

Abstract
Dendritic cells (DCs)1 competent to express the regulatory enzyme indoleamine 2,3 dioxygenase (IDO) in mice are a small but distinctive subset of DCs. Previously, we reported that high dose systemic CpG treatment to ligate TLR9 in vivo induced functional IDO exclusively in splenic CD19+ DCs, which stimulated resting Foxp3-lineage regulatory T cells (Tregs) to rapidly acquire potent suppressor activity. Here we show that IDO was induced in spleen and peripheral lymph nodes after CpG treatment in a dose dependent manner. Induced IDO suppressed local T cell responses to exogenous antigens and inhibited pro-inflammatory cytokine expression in response to TLR9 ligation. IDO induction did not occur in T cell deficient mice, or in mice with defective B7 or PD-1 co-stimulatory pathways. Consistent with these findings CTLA4 or PD-1/PD-L co-stimulatory blockade abrogated IDO induction, and prevented Treg activation via IDO following high dose CpG treatment. Consequently, CD4+CD25+ T cells uniformly expressed IL-17 shortly after TLR9 ligation. These data support the hypothesis that constitutive interactions from activated T cells or Tregs and IDO-competent DCs via concomitant CTLA4→B7 and PD-1→PD-L signals maintain the default potential to regulate T cell responsiveness via IDO. Acute disruption of these non-redundant interactions abrogated regulation via IDO, providing novel perspectives on the pro-inflammatory effects of co-stimulatory blockade therapies. Moreover, interactions between IDO-competent DCs and activated T cells in lymphoid tissues may attenuate pro-inflammatory responses to adjuvants such as TLR ligands.
Introduction
Cells of the innate immune system collate environmental cues from pathogen and damage associated molecular patterns (PAMPs and DAMPs, respectively) in tissues via pattern recognition receptors such as TLRs. Amongst innate immune cells, DCs are uniquely specialized to collate pattern recognition signals, and to acquire, process and present antigens to elicit helper/effector T cell responses via MHC-peptide/TCR interactions and concomitant co-stimulatory signals (B7/CD28). The pivotal role of DCs in controlling T cell responses is exemplified by tolerogenic DC subsets that actively suppress (regulate) T cell responses to antigens they present (1). Thus, in some physiologic settings DCs promote tolerogenic outcomes by presenting antigens to T cells in the context of negative co-stimulation via CTLA4 and PD-1 pathways (2). Rare DCs expressing the inducible enzyme IDO also suppress innate and adaptive immune responses that can overcome the T cell stimulatory properties of other DCs in local environments such as inflamed lymph nodes to create immune privilege (3). Thus, distinct DC subsets exhibit considerable functional heterogeneity and DCs with T cell immune stimulatory and inhibitory functions have been described.
Previously we reported that murine DCs competent to express IDO in physiologic settings were a rare DC population exhibiting attributes of both B cells (such as CD19) and DCs (4, 5). CD19+ DCs in inflamed lymph nodes draining local sites of tumor growth, and B220+ DCs in lymph nodes draining skin exposed to tumor promoters possessed potent and dominant T cell regulatory properties via IDO, due to their ability to activate and stabilize Treg suppression at these sites (6, 7). Splenic CD19+ DCs do not express IDO during homeostasis, but CD19+ DCs were the only DC subset to express functional IDO in mice following treatment with soluble CTLA4 (CTLA4-Ig) and CpGs (>50μg, i/v) that ligate B7 and TLR9, respectively (8, 9). Thus FACS-sorted CD19+ DCs from CpG-treated mice mediated potent IDO-dependent T cell suppression, while sorted CD19neg DCs stimulated robust T cell proliferation that was not enhanced in the presence of IDO inhibitors. Autocrine or paracrine signaling via IFN type 1 receptors (IFNAR) was essential for IDO induction in CD19+ DCs following B7 or TLR9 ligation (10). Here we show that high dose CpG treatment induced dominant regulatory responses via IDO in all peripheral lymphoid tissues, and describe the cellular and molecular factors required for this rapid response to in vivo TLR9 ligation.
Materials and Methods
Mice
All mice were bred in a specific pathogen-free facility and the local (GHSU) Institutional Animal Care and Use Committee approved all procedures involving mice. OT-1 (B6), A1 (CBA) and BM3 (CBA) TCR transgenic mice used as sources of responder T cells in suppression assays were described previously (11). Mice deficient in PD-1 and PD-[L1+L2] genes (12) and Foxp3-GFP knock-in mice (13) were described previously and colonies were maintained at GHSU. Mice with defective RAG-1, TCRβγ, and B7.1/2 genes were purchased from the Jackson Laboratory (Bar Harbor, ME). Except where indicated all mice had B6 backgrounds.
CpG Oligonucleotides
CpG-B (#1826, TCCATGACGTTCCTGACGTT), and CpG-C (#2395, TCGTCGTTTTCGGCGCGCGCCG) with fully phosporothioate backbones were a gift of Coley Pharmaceuticals (Ottawa, Canada). Mice were injected with CpGs (25μg or 100μg/mouse, i/v) as described (9, 14), and spleens were harvested 24 (gene-deficient mice) or 42 (mAb treated mice) hours after CpG treatment; untreated and isotype mAb pre-treated B6 mice were included as positive controls for T cell suppression in every experiment.
1-methyl-[D]-tryptophan
(D-1MT). D-1MT (clinical grade, a gift of Newlink Genetics Inc.) was prepared as a 20mM stock solution in 0.1M NaOH, adjusted to pH 7.4, and stored at −20°C protected from light. D-1MT was added to MLRs at a final concentration of 200μM (9, 14).
mAb blockade
mAbs specific for mouse TGFβ (R&D Systems #1835, clone 1D11), CTLA4 (BD Biosciences, #553719), PD-1 (CD273, BD Biosciences, #55189, clone J43,), PD-L1 (clone MIH7) and PD-L2 (clone TY25) were injected (i/p, 100μg or 500μg anti-TGFβ mAb) 6 hours before CpG treatment. PD-L1 and PD-L2 mAbs were described previously (6). Control mice were pre-treated with isotype-matched mAbs with irrelevant specificities.
MACS enrichment
MACS enrichment for splenic DCs (CD11c+) and Tregs (CD4+CD25+) was performed according to manufacturer’s instructions (Magnetic Cell Separation Technology (MACS) – Miltenyi Biotec Inc., Auburn, CA), except that cells were incubated with mAb-beads at room temperature, and DC-bead conjugates were passed over columns twice. As described previously, IDO-expressing CD19+ DCs (7, 9) and IDO-activated Tregs (6, 14) mediate dominant suppression ex vivo that overcomes the T cell stimulatory effects of a large excess of other DCs and activated T cells, respectively, such that it is not necessary to purify these cell types by flow cytometric methods to detect their T cell suppressive functions. Typically, MACS-separation resulted in ~25-fold enrichment of CD19+ DCs (~5% of selected cells) and selected CD4+CD25+ Tregs were >80% pure.
Analytical flow cytometry
Cells were stained with the following antibodies (CD4-FITC [553047], CD8a-PE [553033], CD11c-PE [557401], CD19-PerCP [551001], CD25-PE [553866], B220-FITC [553088], Thy1.1-PerCP [557266]; (all from Pharmingen-BD-Biosciences, San Jose, CA) and analyzed using a FACS-Calibur or FACS-Canto flow cytometer (Becton-Dickinson). Staining to detect intracellular IL-17 and IFNγ was performed on erythrocyte-free spleen cell suspensions by staining for surface antigens (CD4) then fixing, permeabilizing and staining to detect cytokines overnight at 4°C. as described (14).
Immunohistochemistry
Sections were stained for IDO using a rabbit polyclonal Ab generated using a synthetic IDO peptide as described (15).
Kynurenine detection
Kynurenine was detected by HPLC analysis of lymphoid tissue lysates as described (16). In brief, tissues were homogenized in PBS, supernatants were de-proteinated in TCA, and 50μl used for HPLC (C18 reverse phase column).
In vivo OT-2 T cell responses to OVA
MACS-enriched splenic OT-2 (Thy1.1+) T cells were injected (4–8×106/mouse, i/v) into B6 mice, and mice were treated with CpGs (100μg) one day later; some mice were also given oral D-1MT (2mg/ml) one day before CpG treatment as described (17). Mice were immunized with chicken ovalbumin (OVA, 100μg in CFA) one day after CpG treatment, and draining (inguinal) lymph nodes (dLNs) were collected 4 days after OVA immunization. OT-2 T cells (CD4+Thy1.1+) in dLNs were detected by FACS. LN cells were stimulated ex vivo with (PMA, ionomycin, brefeldin for 4 hours), stained (CD4, Thy1.1), fixed (cytofix/cytoperm, BD Bioscience), and then stained to detect intracellular IFNγ.
DC and Treg suppression assays
Suppressor activity of splenic DCs was assessed by culturing graded numbers of MACS-enriched CD11c+ DCs (0.6–5×104/well) with MACS enriched CD8+ responder T cells (5x104/well) from BM3 or OT-1 (plus OVA peptide, SIINFEKL) TCR transgenic mice with/without D-1MT (200μM final) as described (18). Suppressor activity of splenic Tregs was assessed by culturing graded numbers of MACS-enriched CD4+CD25+ Tregs (1.25–10×103/well) with responder A1 (H-Y-specific) CD4+ T cells (5x104/well), APCs (2x103/well, female CBA) and male (H-Y) peptide (10μM final) as described (6, 14). Thymidine incorporation by responder T cells (in triplicate) was assessed after 72hrs (96hrs. for BM3 responders). As described previously, DCs or Tregs from untreated control mice failed to suppress T cell proliferation, and suppression mediated by rare IDO-expressing CD19+ DCs or IDO-activated Tregs pre-dominated over the T cell stimulatory effects of other DCs or other T cells in cultures (7, 14). DCs (or Tregs) from CpG-treated B6 mice were included as positive controls for suppression in all experiments.
Statistical Analysis
Analyses of T cell proliferation data to detect suppression were performed using Student’s t-test to compare means (+/−1sd) from triplicate cultures within a group. P values of <0.1 were considered significant.
Results
Induced IDO blocks pro-inflammatory responses to CpGs
Previously, we reported that systemic treatment of mice with relatively high doses of TLR9 ligands (CpGs) induced functional IDO exclusively in splenic CD19+ DCs (9). When induced to express IDO, CD19+ DCs blocked IL-6 production by pDCs and activated splenic Tregs (14). Regulatory responses in this model were critically dependent on the dose of CpGs. Thus, selective IDO expression by peri-follicular CD19+ DCs was observed in mice treated with high CpG doses (>50μg), but no IDO+ cells were detected in spleens of mice treated with lower CpG doses (25μg, Fig. 1A). Consistent with these outcomes, the IDO metabolite kynurenine was detected in lysates of spleens from mice treated with high (100μg) but not low (25μg) CpG doses, (Fig. 1B, left panel). Similarly, kynurenine was detected in lysates of peripheral LNs of mice treated with high but not low CpG doses (Fig. 1B, right panel). Thus systemic TLR9 ligands induced IDO activity in peripheral lymphoid tissues, and IDO induction was critically dependent on the dose of CpGs administered. These data showed that IDO induction is finely tuned to the potency of TLR9 signals in all peripheral lymphoid tissues.
Figure 1. IDO co-induced by high dose CpG treatment blocks in vivo T cell responses.

B6 mice were injected (i/v) with CpGs and 24 hours later spleen and peripheral LNs were analyzed to detect IDO (A) and kynurenine (B). A. Spleen sections were stained to detect IDO. Original magnifications, x200 (top right, x1000). f, follicles. B. HPLC analyses to detect kynurenine in tissue lysates from spleen and pooled inguinal/brachial (ing/bra) LNs of untreated mice, and mice treated with 100μg or 25μg CpG. C. Numbers of OVA-specific (Thy1.1+CFSE+) OT-2 T cells in inguinal LNs (dLNs) of B6 mice 4 days after CFA/OVA immunization and pre-treatment with CpGs (100μg, i/v) and oral IDO inhibitor (D-1MT). The proportions of dLN OT-2 T cells that expressed intracellular IFNγ after re-stimulation are indicated (open bars). Data are representative of experiments performed two or more times.
To elucidate potential T cell regulatory effects of TLR9-induced IDO we evaluated local T cell responses to ovalbumin following immunization (OVA+CFA, s/c) of B6 mice harboring small cohorts of marked (Thy1.1+) OVA-specific CD4 T cells from OT-2 TCR transgenic mice. Absolute numbers of OT-2 T cells present in inguinal LNs draining the site of OVA/CFA immunization were consistently lower (~five-fold) in mice pre-treated with high doses (100μg) of CpGs (Fig. 1C). Moreover, the fraction of dLN OT-2 T cells that expressed intracellular IFNγ (after re-stimulation ex vivo) was substantially lower in mice pre-treated with high dose CpGs (open bars in Fig. 1C). However, local OT-2 responses were restored to levels seen in control (OVA immunized) mice when oral IDO inhibitor 1-methyl-[D]-tryptophan (D-1MT) was given before CpG pre-treatment. Thus IDO induced by CpG treatment dominantly regulated OT-2 clonal expansion and differentiation in local dLNs following OVA immunization.
IDO-competence of splenic DCs is T cell dependent
To assess requirements for TLR9-mediated induction of IDO in splenic DCs we measured the T cell stimulatory properties of DCs (MACS-enriched CD11c+) from CpG-treated mice lacking defined lymphocyte populations. Splenic DCs from CpG-treated RAG-1-KO mice, that lack T and B cells, stimulated robust proliferation of OT-1 T cells in the presence of OVA peptide, and proliferation was not enhanced when IDO inhibitor (D-1MT) was present during culture (Fig. 2A, closed and open bars, respectively). As expected, splenic DCs from control CpG-treated B6 mice stimulated weak OT-1 proliferation that was enhanced significantly by adding D-1MT, revealing that IDO suppressed DC T cell stimulatory functions by 80% (Fig. 2A). DCs from CpG-treated TCRβγ-KO mice, which have B cells but lack T cells, also failed to suppress ex vivo OT-1 T cell responses via IDO (Fig. 2B). In a previous study, we reported that IDO-competent DCs developed and functioned normally in spleens of B cell deficient (μMT-KO, JH-KO) mice despite their close relationship to B cells (5). Thus the ability of splenic DCs to express functional IDO in response to TLR9 ligands was abrogated when T cells were developmentally ablated, suggesting that IDO-competence in splenic DCs depended on interactions with T cells.
Figure 2. IDO-competence in DCs is T cell dependent.
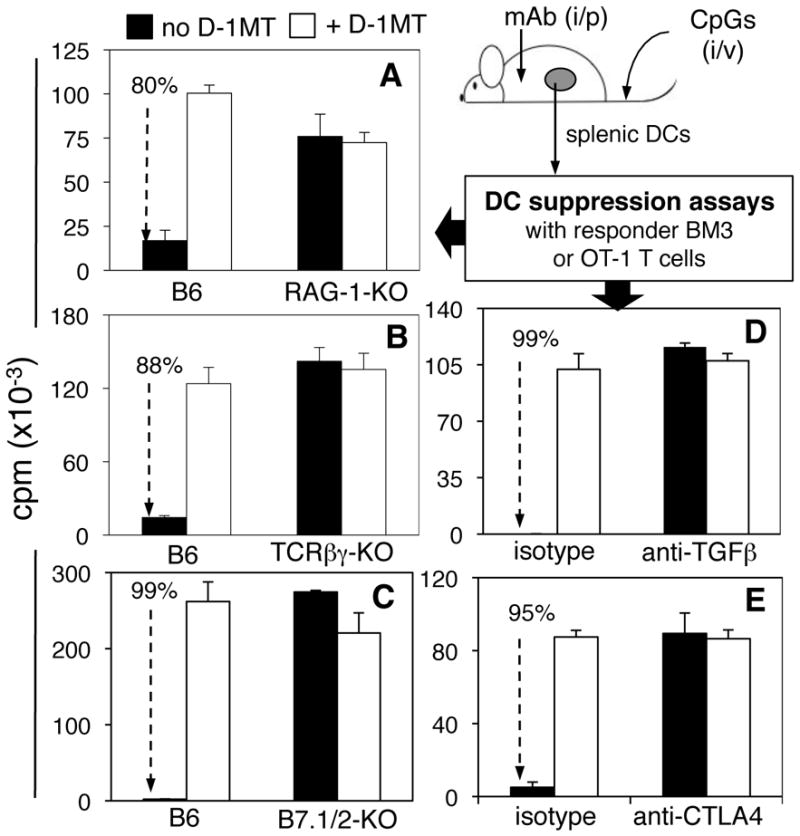
B6 and gene-deficient mice were treated with CpGs (100μg, i/v) and 24hrs later MACS-enriched splenic DCs (6–50 ×103/well) were cultured with responder T cells (5x104/well) from BM3 (A, B) or OT-1 +OVA peptide (C–E) TCR transgenic mice +/−IDO inhibitor (D-1MT). In some cases B6 mice were pre-treated with 500μg anti-TGFβ (D), 100μg anti-CTLA4 (E) mAbs or isotype-matched mAbs 6 hours before CpG treatment, and DCs were isolated 42hrs. later. Thymidine incorporation was assessed after 72hrs (A, B) or 96 (C–E) hrs. Data shown (25 x103 DCs/well) are the means of triplicate cultures and are representative of experiments performed two or more times with graded numbers of DCs in each experiment. Dotted arrows and percentages highlight significant IDO-dependent suppression mediated by DCs from B6 mice (A–C), and B6 mice treated with isotype-matched mAbs (D, E) before CpG treatment.
Next, we identified molecular signals that maintain IDO-competence in splenic DCs. Like T cell-deficient mice, mice with defective B7.1 and B7.2 (CD80, CD86) genes (B7.1/2-KO mice) yielded splenic DCs that stimulated robust OT-1 T cell responses after high dose CpG treatment, and D-1MT had no effect on elicited responses (Fig. 2C). Thus intact co-stimulatory pathways via B7.1 and B7.2 were essential for DCs to acquire competence to express IDO in response to TLR9 ligands.
Previous studies revealed that TGFβ (19) and cells expressing surface CTLA4 (11, 20) stimulated DCs to express IDO. To test if TGFβ and CTLA4 signals were required for IDO-competence to TLR9 ligands we used mAbs (i/p) to block TGFβ and CTLA4 signaling during responses to CpGs. Treatments with anti-TGFβ (500μg) and anti-CTLA4 (CD152, 100μg) mAbs 6 hours before CpG treatment yielded splenic DCs with potent T cell stimulatory properties that were not enhanced by IDO inhibitor (Figs, 2D, E, respectively). In each case, DCs from B6 mice treated with isotype-matched mAbs treatment mediated potent suppression via IDO due to TLR9 ligation. Thus non-redundant signals via TGFβ and co-stimulatory pathways were essential for regulatory responses via IDO in splenic DCs following TLR9 ligation. Moreover, these signals were required constitutively as acute ablation of either pathway abrogated regulatory responses via IDO.
IDO-competence in splenic DCs is PD-1 and PD-L dependent
PD-1 and its ligands PD-L1 and PD-L2 mediate negative co-stimulation that blocks T cell effector functions and promotes T cell death. To test if PD-1/PD-L interactions were required for IDO-competence in splenic DCs we evaluated the effects of PD-1/PD-L ablation on IDO induction in DCs after CpG treatment. Pre-treatment with anti-PD-1 blocking mAb (Fig. 3A) and PD-1 gene ablation (Fig. 3B) eliminated T cell regulatory responses via IDO in DCs. Likewise, pre-treatment with mixtures (50:50) of both anti-PD-L1 and anti-PD-L2 mAbs (Fig. 3C) and genetic ablation of both PD-L1 and PD-L2 genes (PD-[L1+L2]-KO mice, Fig 3D) abrogated T cell regulatory responses via IDO to CpG treatment. Thus, intact PD-1 and PD-L1/2 pathways were essential for DCs to acquire T cell suppressor activity via IDO after TLR9 ligation. Analyses of splenic DCs in mice with defective PD-1 and PD-L1/PD-L2 genes revealed no significant differences in the relative proportions of DCs amongst splenocytes and CD19+ DCs amongst total splenic DCs (Fig. S1), suggesting that the profound functional changes in DC responses to TLR9 ligation in these mice were not caused by failure of CD19+ DCs to develop per se. In accordance with previous reports (21), flow cytometric analyses of PD-1 expression by splenic CD4+ T cells and of PD-L1 and PD-L2 expression by splenic DCs revealed that ~50% of resting splenic Tregs and CD4+ T cells expressed PD-1 in untreated mice, while almost all DCs expressed PD-L1, and no DCs expressed PD-L2 (Fig. S2). Moreover, PD-1, PD-L1 and PD-L2 expression patterns on splenic CD4+ cells and DCs did not change substantially 24 hours after CpG treatment (data not shown).
Figure 3. PD-1/PD-L interactions mediate IDO-competence in DCs.
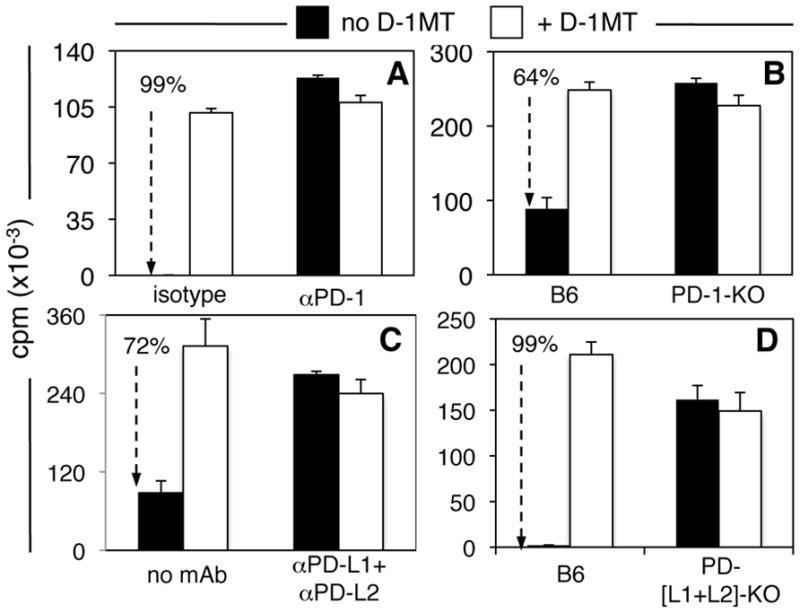
A, C. Mice (B6) were pre-treated with 100μg (i/p) anti-PD-1 mAbs (A) and anti-PD-L1/PD-L2 mAbs (C) or isotype-matched mAbs 6hrs before CpG treatment (100μg, i/v). 42hrs after CpG treatment MACS-enriched splenic DCs (2.5x103/well) were cultured with OT-1 T cells, OVA peptide, +/−D-1MT. B, D. PD-1-KO (B) and PD-[L1+L2]-KO (D) were treated with CpGs and 24hrs later splenic DCs were placed in suppression assays. Thymidine incorporation was evaluated after 72 hours. Dotted arrows with percentages highlight IDO-dependent suppression. Data shown are the means of triplicate cultures and are representative of experiments performed on two or more occasions with graded numbers of DCs (6–50×103/well) in each experiment.
Loss of IDO-dependent T cell suppressive functions may arise due to transcriptional or post-translational blockade of gene expression or IDO enzyme activity, respectively. We stained spleen sections to assess if DCs expressed IDO protein after CpG treatment. No IDO-expressing DCs were detected in spleens from RAG-1-KO mice (Fig. 4B), and from B6 mice pre-treated with anti-PD-1 mAb (Fig. 4C), and mixtures (50:50) of anti-PD-L1 and anti-PD-L2 mAbs (Fig. 4D). As expected pre-treatment with isotype-matched mAbs had no effect on CpG-induced IDO expression by DCs in peri-follicular regions (Fig. 4A). These outcomes were consistent with outcomes from DC suppression assays (Figs. 2, 3), indicating that signals required for IDO-competence in splenic DCs regulated IDO gene expression.
Figure 4. TLR9-mediated IDO induction is PD-1/PD-L dependent.
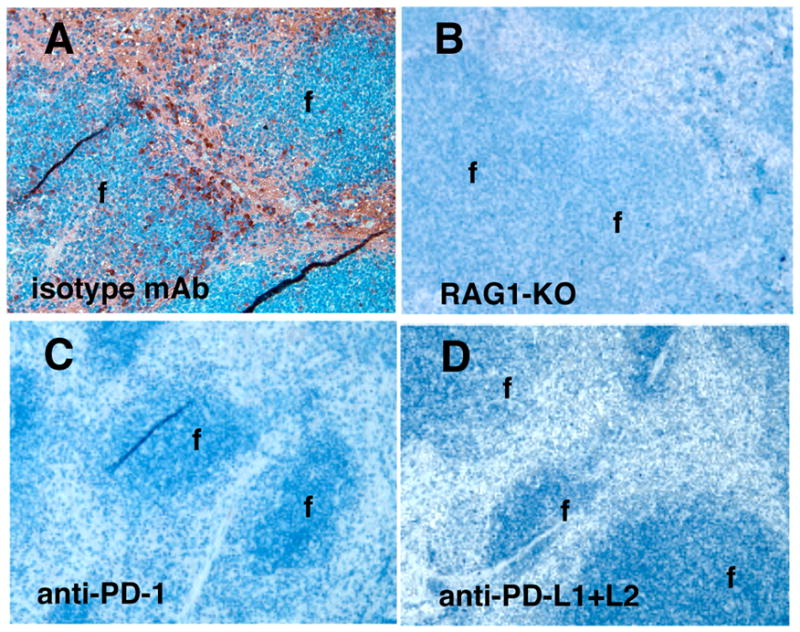
A, C, D. B6 mice were pre-treated with mAbs (100μg, i/p) 6hrs. before CpG treatment then spleens were removed 42hrs. later to detect IDO. B. RAG-1-KO mice were treated with CpGs and spleens were removed 24hrs. later then stained to detect IDO; original magnifications, x200 (f, follicles).
Treg activation after TLR9 ligation depends on co-stimulation to induce IDO in DCs
Previously we reported that splenic Tregs activated rapidly (within 12–18hrs) after high dose CpG treatment due to IDO induction in CD19+ DCs (14). To test if TLR9-induced Treg activation was abrogated by interventions that prevented IDO induction we assessed the functional status of splenic Tregs using an assay that detects suppressor activity of Tregs after activation in vivo (6, 14). Graded numbers of MACS-enriched Tregs from CpG-treated mice were added to cultures containing responder T cells from A1 TCR transgenic mice (CD4, male H-Y-specific, H-2Ek restricted), stimulatory APCs (CBA female) and cognate (male H-Y) peptide; in this assay, Tregs must be activated in vivo as Tregs do not activate during culture (6, 14). Consistent with our previous findings (14), induced Treg suppressor activity was potent because 1250 Tregs from CpG-treated B6 mice suppressed A1 T cell proliferation completely (Treg:Teff = 1:40, Fig. 5A, closed symbols). In addition, Treg suppression was abrogated completely in parallel cultures containing a cocktail of anti-PD-1, anti-PD-L1 and anti-PD-L2 blocking mAbs (data not shown); PD-1/PD-L-dependent suppression ex vivo is a hallmark feature of IDO-activated Tregs from tumor draining lymph nodes, and from spleen after TLR9 ligation (6, 14). Tregs from untreated B6 mice did not mediate suppression ex vivo (data not shown), confirming that suppressive functions were acquired in vivo following CpG treatment, not during culture.
Figure 5. Treg activation after TLR9 ligation is dependent on co-stimulation.
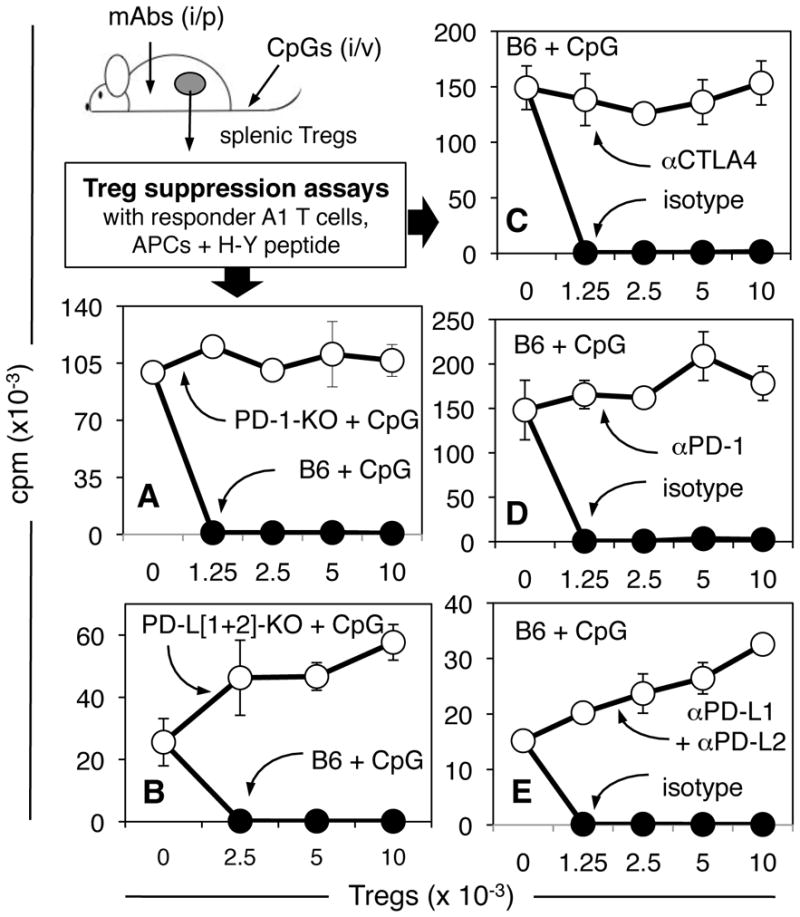
A–C. B6 mice were pre-treated with anti-CTLA4, anti-PD-1 mAbs, or anti-PD-L1 + anti-PD-L2 (50:50) or isotype-matched mAbs (100μg), and treated with CpGs 6 hours later. 42 hours after CpG treatment graded numbers of MACS-enriched (CD4+CD25+) splenic Tregs were added to cultures containing H-Y specific CD4 responder T cells (5x104), APCs (female CBA) and H-Y peptide. Thymidine incorporation was assessed after 72 hours. D, E. PD-1-KO, PD-[L1+L2]-KO and B6 control mice were treated with CpGs and 24 hours later splenic Tregs were assayed for suppressor activity. Data are the means of triplicate cultures (+/−1sd) and were replicated two or more times.
In striking contrast to the potent suppressor functions of Tregs from CpG-treated B6 mice, Tregs from CpG-treated PD-1-KO mice did not suppress A1 T cell proliferation, even when eight times more (104) Tregs were added to cultures (Fig. 5A, open symbols). Tregs from CpG-treated PD-[L1+L2]-KO mice also failed to mediate suppression (Figs. 5B). Moreover, acute blockade of PD-1 and PD-L1+L2 interactions by injecting mAbs just before CpG treatment prevented splenic Tregs from acquiring potent suppressor activity (Figs. 5D, 5E, respectively). Likewise, Tregs from mice pre-treated with anti-CTLA4 mAb before CpG-treatment did not mediate suppression (Fig. 5C). In each case, pre-treatment with isotype-matched mAbs had no effect on activation of Treg suppression following TLR9 ligation. These data support the hypothesis that molecular pathways required for IDO induction in DCs are also essential for Treg activation after TLR9 ligation.
Co-stimulatory blockade to prevent IDO induction allows CpGs to activate CD4 T cells
TLR9 ligands promote rapid production of pro-inflammatory cytokines by innate immune cells and activated effector T cells. Previously, we reported that IDO co-induced after high dose CpG treatment blocked TLR9-induced IL-6 expression by pDCs and prevented re-programming of Foxp3-lineage Tregs to express the pro-inflammatory cytokines IL-17, IFNγ, IL-2 and TNFα (14). To test the hypothesis that co-stimulatory signals block pro-inflammatory cytokine expression in CD4 T cells we assessed expression of intracellular IL-17 in gated splenic CD4+ T cells following high dose CpG treatment. As reported previously, high dose CpG treatment did not stimulate CD4 T cells to express IL-17 in IDO-sufficient B6 mice (Fig. 6A), whereas rapid and uniform IL-17 expression was observed in CD4+CD25+ T cells in CpG-treated IDO1-deficient (IDO1-KO) mice (Fig. 6B). Pre-treating B6 mice with anti-PD-1 (Fig. 6C) or mixtures of anti-PD-L1+anti-PD-L2 mAbs (Fig. 6D) also led to rapid and uniform IL-17 expression by CD4+CD25+ T cells. Thus PD-1/PD-L interactions were essential to prevent CD4+CD25+ T cells expressing IL-17 after high dose CpG treatment. Pre-treating Foxp3-GFP knock-in mice with anti-CTLA4 mAb resulted in uniform IL-17 and IFNγ expression by Foxp3+ (GFP+) CD4+ Tregs after high dose CpG treatment (Fig. 6E, F, respectively). Pre-treatment with isotype-matched irrelevant mAbs did not result in CpG-induced IL-17 and IFNγ expression by Tregs. Thus co-stimulatory signaling via CTLA4/B7 and PD-1/PD-L pathways prevented pro-inflammatory cytokine expression by CD4+CD25+ T cells and Tregs in IDO-sufficient mice.
Figure 6. Co-stimulatory blockade allows cytokine expression by activated CD4 cells and Tregs following high dose CpG treatment.

A–D. B6 mice were treated with anti-PD-1 (C), anti-PD-L1+PD-L2 (D) or isotype-matched (A) mAbs (100μg/mAb, i/p) 6hrs before high dose CpG treatment. Control IDO1-KO mice were also treated with CpGs (B). After 42hrs. splenocytes were stained and analyzed to detect intracellular IL-17. Dot plots show gated CD4 splenocytes; percentages are the proportions of CD4+CD25+ cells expressing IL-17. E, F. Foxp3-GFP mice were treated with anti-CTLA4 or isotype-matched mAbs (100μg, i/p) and treated with CpGs as above. Splenocytes were analyzed to detect intracellular IL-17 and IFNγ. Histograms show cytokine staining profiles for gated GFP+CD4+ cells. Data are representative of experiments performed on two or more occasions.
Discussion
Previously, we reported that systemic administration of relatively high doses of CpGs induced splenic CD19+ DCs to express functional IDO; consequently, CD19+ DCs acquired potent T cell regulatory properties, including the ability to activate Tregs rapidly (5, 9, 14). Here we show that high dose CpG treatment induced IDO activity in peripheral lymphoid tissues, which suppressed T cell responses to exogenous antigens. Moreover, IDO induction in DCs was dependent on the presence of T cells and intact co-stimulatory pathways, and co-stimulatory blockade enhanced pro-inflammatory responses to high dose CpG treatment by activated CD4 T cells and Tregs. These findings reveal that the competence of DCs to express IDO and acquire regulatory phenotypes is critically dependent on the potency of TLR9 ligation and on molecular signals from T cells and Tregs.
Splenic DCs from T cell deficient mice were not competent to acquire regulatory functions via IDO. In contrast, IDO-competent DCs were present and responded normally to high dose CpG treatment in spleens of B cell deficient mice, even though CD19+ DCs and B cells share multiple attributes and have common B cell lineage etiologies (5). The requirement for T cells to establish IDO competence in DCs suggests that IDO-mediated regulation may have evolved primarily to regulate T cell responses.
Few T cells express both PD-1 and surface CTLA4, especially during homeostasis (21, 22). Hence few T cell subsets are capable of interacting constitutively with CD19+ DCs to induce and stabilize IDO-competence, though the possibility that different T cells provide signals via PD-1 and CTLA4 in physiologic settings cannot be excluded. During homeostasis T cells are one of the few cell types to express PD-1, while all T cell subsets and many other cell types express high levels of PD-L1, and no cells express PD-L2 (21). About 50% of splenic T cells and Tregs expressed PD-1, and almost all splenic DCs – including CD19+ DCs – expressed PD-L1 (Fig. S2). Thus, our findings support the hypothesis that interactions between PD-1+ T cells or Tregs and PD-L1+ CD19+ DCs are critical to maintain IDO-competence in DCs. This model is consistent with studies showing that PD-L1 controls the development and regulatory functions of peripheral Tregs (12), and that IL-10 and negative co-stimulatory B7 ligands (B7-H1/4) maintain regulatory phenotypes of human Tregs (23) though a role for IDO in stabilizing these regulatory phenotypes was not described. Inflammatory cytokines such as interferons, IL-2, TNFα and IL-10 released after TLR9 ligation expand the range of cell types expressing PD-1, PD-L1 and PD-L2 (21). Thus opportunities for different cell types to interact via the PD-1/PD-L pathway to stimulate IDO expression in DCs are increased in settings of inflammation.
Naïve T cells do not express CTLA4, but activated T cells do express CTLA4, though CTLA4 is intracellular and is not present at the (outer) cell surface. In contrast, many resting Tregs express surface CTLA4. Interactions between IDO-competent DCs and cells expressing surface CTLA4, including cloned CD4 T cells with regulatory functions, induced DCs to express functional IDO in culture (11, 20). Thus ‘reverse’ signaling via the CTLA4→B7 pathway was sufficient to induce DCs to express IDO ex vivo. However, IDO enzyme activity is not detected in lymphoid tissues during homeostasis, suggesting that constitutive interactions between activated T cells and/or Tregs are insufficient to trigger IDO activity in physiologic settings, and that additional signals are needed to attain signaling thresholds required for functional IDO expression in CD19+ DCs. Thus our findings support the hypothesis that resting Tregs and/or activated T cells expressing PD-1 and surface CTLA4 mediate constitutive, sub-threshold signaling that maintains the default tolerogenic potential of CD19+ DCs via IDO.
IFNα produced after treatments with CTLA4-Ig and high dose CpG to ligate B7 and TLR9 is one way that signaling thresholds may be breached to promote regulatory outcomes via IDO in physiologic settings (10). Autocrine IFNα signaling (via IFNAR/STAT1) was obligatory to induce CD19+ DCs to express IDO after in vivo B7 or TLR9 ligation while IFNγ signaling was not required for these regulatory responses (8, 9). Additional requirements for concomitant, non-redundant signals from T cells via co-stimulatory pathways reveal that signaling via IFN type I receptors (IFNAR) alone is not sufficient for IDO-competence in CD19+ DCs. Belladonna and colleagues reported that the IDO-mediated tolerogenic phenotype of splenic CD8α+ DCs was also dependent on autocrine TGFβ signaling (19); our finding that anti-TGFβ mAb treatment blocked TLR9-mediated IDO induction in splenic DCs is consistent with this study since CD19+ DCs co-express CD8α (8). Thus, IDO-competence in CD19+ DCs is dependent on constitutive, non-redundant interactions with T cells expressing CTLA4 and PD-1, as well as TGFβ. It is not clear if acute or developmental ablation of upstream signals that maintain IDO-competence of CD19+ DCs attenuates cell autonomous responses to TLR9 ligands or to IFNα produced by CD19+ DCs and pDCs following TLR9 ligation. This point notwithstanding, our findings support the hypothesis that the tolerogenic potential of splenic DCs via IDO is finely attuned to constitutive environmental signals from T cells and regulatory cytokines. Acute loss of signals that maintain the IDO-competent functional status of CD19+ DCs led to pro-inflammatory responses to TLR9 ligands by T cells and Tregs.
DCs expressing IDO are not the only regulatory mechanism that stabilizes the default regulatory status of Tregs during homeostasis as genetic IDO gene ablation in mice did not lead to spontaneous autoimmunity (15). However developmental ablation of IDO1 and acute pharmacologic IDO inhibition led to enhanced innate and T cell immunity in local settings of tumor growth, skin inflammation that promotes tumor progression, chronic infections, autoimmunity and pregnancy (7, 24–26). Thus findings from the current study provide novel insights into a natural immune regulatory mechanism that contributes to a wide range of inflammatory syndromes of clinical significance. Based on these findings we hypothesize that experimental immunotherapies targeting regulatory CTLA4 and PD-1/PD-L pathways may interfere with IDO-dependent processes that promote the tolerogenic potential of DCs and Tregs in settings of local inflammation. Moreover, the bifunctional potential of resting Tregs complicates their therapeutic use (26). Thus Treg ablation early in mucosal herpes simplex virus infection unexpectedly led to accelerated fatal infection in mice (27), and Treg adoptive transfer into T cell deficient mice accelerated, rather than slowed skin allograft rejection (28). Also reprogrammed Tregs – not naïve CD4+ T cells - were the obligate source of help to prime CD8+ T cells in naïve mice after OVA vaccination (29). In this latter model, CpGs were essential to induce Treg re-programming in lymph nodes draining local sites of vaccine administration, suggesting that this dosing and delivery regimen avoided stimulating the IDO-dependent regulatory responses we observed in the spleen after high dose, systemic CpG treatment. These examples emphasize the critical importance of identifying mechanisms that control how the bifunctional potential of Tregs manifests in physiologic settings of inflammation relevant to disease syndromes and immunotherapy.
In summary, our findings highlight the tight mechanistic coupling between CD19+ DCs and activated T cells and Tregs needed to maintain tolerogenic potential via IDO in physiologic settings. Though both cell types are relatively rare in physiologic settings, induction of IDO-dependent regulatory phenotypes resulted in dominant local suppression of T cell responses to exogenous antigens in peripheral lymphoid tissues draining local sites of immunization. Thus IDO-mediated regulatory responses to high dose CpG treatment overcame the well known pro-inflammatory effects of CpGs that justify their use as immune adjuvants. Numbers of CD19+ DCs may increase at sites of chronic inflammation associated with potent IDO-dependent suppression and Treg activation such as lymph nodes draining sites of tumor growth and topical exposure to tumor promoters (4, 7). However in our model of acute IDO induction in spleen following high dose CpG treatment numbers of CD19+ DCs and Tregs did not change substantially suggesting that collaborations between these rare cell types dominantly regulates potential pro-inflammatory responses by other DCs and T cell subsets following TLR9 ligation.
Supplementary Material
Acknowledgments
This study was supported by NIH grant support to ALM (AI63402, AI75165), AHS (P01 AI 56299) BRB (AI056299, AI34495, CA72669, HL56067) & DHM (CA096651 and CA103320).
This paper is dedicated to the memory of Doris McCool who managed the mouse colonies that were the source of most mice used in this study. We thank Liesl Desevilla and Diane Addis for expert technical assistance. We also thank Coley Pharmaceuticals and NewLink Genetics Inc. for the generous gifts of CpG oligodeoxynucleotides and D-1MT, respectively.
Footnotes
Non-standard abbreviations used: DCs, dendritic cells; indoleamine 2,3 dioxygenase, (IDO); Tregs, regulatory (Foxp3-lineage) CD4 T cells;; Programmed Death-1/ligand, PD-1/PD-L; D-1MT, 1-methyl-[D]-tryptophan; GCN2, general control-non-derepressible-2;
References
- 1.Cobbold SP, Adams E, Nolan KF, Regateiro FS, Waldmann H. Connecting the mechanisms of T-cell regulation: dendritic cells as the missing link. Immunol Rev. 2010;236:203–218. doi: 10.1111/j.1600-065X.2010.00913.x. [DOI] [PubMed] [Google Scholar]
- 2.Sharpe AH. Mechanisms of costimulation. Immunol Rev. 2009;229:5–11. doi: 10.1111/j.1600-065X.2009.00784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol. 2008;8:74–80. doi: 10.1038/nri2233. [DOI] [PubMed] [Google Scholar]
- 4.Munn DH, Sharma MD, Hou D, Baban B, Lee J, Antonia SJ, Messina JL, Chandler P, Koni PA, Mellor AL. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. 2004;114:280–290. doi: 10.1172/JCI21583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson BA, 3rd, Kahler DJ, Baban B, Chandler PR, Kang B, Shimoda M, Koni PA, Pihkala J, Vilagos B, Busslinger M, Munn DH, Mellor AL. B-lymphoid cells with attributes of dendritic cells regulate T cells via indoleamine 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2010;107:10644–10648. doi: 10.1073/pnas.0914347107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, Munn DH. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117:2570–2582. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller AJ, Sharma MD, Chandler PR, DuHadaway JB, Everhart ME, Johnson BA, Kahler DJ, Pihkala J, Soler AP, Munn DH, Prendergast GC, Mellor AL. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc Natl Acad Sci U S A. 2008;105:17073–17078. doi: 10.1073/pnas.0806173105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baban B, Hansen AM, Chandler PR, Manlapat A, Bingaman A, Kahler DJ, Munn DH, Mellor AL. A minor population of splenic dendritic cells expressing CD19 mediates IDO-dependent T cell suppression via type I IFN signaling following B7 ligation. Int Immunol. 2005;17:909–919. doi: 10.1093/intimm/dxh271. [DOI] [PubMed] [Google Scholar]
- 9.Mellor AL, Baban B, Chandler PR, Manlapat A, Kahler DJ, Munn DH. Cutting Edge: CpG Oligonucleotides Induce Splenic CD19+ Dendritic Cells to Acquire Potent Indoleamine 2,3-Dioxygenase-Dependent T Cell Regulatory Functions via IFN Type 1 Signaling. J Immunol. 2005;175:5601–5605. doi: 10.4049/jimmunol.175.9.5601. [DOI] [PubMed] [Google Scholar]
- 10.Manlapat AM, Kahler DJ, Chandler PR, Munn DH, Mellor AL. Cell autonomous control of interferon type I expression by indoleamine 2,3-dioxygenase in regulatory CD19+ dendritic cells. Eur J Immunol. 2007;37:1064–1071. doi: 10.1002/eji.200636690. [DOI] [PubMed] [Google Scholar]
- 11.Mellor AL, Chandler PR, Baban B, Hansen AM, Marshall B, Pihkala J, Waldmann H, Cobbold SP, Adams E, Munn DH. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int Immunol. 2004;16:1391–1401. doi: 10.1093/intimm/dxh140. [DOI] [PubMed] [Google Scholar]
- 12.Francisco LM, V, Salinas H, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 14.Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, Mellor AL. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol. 2009;183:2475–2483. doi: 10.4049/jimmunol.0900986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baban B, Chandler P, McCool D, Marshall B, Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase expression is restricted to fetal trophoblast giant cells during murine gestation and is maternal genome specific. J Reprod Immunol. 2004;61:67–77. doi: 10.1016/j.jri.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Laich A, Neurauter G, Widner B, Fuchs D. More rapid method for simultaneous measurement of tryptophan and kynurenine by HPLC. Clin Chem. 2002;48:579–581. [PubMed] [Google Scholar]
- 17.Hou D, Muller AJ, Sharma M, Mellor AL, Prendergast GC, Munn DH. Stereoisomers of 1-methyl-tryptophan as pharmacologic inhibitors of indoleamine 2,3- dioxygenase. Cancer Research. 2007;67:792–801. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- 18.Mellor AL, Baban B, Chandler P, Marshall B, Jhaver K, Hansen A, Koni PA, Iwashima M, Munn DH. Cutting edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J Immunol. 2003;171:1652–1655. doi: 10.4049/jimmunol.171.4.1652. [DOI] [PubMed] [Google Scholar]
- 19.Belladonna ML, Volpi C, Bianchi R, Vacca C, Orabona C, Pallotta MT, Boon L, Gizzi S, Fioretti MC, Grohmann U, Puccetti P. Cutting edge: Autocrine TGF-beta sustains default tolerogenesis by IDO-competent dendritic cells. J Immunol. 2008;181:5194–5198. doi: 10.4049/jimmunol.181.8.5194. [DOI] [PubMed] [Google Scholar]
- 20.Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, Belladonna ML, Fioretti MC, Alegre ML, Puccetti P. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4:1206–1212. doi: 10.1038/ni1003. [DOI] [PubMed] [Google Scholar]
- 21.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yi H, Zhen Y, Jiang L, Zheng J, Zhao Y. The phenotypic characterization of naturally occurring regulatory CD4+CD25+ T cells. Cell Mol Immunol. 2006;3:189–195. [PubMed] [Google Scholar]
- 23.Kryczek I, Wei S, Zou L, Zhu G, Mottram P, Xu H, Chen L, Zou W. Cutting edge: induction of B7-H4 on APCs through IL-10: novel suppressive mode for regulatory T cells. J Immunol. 2006;177:40–44. doi: 10.4049/jimmunol.177.1.40. [DOI] [PubMed] [Google Scholar]
- 24.Johnson BA, 3rd, Baban B, Mellor AL. Targeting the immunoregulatory indoleamine 2,3 dioxygenase pathway in immunotherapy. Immunotherapy. 2009;1:645–661. doi: 10.2217/IMT.09.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makala LH, Baban B, Lemos H, El-Awady AR, Chandler PR, Hou DY, Munn DH, Mellor AL. Leishmania major attenuates host immunity by stimulating local indoleamine 2,3-dioxygenase expression. J Infect Dis. 2011;203:715–725. doi: 10.1093/infdis/jiq095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mellor AL, Munn DH. Physiologic control of the functional status of foxp3+ regulatory T cells. J Immunol. 2011;186:4535–4540. doi: 10.4049/jimmunol.1002937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lund JM, Hsing L, Pham TT, Rudensky AY. Coordination of early protective immunity to viral infection by regulatory T cells. Science. 2008;320:1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vokaer B, Van Rompaey N, Lemaitre PH, Lhomme F, Kubjak C, Benghiat FS, Iwakura Y, Petein M, Field KA, Goldman M, Le Moine A, Charbonnier LM. Critical role of regulatory T cells in Th17-mediated minor antigen-disparate rejection. J Immunol. 2010;185:3417–3425. doi: 10.4049/jimmunol.0903961. [DOI] [PubMed] [Google Scholar]
- 29.Sharma MD, Hou DY, Baban B, Koni PA, He Y, Chandler PR, Blazar BR, Mellor AL, Munn DH. Reprogrammed foxp3(+) regulatory T cells provide essential help to support cross-presentation and CD8(+) T cell priming in naive mice. Immunity. 2010;33:942–954. doi: 10.1016/j.immuni.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.