

Regulatory approval of oncology drugs is the cornerstone of the development process and approval trends will ultimately affect clinicians and patients. This review examines changes in aspects of drug development over the lifespan of chemotherapy to provide insight into future trends.
Keywords: Drug therapy, Neoplasms, U.S. Food and Drug Administration, Biological markers, Clinical trial
Abstract
Background.
Regulatory approval of oncology drugs is the cornerstone of the development process and approval characteristics shape eventual utilization. Approval trends and characteristics provide valuable information for drug developers and regulators and ultimately affect clinicians and patients.
Methods.
Indication characteristics were tabulated for drugs approved by the U.S. Food and Drug Administration (FDA) for systemic therapy of malignancies from 1949 through October 2011. Variables included time to approval, initial/supplemental indication, tumor type, stage of disease, specification of protein expression or genetic information, drug class, trial design, concomitant agent, trial size, and endpoint.
Results.
A total of 121 unique anticancer agents, including 242 unique indications, were approved. The number of trials for each indication has decreased; however, trial size has increased and more randomized controlled trials have been performed. Trial designs have increasingly used time-to-event endpoints and rarely have used symptom-based primary endpoints. Approvals have been primarily single agent, with less emphasis on palliative treatments and increasing emphasis on advanced disease stages and requirements for prior therapy. Molecular specifications in labels have increased, but they are present in less than 30% of recent indications and are not associated with shorter approval times.
Conclusion.
Approval of oncology agents is occurring in increasingly more challenging settings, suggesting gaps between eventual practice and development in potentially suboptimal indications. Molecular specifications promise to enhance development, yet widespread use in label indications has not yet been achieved.
Implications for Practice:
Regulatory approval of oncology drugs is the cornerstone of the development process and approval characteristics shape eventual utilization. Approval trends and characteristics provide valuable information for drug developers and regulators, and ultimately impact clinicians and patients. This review found that approval of oncology agents is occurring in increasingly more challenging settings, suggesting gaps between eventual practice and development in potentially sub-optimal indications. Molecular specifications promise to enhance development, yet widespread use in label indications has not yet been achieved.
Introduction
Cancer continues to be a major cause of morbidity and mortality in our society. Although local interventions, including surgery, radiotherapy, and adjuvants are highly effective in early disease, there remains huge unmet need in more advanced stages. Pharmaceuticals make up the mainstay of therapy for advanced cancer. Yet, the ability to develop new anticancer agents is profoundly inefficient. With only 3%–25% of agents entering clinical Investigational New Drug (IND) development actually gaining regulatory approval, and the majority of development costs being spent on drugs that never become approved, there is a dire need to improve efficiency [1–5]. Regulatory approval of oncology drugs is the cornerstone of the development process and approval characteristics shape eventual utilization. Approval trends and characteristics provide valuable information for drug developers and regulators and ultimately affect clinicians and patients. To understand characteristics of successful products, we performed a comprehensive review of approved systemic anticancer agents.
Key strategies to improve efficiency have made incremental advances, including predictive biomarkers [6–8], clinical trial design [9, 10], efficiency in regulatory approval [11, 12], accelerated approval, access to unapproved anticancer agents, membership and voting rights to patients and patient representatives on U.S. Food and Drug Administration (FDA) advisory committees, and reducing the need for IND applications for investigator-initiated studies for marketed agents in nonlabeled cancer indications [13–17].
Cancer chemotherapy has evolved significantly over the past five decades. This evolution has involved many new classes of agents ranging from biologics to immune-stimulating agents to kinase inhibitors. During this time, the sophistication of clinical trial design and interpretation of data have also improved dramatically. A key paradigm currently emphasizes targeted agents and predictive/biomarkers. The current review tracks changes in aspects of drug development over the lifespan of chemotherapy and will give insight into future trends.
Methods
Drug Selection
This study includes all drugs approved by the FDA for systemic therapy of malignancies from 1949 through October 2011, including new drug applications, biologic licensing applications, and supplemental applications. Drugs that were initially approved for a noncancerous condition were also included. Indications for benign tumors or precancerous conditions were excluded from the analysis. In addition, only drugs that are used as systemic chemotherapy were included. Creams and other locally administered drugs were excluded. Generics were only included if they were approved for a unique cancer indication. Approvals for new formulations of a drug for a comparable indication were not included.
Data Collection
Data were collected from drug labels using the FDA website and the Physician Desk Reference (PDR). When the original or new indication label was not available on the FDA website, data were obtained from the PDR of the corresponding year.
Each unique indication is counted for each drug, including indications approved at the same time as long as substantive data support each indication. Approvals for alternate formulations or generic forms of a particular active pharmaceutical ingredient (API) were not counted if the approval was for an existing indication for that API. If a drug was originally approved for a noncancerous condition, the first approval for cancer was counted as the initial indication; any additional indications for cancer were considered supplemental. Indications involving corticosteroids, adjunctive agents, or agents used primarily for local therapy were not included.
Tumor type represents the type of cancer specified in the indication. Indications that either did not specify a tumor type or listed several types, were classified as multiple unless the label included specific clinical studies that demonstrated efficacy for each. Tumor type classifications were initially captured exactly as written in the indication and were grouped into specified categories for the purpose of presentation.
Stage classification was based on the indication and was assigned to one of four categories: local/adjuvant, advanced/metastatic, palliative, or prophylactic. Leukemia and myelodysplastic syndrome indications were excluded from the stage classification analysis. The local/adjuvant category includes indications that specify the following: adjuvant therapy, local, early, locally or regionally advanced, and operable cancer. The palliative category includes all indications that specify palliative therapy or treatment, even if it also specifies metastatic disease.
Prior treatment data were based on the indication and assigned to one of three categories: chemotherapy, surgery or radiotherapy, or no prior therapy. Indications that specified adjuvant therapy were included in the surgery or radiotherapy category. Indications indicating both no prior therapy and prior therapy were included in the “no prior therapy” category. Indications that specify prior therapy without mentioning what type were assigned a category based on currently accepted treatments for that cancer. Molecular specification refers to indications that specify genetic or protein parameters that guide the use of the agent.
Data for the single-agent/combination category were based on specification within the indication of use of the drug as a single agent or in combination with other agents. Indications that did not specify use in combination therapy were classified as single-agent therapy. In situations where both single-agent and combination use was specified, this was counted as such, unless stand-alone clinical trial data were sufficient to designate two separate indications in this analysis.
Data on study size, design, and endpoints used for regulatory approval were obtained from the drug label. The studies considered in this analysis were those that were the basis of FDA approval for each indication. Information on clinical studies was not consistently presented in many pre-1990 labels, and therefore data during this period were not included in some of the analyses. For endpoint evaluation, only one trial (the largest or most relevant to the initial indication) was considered for each indication. Additionally, when the primary study endpoint was not clearly articulated and multiple endpoints met statistical criteria, the endpoint with highest priority was taken for this analysis, using the following hierarchy: survival (e.g., overall survival, median survival, 3-year survival), symptom (e.g., clinical benefit response), time-to-event (progression-free survival, time to progression, time to recurrence, incidence), and response rate (e.g., tumor or other surrogate response rate).
Data Analysis
Data were analyzed to determine trends over time by categorizing the indications into the following periods: pre-1990; 1990–1999; 2000–2005; and 2006–2011. The pre-1990 data were grouped together for several reasons, including lower frequency of oncology drug approvals; granularity of trends during that period are less relevant now and data are less available on the FDA website.
Results
General Approval Characteristics
A total of 121 unique anticancer agents, including 242 unique indications, approved by the FDA for systemic therapy of cancer were evaluated, with approval dates ranging from 1949 to 2011 (Table 1). Considering multiple indications at the time of initial approval, 136 indications have been specified. Sixty-four agents received supplemental approvals, including 106 unique supplemental indications. The proportion of supplemental indications has increased in the most recent period (54% of all approved indications) versus 52% and 34% respectively for the next two most recent periods. Monotherapy approvals were found to be more common than combination approvals, particularly in the initial indications but also in supplemental indications (Fig. 1). This finding has been maintained over time.
Table 1.
Tabulation of indications and drug classes
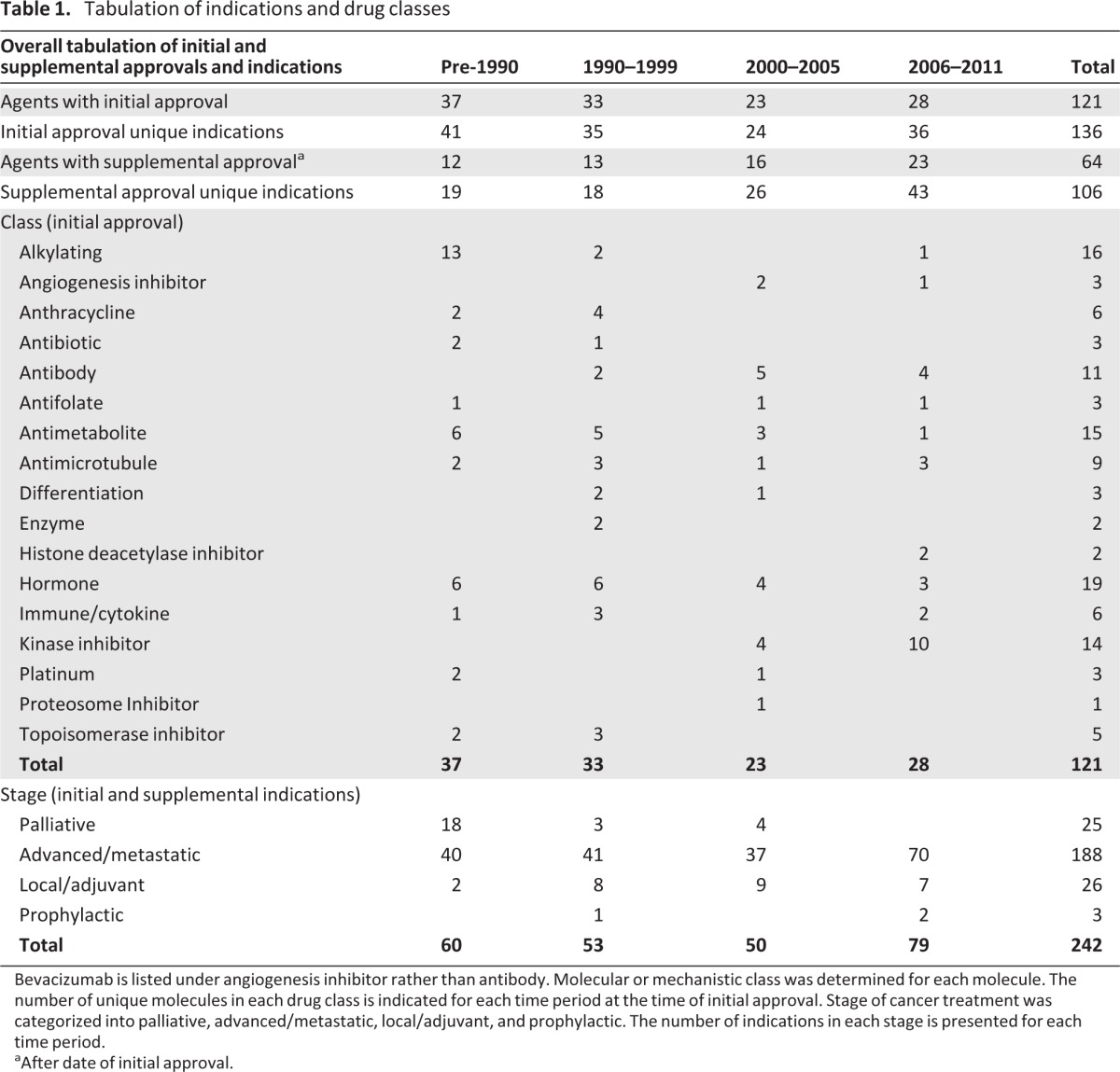
Bevacizumab is listed under angiogenesis inhibitor rather than antibody. Molecular or mechanistic class was determined for each molecule. The number of unique molecules in each drug class is indicated for each time period at the time of initial approval. Stage of cancer treatment was categorized into palliative, advanced/metastatic, local/adjuvant, and prophylactic. The number of indications in each stage is presented for each time period.
aAfter date of initial approval.
Figure 1.

Single-agent and combination approvals. The proportion of approvals involving a single agent (black), combinations of more than one agents (grey), and both single-agent and combination use designated in the same indication (striped). Initial (A) and supplemental (B) approvals are presented. In situations where substantive data independently supported both single-agent and combination use, separate indications were designated.
Review of the suggested first-line treatments from the National Comprehensive Cancer Network for the 12 most common malignancies in the U.S., including certain indication subsets, revealed that 14 of the 21 (67%) indications had combinations listed as the treatment of choice for good-performance status patients, yet only 8 of 18 (44%) of the drugs listed in these combinations were first approved in a combination [18, 19]. This discordance is primarily with older oncology agents and suggests that optimal use of many oncology agents is not articulated in the initial approval.
The proportion of approvals that were accelerated approvals peaked in the early 2000s and is now utilized in about a quarter of approvals (supplemental online Fig. 1A, 1B). There is a clear trend toward faster approval times, with the median plateauing at around 6 months (supplemental online Fig. 1C–E). There was no clear association between faster approval times and use of accelerated approval (Table 2).
Table 2.
Correlation between development parameters and approval time
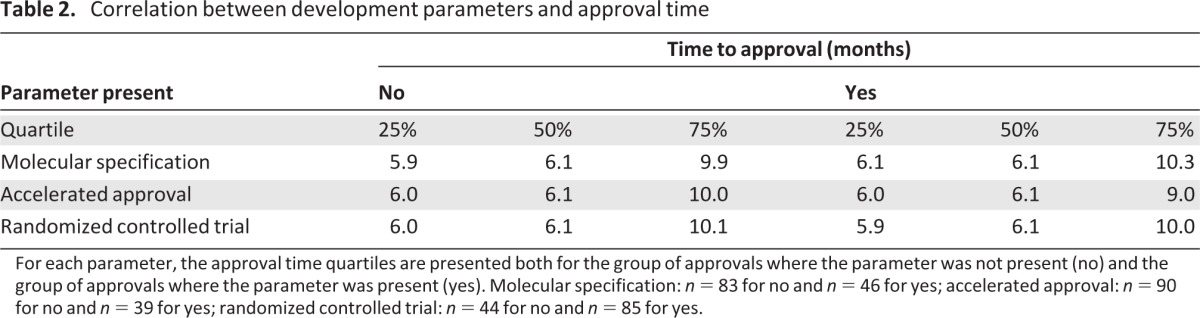
For each parameter, the approval time quartiles are presented both for the group of approvals where the parameter was not present (no) and the group of approvals where the parameter was present (yes). Molecular specification: n = 83 for no and n = 46 for yes; accelerated approval: n = 90 for no and n = 39 for yes; randomized controlled trial: n = 44 for no and n = 85 for yes.
Types of Agents and Malignancies
Initial approvals for alkylating agents, anthracyclines, and topoisomerase inhibitors have been declining over time, whereas antibodies and kinase inhibitors have increased in the past decade (Table 1), making up 15% and 36%, respectively, of all approved molecules in the most recent 6 years.
Breast, prostate, and hematologic malignancies are the most common indications (supplemental online Table 1). There has been a shift away from indications specifying multiple malignancies. Indications in certain malignancies peaked during different time periods; for example germ cell, central nervous system, and gynecologic malignancies peaked in the pre-1990s, breast in the 1990s, colon and lung in the 1990s and early 2000s, and renal and skin in the most recent period. The stage of disease was largely advanced/metastatic disease.
Details of Indications
Important aspects of indications have shifted over time. The requirement for prior treatment has increased recently (Fig. 2). Most pre-1990 initial indications do not specify any prior treatment as part of the approved indication; prior chemotherapy is specified in 67% and 58% of initial indications in the two most recent 6-year periods, respectively. Supplemental indications are much more likely to indicate no prior chemotherapy or an adjuvant setting. Likewise, considering tumor stage, the majority of indications involved advanced/metastatic disease; this increased over time, with 89% of all indications in the most recent 6 years specifying this requirement for prior therapy (Table 1). This trend reflects the fact that many drugs now are first studied in patients with an advanced disease process and where effective standard therapies have been available and have been exhausted.
Figure 2.

Prior therapy. The proportion of initial (A) and supplemental (B) approvals listing no prior therapy (grey), prior chemotherapy (black), or prior surgery or radiotherapy (striped) for each time period is presented.
Designation of indications as palliative was more common early in oncology drug development history, but it has not occurred recently. Prophylactic indications have primarily occurred recently but remain rare. With regard to tumor characteristics, there has been a significant rise in protein and genetic specifications over time, reflecting the advancing science of molecular classifications and the availability of well qualified in vitro diagnostics; however, the majority of indications still have no such specification (Fig. 3). Specification of genotypic information is increasing most rapidly, with 27% of initial approvals specifying this in the most recent 6 years, compared with 8% in the 6 years before that and only 3% in the preceding decade. Molecular specification did not appear to be associated with slower or faster approval time (Table 2).
Figure 3.

Molecular specification. The proportion of initial approvals listing protein expression (black), a genetic parameter (striped), or no specification of molecular phenotype or genotype (grey) in the indication is presented. Abbreviation: Spec, specification.
Study Design
The number of trials for each indication has decreased since 1990, with 83% of initial indications in the most recent 6 years being based on a single trial (supplemental online Fig. 2). In contrast, trial design has improved. Trial size has consistently increased, with a median of 356 patients in the most recent 6 years, 303 patients in the 6 years prior to that, and 229 patients in the decade prior to that (supplemental online Table 2).
Furthermore, slightly more randomized controlled trials (RCTs) have been performed, with 67% of approvals (initial and supplemental) in the most recent 6 years involving an RCT (supplemental online Fig. 2), compared with 65% in the 6 years prior to that and 61% in the preceding decade. The proportion of indications with RCTs that used placebo were as follows: 4/31 (13%) in 1990–1999, 6/32 (19%) in 2000–2005, and 14/53 (26%) in 2006–2011. There was not a clear relationship between use of RCTs and approval time (Table 2), although the complexity of RCTs, including design and use of active control arms, likely confounds this analysis.
Another example of oncology drug approval occurring in less-than-ideal situations is the observation that most drugs are initially approved in advanced disease, palliative settings, and/or in later lines of treatment when cancer is biologically much more difficult to treat. Reasons for this trend likely include the need for larger studies, longer time to endpoints, and the requirement that a new drug be superior or not inferior to existing drugs.
The primary endpoints for studies supporting cancer indications usually involved time-to-event or response, while survival was less common and symptom palliation was rarely the endpoint of interest (Fig. 4). The use of time-to-event has increased over time, with 43% of recent approvals (2006–2011) using such an endpoint compared to 33% in the 6 years prior to that (2000–2005) and only 13% in the decade prior to that (1990–1999).
Figure 4.

Primary endpoints for indications. Primary endpoint categories of time to event (grey), response rate (striped), symptom based (white), and survival (black) are presented for time periods from 1990 through 2011. Abbreviation: RR, response rate.
Discussion
Evaluation of FDA approval trends over the history of effective anticancer therapy reveals a number of notable findings, some of which are expected and others somewhat surprising. Approvals cover a broad range of indications and involve agents with a wide variety of mechanisms of action. Advancing technology and discovery of novel classes of drugs appear to correlate with shifts in approval trends. Significant trends included more cell signaling targets, more advanced disease, more prior therapy in the case of initial approvals, larger trials, more RCTs (but fewer numbers of trials), faster approval times, and greater use of molecular specification.
A concerning observation in this evaluation is that oncology drug approvals occur largely in clinical situations that may not be suited to the optimal application of a drug. This raises the question of whether potentially valuable drugs are being overlooked or are failing to achieve initial approval. From a biological perspective, one might expect many more drugs to have activity in situations where tumor burden is low, where treatment resistance is low, and when combined with other complementary agents. Yet, approval patterns are just the opposite. The majority of initial oncology drug approvals involve trials testing single agents in advanced-disease settings. Even in supplemental approvals, single-agent studies dominate. There are only a handful of examples where a drug is approved in a combination, but not as single agent. Such trials are more complex and typically require an active control comparator. This is despite numerous examples in oncology where combination studies demonstrate superior efficacy over single agents, perhaps due to the multiple derangements that are now recognized to exist in any given malignancy [20]. Reasons for the high rate of single-agent approvals include technical and regulatory challenges of demonstrating safety and efficacy of a drug when combined with other active agents.
Another example of oncology drug approval occurring in less-than-ideal situations is the observation that most drugs are initially approved in advanced disease, palliative settings, and/or in later lines of treatment when cancer is biologically much more difficult to treat. Reasons for this trend likely include the need for larger studies, longer time to endpoints, and the requirement that a new drug be superior or not inferior to existing drugs. Strategies to reduce the chance of overlooking a valuable drug might include novel study designs, better predictive models, and perhaps changes in regulatory approach. “Window” studies evaluating a new drug early in the sequence of treatments can allow safe initial assessment of clinical activity in such settings [21]. Increased use of small well-designed randomized studies may enhance early recognition of agents or combinations [9, 22–26]. The use of preclinical models designed to differentiate activity in various clinical settings has had relatively poor correlation with clinical utility, although ongoing efforts to improve predictability of preclinical evaluations may prove beneficial [27–31]. Finally, regulatory paradigms could be shifted, perhaps lowering the threshold for a drug to be considered efficacious, focusing more on individuals rather than large heterogeneous populations when assessing efficacy, and, upon lowering threshold for initial drug approval, shift resources to postmarketing setting to ensure safety and efficacy as experience grows. These changes would reduce the costs and technical hurdles for initial approval and allow a broader spectrum of oncology drugs to be approved.
As our understanding of cancer biology and the human genome expand, one approach to improving cancer treatment efficiency is to develop predictive biomarkers that allow selection of a sensitive patient population. This topic has received much attention from drug developers, academic researchers, and regulators, including significant work to facilitate incorporation of predictive biomarkers [8, 14, 32–34]. Although the incorporation of molecular specification into the label is clearly increasing, it is surprising to find that less than 30% of cancer drug approvals in the past 6 years mentioned genetic or protein expression in the labeled indication. One limitation may be challenges in demonstrating clinical usefulness of companion diagnostics [35]. It is also interesting that molecular specification did not affect approval times, perhaps balancing greater complexity with improved definition of populations.
As recognition of the importance of well-designed clinical trials is increasing, the proportion of studies that are randomized and placebo-controlled is also increasing, as is the study size. However, a significant fraction of approvals continue to be based on nonrandomized studies. Potential drivers for this include FDA initiatives of accelerated approval [15], which accounted for a quarter of initial oncology drug approvals over the past decade, and the Orphan Drug Act [36, 37]. Such approvals appear to be robust, as Tsimberidou et al. evaluated 31 oncology drugs approved by single-arm studies and found that all but one retained marketing approval, and only one additional such drug was withdrawn since that report [38, 39]. Advancing further creative clinical trial design will continue to improve efficiency and accuracy of oncology drug development [9, 10, 23–26]. We also found a clear trend toward reduced approval time in more recent periods, possibly related to implementation of the Prescription Drug User Fee Act first enacted in 1992 and perhaps to greater interaction during the design and implementation of trials between sponsors and FDA.
Our evaluation focuses on oncology drug approvals in the U.S. and the role of the FDA, so it should be noted that oncology approvals in other regulatory settings might reveal different results [40–43].
See www.TheOncologist.com for supplemental material available online.
Supplementary Material
Author Contributions
Conception and design: Robert E. Martell, David Sermer, Kenneth Getz, Kenneth I. Kaitin
Collection and/or assembly of data: Robert E. Martell, David Sermer, Kenneth Getz, Kenneth I. Kaitin
Data analysis and interpretation: Robert E. Martell, David Sermer, Kenneth Getz, Kenneth I. Kaitin
Manuscript writing: Robert E. Martell, David Sermer, Kenneth Getz, Kenneth I. Kaitin
Final approval of manuscript: Robert E. Martell, David Sermer, Kenneth Getz, Kenneth I. Kaitin
Disclosures
The authors indicated no financial relationships.
References
- 1.Desdouits F, Delaporte L, Parnis S, et al. New York: Bionest Partners; 2007. Up or Out in Oncology? [Google Scholar]
- 2.DiMasi JA, Grabowski HG. Economics of new oncology drug development. J Clin Oncol. 2007;25:209–216. doi: 10.1200/JCO.2006.09.0803. [DOI] [PubMed] [Google Scholar]
- 3.Lou K, de Rond M. The “not invented here” myth. Nat Rev Drug Discov. 2006;5:451–452. doi: 10.1038/nrd2063. [DOI] [PubMed] [Google Scholar]
- 4.Gilbert J, Henske P, Singh A. Rebuilding big pharma's business model. In Vivo, The Business and Medicine Report. 2003;21:1–10. [Google Scholar]
- 5.DiMasi JA, Feldman L, Seckler A, Wilson A. Trends in risks associated with new drug development: Success rates for investigational drugs. Clin Pharmacol Ther. 2010;87:272–277. doi: 10.1038/clpt.2009.295. [DOI] [PubMed] [Google Scholar]
- 6.Alymani NA, Smith MD, Williams DJ, Petty RD. Predictive biomarkers for personalised anti-cancer drug use: Discovery to clinical implementation. Eur J Cancer. 2010;46:869–879. doi: 10.1016/j.ejca.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 7.de Bono JS, Ashworth A. Translating cancer research into targeted therapeutics. Nature. 2010;467:543–549. doi: 10.1038/nature09339. [DOI] [PubMed] [Google Scholar]
- 8.Taube SE, Clark GM, Dancey JE, et al. A perspective on challenges and issues in biomarker development and drug and biomarker codevelopment. J Natl Cancer Inst. 2009;101:1453–1463. doi: 10.1093/jnci/djp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ratain MJ, Sargent DJ. Optimising the design of phase II oncology trials: The importance of randomisation. Eur J Cancer. 2009;45:275–280. doi: 10.1016/j.ejca.2008.10.029. [DOI] [PubMed] [Google Scholar]
- 10.Sylvester R, Van Glabbeke M, Collette L, et al. Statistical methodology of phase III cancer clinical trials: Advances and future perspectives. Eur J Cancer. 2002;38:S162–S168. doi: 10.1016/s0959-8049(01)00442-7. [DOI] [PubMed] [Google Scholar]
- 11.Sridhara R, Johnson JR, Justice R, et al. Review of oncology and hematology drug product approvals at the US Food and Drug Administration between July 2005 and December 2007. J Natl Cancer Inst. 2010;102:230–243. doi: 10.1093/jnci/djp515. [DOI] [PubMed] [Google Scholar]
- 12.Lanthier ML, Sridhara R, Johnson JR, et al. Accelerated approval and oncology drug development timelines. J Clin Oncol. 2010;28:e226–e227. doi: 10.1200/JCO.2009.26.2121. [DOI] [PubMed] [Google Scholar]
- 13.Johnson JR, Temple R. Food and Drug Administration requirements for approval of new anticancer drugs. Cancer Treat Rep. 1985;69:1155–1159. [PubMed] [Google Scholar]
- 14.Yap TA, Sandhu SK, Workman P, de Bono JS. Envisioning the future of early anticancer drug development. Nat Rev Cancer. 2010;10:514–523. doi: 10.1038/nrc2870. [DOI] [PubMed] [Google Scholar]
- 15.Johnson JR, Ning YM, Farrell A, et al. Accelerated approval of oncology products: The Food and Drug Administration experience. J Natl Cancer Inst. 2011;103:636–644. doi: 10.1093/jnci/djr062. [DOI] [PubMed] [Google Scholar]
- 16.Fine BM, Amler L. Predictive biomarkers in the development of oncology drugs: A therapeutic industry perspective. Clin Pharmacol Ther. 2009;85:535–538. doi: 10.1038/clpt.2009.9. [DOI] [PubMed] [Google Scholar]
- 17.Patterson SD, Cohen N, Karnoub M, et al. Prospective-retrospective biomarker analysis for regulatory consideration: White paper from the industry pharmacogenomics working group. Pharmacogenomics. 2011;12:939–951. doi: 10.2217/pgs.11.52. [DOI] [PubMed] [Google Scholar]
- 18.National Comprehensive Cancer Network. NCCN Guidelines. [Accessed June 21, 2010]. Available at http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site.
- 19.American Cancer Society. Cancer Facts & Figures 2012. Atlanta, GA: American Cancer Society; 2012. [Google Scholar]
- 20.Kwak EL, Clark JW, Chabner B. Targeted agents: The rules of combination. Clin Cancer Res. 2007;13:5232–5237. doi: 10.1158/1078-0432.CCR-07-1385. [DOI] [PubMed] [Google Scholar]
- 21.Glimelius B, Lahn M. Window-of-opportunity trials to evaluate clinical activity of new molecular entities in oncology. Ann Oncol. 2011;22:1717–1725. doi: 10.1093/annonc/mdq622. [DOI] [PubMed] [Google Scholar]
- 22.Sharma MR, Stadler WM, Ratain MJ. Randomized phase II trials: A long-term investment with promising returns. J Natl Cancer Inst. 2011;103:1093–1100. doi: 10.1093/jnci/djr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suman VJ, Dueck A, Sargent DJ. Clinical trials of novel and targeted therapies: Endpoints, trial design, and analysis. Cancer Invest. 2008;26:439–444. doi: 10.1080/07357900801971057. [DOI] [PubMed] [Google Scholar]
- 24.Thall PF. A review of phase 2–3 clinical trial designs. Lifetime Data Anal. 2008;14:37–53. doi: 10.1007/s10985-007-9049-x. [DOI] [PubMed] [Google Scholar]
- 25.LoRusso PM, Anderson AB, Boerner SA, Averbuch SD. Making the investigational oncology pipeline more efficient and effective: Are we headed in the right direction? Clin Cancer Res. 2010;16:5956–5962. doi: 10.1158/1078-0432.CCR-10-1279. [DOI] [PubMed] [Google Scholar]
- 26.Chabner BA. Early accelerated approval for highly targeted cancer drugs. N Engl J Med. 2011;364:1087–1089. doi: 10.1056/NEJMp1100548. [DOI] [PubMed] [Google Scholar]
- 27.Cook N, Jodrell DI, Tuveson DA. Predictive in vivo animal models and translation to clinical trials. Drug Discov Today. 2012;17:253–260. doi: 10.1016/j.drudis.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 28.Damia G, D'Incalci M. Contemporary pre-clinical development of anticancer agents: What are the optimal preclinical models? Eur J Cancer. 2009;45:2768–2781. doi: 10.1016/j.ejca.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 29.Johnson JI, Decker S, Zaharevitz D, et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. 2001;84:1424–1431. doi: 10.1054/bjoc.2001.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma SV, Haber DA, Settleman J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat Rev Cancer. 2010;10:241–253. doi: 10.1038/nrc2820. [DOI] [PubMed] [Google Scholar]
- 31.Suggitt M, Bibby MC. 50 years of preclinical anticancer drug screening: Empirical to target-driven approaches. Clin Cancer Res. 2005;11:971–981. [PubMed] [Google Scholar]
- 32.Khleif SN, Doroshow JH, Hait WN. AACR-FDA-NCI Cancer Biomarkers Collaborative consensus report: Advancing the use of biomarkers in cancer drug development. Clin Cancer Res. 2010;16:3299–3318. doi: 10.1158/1078-0432.CCR-10-0880. [DOI] [PubMed] [Google Scholar]
- 33.Dancey JE, Dobbin KK, Groshen S, et al. Guidelines for the development and incorporation of biomarker studies in early clinical trials of novel agents. Clin Cancer Res. 2010;16:1745–1755. doi: 10.1158/1078-0432.CCR-09-2167. [DOI] [PubMed] [Google Scholar]
- 34.Zwierzina H. Biomarkers in drug development. Ann Oncol. 2008;19:v33–v37. doi: 10.1093/annonc/mdn309. [DOI] [PubMed] [Google Scholar]
- 35.Kaitin K. Tufts Center for the Study of Drug Development Impact Report. Boston, MA: Tufts University; 2011. Lack of clinically useful diagnostics hinder growth in personalized medicines; p. 13. [Google Scholar]
- 36.Kesselheim AS, Myers JA, Avorn J. Characteristics of clinical trials to support approval of orphan vs nonorphan drugs for cancer. JAMA. 2011;305:2320–2326. doi: 10.1001/jama.2011.769. [DOI] [PubMed] [Google Scholar]
- 37.Hirschfeld S, Pazdur R. Oncology drug development: United States Food and Drug Administration perspective. Crit Rev Oncol Hematol. 2002;42:137–143. doi: 10.1016/s1040-8428(02)00008-2. [DOI] [PubMed] [Google Scholar]
- 38.Tsimberidou AM, Braiteh F, Stewart DJ, Kurzrock R. Ultimate fate of oncology drugs approved by the us food and drug administration without a randomized Trial. J Clin Oncol. 2009;27:6243–6250. doi: 10.1200/JCO.2009.23.6018. [DOI] [PubMed] [Google Scholar]
- 39.U.S. Food and Drug Administration. Pfizer voluntarily withdraws cancer treatment Mylotarg from U.S. market. [Accessed June 21, 2010]. Available at http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm216448.htm.
- 40.Trotta F, Leufkens HG, Schellens JH, et al. Evaluation of oncology drugs at the European Medicines Agency and US Food and Drug Administration: When differences have an impact on clinical practice. J Clin Oncol. 2011;29:2266–2272. doi: 10.1200/JCO.2010.34.1248. [DOI] [PubMed] [Google Scholar]
- 41.Brown JS, Bienz-Tadmor B, Lasagna L. Availability of anticancer drugs in the United States, Europe, and Japan from 1960 through 1991. Clin Pharmacol Ther. 1995;58:243–256. doi: 10.1016/0009-9236(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 42.Apolone G, Joppi R, Bertele V, Garattini S. Ten years of marketing approvals of anticancer drugs in Europe: Regulatory policy and guidance documents need to find a balance between different pressures. Br J Cancer. 2005;93:504–509. doi: 10.1038/sj.bjc.6602750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tafuri G, Leufkens HG, Laing R, Trotta F. Therapeutic indications in oncology: Emerging features and regulatory dynamics. Eur J Cancer. 2010;46:471–475. doi: 10.1016/j.ejca.2009.11.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.