. suggest that such phenomena may occur within natural microbiomes.73 These investigators screened the microbiome from a New Mexico cave that has been isolated from all human, animal or water contact for over 4 million years for intrinsic antibiotic resistance. Surprisingly, amongst low G + C Gram-positive bacteria isolated from the cave, a broad range of antibiotic resistances were identified, including towards DAP. Of interest, the mechanism of DAP-R was novel, and involved inactivation by hydrolytic cleavage of the ester bond between the threonine and kynurenine residues, resulting in ring-opening inactivation. Whether the “ancient” development of DAP-R in the absence of exposure to this “modern” agent results from natural production of DAP-like molecules by part of this archaic microbiome remains to be determined.
A summary of the putative role of CM modifications in the DAP-R phenotype in S. aureus can be found in Table 1.
Table 1.
Contributing CM mechanisms in S. aureus associated with the DAP-R phenotype
| Parameter | Mechanism(s) |
|---|---|
| Increased relative positive surface charge | mprF SNPs with gain-in-function of L-PG synthesis and/or outer CM flipping |
| Altered CM order | Extremes of rigidity or fluidity |
| Increased CM pigment production | Excess CM rigidity related to overproduction of staphyloxantin |
| Resistance to depolarization and/or permeabilization | Reduced capacity to initiate bactericidal pathways (e.g., small molecule leakage) |
| Reduced CM PG content | Altered ability to oligomerize DAP within the CM; Reduced PG:CL CM "docking sites" for DAP |
Role of DAP cell envelope perturbations—implications for DAP's mechanisms of action and DAP-R phenotype
Early work on the mechanism of action of DAP argued for lipoteichoic acid74 and, in particular, cell wall biosynthesis as target pathways.75,76 The latter conclusion was based on results from precursor incorporation assays and analysis of internal precursor pools following DAP exposures. Thus, the ultimate soluble CW precursor, UDP-N-acetyl-muramic acid-pentapeptide, was not found to accumulate intracellularly (a typical feature of antibiotics inhibiting subsequent CM-bound steps of peptidoglycan synthesis). Therefore, it was argued that some of the very early intracellular steps involved in the conversion of glucosamine-6-phosphate to UDP-GlcNAc, catalyzed by the sequential action of GlmS, GlmU, and GlmM, would be inhibited. DAP was assumed not to enter the cell,74 and therefore, a direct inhibition of these enzymes by DAP was excluded, although a regulatory effect on peptidoglycan biosynthesis was not considered. More recently, a transcriptional profiling study identified differential expression of 32 cell envelope-related genes in both MRSA and MSSA strains following DAP challenge.77 These profiles were compared to those induced by (i) specific CM-targeting agents, such as CCCP, (ii) CW-specific agents, such as oxacillin and vancomycin, and (iii) an agent (nisin) that targets both the CM and CW. These investigators found that DAP induced the expression of genes that paralleled both those induced by the above CM-targeting agents, but also 26 genes reported as prominent members of the “CW stress stimulon,” including vraRS, pbp2, prsA, and tcaA.78,79 Interestingly, an in-depth transcriptomic and proteomic analysis in B. subtilis, comparing responses induced by exposure to DAP and the structurally related lipopeptide, friulimicin, identified the cell envelope as the site of action of both lipopeptides, although major mechanistic differences between the two compounds were suggested.80 In this regard, there was a dramatic difference in the LiaRS response (analogues to the VraRS TCS in S. aureus), which was heavily induced by DAP, but not friulimicin. In contrast, friulimicin specifically targets the lipid carrier, undecaprenol-phosphate.81
In addition, DAP induces cell envelope stress responses that closely parallel those of bacitracin78, further supporting the notion of a combined CM–CW mechanism; these latter data mirror similar findings related to other CM-targeting glycopeptides, such as telavancin and teicoplanin.82 Furthermore, Fischer et al.83 recently compared the transcriptomic and proteomic profiles of a DAP-S/DAP-R MSSA strain pair. Of note, a number of genes involved in CW metabolism were upregulated in the DAP-R isolate as compared to the DAP-S strain, including the hydrolases/amidases lytN and lytH, the WTA biosynthesis enzymes tagA and tagG, pbp2 and pbp4, and yycI and yycJ, encoded in an operon together with the essential TCS yycFG (also termed vicRK and walRK). The putative functional consequences of these alterations in CW-associated gene expressions in this latter strain-pair were further assessed by Bertsche et al.84, showing that the thickened CW phenotype in the DAP-R strain was likely related to an upregulation of the tag operon, correlating with the excess production of WTA. Moreover, the enhanced positive surface charge phenotype of this latter DAP-R strain was explicable on the basis of upregulation of dlt expression, and a resultant increase in the D-alanylation of this excess WTA. It is also possible that DAP-R could have been due in part to more dense “packing” of the CW architecture due to excess WTA, limiting DAP access through the CW.85 In contrast, Boyle-Vavra et al. performed comparative genome pyrosequencing of an isogenic pair of USA800 MRSA strains obtained before and after DAP therapy in a patient with recurrent bacteremia, and found neither thick CW phenotypes nor sequence or transcriptional profiling differences pointing to CW perturbations.86
Recent data from our own laboratories have provided additional information that lend credence to the role of the CW in DAP-R. Thus, Yang et al. noted that expression of dlt (responsible for WTA D-alanylation) was enhanced in a DAP-R clinical MSSA isolate; a SNP in mprF was also noted, although its expression profiles and L-PG production and flipping were at parental levels.50 Such controversy is in line with the notion of pleiotropic (and perhaps strain-specific) DAP effects on CW and CM, with concomitantly diverse DAP-R mechanisms involving the global structure and function of the entire cell envelope.
A number of studies have documented a thickened CW phenotype in comparing DAP-S parental strains with their respective DAP-R variants, rather reminiscent of VISA strains.51,53,55,61,66,70,g This phenotype has been observed both among DAP-R strains derived by serial in vitro passage, as well as during DAP treatment in vivo. 55,56,61,f
Interestingly, a gene belonging to the CW stress stimulon, cwrA (cell wall–responsive antibiotics; SA2343), was found to be both highly upregulated in several clinical VISA strains87, and also upregulated upon DAP challenge77. Using a cwrA-lux-reporter fusion, cwrA was clearly induced by DAP and CW-active agents, vancomycin, bacitracin, and penicillin, but not by exposure to compounds which interfered with DNA-, RNA-, protein or fatty acid biosynthesis, or by CM-disrupting agents.88 The exact function of the CM-spanning CwrA, is so far unknown. It appears to counteract CW damage, and was additionally found to be upregulated 100- to 500-fold when genes of the mevalonate pathway are downregulated.89 The mevalonate pathway is the only route to isoprenoid synthesis in staphylococci, also providing the direct precursor (IPP) to undecaprenol-pyrophosphate synthesis, the essential lipid carrier for peptidoglycan, WTA and capsule biosynthesis (Fig. 3). In line with these findings, transcriptional profiling of S. aureus treated with DAP showed a significant upregulation of genes of the mevalonate pathway (mvaK1, mvaK2, mvaD) leading to the formation of isopentenylpyrophosphate (IPP). IspA, coding for farnesylpyrophosphate synthase, catalyzing the subsequent conversion of IPP to farnesylpyrophosphate (FPP) was also found to be upregulated (unpublished data). Thus, this ΔcwrA mutant is characterized by a clumping phenotype, while its transcriptomic analyses showed the upregulation of the dlt operon, sceD, lacA-G, ssaA, and lytM. Interestingly, this ΔcwrA mutant was characterized by a thickened CW, comparable to several DAP-R mutants.88
Figure 3.
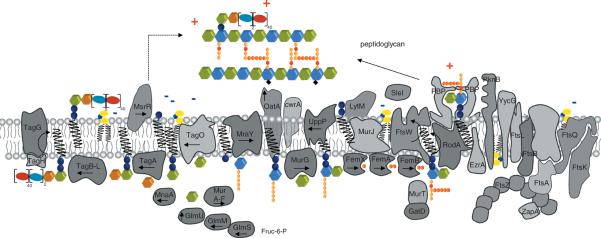
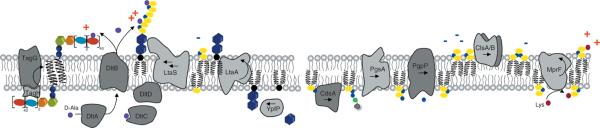
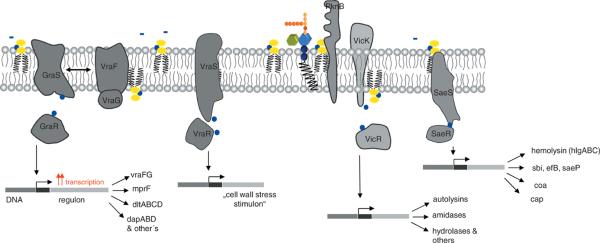
(A) Cartoon of enzymes and bactroprenol-bound substrates comprising the partially overlapping machineries for biosynthesis of CW teichoic acids (left), peptidoglycan (middle) and for cell division (right). CM areas in which these pathways take place are rich negatively-charged bactoprenol-phosphate/pyrophosphate and PG (indicated in yellow), and thus attract DAP and other CAPs. (B) Biosynthetic pathways generating negatively-charges lipids and CW components, and reactions involved in modulation of the surface charge by D-alanylation and lysinylation (positive charges indicated in red) resulting in reduced DAP activity and DAP-R development. (C) Sensor systems involved in controlling CW integrity and proposed regulons; mutations in yycG (vicK) and vraS frequently observed in DAP-R mutants may reduce precision in CW structure and function, and allow for growth of impaired, but viable cells.
Apart from the strong induction of vraRS by DAP, results from our studies and those of other labs, also clearly link additional signal transduction regulatory pathways to DAP-R and DAP's mechanism of action. The essential yycFG TCS has been shown to contribute to DAP-R in both clinical and laboratory-derived DAP-R mutants36,61, and YycG kinase has even been suggested to be a direct target of DAP.90 Friedman et al.36 identified SNPs in yycG, resulting in amino acid substitutions affecting the cytoplasmic PAS (R236C) and HAMP (S221) domains of the histidine kinase. Whether these mutations positively or negatively affect their enzymatic activity, or if they play a decisive role in DAP's mechanism is not known.
YycFG is felt to directly or indirectly regulate fatty acid biosynthesis and to modulate fatty acid chain length, thereby altering CM composition.91 In S. aureus, YycFG has been shown to regulate nine genes involved in cell envelope and lipid metabolism, including atl, lytM, sceD, isaA, ssaA, SA0620, SA2353, SA2097, and SA0710.92–94
The YycFG TCS has further been implicated in the control of CW biosynthesis turnover by “sensing” different levels of the CW building block, lipid II,94 although biochemical evidence to support such a function is lacking. Nevertheless, data from several recent reports support the notion that the YycFG TCS plays a fundamental role in CW metabolism. This is also in good agreement with the localization of YycG to the cell division site95 and its proposed interaction with the cell division protein, FtsZ.45 DAP also preferentially localizes to the septum45, the site of cell division, which has to be highly synchronized with CW biosynthesis and other cell envelope biosynthetic pathways (Fig. 3). Of interest, several reports also link mutations in YycFG to the VISA phenotype96,97.
Interestingly, Friedman et al. isolated a clinical DAP-R mutant that contained a single nucleotide insertion leading to a frameshift that might result in a loss of function of YycG.37 Since this TCS has been reported to be essential in S. aureus, the authors suggested that phosphorylation of the YycF response regulator might be taken over by another non-cognate, unrelated kinase—for example, the Ser-Thr kinase PknB. More recently it was further shown that YycFG-depleted cells are characterized by a thickened CW and aberrant septum formation, and that overexpression of ssaA and lytM, involved in cleaving of the cross-bridges of adjacent stem peptides, which is supposed to result in peptidoglycan relaxation, restored cell viability of a yycFG (walRK) mutant.98
The TCS, SaeRS, has also been found to be up-regulated when S. aureus is exposed to DAP, whereas a serial passage DAP-R mutant (in the absence of DAP challenge) down-regulates SaeRS (unpublished results). This sensor system has also been found to be upregulated by β-lactams and vancomycin exposures.99 The SaeRS system controls a number of major virulence factors, such as hla and hlb (encoding α-and β-hemolysin), coa (encoding coagulase), fnbA (encoding fibronectin binding protein A) and the capA-P operon (encoding capsule formation) (Fig. 3).100–102 The autoregulated, CM-integrated kinase, SaeS, lacks typical extra- or intracellular signalling domains, and has been discussed to sense alterations of the CM.103 Recently, SaeS has been suggested to sense and recognize specific lipids, lipid patches, as well as changes in CM dynamics (like fluidity/rigidity) and surface charge. In an ΔmprF mutant, CM proteome analysis identified a significant decrease in SaeS expression, suggesting a direct interaction with L-PG metabolism.104 In this same study, L-PG depletion also affected the concentration of two members of the LytR-CPsA-Psr family, msrR (SA1195) and SA0908, respectively. These proteins have very recently been implicated in catalyzing the linkage of WTA and capsule precursors to the peptidoglycan network;105,106 moreover, growth defects that occur upon depletion of these proteins are restored in a ΔtagO background.107
Alteration of CM dynamics might therefore trigger a cascade of regulatory events, leading to pleiotropic effects, emanating in multiple cell envelope biosynthetic pathways, including peptidoglycan, WTA, LTA, and lipid metabolism. These potential interrelationships are highlighted in Figures 3A–C.
Finally, very recent data from Pogliano et al. support an additional putative DAP mechanism primarily by acting on the CM, resulting in the delocalization of proteins involved in cell division and CW synthesis, associated with dramatic CM defects.h
Approach to therapy of DAP-R S. aureus infections
There are two basic ways to examine therapeutic strategies in DAP-R S. aureus strains: (i) prevention of the emergence of DAP-R in DAP-S strains; and (ii) treatment of established DAP-R infections. These strategies are summarized below.
Circumvention of DAP-R
Complicating the analysis of the literature, there have been numerous DAP-R prevention strategies assessed in multiple model systems, including: standard in vitro media studies; PK-PD chamber models, with or without simulated endocarditis vegetations; hollow-fiber PK-PD in vitro models; in vitro biofilm models; and a variety of in vivo animal models (soft tissue; osteomyelitis; and endocarditis). Moreover, the range of DAP drug-dosing and/or DAP combination therapy regimens tested to prevent emergence of DAP-R has been broad. It should be emphasized that there is a relatively large literature on combination therapy with DAP plus second agents for the in vitro killing of DAP-S strains (not the topic of this review). These investigations predominantly feature DAP plus either rifampin, gentamicin or β-lactams (especially ampicillin and its congeners); in general, these studies show either an additive or synergistic effect against a substantial proportion of MSSA and MRSA strains, with no antagonism noted (reviewed in detail elsewhere).108,109 The majority of the in vitro PK-PD models appear to favor DAP-rifampin or DAP-gentamicin combination strategies to both enhance S. aureus killing and prevent emergence of DAP-R variants. In addition, the use of DAP-clarithromycin combinations appears particularly effective in biofilm infection models of S. aureus. Of note, recent studies by Rose et al. and Bertiet al., in an in vitro–simulated endocarditis model, have suggested that use of higher-dose DAP-alone regimens (e.g. the equivalent of human-like 10 mg/kg/d dosing), as well as combinations of DAP-clarithromycin or DAP-oxacillin, can forestall emergence of DAP-R.110,111,i These data-sets were somewhat validated by Sakoulas et al. in the rat endocarditis model;112 the study showed higher-dose DAP prevented emergence of DAP hetero-resistance, as measured by rightward shifts in population analysis curves when comparing 4 and 6 mg/kg human PK-equivalent dosing.
The use of DAP combination therapies to prevent the emergence of DAP-R during the therapy of DAP-S infections has not been systematically studied. However, there are several investigations in three distinct animal models (soft tissue; endocarditis; and osteomyelitis) which have lent credence to the notion that addition of rifampin to DAP may potentially mitigate the development of DAP-R during therapy. Saleh-Mghir et al. performed a seminal study in the treatment of a silicone elastomer prosthetic knee infection model in rabbits, given either DAP or DAP plus rifampin.71 Aside from its enhanced efficacy as compared to DAP monotherapy, the combination regimen was able to completely prevent emergence of MRSA strains with elevated DAP MICs, whereas 50% of knee joint MRSA isolated at time of sacrifice following DAP therapy alone exhibited this latter phenotype. Lefebrve et al. in a more acute non-prothetic joint osteomyelitis model found very similar outcomes in terms of efficacy enhancement and prevention of emergence of DAP-R variants with combined DAP-rifampin regimens as compared to monotherapy.113 Finally, Cirioni et al. utilized a S. aureus subcutaneous vascular graft biofilm pouch infection model in rats treated with DAP and with catheter-impregnated rifampin.114 As in the osteomyelitis studies above, DAP-rifampin combination therapy resulted in increased S. aureus clearances from the site of infection, as well as prevention of evolution of rifampin-R in vivo and DAP-R mutants in vitro.
In conclusion, the weight of in vitro and animal model studies would support the addition of rifampin to DAP therapy, especially during long-term treatment with this agent in “high-inoculum” infections (e.g., endocarditis; osteomyelitis) to circumvent DAP-R variants from developing. Whether higher-dose DAP alone (8–12 mg/kg/d in humans) could achieve a similar outcome remains to be determined.
Optimal therapy of infections caused by DAP-R S. aureus strains
There are no definitive clinical trials to guide clinicians in the treatment of established DAP-R infections. Most such clinical data come from isolated case reports, especially in cases of endocarditis. A variety of alternative therapies have been attempted, including linezolid, vancomycin-gentamicin, nafcillin-gentamicin, trimethoprim-sulfamethoxazole, quinupristin-dalfopristin and DAP-β-lactam combinations. In many such endocarditis cases, valvular surgery was eventually required for radical cure of infection.
One important approach to the in vivo management of such infections has been evaluated utilizing the experimental endocarditis model: high-dose DAP. Chambers et al. examined the therapy of rabbit aortic endocarditis caused by a DAP-R MRSA isolate from a DAP-treated patient with tricuspid endocarditis who failed treatment. These authors assessed two DAP drug-regimens, 12 and 18 mg/kg/d which provided human-like PK-PD dosing paralleling 6 and 10 mg/kg/d strategies, respectively.18 The high-dose DAP regimen (18 mg/kg/d), but not the low-dose regimen (12 mg/kg), was effective in reducing MRSA densities in all target tissues in this model (vegetations, kidneys, and spleen). It should be emphasized, however, that the high-dose regimen did not affect complete MRSA clearances in any target organ in any animal.
Telavancin is a novel glycolipopeptide agent which has a dual function mechanism of action, including a vancomycin-like effect on CW synthesis, as well as a CM depolarizing property.115 This agent possesses very good activity against DAP-S and DAP-R S. aureus strains116 and is highly active in an in vitro PK-PD chamber model against DAP-R strains.117 It has also been shown to have excellent efficacy in several models of aortic endocarditis due to MSSA, MRSA, and VISA strains.118,119 Our laboratory has recently completed a similar study in rabbits examining the efficacy of telavancin in experimental aortic endocarditis due to a DAP-R MRSAj. Of note, this agent was highly effective at reducing MRSA densities in all relevant target organs, essentially sterilizing these sites and preventing post-therapy relapses. Lastly, there has been relatively little clinical experience with telavancin to treat patients with S. aureus endocarditis caused by DAP-R strains, or in clinical scenarios in which DAP therapy has failed.120,121 In addition, the clinical availability of this agent for patient use remains problematic.
Among other newer anti-S. aureus agents, particularly active against MRSA in vitro, both ceftaroline and oritavancin have shown promising activity against DAP-R strains.122,123 However, there is no current in vivo documentation of such efficacy in relevant animal models, especially in experimental endocarditis.
One of the more intriguing recent approaches to the treatment of both persistent DAP-S, as well a s DAP-R S. aureus infections has been the use of combined therapy with DAP plus antistaphylococcal β-lactams (e.g., oxacillin or nafcillin). Houck and Rand had previously documented the potential for in vitro synergy between DAP and such β-lactams.124 Dhand et al. collected 7 contemporary patients with persistent or relapsing MRSA bacteremia despite DAP therapy.57 All initial pretherapy isolates were DAP-S. Relapse isolates from one of the three patients with serial MICs performed became DAP-R in vitro. Six of the 7 patients eventually received DAP-nafcillin therapy, while one patient was given DAP-oxacillin therapy. Six of the 7 patients experienced clinical cures on such treatment. In vitro analyses revealed (i) synergistic killing between DAP plus oxacillin as compared to DAP alone, (ii) enhancement of DAP binding with pretreatment to nafcillin, and (iii) reduction in the organism's relative positive surface charge by pre-exposures to such β-lactams. The mechanism(s) of these observations remains to be elucidated. DAP-R MRSA strains frequently demonstrate the so-called “see-saw” effect in which susceptibility to oxacillin or nafcillin increases as DAP susceptibility falls. This phenomenon was not observed in this investigation. The authors proposed that β-lactam-induced release of wall lipotechoic acid (positively-charged) may have contributed to the reduction in the strain's relative positive surface charge, fostering an enhancement of DAP binding.
Summary
The mechanisms of DAP-R in S. aureus appear to be quite diverse and involve both CM and CW phenotypic changes. DAP-R strains often accumulate single nucleotide polymorphisms in several trademark gene loci, especially involving mprF and yycFG. In addition, other perturbations of the CM have been identified in DAP-R strains including: extremes in CM order; resistance to CM depolarization and permeabilization; and reduced surface binding of DAP. Moreover, modifications of the CW appear to also contribute to DAP-R, including enhanced expression of the dlt operon (involved in D-alanylation of CW teichoic acids) and progressive CW thickening. Which of the CM and/or CW perturbations are actually causal in the DAP-R phenotype remains to be clarified. Clinical strategies to circumvent the emergence of DAP-R in vivo are under study, but early investigations point to high-dose DAP therapy with or without adjunctive rifampin, clarithromycin, or oxacillin as promising alternatives.
Acknowledgments
Some of the contents of this review have been partially supported by a research grant from the National Institutes of Health (NIAID), Grant number RO1-AI-039108-14 to ASB. Also, TS and HGS gratefully acknowledge support from the German Research Foundation (grants SA292/13-1 and SCHN 1284/1-2)
Footnotes
Conflicts of interest The authors declare no conflicts of interest.
See also Sader, H.S., Fey, P.D., Fish, D.N. et al.. 2009. Evaluation of vancomycin and daptomycin potency trends (`MIC creep') against methicillin-resistant Staphylococcus aureus isolates collected in nine US medical centers from 2002 to 2006. Antimicrob Agent Chemother 53:4127–4132; and Sader, H.S., Moet, G.J., Farrell, D.J. et al.. 2011. Antimicrobial susceptibility of daptomycin and comparator agents tested against methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci: trend analysis of a 6-year period in US medical centers (2005–2010). Diagn Microbiol Infect Dis 70:412–416.
See also Fattouh, N., Chung, P., Ostrowsky, B. et al.. Treatment and outcomes of patients with infections associated with daptomycin non-susceptible Staphylcoccus aureus. Fifty-first Interscience Conference on Antimicrobial Agents and Chemotherapy; San Francisco, CA; Sept 2012. Abstract K-1634.
See also Jung, D., Rozek, A., Okon, M. et al.. 2004. Structural transitions as determinants of the action of the calcium-dependent antibiotic daptomycin. Chemistry and Biology 11: 949–957; and Koeth, L and Thorne, G. 2010. Daptomicin in vitro susceptibility methodology: a review of methods, including determination of calcium in testing media. Clin Microbiol Newsletter 32:161–169.
See Slavetinsky CJ, Peschel A, Ernst CM. 2012. Alanyl-phosphatidylglyercol and lysyl-phosphatidylglycerol are translocated by the same MprF flippases and have similar capacities to protect against the antibiotic daptomycin in Staphylococcus aureus. Antimicrob Agents Chemother 56:3492–3497. Epub April 9, 2012.
See Petersen, P.J., Bradford, P.A., Weiss, W.J., et al.. 2002. In vitro and in vivo activities of tigecycline (GAR-936), daptomycin and comparative antimicrobial agents against glycopeptides intermediate Staphylococcus aureus and other resistant gram-positive pathogens. Antimicrob Agents Chemother 46: 2595–2601.
See Mishra, N.M., A. Rubio, C.C. Nast, et al. 2012. Differential adaptations of methicillin-resistant Staphylococcus aureus to serial in vitro passage in daptomycin: evolution of daptomycin resistance and the role of membrane carotenoid content and fluidity. Intl J of Microbiol 683450. Epub 2012 Aug 16
Also see Camargo, I.L., Neoh, H.M., Cui, L., et al.. 2008. Serial daptomycin selection generates daptomycin-nonsusceptible Staphylococcus aureus strains with a heterogeneous vancomycin-intermediate phenotype. Antimicrob Agents Chemother 52: 4289–4299.
Pogliano, J., Pogliano, N., Silverman, J.A.. 2012. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J. Bacteriol. 194: 4494–4504.
Also see Berti, A.D., Wergin, J.E., Girdaukas, G.G., et al. 2012. Altering the proclivity towards daptomycin resistance in methicillin-resistant Staphylococcus aureus using combinations with other antibiotics. Antimicrob Agents Chemother 56:5046–5053.
see: Xiong YQ, Hady WA, Bayer AS et al. 2012. Telavancin in therapy of experimental arotic valve endocarditis in rabbits due to daptomycin-nonsusceptible methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 56:5528–5533.
REFERENCES
- 1.Liu C, Bayer A, Cosgrove SE, et al. Infectious Diseases Society of America (IDSA) Clinical practice guidelines by the IDSA for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52:e18–55. doi: 10.1093/cid/ciq146. [DOI] [PubMed] [Google Scholar]
- 2.Tong SYC, Chen Luke F, Fowler VG. Colonization, pathogenicity, host susceptibility, and therapeutics for Staphylococcus aureus: what is the clinical relevance? Sem in Immunopatholo. 2012;34:185–200. doi: 10.1007/s00281-011-0300-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chambers HF, DeLeo FR. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol. 2009;7:629–641. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vancomycin-resistant Staphylococcus aureus – Pennsylvania. MMWR (Morb Mortal Wkly Rep. 2002;51:565–567. 2002. [PubMed] [Google Scholar]
- 5.Liu C, Graber CJ, Karr M, et al. A population-based study of the incidence and molecular epidemiology of methicillin-resistant Staphylococcus aureus disease in San Francisco 2004–2005. Clin Infect Dis. 2008;46:1637–1646. doi: 10.1086/587893. [DOI] [PubMed] [Google Scholar]
- 6.Howden BP, Davies JK, Johnson PD, et al. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: Resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev. 2010;23:99–139. doi: 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lodise TP, Graves J, Evans A, et al. Relationship between vancomycin MIC and failure among patients with methicillin-resistant Staphylococcus aureus bacteremia treated with vancomycin. Antimicrob Agents Chemother. 2008;52:3315–20. doi: 10.1128/AAC.00113-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haque NZ, Zuniga LC, Peyrani P, et al. Improving Medicine through Pathway Assessment of Critical Therapy of Hospital-Acquired Pneumonia (IMPACT-HAP) Investigators. Relationship of vancomycin minimum inhibitory concentration to mortality in patients with methicillin-resistant Staphylococcus aureus hospital-acquired, ventilator-associated, or health-care-associated pneumonia. Chest. 2010;138:1356–62. doi: 10.1378/chest.09-2453. [DOI] [PubMed] [Google Scholar]
- 9.Yoon YK, Kim JY, Park DW. Predictors of persistent methicillin-resistant Staphylococcus aureus bacteraemia in patients treated with vancomycin. J Antimicrob Chemother. 2010;65:1015–1018. doi: 10.1093/jac/dkq050. [DOI] [PubMed] [Google Scholar]
- 10.Sakoulas G, Moise-Broder PA, Schentag JJ, et al. Relationship of MIC and bactericidal activity to efficacy of vancomycin for treatment of methicillin-resistant Staphylococcus aureus bacteremia. J Clin Microbiol. 2004;42:2398–402. doi: 10.1128/JCM.42.6.2398-2402.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hidayat LK, Hsu D, Quist R, et al. High-dose vancomycin therapy for methicillin-resistant Staphylococcus aureus infections: Efficacy and toxicity. Arch Intern Med. 2006;66:2138–2144. doi: 10.1001/archinte.166.19.2138. [DOI] [PubMed] [Google Scholar]
- 12.Maclayton DO, Suda KJ, Coval KA, et al. Case-control study of the relationship between MRSA bacteremia with a vancomycin MIC of 2 microg/mL and risk factors, costs, and outcomes in inpatients undergoing hemodialysis. Clin Ther. 2006;28:1208–1216. doi: 10.1016/j.clinthera.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 13.Neoh HM, Hori S, Komatsu M, et al. Impact of reduced vancomycin susceptibility on the therapeutic outcome of MRSA bloodstream infections. Ann Clin Microbiol Antimicrob. 2007;6:13. doi: 10.1186/1476-0711-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soriano A, Marco F, Martínez JA, et al. Influence of vancomycin minimum inhibitory concentration on the treatment of methicillin-resistant Staphylococcus aureus bacteremia. Clin Infect Dis. 2008;46:193–200. doi: 10.1086/524667. [DOI] [PubMed] [Google Scholar]
- 15.Musta AC, Riederer K, Shemes S, et al. Vancomycin MIC plus heteroresistance and outcome of methicillin-resistant Staphylococcus aureus bacteremia: Trends over 11 years. J Clin Microbiol. 2009;47:1640–1644. doi: 10.1128/JCM.02135-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang JL, Wang JT, Sheng WH, et al. Nosocomial methicillin-resistant Staphylococcus aureus (MRSA) bacteremia in Taiwan: Mortality analyses and the impact of vancomycin, MIC = 2 mg/L, by the broth microdilution method. BMC Infect Dis. 2010;10:159. doi: 10.1186/1471-2334-10-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi EY, Huh JW, Lim CM, et al. Relationship between the MIC of vancomycin and clinical outcome in patients with MRSA nosocomial pneumonia. Intensive Care Med. 2011;37:639–647. doi: 10.1007/s00134-011-2130-7. [DOI] [PubMed] [Google Scholar]
- 18.Chambers HF, Basuino L, Diep BA, et al. Relationship between susceptibility to daptomycin in vitro and activity in vivo in a rabbit model of aortic valve endocarditis. Antimicrob Agents Chemother. 2009;53:1463–1467. doi: 10.1128/AAC.01307-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vikram HR, Havill NL, Koeth LM, et al. Clinical progression of methicillin-resistant Staphylococcus aureus vertebral osteomyelitis associated with reduced susceptibility to daptomycin. J Clin Microbiol. 2005;43:5384–53387. doi: 10.1128/JCM.43.10.5384-5387.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Julian K, Kosowska-Shick K, Whitener C, et al. Characterization of a daptomycin-nonsusceptible, vancomycin-intermediate Staphylococcus aureus strain in a patient with endocarditis. Antimicrob Agents Chemother. 2007;51:3445–3448. doi: 10.1128/AAC.00559-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayden MK, Rezai K, Hayes K, et al. Development of daptomycin resistance in vivo in methicillin-resistant Staphylococcus aureus. J Clin Microbiol. 2005;43:5285–5287. doi: 10.1128/JCM.43.10.5285-5287.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones T, Yeaman MR, Sakoulas G, et al. Failures in clinical treatment of Staphylococcus aureus infection with daptomycin are associated with alterations in surface charge, membrane phospholipid asymmetry and drug binding. Antimicrob Agents Chemother. 2008;52:269–278. doi: 10.1128/AAC.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murthy MH, Olson ME, Wickert RE, et al. Daptomycin non-susceptible methicillin-resistant Staphylococcus aureus USA 300 isolate. J Med Microbiol. 2008;57:1036–1038. doi: 10.1099/jmm.0.2008/000588-0. [DOI] [PubMed] [Google Scholar]
- 24.Fowler VG, Jr, Boucher HW, Corey GR, et al. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N Engl J Med. 2006;355:653–665. doi: 10.1056/NEJMoa053783. [DOI] [PubMed] [Google Scholar]
- 25.Cunha BA, Pherez FM. Daptomycin resistance and treatment failure following vancomycin for methicillin-resistant Staphylococcus aureus mitral valve acute bacterial endocarditis. Eur J Clin Microbiol Infect Dis. 2009;28:831–833. doi: 10.1007/s10096-008-0692-2. [DOI] [PubMed] [Google Scholar]
- 26.Sharma M, Riederer K, Chase P, et al. High rate of decreasing daptomycin susceptibility during the treatment of persistent Staphylococcus aureus bacteremia. Eur J Clin Microbiol Infect Dis. 2008;27:433–437. doi: 10.1007/s10096-007-0455-5. [DOI] [PubMed] [Google Scholar]
- 27.Muraih JK, Pearson A, Silverman J, et al. Oligomerization of daptomycin on membranes. Biochim Biophys Acta. 2011;1808:1154–1160. doi: 10.1016/j.bbamem.2011.01.001. Biochim Biophys Acta. [DOI] [PubMed] [Google Scholar]
- 28.Ho SW, Jung D, Calhoun JR, et al. Effect of divalent cations on the structure of the antibiotic daptomycin. Eur Biophy J. 2008;37:421–433. doi: 10.1007/s00249-007-0227-2. [DOI] [PubMed] [Google Scholar]
- 29.Strauss SK, Hancock REW. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: comparison with cationic antimicrobial peptides and lipopeptides. Biochim Biophys Acta. 2006;1758:1215–1223. doi: 10.1016/j.bbamem.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 30.Scott HRP, Baek S-B, Jung D, et al. NMR structure of the antibiotic lipopeptide daptomycin in DHPC micelles. Biochim Biophys Acta. 2007;1768:3116–3126. doi: 10.1016/j.bbamem.2007.08.034. [DOI] [PubMed] [Google Scholar]
- 31.Grunewald J, Sieber SA, Mahlert MA, et al. Synthesis and derivatization of daptomycin: A chemoenzymatic route to acidic lipopeptide antibiotics. J Am Chem Soc. 2004;126:17025–17031. doi: 10.1021/ja045455t. [DOI] [PubMed] [Google Scholar]
- 32.Jung D, Rozek A, Okon M, et al. Structural transitions as determinants of the action of the calcium-dependent antibiotic, daptomycin. Chem Biol. 2004;11:949–957. doi: 10.1016/j.chembiol.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 33.Kopp F, Grunewald J, Mahlert C, et al. Chemoenzymatic design of acidic lipopeptide hybrids: new insights into the structure-activity relationship of daptomycin and A54145. Biochemistry. 2006;45:10474–10481. doi: 10.1021/bi0609422. [DOI] [PubMed] [Google Scholar]
- 34.Cotroneo N, Harris R, Perlmutter N, et al. Daptomycin exerts bactericidal activity without lysis of Staphylococcus aureus. Antimicrob Agents Chemother. 2008;52:2223–2225. doi: 10.1128/AAC.01410-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mascio TM, Adler JD, Silverman JA. Bactericidal action of daptomycin against stationary-phase and nondividing Staphylococcus aureus cells. Antimicrob Agents Chemother. 2007;51:4255–4260. doi: 10.1128/AAC.00824-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedman L, Adler JD, Silverman JA. Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006;50:2137–2145. doi: 10.1128/AAC.00039-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang SJ, Xiong YQ, Dunman PM, et al. Regulation of mprF in daptomycin-nonsusceptible Staphylococcus aureus. Antimicrob Agents Chemother. 2009;53:2636–2637. doi: 10.1128/AAC.01415-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaatz GW, Lundstrom TS, Seo SM. Mechanisms of daptomycin resistance in Staphylococcus aureus. Int J Antimicrob Agents. 2006;28:280–287. doi: 10.1016/j.ijantimicag.2006.05.030. [DOI] [PubMed] [Google Scholar]
- 39.Patel D, Husain M, Vidaillac C, et al. Mechanisms of in vitro-selected daptomycin non-susceptibility in Staphylococcus aureus. Int J Antimicrob Agents. 2011;38:442–446. doi: 10.1016/j.ijantimicag.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 40.Ernst CM, Peschel A. Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids. Mol Microbiol. 2011;80:290–299. doi: 10.1111/j.1365-2958.2011.07576.x. [DOI] [PubMed] [Google Scholar]
- 41.Ernst CM, Staubitz P, Mishra NN, et al. The bacterial defensin resistance protein, MprF, consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PloS Pathog. 2009;5:e1000660. doi: 10.1371/journal.ppat.1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldmann AJ, Ernst CM, Peschel A, et al. Multiple peptide resistance factor (MprF)-mediated resistance of Staphylococcus aureus against antimicrobial peptides coincides with a modulated peptide interaction with artificial membranes comprising lysyl-phosphatidylglycerol. J Biol Chem. 2011;286:18692–18700. doi: 10.1074/jbc.M111.226886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kilelee E, Pokorny A, Yeaman MR, et al. Lysyl-PG attenuates membrane perturbation rather than surface association of the platelet cationic antimicrobial peptide 6W-RP-1 in a model membrane system – implications for daptomycin resistance. Antimicrob Agents Chemother. 2010;54:4476–4479. doi: 10.1128/AAC.00191-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hachmann A-B, Sevim E, Gaballa A, et al. Reduction in membrane phosphatidylglycerol content leads to daptomycin resistance in Bacillus subtilis. Antimicrob Agents Chemother. 2011;55:4326–4337. doi: 10.1128/AAC.01819-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hachmann A-B, Angert ER, Helmann JD. Genetic analysis of factors affecting susceptibility of Bacillus subtilis to daptomycin. Antimicrob Agents Chemother. 2009;53:1598–1609. doi: 10.1128/AAC.01329-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peleg AY, Miyakis S, Ward DV, et al. Whole genome characterization of the mechanisms of daptomycin resistance in clinical and laboratory derived isolates of Staphylococcus aureus. PloS One. 2012;7:e28316. doi: 10.1371/journal.pone.0028316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muraih JK, Harris J, Taylor SD, et al. Characterization of daptomycin oligimerization with perylene excimer fluorescence: stoichiometric binding of phosphatidylglycerol triggers oligomer formation. Bichemica et Biophysica Acta. 2012;1818:673–678. doi: 10.1016/j.bbamem.2011.10.027. [DOI] [PubMed] [Google Scholar]
- 48.Rubio A, Conrad M, Haselbeck RJ, et al. Regulation of mprF by antisense RNA restores daptomycin susceptibility to daptomycin-resistant isolates of Staphylococcus aureus. Antimicrob Agents Chemother. 2011;55:364–367. doi: 10.1128/AAC.00429-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang S-J, Mishra NN, Rubio A, et al. The causal role of single nucleotide polymorphisms with the mprF gene of Staphylococcus aureus in daptomycin resistance. 51st ICAAC; Chicago, IL. Sep, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang SJ, Kreiswirth BN, Sakoulas G, et al. Enhanced expression of dltABCD, but not mprF, is associated with development of daptomycin nonsusceptibility in a clinical endocarditis isolate of Staphylococcus aureus. J Infect Dis. 2009;200:1916–1920. doi: 10.1086/648473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patel JB, Jevitt LA, Hageman J, et al. An association between reduced susceptibility to daptomycin and reduced susceptibility to vancomycin in Staphylococcus aureus. Clin Infect Dis. 2006;42:1652–1653. doi: 10.1086/504084. [DOI] [PubMed] [Google Scholar]
- 52.Mwangi MM, Wu SW, Zhou Y, et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc Nat Acad Sci (USA) 2007;104:9451–9456. doi: 10.1073/pnas.0609839104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pillai SK, Gold HS, Sakoulas G, et al. Daptomycin nonsusceptibility in Staphylococcus aureus with reduced vancomycin susceptibility is independent of alterations in mprF. Antimicrob Agents Chemother. 2007;51:2223–2225. doi: 10.1128/AAC.00202-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakoulas G, Adler J, Thauvin-Eliopoulos C, et al. Induction of daptomycin heterogeneous susceptibility in Staphylococcus aureus by exposure to vancomycin. Antimicrob Agents Chemother. 2006;50:1581–1585. doi: 10.1128/AAC.50.4.1581-1585.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cui L, Toninaga E, Neoh H-M, et al. Correlation between reduced daptomycin susceptibility and vancomycin resistance in vancomycin-intermediate Staphylococcus aureus. Antimicrob Agents Chemother. 2006;50:1079–1082. doi: 10.1128/AAC.50.3.1079-1082.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mishra NN, Yang SJ, Sawa A, et al. Analysis of cell membrane characteristics of in vitro-selected daptomycin-resistant strains of MRSA. Antimicrob Agents Chemother. 2009;53:2312–2318. doi: 10.1128/AAC.01682-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dhand A, Bayer AS, Pogliano J, et al. Use of anti-staphylococcal beta-lactams to increase daptomycin activity in eradicating persistent bacteremia due to methicillin-resistant Staphylococcus aureus (MRSA): Role of enhanced daptomycin binding. Clin Infect Dis. 2011;53:158–163. doi: 10.1093/cid/cir340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bayer AS, Prasad R, Chandra J, et al. In vitro resistance of Staphylococcus aureus to thrombin-induced microbicidal protein is associated with alterations in membrane fluidity. Infect Immun. 2000;68:3548–3553. doi: 10.1128/iai.68.6.3548-3553.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kupferwasser LI, Skurray RA, Brown MH, et al. Staphylococcus aureus resistance to the cationic peptide, thrombin-induced platelet microbicidal protein-1 is encoded by the multiresistance plasmid pSK1. Antimicrob Agents Chemother. 1999;43:2395–2399. doi: 10.1128/aac.43.10.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bayer AS, Kupferwasser LI, Brown MH, et al. Low-level Staphylococcus aureus resistance to thrombin-induced platelet microbicidal protein-1 (tPMP-1) in vitro associated with qacA gene carriage is independent of multidrug efflux pump activity. Antimicrob Agents Chemother. 2006;50:2448–2454. doi: 10.1128/AAC.00028-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mishra NN, McKinnell J, Yeaman MR, et al. In vitro cross-resistance of daptomycin and host defense cationic antimicrobial peptides in clinical methicillin-resistant Staphylococcus aureus (MRSA) isolates. Antimicrob Agents Chemother. 2011;55:4012–4018. doi: 10.1128/AAC.00223-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu CI, Liu GY, Song Y, et al. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science. 2008;319:1391–1394. doi: 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mishra NN, Liu GY, Yeaman MR, et al. Carotenoid-related alteration of cell membrane fluidity impacts Staphylococcus aureus susceptibility to host defense peptides. Antimicrob Agents Chemother. 2011;55:526–531. doi: 10.1128/AAC.00680-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Holt DC, Holden MTG, Tong SYC. A very early-branching Staphylococcus aureus lineage lacking the carotenoid pigment staphyloxanthin. Genome Biol Evol. 2011;3:881–895. doi: 10.1093/gbe/evr078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yeaman MR, Bayer AS, Koo S-P, et al. Platelet microbicidal proteins differentially permeabilize and depolarize the Staphylococcus aureus cytoplasmic membrane. J Clin Invest. 1998;101:178–187. doi: 10.1172/JCI562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang S-J, Nast CC, Mishra N, et al. Cell wall thickening is not a universal accompaniment of the daptomycin non-susceptibility phenotype in Staphylococcus aureus: evidence for multiple resistance mechanisms. Antimicrob Agents Chemother. 2010;54:3079–3085. doi: 10.1128/AAC.00122-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Leeuw E, Li C, Zeng P, et al. interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett. 2010;584:1543–1548. doi: 10.1016/j.febslet.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sass V, Pag U, Tossi A, et al. Mode of action of human beta-defensin 3 against Staphylococcus aureus and transcriptional analysis of responses to defensin challenge. Int J Med Microbiol. 2008;298:619–633. doi: 10.1016/j.ijmm.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 69.Sass V, Schneider T, Wilmes M, et al. Human beta-defensin 3 inhibits cell wall biosynthesis in staphylococci. Infect Immun. 2010;78:2773–2800. doi: 10.1128/IAI.00688-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mishra NM, Bayer AS, Moise PA, et al. Reduced susceptibility to host defense cationic peptides and daptomycin co-emerges among methicillin-resistant Staphylococcus aureus (MRSA) in daptomycin-naïve bacteremic patients. J Infect Dis. 2012;206:1160–1167. doi: 10.1093/infdis/jis482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saleh-Mghir A, Muller-Serieys C, Dinh A, et al. Adjunctive rifampin is crucial for optimizing daptomycin efficacy against rabbit prosthetic joint infection due to methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2011;55:4589–4593. doi: 10.1128/AAC.00675-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mishra NN, Bayer AS, Cremieux A-C, et al. Co-evolution of increasing daptomycin MICs and resistance to host defense peptides in rabbits with MRSA prosthetic joint infections. 51st ICAAC; Chicago, IL. Sep, 2011. [Google Scholar]
- 73.Bhullar K, Waglechner N, Pawlowski A, et al. Antibiotic resistance is prevalent in an isolated cave microbiome. PloS One. 2012;7:1–11. doi: 10.1371/journal.pone.0034953. e34953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Canepari P, Boaretti M, Lleo MM, Satta G. Lipoteichoic acid as a new target for activity of antibiotics: mode of action of daptomycin (LY146032) Antimicrob Agents Chemother. 1990;34:1220–1226. doi: 10.1128/aac.34.6.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Allen NE, Hobbs JN, Alborn WE. Inhibition of peptidoglycan biosynthesis in gram-positive bacteria by LY146032. Antimicrob Agents Chemother. 1981;31:1093–1099. doi: 10.1128/aac.31.7.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mengin-Lecreulx D, Allen NE, Hobbs JN, et al. Inhibition of peptidoglycan biosynthesis in Bacillus megaterium by daptomycin. FEMS Microbiol Lett. 1990;57:245–248. doi: 10.1016/0378-1097(90)90074-z. [DOI] [PubMed] [Google Scholar]
- 77.Muthaiyan A, Silverman JA, Jayaswal RK, et al. transcriptional profiling reveals that daptomycin induces the Staphylococcus aureus cell wall stress stimulon and genes responsive to membrane depolarization. Antimicrob Agent Chemother. 2008;52:980–990. doi: 10.1128/AAC.01121-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Utaida S, Dunman PM, Macapagal D, et al. Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology. 2003;149:2719–2732. doi: 10.1099/mic.0.26426-0. [DOI] [PubMed] [Google Scholar]
- 79.Kuroda M, Kuroda H, Oshima T, et al. Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol Microbiol. 2003;49:807–821. doi: 10.1046/j.1365-2958.2003.03599.x. [DOI] [PubMed] [Google Scholar]
- 80.Wecke T, Zühlke D, Mäder U, et al. Daptomycin versus friulimicin B: in-depth profiling of Bacillus subtilis cell envelope stress responses. Antimicrob Agents Chemother. 2009;53(4):1619–1623. doi: 10.1128/AAC.01046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schneider T, Gries K, Josten M, et al. The lipopeptide antibiotic friulimicin B inhibits cell wall biosynthesis through complex formation with bactoprenol phosphate. Antimicrob Agents Chemother. 2009;53(4):1610–1618. doi: 10.1128/AAC.01040-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Song Y, Lunde CS, Benton BM, et al. Further insights into the mode of action of the lipoglycopeptide telavancin through global gene expression studies. Antimicrob Agents Chemother. 2012;56(6):3157–3164. doi: 10.1128/AAC.05403-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fischer A, Yang S-J, Bayer AS, et al. Daptomycin resistance mechanisms in clinically derived Staphylococcus aureus strains assessed by a combined transcriptomics and proteomics approach. J Antimicrob Chemother. 2011;66:1696–1711. doi: 10.1093/jac/dkr195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bertsche U, Weidenmaier C, Kuehner D, et al. Correlation of daptomycin-resistance in a clinical Staphylococcus aureus strain with increased cell wall teichoic acid production and d-alanylation. Antimicrob Agents Chemother. 2011;55:3922–3928. doi: 10.1128/AAC.01226-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gutberlet T, Frank J, Bradaczek H, et al. Effect of lipoteichoic acid on thermotropic membrane properties. J Bacteriol. 1997;179:2879–2883. doi: 10.1128/jb.179.9.2879-2883.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boyle-Vavra S, Jones M, Gourley BL, et al. Comparative genome sequencing of an isogenic pair of USA800 clinical methicillin-resistant Staphylococcus aureus isolates obtained before and after daptomycin treatment failure. Antimicrob Agents Chemother. 2011;55:2018–2025. doi: 10.1128/AAC.01593-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McAleese F, Wu SW, Sieradzki K, et al. Overexpression of genes of the cell wall stimulon in clinical isolates of Staphylococcus aureus exhibiting vancomycin-intermediate-S. aureus-type resistance to vancomycin. J Bacteriol. 2006;188:1120–1133. doi: 10.1128/JB.188.3.1120-1133.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Balibar CJ, Shen X, McGuire D, Yu D, McKenney D, Tao J. cwrA, a gene that specifically responds to cell wall damage in Staphylococcus aureus. Microbiology. 2010;156(Pt 5):1372–83. doi: 10.1099/mic.0.036129-0. [DOI] [PubMed] [Google Scholar]
- 89.Balibar CJ, Shen X, Tao J. The mevalonate pathway of Staphylococcus aureus. J Bacteriol. 2009;191(3):851–61. doi: 10.1128/JB.01357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Baltz RH. Daptomycin: mechanisms of action and resistance, and biosynthetic engineering. Curr Opin Chem Biol. 2009;13:144–151. doi: 10.1016/j.cbpa.2009.02.031. Review. [DOI] [PubMed] [Google Scholar]
- 91.Mohedano ML, Overweg K, de la Fuente A, et al. Evidence that the essential response regulator YycF in Streptococcus pneumoniae modulates expression of fatty acid biosynthesis genes and alters membrane composition. J Bacteriol. 2005;187:2357–2367. doi: 10.1128/JB.187.7.2357-2367.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dubrac S, Msadek T. Identification of genes controlled by the essential YycG/YycF two-component system of Staphylococcus aureus. J Bacteriol. 2004;186:1175–1181. doi: 10.1128/JB.186.4.1175-1181.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dubrac S, Boneca IG, Poupel O, et al. New insights into the WalK/WalR (YycG/YycF) essential signal transduction pathway reveal a major role in controlling cell wall metabolism and biofilm formation in Staphylococcus aureus. J Bacteriol. 2007;189:8257–8269. doi: 10.1128/JB.00645-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dubrac S, Bisicchia P, Devine KM, et al. A matter of life and death: cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol Microbiol. 2008;7:1307–1322. doi: 10.1111/j.1365-2958.2008.06483.x. [DOI] [PubMed] [Google Scholar]
- 95.Fukushima T, Szurmant H, Kim EJ, et al. A sensor histidine kinase co-ordinates cell wall architecture with cell division in Bacillus subtilis. Mol Microbiol. 2008;69:621–632. doi: 10.1111/j.1365-2958.2008.06308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Howden BP, McEvoy CR, Allen DL, et al. Evolution of multidrug resistance during Staphylococcus aureus infection involves mutation of the essential two component regulator WalKR. PLoS Pathog. 2011;7(11):e1002359. doi: 10.1371/journal.ppat.1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jansen A, Turck M, Szekat C, et al. Role of insertion elements and yycFG in the development of decreased susceptibility to vancomycin in Staphylococcus aureus. Int J Med Microbiol. 2007;297:205–215. doi: 10.1016/j.ijmm.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 98.Delaune A, Poupel O, Mallet A, et al. Peptidoglycan crosslinking relaxation plays an important role in Staphylococcus aureus WalKR-dependent cell viability. PLoS One. 2011;6(2):e17054. doi: 10.1371/journal.pone.0017054. 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kuroda H, Kuroda M, Cui L, et al. Subinhibitory concentrations of beta-lactam induce haemolytic activity in Staphylococcus aureus through the SaeRS two-component system. FEMS Microbiol Lett. 2007;268:98–105. doi: 10.1111/j.1574-6968.2006.00568.x. [DOI] [PubMed] [Google Scholar]
- 100.Rogasch K, Rühmling V, Pané-Farré, et al. Influence of the two-component system SaeRS on global gene expression in two different Staphylococcus aureus strains. J. Bacteriol. 2006;188:7742–7758. doi: 10.1128/JB.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Steinhuber A, Goerke C, Bayer MG, et al. Molecular architecture of the regulatory locus sae of Staphylococcus aureus and its impact on expression of virulence factors. J. Bacteriol. 2003;185:6278–6286. doi: 10.1128/JB.185.21.6278-6286.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Geiger T, Goerke C, Mainiero M, et al. The virulence regulator Sae of Staphylococcus aureus: promoter activities and response to phagocytosis-related signals. J. Bacteriol. 2008;190:3419–3428. doi: 10.1128/JB.01927-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mascher T. Intramembrane-sensing histidine kinases: a new family of cell envelope stress sensors in Firmicutes bacteria. FEMS Microbiol. Lett. 2006;264:133–144. doi: 10.1111/j.1574-6968.2006.00444.x. [DOI] [PubMed] [Google Scholar]
- 104.Sievers S, Ernst CM, Geiger T, et al. Changing the phospholipid composition of Staphylococcus aureus causes distinct changes in membrane proteome and membrane-sensory regulators. Proteomics. 2010;10:1685–1693. doi: 10.1002/pmic.200900772. [DOI] [PubMed] [Google Scholar]
- 105.Kawai Y, Marles-Wright J, Cleverley RM, et al. A widespread family of bacterial cell wall assembly proteins. EMBO J. 2011;30:4931–4941. doi: 10.1038/emboj.2011.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Eberhardt A, Hoyland CN, Vollmer D, et al. Attachment of capsular polysaccharide to the cell wall in Streptococcus pneumoniae. Microb Drug Resist. 2012;18:240–255. doi: 10.1089/mdr.2011.0232. [DOI] [PubMed] [Google Scholar]
- 107.Dengler V, Meier PS, Heusser R, et al. Deletion of hypothetical wall teichoic acid ligases in Staphylococcus aureus activates the cell wall stress response. FEMS Microbiol Lett. 2012 May 28;333:109–120. doi: 10.1111/j.1574-6968.2012.02603.x. [DOI] [PubMed] [Google Scholar]
- 108.Steenbergen JN, Mohr JF, Thorne GM. Effects of daptomycin in combination with other antimicrobial agents: a review of in vitro and animal model studies. J Antimicrob Agents Chemother. 2009;64:1130–1138. doi: 10.1093/jac/dkp346. [DOI] [PubMed] [Google Scholar]
- 109.Nadrah K, Strle F. Antibiotic combinations with daptomycin for the treatment of Staphylococcus aureus infections. Chemother Res Pract. 2011;2011:619321. doi: 10.1155/2011/619321. Epub 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rose WE, Rybak MJ, Kaatz GW. Evaluation of daptomycin treatment of Staphylococcus aureus bacterial endocarditis: an in vitro and in vivo simulation using historical and current dosing strategies. J Antimicrob Chemother. 2007;60:334–340. doi: 10.1093/jac/dkm170. [DOI] [PubMed] [Google Scholar]
- 111.Rose WE, Leonard SN, Rybak MJ. Evaluation of daptomycin pharmacodynamics and resistance at various dosage regiments against Staphylococcus aureus isolates with reduced susceptibilities to daptomycin in an in vitro pharmacodynamic model with simulated endocardial vegetations. Antimicrob Agents Chemother. 2008;52:3061–3067. doi: 10.1128/AAC.00102-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sakoulas G, Eliopoulos GM, Alder J, et al. Efficacy of daptomycin in experimental endocarditis due to methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2003;47:1714–1718. doi: 10.1128/AAC.47.5.1714-1718.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lefebvre M, Jacqueline C, Amador G, et al. Efficacy of daptomycin combined with rifampicin for the treatment of experimental methicillin-resistant Staphylococcus aureus (MRSA) acute osteomyelitis. Intl J Antimicrob Agents. 2010;36:542–544. doi: 10.1016/j.ijantimicag.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 114.Cirioni O, Mocchegiani F, Ghiselli R, et al. Daptomycin and rifampin alone and in combination prevent vascular graft biofilm formation and emergence of antibiotic resistance in a subcutaneous rat pouch model of staphylococcal infection. Eur J Vasc Endovasc Surg. 2010;40:817–822. doi: 10.1016/j.ejvs.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 115.Lunde CS, Hartouni SR, Janc JW, et al. Telavancin disrupts the functional integrity of the bacterial membrane through targeted interaction with the cell wall precursor Lipid II. Antimicrob Agents Chemother. 2009;53:3375–3383. doi: 10.1128/AAC.01710-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Krause KM, Renelli M, Difuntorum S, et al. In vitro activity of telavancin against resistant gram-positive bacteria. Antimicrob Agents Chemother. 2008;52:2647–2652. doi: 10.1128/AAC.01398-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Steed ME, Vidaillac C, Rybak MJ. Evaluation of telavancin activity versus daptomycin and vancomycin against daptomycin-nonsusceptible Staphylococcus aureus in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother. 2012;56:955–959. doi: 10.1128/AAC.05849-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Madrigal AG, Basuino L, Chambers HF. Efficacy of telavancin in a rabbit model of aortic valve endocarditis due to methicillin-resistant Staphylococcus aureus or vancomycin-intermediate Staphylococcus aureus. Antimicrob Agents Chemother. 2005;49:3163–3165. doi: 10.1128/AAC.49.8.3163-3165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Miro JM, Garcia-de-la-Maria C, Amero Y, et al. Efficacy of telavancin in the treatment of experimental endocarditis due to glycopeptide-intermediate Staphylococcus aureus. Antimicrob Agents Chemother. 2007;51:2373–2377. doi: 10.1128/AAC.01266-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Joson J, Grover C, Downer C, et al. Successful treatment of methicillin-resistant Staphylococcus aureus mitral valve endocarditis with sequential linezolid and telavancin monotherapy following daptomycin failure. J Antimicrob Chemother. 2011;66:2186–2188. doi: 10.1093/jac/dkr234. [DOI] [PubMed] [Google Scholar]
- 121.Marcos LA, Camins BC. Successful treatment of vancomycin-intermediate Staphylococcus aureus pacemaker lead infective endocarditis with telavancin. Antimicrob Agents Chemother. 2010;54:5376–5378. doi: 10.1128/AAC.00857-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vidaillac C, Parra-Ruiz J, Rybak MJ. In vitro time-kill analysis of oritavancin against clinical isolates of methicillin-resistant Staphylococcus aureus with reduced susceptibility to daptomycin. Diagn Microbiol Infect Dis. 2011;71:470–473. doi: 10.1016/j.diagmicrobio.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 123.Steed M, Vidaillac C, Rybak MJ. Evaluation of ceftaroline activity versus daptomycin against daptomycin-nonsusceptible methicillin-resistant Staphylococcus aureus strains in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother. 55:3522–3526. doi: 10.1128/AAC.00347-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rand KH, Houck HJ. Synergy of daptomycin with oxacillin and other beta-lactams against methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2004;48:2871–2875. doi: 10.1128/AAC.48.8.2871-2875.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]