

Abstract
Control of blood vessel tone is central to vascular homeostasis. Here, we show that metabolism of tryptophan to kynurenine by indoleamine 2,3-dioxygenase (IDO) expressed in endothelial cells contributes to arterial vessel relaxation and the control of blood pressure. Infection of mice with malarial parasites (Plasmodium berghei), and experimental induction of endotoxemia, caused endothelial expression of IDO, resulting in decreased plasma tryptophan, increased kynurenine, and hypotension. Pharmacological inhibition of IDO increased blood pressure in systemically inflamed mice, but not in mice deficient for IDO or interferon-γ, which is required for IDO induction. Tryptophan dilated pre-constricted porcine coronary arteries only if active IDO and an intact endothelium were both present. Kynurenine dose-dependently decreased blood pressure in spontaneously hypertensive rats, inhibited contraction of arteries, and relaxed pre-constricted rings endothelium-independently. Arterial relaxation by kynurenine was mediated by activation of the adenylate and soluble guanylate cyclase pathways.
Systemic inflammation such as sepsis is characterized by arterial hypotension and systemic inflammatory response syndrome1. The associated increase in circulating pro-inflammatory cytokines, including interferon-γ (IFNγ), leads to the unchecked production of effector molecules such as reactive oxygen and nitrogen species that themselves can contribute to pathology1. Nitric oxide (NO) is one such effector molecule. Synthesis of large amounts of NO by inducible nitric oxide synthase (iNOS) is implicated in the hypotension that accompanies sepsis2. This pathologic situation contrasts with normal vascular homeostasis, in which small amounts of NO formed by endothelial nitric oxide synthase relax blood vessels3. IFNγ or lipopolysaccharide (LPS) induce iNOS in vascular cells4, generating excessive NO, which results in profound vasodilation with hypotension. Treatment with NO synthesis inhibitors improves hemodynamic parameters in animal models of endotoxemia5. Mice lacking iNOS are largely, but not completely, protected from LPS-induced hypotension6.
Increased production of NO also has been demonstrated in human sepsis7. Inhibition of NO synthesis can promote the resolution of shock and temporarily improve hemodynamic variables, including an increase in mean arterial blood pressure (BP)8. However, inhibition of NO synthesis does not improve 28-day survival8 or consistently yield long-term benefits to vascular health9. Therapies aimed at targets other than iNOS also have failed to improve outcomes1.
The above findings along with a recent report10 indicate that additional pathway(s) may be involved in the regulation of vascular tone during acute systemic inflammation. In this regard, it is noteworthy that the metabolism of arginine to NO is functionally related to that of tryptophan (Trp) initiated by indoleamine 2,3-dioxygenase (IDO) (Supplementary Fig. 1 online). Both pathways are implicated in anti-microbial activities, anti-tumor defence, neuropathology, and immune regulation11. Similar to iNOS, IDO is not normally expressed in cells12 and requires induction via IFNγ13 or TNFα. Once induced, IDO converts Trp to N-formyl-kynurenine, which decomposes spontaneously to formate and kynurenine (Kyn). Depending on the cell type involved, Kyn may be metabolized further along the kynurenine pathway (Supplementary Fig. 1 online).
Despite the similarity between iNOS and IDO, a direct vasomotor regulatory role for kynurenine pathway metabolites is not known. Of relevance, during systemic inflammation, vascular endothelial cells are the primary site for IDO expression in vivo14,15. Furthermore, IFNγ induces IDO in vitro in various vascular cells16,17. Therefore, we examined the induction and potential function of IDO and Trp metabolism in blood vessels under conditions of systemic inflammation.
RESULTS
IDO contributes to the regulation of BP
We tested the role of IDO in the regulation of BP in three models of systemic inflammation. Infection of C57BL/6J mice with Plasmodium berghei ANKA (PbA) leads to systemic inflammation18 and the expression of IDO protein in vascular endothelial cells14,15,19. This included resistance vessels, such as arterioles in kidney, as assessed by immunohistochemistry five days post-infection (Figs. 1a-c). Expression of IDO was predominantly selective for endothelial cells19 (Fig. 1a). Systemic IDO expression translated into functional activity, as plasma concentrations of Trp were decreased (Fig. 1d) and Kyn increased significantly (Fig. 1e). Up-regulation of IDO contributed to hypotension (Fig. 1f), which is known to be associated with severe and cerebral malaria in humans20 and mice21. Administration of the selective IDO inhibitor 1-methyl-D-tryptophan (1-Me-Trp)22 restored normal systolic blood pressure (SBP) in infected, but not uninfected, mice (Fig. 1f).
Fig. 1. IDO pathway contributes to the regulation of BP in PbA-infected mice.

(a-c) Expression of IDO in endothelial cells of resistance arteries in kidney. IDO expression was assessed by immunohistochemistry in non-infected mice and in mice 5 days post-infection with PbA (bar = 40 μm). IDO was expressed strongly in endothelial cells of PbA-infected mice (a) but was absent in non-infected mice (b) and in corresponding isotype control sections of PbA-infected mice (c). (d) Decreased plasma Trp and (e) increased Kyn concentration in PbA-infected Ido+/+ not Ido-/- mice. (f-g) SBP determined by tail cuff method in conscious C57BL/6J wild type (f) or Ido-/- (g) mice with or without PbA infection, before and after i.p. injection of 1 mL 20 mM 1-Me-Trp. Results in d-g represent mean±SEM, with the number of animals (N) used for each treatment indicated. *P<0.05 versus uninfected control (d, f) or before 1-Me-Trp injection in PbA-infected mice (g).
The role of IDO in the regulation of BP was confirmed in mice during PbA infection. Infection had no significant impact on plasma Trp and Kyn in Ido-/- mice (Figs. 1d and e), and infusion of 1-Me-Trp failed to increase SBP in infected mice that lacked IDO (Fig. 1g) or IFNγ (Supplementary Fig. 2 online) and therefore did not express endothelial IDO19. Similar results were observed when BP was measured in unconscious mice (Supplementary Fig. 2 online). Nonetheless, infection also caused hypotension in Ido-/- mice (Fig. 1g). As there is both redundancy in the regulation of BP23,24 and interaction between IDO and NOS11, we compared the relative contribution of NOS to the regulation of BP in wild type versus Ido-/- mice. In uninfected animals, L-NAME increased BP similarly in both types of mice, whereas the NOS inhibitor increased BP to a greater extent in infected Ido-/- than wild type mice (Supplementary Fig. 3 online). Separate studies with mice deficient in iNOS confirmed that iNOS contributes to the regulation of BP in PbA infection (Supplementary Fig. 4 online). Together, these results suggest that NOS compensates for IDO in the regulation of BP in PbA-infected Ido-/- mice.
Similar to PbA infection, IDO was expressed in endothelial cells of resistance vessels from mice suffering from endotoxemia (Fig. 2a-d) or infection with P. berghei K173 (PbK) (Supplementary Fig. 5a,b online), a strain of malaria that causes severe disease but no cerebral complications25. Again, this translated into functional enzyme activity, as shown by the changes in plasma Trp and Kyn concentration (Fig. 2e, Supplementary Fig. 5c online), and was associated with hypotension (Fig. 2f, Supplementary Fig. 5d online). Also similar to PbA infection, infusion of 1-Me-Trp restored SBP in septic (Fig. 2f) and PbK-infected mice (Supplementary Fig. 5d online). Different to the situation with PbA, however, the extent of hypotension was attenuated in Ido-/- mice early in endotoxemia (74.3 ± 1.5 versus 81.9 ± 1.7 mmHg at 16 h post-LPS in wild type versus Ido-/- mice, P<0.01); SBP was comparable in Ido-/- and wild type mice at later stages, as it returned to normal values (Fig. 2g). The drop in SBP was mirrored by the increased ratio of plasma Kyn/Trp, an indicator of IDO activity (Fig 2h, Supplementary Fig. 6, online). Tissue mRNA of IDO was induced 2 h after LPS and reached maximal levels at 5-10 h after LPS (Supplementary Fig. 7, online). Furthermore, pharmacological inhibition of IDO immediately before or up to 8 h after LPS injection attenuated the extent of hypotension (Fig. 2i). As iNOS participates in LPS-induced hypotension in mice, we determined the effect of IDO inhibition in iNos-/- versus wild type mice. LPS significantly lowered SBP in both types of mice (Fig. 2j). While the extent of hypotension was more pronounced in wild type than iNos-/- mice (Fig. 2j), the relative contribution of IDO to this decrease in SBP was comparable in the two mouse genotypes (Fig. 2k). To confirm the role of the IDO in the regulation of BP, we employed telemetry instead of the tail-cuff method. As implantation of the transmitter required for telemetry itself represented an inflammatory challenge to the animal, including induction of IDO (Supplementary Fig. 8 online), we compared mean arterial pressure (MAP) in transmitter-implanted wild type versus Ido-/- mice. MAP was higher in Ido-/- compared with wild type mice (Supplementary Fig. 9 online). Together, these results indicate that IDO contributes to the regulation of BP during systemic inflammation, and that IDO deficiency can manifest as a phenotypic difference in BP.
Fig. 2. IDO contributes to the regulation of BP in LPS-induced endotoxemia.
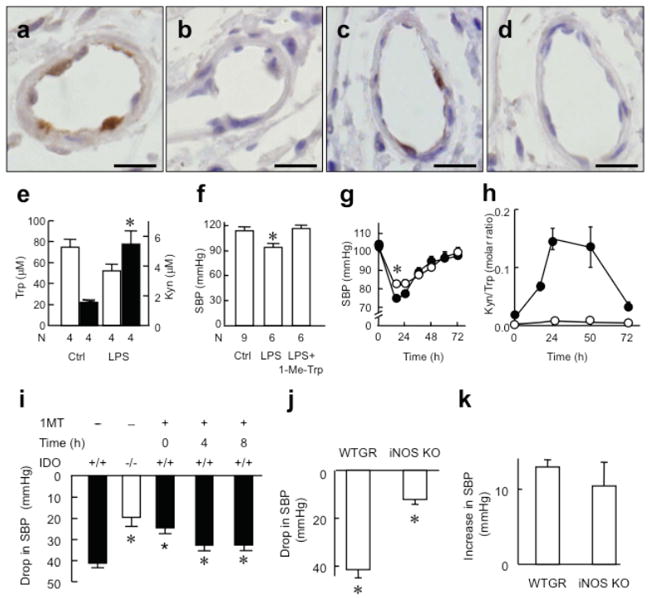
Expression of IDO in endothelial cells of resistance arteries of kidney (a,b) and intestine (c,d) assessed by immunohistochemistry in C57BL/6J mice 48 h after treatment with LPS (7.5 mg/kg, i.p.) (bar = 10 μm). IDO was expressed in endothelial cells of endotoxemic mice (a,c) but staining was absent in isotype controls (b,d). (e) Plasma concentration of Trp (open columns) and Kyn (filled columns) in endotoxemic mice 48 h after i.p. injection of LPS. *P<0.05 versus control. (f) SBP in endotoxemic mice 48 h after LPS injection. *P<0.05 versus both control and LPS+1-Me-Trp. (g) SBP in wild type (o) and Ido-/- mice (●) after LPS injection (n=12-30 for each time point). *P<0.01, wild type versus Ido-/- mice. (h) Ratio of plasma Kyn/Trp, a marker of IDO activity, in wild type and Ido-/- mice (n=3-5 for each time point) after LPS injection. (i) Drop in SBP caused by LPS in wild type versus Ido-/- mice, and in wild type mice co-administered with 1-Me-Trp (50 mg/kg, i.p.) at 0, 4, 8 h post LPS. (j) Drop in SBP of wild type (WTGR) and iNos-/- mice 24 h after LPS (n=4 animals per group). *P<0.05 versus untreated mice. (k) Increase in SBP by 1-Me-Trp in mice at 24 h after LPS injection (n=4 animals per group).
IFNγ-induced endothelial IDO converts Trp to Kyn
Congruous with previous observations that IFNγ is a principal inducer of IDO in vivo26 in malaria and in vitro13, IFNγ dose- and time-dependently induced IDO mRNA (Supplementary Fig. 10 online) and protein in porcine aortic endothelial cells (Figs. 3a and c). Additionally, IFNγ induced IDO activity (Figs. 3b and d). IDO remained active for at least 48 h, as judged by the accumulation of Kyn in the media (Fig. 3d). Consumption of Trp resulted in near stoichiometric accumulation of Kyn (Figs. 3b and d), indicating that Kyn is the major metabolite produced by IFNγ-treated porcine aortic endothelial cells. Comparable results were obtained with IFNγ-primed human umbilical venous (Supplementary Fig. 11 online) and rabbit aortic endothelial cells (data not shown). Therefore, induction of IDO is likely a general response of endothelial cells to this pro-inflammatory cytokine.
Fig. 3. IFNγ-dependent expression of IDO in endothelial cells and its associated conversion of Trp to Kyn.

(a-d) IFNγ induced expression of IDO protein and activity in porcine aortic endothelial cells. Cells were incubated for 20 h in the presence of Trp (200 μM) and the indicated concentration of IFNγ (a, b) or with 250 ng IFNγ/mL for the indicated times (c, d). Cells were harvested. Expression of IDO (46 KDa) and β-actin (42 KDa) was assessed by Western blot and quantified by densitometry (a, c). The concentration of Trp (circles) and Kyn (squares) in the supernate was determined (b, d) in the presence (filled symbols) and absence (open symbols) of cells. (e-f) Production of Kyn by porcine coronary arteries. Fresh porcine circumflex arteries were incubated at 37 °C for up to 24 h in M199 containing 200 μM Trp. (e) Arteries with intact endothelium (EC) were incubated with 0 (❍), 200 (▲) or 400 ng IFNγ/mL (■), the released Kyn determined and its concentration in the coronary lumen calculated taking into account the ratio of luminal versus medium volume. (f) Arteries with intact or denuded EC were incubated for 24 h in the absence or presence of 400 ng IFNγ/mL or the IDO inhibitor 1-Me-Trp (1 mM). Results show mean±SEM of N=3 (a-d) or 4 (e-f) independent experiments. Where error bars are not shown, symbol is larger than the error. *P<0.01 versus control (e) or all other groups (f); †P<0.01 versus 200 ng IFNγ/mL (e).
IFNγ also induced IDO activity dose- and time-dependently in isolated porcine coronary arteries, which caused the luminal concentration of Kyn to reach the millimolar range (Fig. 3e). Other kynurenine pathway metabolites, 3-hydroxykynurenine, kynurenic acid, 3-hydroxyanthranilate and quinolinic acid, were not detected (not shown). IFNγ-induced Kyn originated predominantly from endothelial cells, since removal of the endothelium decreased the accumulated Kyn by ~65% (from 1.20 ± 0.20 to 0.40 ± 0.03 mM, P<0.01) (Fig. 3f). Kyn production required active IDO, as inclusion of 1-Me-Trp (1 mM) during IFNγ pre-treatment prevented Kyn accumulation (Fig. 3f). Thus, IFNγ-induced Trp metabolism by coronary arteries is restricted mostly to endothelial cells, and these cells are primarily responsible for the production of large quantities of Kyn.
The IDO pathway regulates vascular tone via conversion of Trp to Kyn
Pre-constricted porcine coronary arteries responded to addition of Trp by relaxation (Fig. 4a). This depended on IDO activity, because IFNγ was required for relaxation during the 20 h pretreatment, while 1-Me-Trp inhibited relaxation (Fig. 4a). Arteries pretreated with IFNγ and 1-Me-Trp responded normally to the NO-generating drug sodium nitroprusside (10 μM) (96.5 ± 2.1% relaxation compared with that seen with arteries prior to pretreatment, n = 4). This demonstrates that their response to endothelium-independent vasodilators was preserved. Despite this, removal of the endothelium greatly decreased relaxation of IFNγ-pretreated vessels in response to Trp (Fig. 4a). These data suggest that IDO-mediated, endothelium-dependent metabolism of Trp produces arterial relaxation.
Fig. 4. The IDO pathway metabolite Kyn regulates vascular tone.

(a) Relaxation of porcine coronary arteries in response to addition of Trp. Fresh porcine circumflex arteries with intact or denuded endothelium (EC) were incubated for 20 h at 37 °C in M199 in the absence or presence of 400 ng IFNγ/mL. Arteries were then removed, washed and incubated with 300 μM N-nitro-l-arginine methyl ester and 10 μM indomethacin for 30 min and pre-contracted with U-46619 to 50-60% of maximum force. Relaxation was determined in response to addition of Trp (8 mM final concentration). *P<0.05 versus EC-denuded and 1-Me-Trp-treated groups. (b) Contraction of porcine coronary artery in response to U-46619 in control preparations (❍) and in preparations pre-treated with 6 mM Kyn for 1 h (●). *P<0.05 versus control. (c) Relaxation of denuded vessel rings (▲) or rings with intact EC in response to added DMSO (❍, vehicle control) or Kyn (●). *P<0.05 versus control. (d, e) Trp, Kyn or sodium nitroprusside (SNP) was injected into spontaneously hypertensive rats via the femoral vein to give final blood concentrations ranging from 0.15 – 1.5 mM (for Kyn and Trp) and 0.15 – 1.5 μM (SNP), with systolic blood pressure being monitored continuously. We assumed a total blood volume of 17 mL for the 250 g rats used. (d) Typical blood pressure responses elicited by 1.5 mM of either Kyn or Trp, or 1.5 μM SNP. (e) Dose-dependent changes in systolic blood pressure following injection of Kyn (●), Trp (Δ) or SNP (❍). The results show mean±SEM of four (a), six (b, c) or three (e) independent experiments.
As endothelial IDO effectively metabolized Trp to Kyn, we next examined whether Kyn could directly modulate vascular tone. Indeed, Kyn significantly attenuated the contractile response of porcine coronary arteries to the thromboxane analog U-46619 (Fig. 4b), as indicated by the right-shift of the dose-response curve. Similar results were obtained with rabbit aortic rings (Supplementary Fig. 12 online). Furthermore, Kyn dose-dependently relaxed pre-constricted porcine coronary arteries (Fig. 4c), rabbit aorta (Supplementary Fig. 13 online) and mouse abdominal aorta (not shown), a response that did not require an intact endothelium. Relaxation was observed at Kyn concentrations ≥300 μM (Fig. 4c), which corresponds to concentrations achieved in vivo in blood vessels of the brain of PbA-infected mice26 and ex vivo in IFNγ-treated porcine coronary arteries (Figs. 3e and f). In contrast, formate (≤5 mM), Trp, kynurenic acid, 3-hydroxykynurenine or 3-hydroxyanthranilic acid (all ≤1 mM), or quinolinic acid (≤ 0.1 mM) were ineffective in dilating vessels (Supplementary Fig. 14 online). Results similar to those observed with porcine coronary arteries were seen with rabbit and mouse aorta (not shown), indicating that while the vessel relaxing property was limited to Kyn, it likely represented a general response of different vascular beds.
Intravenous infusion of Kyn caused a transient decrease in the mean arterial BP of spontaneously hypertensive rats, similar to the effect of sodium nitroprusside (positive control) (Fig. 4d). In contrast, infusion of Trp up to 5 mM final plasma concentration did not affect mean arterial BP (Fig. 4d). Both Kyn and sodium nitroprusside, but not Trp, lowered mean arterial BP in a concentration-dependent manner (Fig. 4e).
Kyn relaxes blood vessels via activation of soluble guanylate cyclase (sGC)
Having established Kyn as a novel endothelium-derived vasodilator, we next asked how it relaxes blood vessels. By examining known pathways involved in the regulation of vascular tone, we observed Kyn to activate the cGMP-dependent pathway, as indicated by its ability to activate purified rat sGC (Fig. 5a), and to increase tissue cGMP (Fig. 5b). Furthermore, Rp-8-CTP-cGMPS (10 μM), a protein kinase G inhibitor, attenuated Kyn-induced relaxation of porcine coronary arteries (Fig. 5c).
Fig. 5. Kyn relaxes blood vessels via the sGC/cGMP/PKG pathway.

(a) Activity of heme-containing (empty bars) and heme-free (filled bars) purified rat sGC in response to vehicle (control), 5 mM Kyn or 10 nM DEA/NO. (b) cGMP content in intact coronary arteries exposed to 1 mM Kyn, 10 μM ODQ or 1 mM Kyn + 10 μM ODQ in the presence of 300 μM nitro-l-arginine methyl ester and 10 μM indomethacin. (c) Response of porcine coronary arteries to 3 mM Kyn after pretreatment for 30 min with 10 μM ODQ or 10 μM Rp-8-CTP-cGMPS (inhibitor of cGMP-dependent protein kinase, PKG). (d) Activity of heme-containing sGC in response to Kyn in the absence (❍) or presence (●) of 10 μM ODQ. Pre-treatment of porcine coronary arteries with 10 μM ODQ (filled bars) at 37 °C for 24 h decreased (e) the content of sGCβ subunit, (f) vasorelaxation to DEA/NO and (g) Kyn, when compared with control (24 h incubation at 37 °C without ODQ, empty bars). (h) Enhancement of phosphorylation of vasodilator-stimulated phosphoprotein (VASP) at Ser239 in human platelets, after 3 min stimulation with 10 mM Kyn or 300 nM DEA/NO. Results show mean±SEM with N as indicated, except (d) and (e) that are representative of three separate experiments. Western blots (e) and (h) are representative of three separate experiments. *P≤0.05 versus respective control.
Further detailed mechanistic studies on the ability of Kyn to activate sGC were prompted by the unexpected observation that 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), an inhibitor of sGC, failed to inhibit the Kyn-induced increase in porcine coronary artery cGMP (Fig. 5b) and subsequent relaxation of these vessels (Fig. 5c) and rabbit aorta (Supplementary Fig. 15 online). In fact, ODQ enhanced the Kyn-induced increase in aortic cGMP (Fig. 5b), as reported previously for BAY 58-2667 (cinaciguat), a therapeutic agent capable of selectively activating the oxidized/heme-free form of sGC via binding to the enzyme’s heme pocket27,28. For NO to activate sGC, its heme must be reduced. ODQ oxidizes heme such that the enzyme becomes refractory to activation by NO27. Kyn activated heme-containing and heme-free purified rat sGC with equal efficacy whereas, as expected, NO only activated the heme-containing enzyme (Fig. 5a). Kyn-dependent activation of heme-containing, purified sGC was enhanced significantly by the presence of ODQ (Fig. 5d). Together, these data explain why ODQ did not inhibit the Kyn-mediated increase in cGMP and vessel relaxation.
To confirm the role of sGC in Kyn-induced vessel relaxation, we pretreated porcine coronary arteries with ODQ (10 μM) at 37 °C for 24 h to down-regulate sGC expression29 prior to functional studies. This pretreatment decreased the β subunit content of sGC (Fig. 5e), and it also significantly decreased vessel relaxation in response to the NO-donor DEA/NO (Fig. 5f) or Kyn (Fig. 5g). Consistent with the implied role of sGC and protein kinase G in Kyn-induced vessel relaxation, Kyn stimulated phosphorylation of serine 239 in vasodilator-stimulated phosphoprotein (VASP) in washed human platelets, similar to the effect of NO (positive control) (Fig. 5h).
Kyn activates adenylate cyclase and this contributes to vessel relaxation
Kyn activated the cAMP pathway, as indicated by its ability to increase the activity of adenylate cyclase isolated from porcine coronary arteries (Fig. 6a). Also, Kyn increased the cAMP content in these arteries (Fig. 6b) and rat aortic smooth muscle cells (Fig. 6c), as well as the phosphorylation of VASP serine 157 in washed human platelets (Fig. 6d) and human MEG-01 cells (Fig. 6e). These properties of Kyn translated into functional activity, as the inhibitor of adenylate cyclase SQ22,536 (100 μM) attenuated Kyn-induced relaxation of porcine coronary arteries (Fig. 6f). In contrast, inhibition of protein kinase A using Rp-8-CTP-cAMPS (30 μM) (Fig. 6f), H-89 (10 μM) (Fig. 6g), or KT 5720 (Supplementary Fig. 16 online) had no material effect on Kyn-induced vessel relaxation. This inability to affect vessel relaxation by the Trp metabolite could not be explained by a lack of efficacy of the drug because H-89 inhibited formation of VASPp-Ser157 (Fig. 6e), a marker of PKA activity30, and it significantly attenuated forskolin-induced relaxation (Fig. 6h). These data, together with a previous report by others31, suggest that activation of adenylate cyclase by Kyn contributes to vessel relaxation via a cAMP-dependent and PKA-independent pathway (Fig. 6i).
Fig. 6. Involvement of the adenylyl cyclase/cAMP pathway in vessel relaxation induced by Kyn.
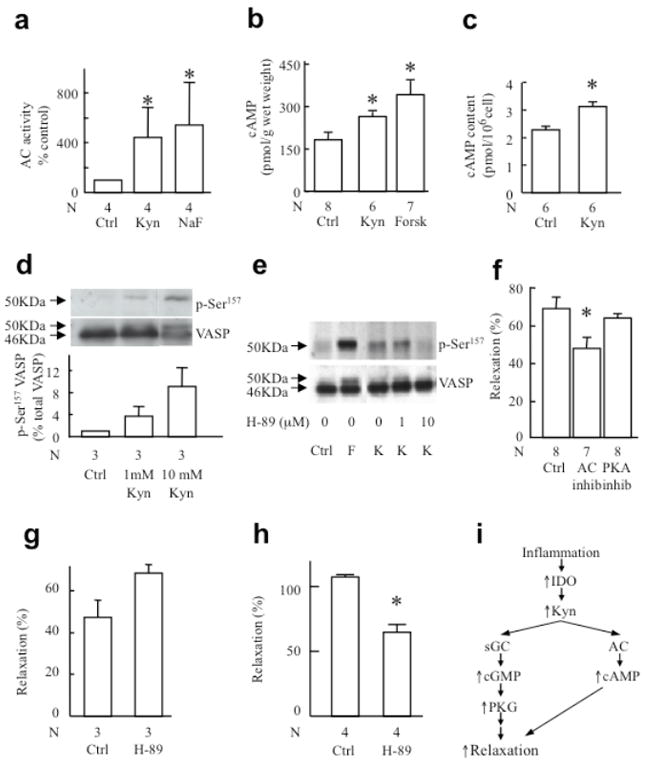
(a) Activation of adenylyl cyclase by 1 mM Kyn or 10 mM NaF. Adenylyl cyclase was enriched as a membrane preparation from porcine coronary arteries and exposed to the agent indicated for 12 min at 37 °C before determination of cAMP. (b) cAMP levels in intact coronary arteries exposed to 1 mM Kyn or 0.6 μM forskolin (positive control) in the presence of isobutylmethylxanthine (inhibitor of phosphodiesterases). (c) cAMP levels in rat aortic smooth muscle cells exposed to vehicle (PBS, control) or 1 mM Kyn for 5 min. (d) Phosphorylation of VASP at Ser157 in human platelet after a 3-min stimulation with 1 or 10 mM Kyn. (e) VASP phosphorylation at Ser157 in MEG-01 cells 3 min after stimulation with 1 mM Kyn (K) in the presence or absence of the PKA inhibitor H-89 (0, 1 and 10 μM). Forskolin (F) was used as a positive control. (f) Response of porcine coronary arteries to 3 mM Kyn after pretreatment for 30 min with 100 μM SQ22,536 (inhibitor of adenylyl cyclase, AC) or 30 μM Rp-8-CTP-cAMPS (inhibitor of cAMP-dependent protein kinase, PKA). (g-h) Relaxation of pre-constricted mouse aortic rings to 3 mM Kyn (g) or 1 μM forskolin (h) after pretreatment for 30 min with 10 μM of the PKA inhibitor H-89. Results show mean±SEM with N as indicated. *P<0.05 versus respective control. (i) Proposed mechanism of inflammation-dependent contribution of Trp metabolism to vessel relaxation during systemic inflammation.
DISCUSSION
Vascular endothelial cells synthesize vasodilators such as NO, prostacyclin, hydrogen peroxide, carbon monoxide and epoxyeicosatrienoic acids, as well as vasoconstrictors, like thromboxane A2, prostaglandin H2 and endothelin-1. These substances play key roles in the local regulation of vascular tone and BP. Our results indicate that Trp metabolism to Kyn via the action of IDO is a novel endothelium-dependent pathway contributing to vascular relaxation and the regulation of BP in systemic inflammation.
Several lines of evidence support our contention that IDO can contribute to BP regulation. First, IDO was expressed in endothelial cells of resistance vessels of mice hypotensive as a result of systemic inflammation, and in these mice inhibition of IDO using 1-Me-Trp restored normal BP. Second, 1-Me-Trp had no effect on BP in animals in the absence of inflammation, or in animals suffering from inflammation but that were deficient in IDO or IFNγ and hence did not express IDO19. Third, administration of the IDO product, Kyn, decreased arterial BP in hypertensive rats, relaxed pre-constricted arteries and attenuated vessel constriction. Fourth, provision of the IDO substrate, Trp, to arteries expressing active IDO caused relaxation that was inhibited by 1-Me-Trp.
Our findings show that endothelial expression of IDO and the associated conversion of Trp to Kyn contribute to vascular tone regulation. Indeed, in the models of systemic inflammation used, IDO induction is predominantly in vascular endothelial cells, indicating that they represent a major source of IDO activity in vivo. Circulating concentrations of key pro-inflammatory cytokines, including IFNγ, increase in systemic inflammation18. IFNγ is a principal inducer of IDO in various cells including those of the blood vessel wall16. Consistent with this report, we found that IFNγ treatment caused a time- and dose-dependent induction of IDO in endothelial cells of different species, and this was associated with the conversion of Trp to Kyn. Therefore, many inflammatorys condition might provide an environment in which IDO-mediated formation of Kyn contributes to the regulation of vascular tone.
Our findings that administered Kyn lowered BP in hypertensive rats and activated isolated sGC suggest that Kyn itself is the vasoactive metabolite of Trp. The levels of Kyn required to achieve these biological activities are in the high micromolar-to-low millimolar range. Such concentrations accumulate in the lumen of both IFNγ-treated porcine coronary arteries and brain blood vessels of mice infected with PbA26, the latter calculated from the tissue Kyn concentration, based on the exclusive localization of IDO in endothelial cells14,15,19, and the knowledge that endothelial cells account for ≤1% of brain mass15. Nevertheless, we cannot exclude the possibility that a Kyn derivative or trace contaminant present in Kyn was responsible for some of the activities observed, although when examined by NMR and mass spectrometry, the commercial Kyn preparations used for the present studies were pure (data not shown). Being a heme ligand32, we also considered the possibility that formate, a co-product of IDO action on Trp, contributes to vessel relaxation. In contrast to Kyn, however, formate (≤ 5 mM) did not relax pre-constricted aortic rings (not shown). Therefore, formate is not a participant in the regulation of vascular tone, similar to the Kyn metabolites, 3-hydroxykynurenine, kynurenic acid, 3-hydroxyanthranilate and quinolinic acid.
Hypotension is a common complication of sepsis and cerebral malaria in humans2,20 and mice6,21. We observed pharmacological inhibition of IDO to restore normal BP in endotoxemic and infected wild type mice, and genetic deletion of IDO to attenuate the drop in BP in mice challenged with LPS. Of potential clinical relevance, attenuation of hypotension was achieved with IDO inhibition commencing as late as 8 h after induction of endotoxemia. Also, a recent study reported pharmacological and genetic blockade of IDO to decrease mortality induced by LPS33, although that study did not investigate a role of IDO in the regulation of BP. Notwithstanding this, however, hypotension was still observed in Ido-/- mice, in both cerebral malaria and endotoxemia. This may reflect redundancy in the regulation of BP. For example, mice deficient in heme oxygenase-1 (that generates carbon monoxide) or soluble epoxide hydrolase (that inactivates epoxyeicosatrienoic acids) show normal basal BP34,35. Interestingly however, both types of mice reveal an attenuated BP response when challenged with LPS35,36, similar to the situation observed in the present study with Ido-/- mice (Fig. 2g). In the case of heme oxygenase-1-deficient mice, increased expression of endothelin-1 was suggested as a compensatory mechanism36, whereas our studies (Supplementary Fig. 3 online) suggest an increase in the contribution of iNOS to hypotension seen in Ido-/- mice. The latter appears to contrast with a recent report indicating decreased bioavailability of NO in PbA infection37. However, that study37 examined blood-related parameters of NO bioavailability, whereas our measure of NO activity was limited to the vessel wall. Also, we observed an increased contribution only in the case of Ido-/- mice, which were not studied by Gramaglia and co-workers37. Compared with iNOS, the contribution of IDO to the drop in BP in endotoxemic mice is smaller (~30 versus ~10 mmHg, respectively). However, IDO may be relatively more important in humans, where iNOS plays a lesser role while IDO activity is more pronounced11.
A common thread of NO-modulated processes in the vasculature is their dependence on cGMP production by the heme-containing sGC. Activation of sGC occurs when NO binds to heme. We provide direct evidence that Kyn can activate all facets of this pathway, resulting in blood vessel relaxation. Surprisingly, Kyn also activated oxidized/heme-free sGC, in sharp contrast to NO, which requires reduced heme to function. Although Kyn is not a strong activator of oxidized/heme-free sGC, it would be expected to cause biologically relevant increases in cGMP. Our findings reveal Kyn as the first natural substance capable of activating oxidized/heme-free sGC, raising the intriguing possibility that IDO-mediated conversion of Trp to Kyn may represent a previously unknown endogenous ‘back-up’ system contributing to vessel relaxation in situations where sGC becomes oxidized/heme-depleted and hence refractory to activation by NO. Oxidative stress associated with inflammation and vascular disease states leads to a form of sGC that is indistinguishable from the in vitro oxidized/heme-free enzyme, such that the physiological existence of oxidized and finally heme-free sGC can no longer be rejected29.
In conclusion, our findings provide novel insights into the regulation of BP that have potentially far-reaching implications. Our work provides a potential novel therapeutic strategy for the regulation of vascular tone in hypertension, through manipulating the tissue activity of IDO and/or concentration of Trp and Kyn. We hypothesize that Trp metabolism to Kyn could be important in systemic inflammation, perhaps particularly in conditions where NO bioactivity is impaired due to dysfunctional sGC.
METHODS
Animals
C57BL/6J (Animal Resources Centre, Perth, Australia) and Ido-/-38 mice, and spontaneous hypertensive rats (Animal Resources Centre) were used. Endotoxemia of mice was induced by i.p. injection of 7.5 mg/kg LPS (0111:B4, Sigma). The local Animal Ethics Committee approved all protocols.
Physiology
SBP was recorded in conscious mice using a computerized, non-invasive tail cuff system (BP 2000 Blood Pressure Analysis System, Visitech Systems, Apex NC) that has been shown to give results that correlate closely with intra-arterial measurements39. An initial 10 inflation-deflation cycle was employed for each mouse to set amplifier and instrument controls. Subsequently, 10 readings were averaged for a single session value. BP was recorded before and 1 h after administration of 1-Me-Trp (200 mg/kg body weight, i.p.). In Fig. 2i, BP was recorded before and during the onset of endotoxaemia with and without 1-Me-Trp (50 mg/kg, i.p.) administered at 0, 4 and 8 h after injection of LPS. Mean arterial BP was recorded in the femoral artery of anaesthetized rats, before and after intravenous infusion of l-Trp, l-Kyn or sodium nitroprusside (Sigma). For these experiments, animals were pre-treated with atropine sulphate (0.5 mg/kg, i.v.) to block alterations of BP mediated by vagal reflex.
To measure vascular reactivity, porcine coronary artery rings were mounted in Krebs buffer in a myobath40. Following contraction with KCl (25, 50, 100 mM), the cumulative concentration-response curve to the thromboxane A2 analog U-46619 (Cayman) was constructed. To measure dilation, rings were pre-contracted with U-46619 (30 nM) to ~50-60% of the maximum response. The cumulative concentration-response curve to bradykinin (Sigma) was constructed and expressed as percentage tension change relative to initial contraction. For experiments using mouse aortas, rings were mounted in Krebs buffer in a multi wire myograph system (Model 610M, Danish Myo Technology, Denmark). Following contraction with 3 consecutive additions of KCl (100 mM), rings were pre-contracted with norepinephrine (100 nM) to ~50-60% of the maximum KCl response. The subsequent response to Kyn (Sigma or Fluka) was recorded.
The above process was repeated using N-nitro-l-arginine methyl ester (300 μM) and indomethacin (10 μM) (Sigma), to inhibit the activities of endothelial nitric oxide synthase and cyclooxygenase, respectively. Additionally, the indicated concentrations of Trp or Kyn were used instead of bradykinin, with or without additional inhibitors. All other conditions were identical to those described above. Vessels were allowed to equilibrate for ≥60 min between exposures to different agents. Where required, rings were denuded of the endothelium, which was confirmed by lack of vascular response to bradykinin. To down-regulate sGC, porcine coronary artery rings were pre-incubated in Krebs solution at 37 °C for 24 h in the presence or absence (control) of 10 μM ODQ (Sigma).
Cell and tissue culture
Endothelial cells were harvested from porcine aortas, obtained from a local abattoir, and cultured with M199 (Invitrogen)41. Rat aortic smooth muscle cells (Cell Applications) were cultured as described41,42. Where indicated, confluent cells were pre-treated with the indicated concentration of porcine IFNγ (Endogen). Human platelet-rich plasma was isolated and washed prior to use as described43. For ex vivo induction of IDO, porcine coronary artery rings were incubated at 37 °C in serum-free M-199 without and with IFNγ for up to 48 h.
Immunohistochemistry
Expression of IDO in arterioles from kidney/intestine collected from mice 5 (PbA19) or 8 (PbK) days post infection, or 48 h post injection with LPS was assessed using a rabbit polyclonal anti-IDO antibody (TransGenic Inc., Japan). Appropriate controls including endogenous biotin block were used.
Biochemical analyses
IDO activity was assessed by the conversion of Trp to Kyn as described previously, with Trp, Kyn, 3-hydroxykynurenine, kynurenic acid and 3-hydroxyanthranilic acid measured by high-performance liquid chromatography (HPLC)26,44,45. Mouse plasma was treated with 5% trichloroacetic acid to precipitate proteins prior to HPLC of Trp and Kyn (as above).
The concentrations of cGMP and cAMP in porcine coronary artery homogenates were assessed using EIA kits (Cayman Chemical) following the manufacturer’s protocols. Prior to homogenization, arterial rings were incubated at 37 °C with the stimulator indicated and in the presence of 300 μM N-nitro-l-arginine methyl ester, 10 μM indomethacin and 100 μM isobutylmethylxanthine (Sigma). Rat sGC was purified from a baculovirus/Sf9 expression system and its activity measured as described27. Heme-free rat sGC was prepared as described27,46. Briefly, the enzyme was incubated with 0.5 % Tween-20 for 5 min at 37°C, which causes loss of the prosthetic heme-group. To avoid reconstitution of sGC with heme, Tween-20 was also present in the test assay. Adenylyl cyclase was enriched as a membrane preparation47 from porcine coronary arteries. Its activity was assessed in the absence or presence of 1 mM Kyn or 10 mM NaF (positive control) at 37 °C for 12 min. cAMP was purified using sequential Dowex and neutral alumina columns before assay.
Western blotting
For IDO, porcine aortic endothelial cell lysates with equal amounts of protein were subjected to Western blotting48. For sGC, Western blotting of porcine coronary artery homogenates using a rabbit polyclonal anti-sGC antibody (Alexis Biochemicals) was employed. To assess vasodilator-stimulated phosphoprotein, platelets or MEG-01 cells (1 vol) were stimulated with the indicated concentration of Kyn or DEA/NO (Cayman) or forskolin (Sigma) for 3 min at 37 °C, treated with stopping solution (0.5 vol 5% glycerol, 6% sodium dodecyl sulfate, 0.0075% bromophenol blue and 200 mM Tris/HCl, pH 6.8), and subjected to Western blotting using either a rabbit polyclonal antibody to total vasodilator-stimulated phosphoprotein (M4, AXXORA, UK) or mouse monoclonal antibodies to phospho-serine 157 and phospho-serine 239 vasodilator-stimulated phosphoprotein (nanoTools, Germany).
Statistics
Data are expressed as mean±SEM. Statistical analyses were performed using the Prism statistical program (GraphPad v3.0). One-way ANOVA and the Student-Newman-Keul’s test were used to evaluate differences between groups. Concentration-response curves were compared by two-way ANOVA for repeated measures. Otherwise, the Mann-Whitney test was used to evaluate differences between treatments. In all cases, P < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank Maura Finnemore, Katherine Choy, Drs Houta Salahifar and Emile Andriambeloson for assistance and preliminary experiments, Dr. Judy Whitworth for the suggestion to test the effect of Kyn on BP in spontaneously hypertensive rats, and Dr. Geoff Head for advice on telemetry. This work was supported by project grants G03S1177 from the National Heart Foundation (NHF) to R.S., 400992 and 401106 from the National Health & Medical Research Council (NHMRC) of Australia to R.S., DP0987074 from the Australian Research Council to N.H., 512469 from NHMRC to N.H. and NHF (OS 98S0008) and ARC (DP0343325) Fellowship to P.K.W. Dr Stocker holds an NHMRC Senior Principal Research Fellowship and University of Sydney Medical Foundation and Professorial Research Fellowships.
References
- 1.Riedemann NC, Guo RF, Ward PA. Novel strategies for the treatment of sepsis. Nature Medicine. 2003;9:517–523. doi: 10.1038/nm0503-517. [DOI] [PubMed] [Google Scholar]
- 2.Gomez-Jimenez J, et al. L-arginine: nitric oxide pathway in endotoxemia and human septic shock. Crit Care Med. 1995;23:253–258. doi: 10.1097/00003246-199502000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Ignarro LJ, Cirino G, Casini A, Napoli C. Nitric oxide as a signaling molecule in the vascular system: an overview. J Cardiovasc Pharmacol. 1999;34:879–886. doi: 10.1097/00005344-199912000-00016. [DOI] [PubMed] [Google Scholar]
- 4.Kröncke KD, Fehsel K, Kolb-Bachofen V. Inducible nitric oxide synthase in human diseases. Clin Exp Immunol. 1998;113:147–156. doi: 10.1046/j.1365-2249.1998.00648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyer J, et al. Reversal of hyperdynamic response to continuous endotoxin administration by inhibition of NO synthesis. J Appl Physiol. 1992;73:324–328. doi: 10.1152/jappl.1992.73.1.324. [DOI] [PubMed] [Google Scholar]
- 6.MacMicking JD, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 7.Ochoa JB, et al. Nitrogen oxide levels in patients after trauma and during sepsis. Ann Surg. 1991;214:621–626. doi: 10.1097/00000658-199111000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bakker J, et al. Administration of the nitric oxide synthase inhibitor NG-methyl-L-arginine hydrochloride (546C88) by intravenous infusion for up to 72 hours can promote the resolution of shock in patients with severe sepsis: results of a randomized, double-blind, placebo-controlled multicenter study (study no. 144-002) Crit Care Med. 2004;32:1–12. doi: 10.1097/01.CCM.0000105118.66983.19. [DOI] [PubMed] [Google Scholar]
- 9.Kirkebøen KA, Strand OA. The role of nitric oxide in sepsis--an overview. Acta Anaesthesiol Scand. 1999;43:275–288. doi: 10.1034/j.1399-6576.1999.430307.x. [DOI] [PubMed] [Google Scholar]
- 10.Cauwels A, Janssen B, Buys E, Sips P, Brouckaert P. Anaphylactic shock depends on PI3K and eNOS-derived NO. J Clin Invest. 2006;116:2244–2251. doi: 10.1172/JCI25426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas SR, Stocker R. Redox reactions related to indoleamine 2,3-dioxygenase and tryptophan metabolism along the kynurenine pathway. Redox Rep. 1999;4:199–220. doi: 10.1179/135100099101534927. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida R, Oku T, Kishida T, Hayaishi O. Induction of pulmonary indoleamine 2,3-dioxygenase by interferon. Proc Natl Acad Sci U S A. 1981;78:129–132. doi: 10.1073/pnas.78.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Werner ER, et al. Interferon-γ-induced degradation of tryptophan by human cells in vitro. Biol Chem Hoppe Seyler. 1987;368:1407–1412. doi: 10.1515/bchm3.1987.368.2.1407. [DOI] [PubMed] [Google Scholar]
- 14.Hansen AM, Driussi C, Turner V, Takikawa O, Hunt NH. Tissue distribution of indoleamine 2,3-dioxygenase in normal and malaria-infected tissue. Redox Rep. 2000;5:112–115. doi: 10.1179/135100000101535384. [DOI] [PubMed] [Google Scholar]
- 15.Ball HJ, McParland B, Driussi C, Hunt NH. Isolating vessels from the mouse brain for gene expression analysis using laser capture microdissection. Brain Res Protoc. 2002;9:206–213. doi: 10.1016/s1385-299x(02)00147-2. [DOI] [PubMed] [Google Scholar]
- 16.Sakash JB, Byrne GI, Lichtman A, Libby P. Cytokines induce indoleamine 2,3-dioxygenase expression in human atheroma-associated cells: implications for persistent Chlamydophila pneumoniae infection. Infect Immun. 2002;70:3959–3961. doi: 10.1128/IAI.70.7.3959-3961.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Owe-Young R, et al. Kynurenine pathway metabolism in human blood-brain-barrier cells: implications for immune tolerance and neurotoxicity. J Neurochem. 2008;105:1346–1357. doi: 10.1111/j.1471-4159.2008.05241.x. [DOI] [PubMed] [Google Scholar]
- 18.Mitchell AJ, et al. Early cytokine production is associated with protection from murine cerebral malaria. Infect Immun. 2005;73:5645–5653. doi: 10.1128/IAI.73.9.5645-5653.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansen AM, et al. Increased expression of indoleamine 2,3-dioxygenase in murine malaria infection is predominantly localised to the vascular endothelium. Int J Parasitol. 2004;34:1309–1319. doi: 10.1016/j.ijpara.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 20.Bruneel F, et al. Shock complicating severe falciparum malaria in European adults. Intensive Care Med. 1997;23:698–701. doi: 10.1007/s001340050396. [DOI] [PubMed] [Google Scholar]
- 21.Chang WL, et al. CD8+-T-cell depletion ameliorates circulatory shock in Plasmodium berghei-infected mice. Infect Immun. 2001;69:7341–7348. doi: 10.1128/IAI.69.12.7341-7348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munn DH, et al. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Connelly L, Madhani M, Hobbs AJ. Resistance to endotoxic shock in endothelial nitric-oxide synthase (eNOS) knock-out mice: a pro-inflammatory role for eNOS-derived no in vivo. J Biol Chem. 2005;280:10040–10046. doi: 10.1074/jbc.M411991200. [DOI] [PubMed] [Google Scholar]
- 24.Quaschning T, et al. Lack of endothelial nitric oxide synthase promotes endothelin-induced hypertension: lessons from endothelin-1 transgenic/endothelial nitric oxide synthase knockout mice. J Am Soc Nephrol. 2007;18:730–740. doi: 10.1681/ASN.2006050541. [DOI] [PubMed] [Google Scholar]
- 25.Neill AL, Hunt NH. Pathology of fatal and resolving Plasmodium berghei cerebral malaria in mice. Parasitology. 1992;105:165–175. doi: 10.1017/s0031182000074072. [DOI] [PubMed] [Google Scholar]
- 26.Sanni LA, et al. Dramatic changes in oxidative tryptophan metabolism along the kynurenine pathway in experimental cerebral and noncerebral malaria. Am J Pathol. 1998;152:611–619. [PMC free article] [PubMed] [Google Scholar]
- 27.Stasch JP, et al. NO- and haem-independent activation of soluble guanylyl cyclase: molecular basis and cardiovascular implications of a new pharmacological principle. Br J Pharmacol. 2002;136:773–783. doi: 10.1038/sj.bjp.0704778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt HHHW, Schmidt PM, Stasch JP. NO- and haem-independent soluble guanylate cyclase activators. In: Hofmann F, Schmidt HHHW, Stasch JP, editors. Handbood of Experimental Pharmacology: cGMP Generators, Effectors and Therapeutic Implications. Vol. 191. Springer Verlag; Berlin: 2009. pp. 309–339. [DOI] [PubMed] [Google Scholar]
- 29.Stasch JP, et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 2006;116:2552–2561. doi: 10.1172/JCI28371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butt E, et al. cAMP- and cGMP-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J Biol Chem. 1994;269:14509–14517. [PubMed] [Google Scholar]
- 31.Lincoln TM, Cornwell TL, Taylor AE. cGMP-dependent protein kinase mediates the reduction of Ca2+ by cAMP in vascular smooth muscle cells. Am J Physiol. 1990;258:C399–C407. doi: 10.1152/ajpcell.1990.258.3.C399. [DOI] [PubMed] [Google Scholar]
- 32.Armstrong RS, Irwin MJ, Wright PE. Resonance Raman evidence for constrained heme structure in soybean leghemoglobin and its derivatives. Biochem Biophys Res Commun. 1980;95:682–689. doi: 10.1016/0006-291x(80)90839-6. [DOI] [PubMed] [Google Scholar]
- 33.Jung ID, et al. Blockade of indoleamine 2,3-dioxygenase protects mice against lipopolysaccharide-induced endotoxin shock. J Immunol. 2009;182:3146–3154. doi: 10.4049/jimmunol.0803104. [DOI] [PubMed] [Google Scholar]
- 34.Wiesel P, et al. Exacerbation of chronic renovascular hypertension and acute renal failure in heme oxygenase-1-deficient mice. Circ Res. 2001;88:1088–1094. doi: 10.1161/hh1001.091521. [DOI] [PubMed] [Google Scholar]
- 35.Luria A, et al. Compensatory mechanism for homeostatic blood pressure regulation in Ephx2 gene-disrupted mice. J Biol Chem. 2007;282:2891–2898. doi: 10.1074/jbc.M608057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiesel P, et al. Endotoxin-induced mortality is related to increased oxidative stress and end-organ dysfunction, not refractory hypotension, in heme oxygenase-1-deficient mice. Circulation. 2000;102:3015–3022. doi: 10.1161/01.cir.102.24.3015. [DOI] [PubMed] [Google Scholar]
- 37.Gramaglia I, et al. Low nitric oxide bioavailability contributes to the genesis of experimental cerebral malaria. Nat Med. 2006;12:1417–1422. doi: 10.1038/nm1499. [DOI] [PubMed] [Google Scholar]
- 38.Mellor AL, et al. Cutting edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J Immunol. 2003;171:1652–1655. doi: 10.4049/jimmunol.171.4.1652. [DOI] [PubMed] [Google Scholar]
- 39.Krege JH, Hodgin JB, Hagaman JR, Smithies O. A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension. 1995;25:1111–1115. doi: 10.1161/01.hyp.25.5.1111. [DOI] [PubMed] [Google Scholar]
- 40.Lau AK, et al. Probucol promotes functional reendothelialization in balloon-injured rabbit aortas. Circulation. 2003;107:2031–2036. doi: 10.1161/01.CIR.0000062682.40051.43. [DOI] [PubMed] [Google Scholar]
- 41.Thomas SR, Chen K, Keaney JF., Jr Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-Kinase-dependent signaling pathway. J Biol Chem. 2002;277:6017–6024. doi: 10.1074/jbc.M109107200. [DOI] [PubMed] [Google Scholar]
- 42.Palmer D, Tsoi K, Maurice DH. Synergistic inhibition of vascular smooth muscle cell migration by phosphodiesterase 3 and phosphodiesterase 4 inhibitors. Circ Res. 1998;82:852–861. doi: 10.1161/01.res.82.8.852. [DOI] [PubMed] [Google Scholar]
- 43.Becker EM, et al. The vasodilator-stimulated phosphoprotein (VASP): target of YC-1 and nitric oxide effects in human and rat platelets. J Cardiovasc Pharmacol. 2000;35:390–397. doi: 10.1097/00005344-200003000-00007. [DOI] [PubMed] [Google Scholar]
- 44.Thomas SR, Mohr D, Stocker R. Nitric oxide inhibits indoleamine 2,3-dioxygenase activity in interferonγ-primed mononuclear phagocytes. J Biol Chem. 1994;269:14457–14464. [PubMed] [Google Scholar]
- 45.Christen S, Peterhans E, Stocker R. Antioxidant activities of some tryptophan metabolites: Possible implication for inflammatory diseases. Proc Natl Acad Sci U S A. 1990;87:2506–2510. doi: 10.1073/pnas.87.7.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt P, Schramm M, Schröder H, Stasch JP. Preparation of heme-free soluble guanylate cyclase. Protein Expr Purif. 2003;31:42–46. doi: 10.1016/s1046-5928(03)00142-6. [DOI] [PubMed] [Google Scholar]
- 47.Crawford A, Hunt NH, Dawborn JK, Michelangeli VP, Martin TJ. Membranes from a transplantable osteogenic sarcoma responsive to parathyroid hormone and prostaglandins: regulation of adenylate cyclase and of hormone metabolism. J Endocrinol. 1978;77:213–224. doi: 10.1677/joe.0.0770213. [DOI] [PubMed] [Google Scholar]
- 48.Thomas SR, et al. Antioxidants inhibit indoleamine 2,3-dioxygenase in IFN- γ - activated human macrophages: posttranslational regulation by pyrrolidine dithiocarbamate. J Immunol. 2001;166:6332–6340. doi: 10.4049/jimmunol.166.10.6332. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.