

Abstract
Mitochondria produce the energy required for proper cardiac contractile function, and cardiomyocytes that exhibit reduced mitochondrial electron transport will have reduced energy production and decreased contractility. Mitochondrial DNA (mtDNA) encodes the core subunits for the protein complexes of the electron transport chain (ETC). Reduced mtDNA abundance has been linked to reduced ETC and the development of heart failure in genetically engineered mice and in human diseases. Nucleoside reverse transcriptase inhibitors (NRTIs) for HIV/AIDS are used in antiretroviral regimens, which cause decreased mtDNA abundance by inhibiting the mitochondrial polymerase, pol γ as a limiting side effect. We explored consequences of AZT exposure on mtDNA abundance in an established transgenic mouse model (TG) in which a cardiac-targeted mutant form of pol γ displays a dilated cardiomyopathy (DCM) phenotype with increased left ventricle (LV) mass and increased LV end diastolic dimension. TG and wild-type littermate mice received 0.22mg/day AZT or vehicle for 35 days and subsequently analyzed for physiological, histological, and molecular changes. After 35 days, Y955C TGs exhibited cardiac fibrosis independent of AZT. Reduced mtDNA abundance was observed in the Y955C mouse; AZT treatment had no effect on that depletion suggesting that Y955C was sufficient to reduce mtDNA abundance maximally. Isolated mitochondria from AZT-treated Y955C hearts displayed reduced mitochondrial energetic function by oximetric measurement. AZT treatment of the Y955C mutation further reduced basal mitochondrial respiration and state IVo respiration. Together, these results demonstrate that defective pol γ function promotes cardiomyopathy, cardiac fibrosis, mtDNA depletion, and reduced mitochondrial energy production.
Keywords: mitochondrial DNA, cardiac fibrosis, heart, polymerase gamma, AZT
Mitochondria produce the energy required for proper cardiac function, providing cardiomyocytes the ATP necessary for contractility and cellular maintenance. Energy is produced via oxidative phosphorylation across the mitochondrial membrane. Mitochondrial energy production is diminished in heart failure, suggesting energy starvation as an important mechanism (1). Mitochondrial DNA (mtDNA) encodes key subunits for the protein complexes of the electron transport chain (ETC), and reduced mtDNA abundance has been linked to the development of heart failure in genetically engineered mice and in human diseases (2–5).
Current therapeutic protocols for HIV/AIDS include antiretroviral combinations (HAART) with nucleoside reverse transcriptase inhibitors (NRTIs) that inhibit HIV reverse transcriptase serving as cornerstones. Although NRTIs effectively inhibit HIV replication, limiting side effects relate to mitochondrial polymerase, pol γ (6–8). By inhibiting pol γ, NRTIs decrease mtDNA abundance, reduce mitochondrial energy production, and promote cellular dysfunction and disease (2). Our findings and those of others strongly implicated dideoxy-NRTIs in the development of mitochondrial and cardiovascular toxicity (4, 9–12). It logically follows that understanding the relationship between NRTIs, mitochondrial dysfunction, and cardiac myocyte dysfunction provides insight into mechanisms of heart failure.
We explored the consequences of AZT exposure on mtDNA abundance in a genetically engineered mouse model that expresses a mutant form of pol γ targeted to the heart with a commonly employed, robust cardiac promoter (2, 3). In human disease, Y955C pol γ mutant is associated with the development of progressive external ophthalmoplegia and other defects (13–15). Biochemically, the Y955C mutation reduces polymerase processivity, creating longer DNA replication times and decreased mtDNA abundance (13–15). The transgenic mice we generated exhibited DCM with reduced cardiac mtDNA abundance. We congenically backcrossed these mice to C57/Bl6 background. Histological, physiological, and molecular analyses were used to determine the individual and combined effects of this transgene and AZT on cardiac function. Results show decreased cardiac contractile function, increased fibrosis, and reduced mitochondrial functional all occur in the Y955C TG. That effect could not be exacerbated by AZT treatment, suggesting that Y955 polymerase function was sufficient alone to cause heart failure.
MATERIALS AND METHODS
Reagents
All reagents were analytical grade and purchased from Sigma Aldrich (St. Louis, MO) unless otherwise indicated.
Mice
Transgenic Y955C FVB/N mice were generated as previously described, with cardiac-specific Y955C transgene expression achieved using the cardiac-specific α-myosin heavy chain promoter (3). FVB/N Y955C mice were backcrossed with wild-type C57Bl/6 mice (Jackson Labs, Bar Harbor, ME) to produce Y955C C57Bl/6 mice, which were bred to the fifth generation prior to experimentation. For genotyping, genomic DNA was extracted from mouse tail clippings, and genotype was determined by PCR. PCR primers utilized are as follows: forward- 5′-CGTAGTCGACGATGAGCCGCCTG-3′, reverse-5′-CTGCGCTGGAAGCTGCTTAGC-3′. All mice were housed at Emory University in accordance with IACUC protocols in an AAALAC certified vivarium according to NIH guidelines.
Mice were weaned at 3 weeks and genotyped, and experiments on mice were started at 8–12 weeks of age. Mice were administered 0.22mg/day AZT (1-[(2R,4S,5S)-4-azido-5-(hydroxymethyl)oxolan-2-yl]-5-methylpyrimidine-2,4-dione) or 1% CMC vehicle by gavage once daily for 35 days. Mice were analyzed for physiological, histological, and molecular changes after 35 days. All treatments and procedures were conducted in in accordance with approved IACUC protocols.
Echocardiography
Physiological parameters of cardiac function were determined using echocardiography. Mice were anesthetized with Avertin (0.25 mg/g of body weight) and weighed to determine body mass. Echocardiography was performed using VisualSonics Vevo 770 (VisualSonics, Toronto, Ontario, CA). Results from two-dimensional M-mode analysis along the short axis (at the level of the largest LV diameter) were used to determine LV mass, LVEDD, and LV fractional shortening. LV mass values were normalized to mouse body weight.
Histology
Following 35 days of treatment, mice were terminated by cervical dislocation under avertin anesthesia, and hearts were removed, sectioned rapidly with a razor blade (2 mm sections), fixed in 10% neurtral buffered formalin and embedded in paraffin. Six micrometer histological sections were processed with Masson’s trichome, and images were collected at both 200X and 400X magnification using a Nikon Eclipse E800M microscope (Nikon, Melville, NY). Histo-morphometric analysis was performed with the experimenter blinded to treatment or transgenic status. Quantitative microscopic analysis of cardiac fibrosis was determined on the stained tissue sections highlighting the collagen fibers blue. Five to ten randomly selected photomicrographs (200X) of each LV sample were digitized. The area of fibrosis was measured using color selection with a set tolerance and converted to a binary image using Adobe Photoshop (Adobe, San Jose, CA). Fibrosis was defined as the fraction of blue within the total cellular area using NIH Image J software (scale = 9.3 pixels/μm). Data are presented as mean ± SEM.
Hydroxyproline Analysis
Hydroxyproline quantitation was performed as a biochemicalm index of collagen abundance as described by others (16). Approximately 10 mg of frozen left ventricle cardiac tissue from each animal was used. Tissue was acid hydrolyzed overnight at 99oC in 4N HCl. Samples were evaporated at 60°C using an vacufuge (Eppendorf, Hauppauge, NY), resuspended in ddH2O, vortexed vigorously, centrifuged at 3000XG for 10 minutes, and the supernatant was retained. Each sample was mixed with 1.5mL ddH2O, 450μL 6mM HCl, 1mL 0.05M chloramine-T, and incubated at room temperature for 20 minutes. To each sample, 1mL 3.15M perchloric acid was added, stood for 5 minutes at room temperature, and 1mL 20% p-dimethylaminebenzaldehyde (p-DABA) was added to the samples. Samples were incubated for 20 minutes at 60°C, cooled to room temperature and loaded (200μL) in triplicate into a UV plate to be read in a SpectraMax 190 microplate reader (Molecular Devices, Sunnydale, CA) at the absorbance of 575nm. Samples were normalized to a standard curve.
For total protein, the ninhydrin assay was used as previously described (17). Hydrolyzed samples above were diluted 1:200 with H2O. The diluted samples were mixed with 10X volume of ninhydrin reagent (200mg ninhydrin, 7.5mL ethylene glycol, 2.5mL 4 N sodium acetate buffer, pH 5.5, 250μL 100mg/mL stannous chloride) followed by incubation at 99°C for 10 minutes. The samples (200μL) were loaded in triplicate into a UV plate to be read in a SpectraMax 190 microplate reader at the absorption of 575nm. Samples absorption was normalized to a standard curve. Hydroxyproline was normalized to total protein and expressed as mg hydroxyproline/mg total protein.
Mitochondrial DNA (mtDNA) and Nuclear DNA Abundance
Methods utilized are similar to those described previously (3). DNA sequences for primers and probes used for quantitation of mitochondrial and nuclear DNA analyzed the COX I gene of the mtDNA and the POLG2 gene of the nuclear DNA. Amplification was performed using the Lightcycler 480 system (Roche).
Mitochondrial Oximetry
Mitochondria were isolated from mouse hearts immediately following sacrifice using differential centrifugation and a commercial mitochondrial isolation kit (Sigma Aldrich). All steps were performed on ice. Heart tissue (~30mg wet weight) was washed in KHB buffer, minced with a razor blade, trypsinized for 3 minutes, centrifuged at 16,000XG for 1 minute, and retrypsinized for 20 minutes following manufacturer’s instructions. Samples were centrifuged at 16,000XG for 1 minute, resuspended in the proprietary extraction buffer, and homogenized using 15–25 strokes of a Dounce homogenizer. Mitochondria were enriched by differential centrifugation according to manufacturer’s protocol, and resuspended in the proprietary storage buffer. An aliquot was quantitated for protein using the Bradford assay, and 5μg of protein was diluted into 1X mitochondrial assay solution (1X MAS: 70mM sucrose, 220mM mannitol, 5mM KH2PO4, 5mM MgCl2, 2mM HEPES, 1mM EGTA, 0.2% BSA, pH 7.2 with 1N KOH) to a final volume of 50μL, placed into a V7 plate (Seahorse Bioscience, Billerica, MA) and centrifuged at 3,400XG for 20 minutes at 4°C. Following centrifugation, 550μL of 1X MAS containing 10mM pyruvate and 5mM malate was added to each well, and the mitochondrial respiration was analyzed in an XF24 flux analyzer (Seahorse Bioscience) using the manufacturer’s protocol. As defined by others, basal respiration is defined as the oxygen consumption rate of the mitochondria immediately following equilibration. State III respiration was achieved by injecting ADP to a final concentration of 2.5mM. State IVo respiration was achieved by injecting oligomycin to a final concentration of 1.0μg/mL. Non-mitochondrial respiration (data not shown) was achieved by injecting rotenone to a final concentration of 2μM at the end and measuring. Oximetric results are provided as pmol O2/min/μg protein.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 5.0 (Graphpad, La Jolla, CA). Each experiment was analyzed using a one-way or two-way ANOVA were appropriate, with a p<0.05 deemed statistically significant. The experiments are displayed as a mean ± SEM, with all experiments performed at least 3 times each.
RESULTS
Transgenic Mice
The Y955C TG congenically expressed in C57Bl/6 had no impact on body weight compared to wild-type littermates over the course of the experiments (Figure 1A). This observation made body weight an acceptable parameter to normalize cardiac echocardiographic measurements. Similarly, AZT exposure had no effect on body weight either alone or in the Y955C TG (Figure 1A). Y955C TG mice exhibited a significant increase in LV mass compared to wild-type mice (Figure 1B). This result in congenic TGs is in agreement with our previous studies using TGs on the FVB/N background (2, 3), suggesting that strain specific effects did not exist for this TG. LV end diastolic dimension (LVEDD) measurements show a moderate increase in LVEDD in the Y955C mouse compared to wild-type mice (Figure 1C). AZT itself does not increase LVEDD, but a statistically significant increase in LVEDD was found in the Y955C-AZT treated mice compared to AZT-treated wild-type. This suggests a combined effect on increasing LVEDD and thus worsening of the dilation. No change in the fractional shortening was seen (Figure 1D), which is also in agreement with previous studies (2, 3). Together, these results demonstrate the important but sometimes neglected point that that the inbred background of the mouse can cause specific effects (18). The inbred strain does not sensitize it to the Y955C transgene and that native pol γ is crucial to maintaining cardiac performance.
Figure 1. Physiological Measurement of Cardiac Function.
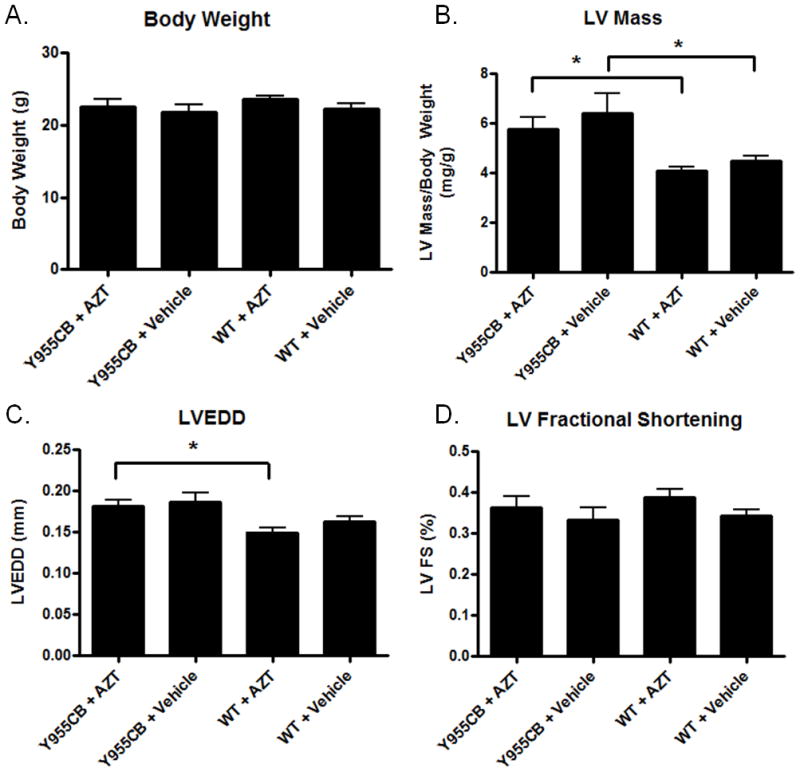
Wild-type and Y955C pol γ transgenic mice were exposed to AZT or vehicle for 35 days. Following treatment, the mice were analyzed for echocardigraphic changes. A) No change in body weight was observed. B) Y955C transgenic mice displayed an increase in LV mass, with AZT having no appreciable effect by itself or in combination with the Y955C transgene. C) LVEDD was increased in Y955C AZT-treated mice compared to AZT-treated wild-type mice. D) There was no significant change in fractional shortening observed in any treatment group compared to controls.
Cardiac Fibrosis
Cardiac fibrosis is an important histological marker of the DCM phenotype (19, 20). Cardiac fibrosis was determined quantitatively and parametrically in mice using two independent methods. Methods were chosen because they offered robust results morphologically and biochemically. Results obtained from each corroborated the other. For morphological studies, paraffin-embedded sections of cardiac tissue stained with Masson’s trichrome highlighted fibrosis microscopically in hearts. Utilizing image analysis, the area of fibrosis from LV tissue sections was defined. The fibrosis pattern appears interstitial and perimyseal (Figure 2). Histological analysis revealed a 3-fold increase in the area of fibrosis in the hearts of Y955C transgenic mice, independent of AZT treatment (Figure 2).
Figure 2. Cardiac Fibrosis.
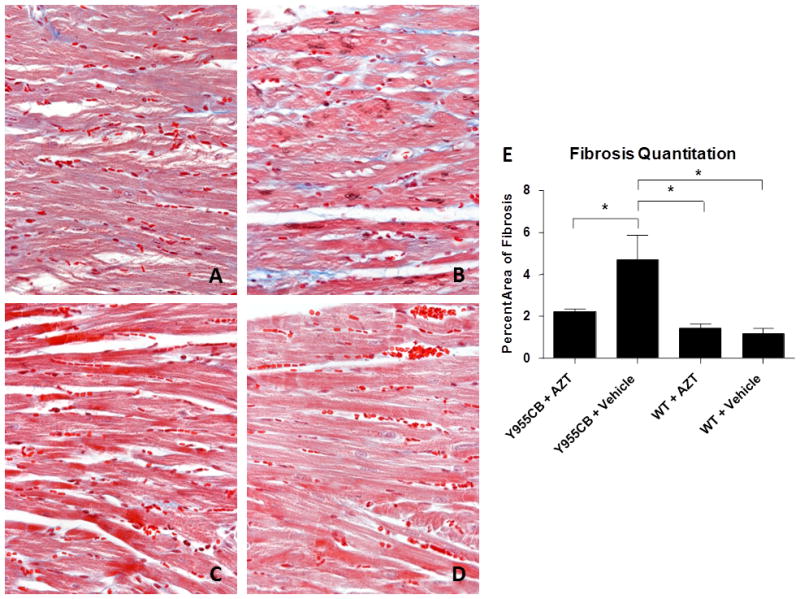
Cardiac fibrosis was evaluated using histological sections stained by Masson’s Trichrome. Presented are representative images (400x) depicting cardiac fibrosis from Y955CB transgenic mice treated with AZT (A), vehicle (B) or wild-type mice treated with AZT (C) or vehicle (D). The level of cardiac fibrosis was determined by calculating the area of blue as a percentage of the total cellular area (E).
We measured the abundance of hydroxyproline in cardiac tissue samples as a parallel biochemical marker of fibrosis (16). Those results were normalized to total protein using ninhydrin quantitation of the same hydrolyzed samples. A 60% increase in hydroxyproline was found in the Y955C LVs compared to wild-type mouse LVs (Figure 3); however, AZT reduced the fibrosis in the Y955C mouse by 50% compared to Y955C with vehicle. These results support the hypothesis of a mitochondrially-driven increase in fibrosis and suggest that AZT treatment may promote a cellular response that reduces fibrosis formation (21–23).
Figure 3. Hydroxyproline Quantitation.
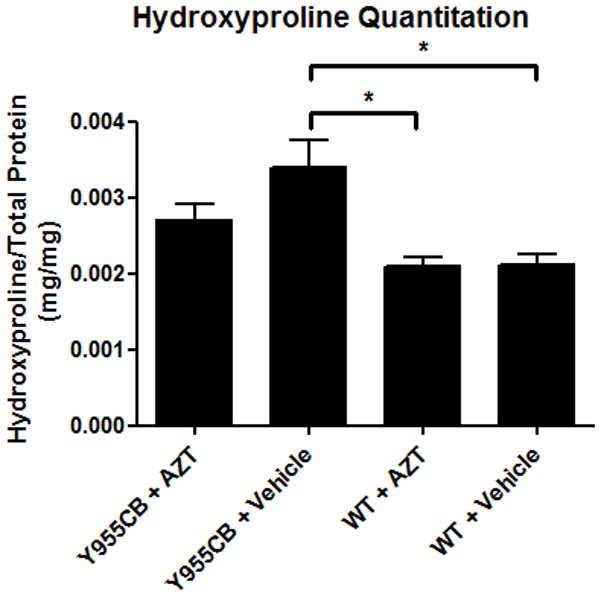
Hydroxyproline was detected using a colormetric assay on hydrolyzed tissue samples, with results normalized to total protein via ninhydrin assay. Y955C mice displayed a significant increase in hydroxyproline. Interestingly, AZT treatment reduced the amount of hydroxyproline, and therefore fibrosis, in the Y955C mice.
mtDNA Abundance
Y955C mice displayed a 50% decrease in mtDNA abundance compared to wild- type controls (Figure 4). In contrast, AZT exposure caused a statistically significant 20% increase in mtDNA abundance, suggesting that the doses utilized in this experiment may provoke a compensatory mechanism. However, the observed increase in mtDNA abundance provided by AZT was blunted by the Y955C transgene. Results indicate the Y955C transgene reduces mtDNA abundance in a robust manner that negates any compensatory mechanism AZT may elicit.
Figure 4. Mitochondrial DNA (mtDNA) Abundance.
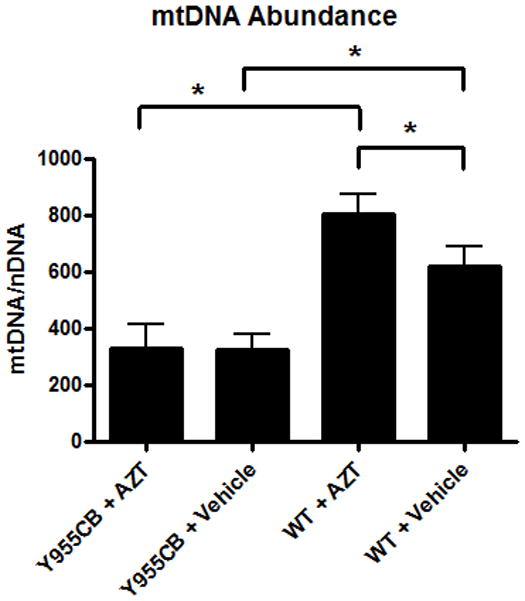
Abundance of mtDNA was quantitated in heart samples from each experimental group. Y955C transgenic mice displayed a 50% decrease in mtDNA abundance compared to wild-type untreated groups. AZT alone elicited a 30% increase in mtDNA abundance compared to vehicle-treated wild-type mice, though Y955C-AZT mice displayed a 50% reduction in mtDNA abundance compare to vehicle-treated wild-type mice.
Mitochondrial Function
Reduced mtDNA abundance will lead to reduced ETC activity by limiting the abundance of ETC polypeptides. In experiments here, preparations of isolated mitochondria from the murine LVs displayed high coupling and good response to ETC-specific compounds. Basal respiration (oxygen consumption of the isolated mitochondria after pyruvate/malate addition but before ADP addition) served as an indicator of mitochondrial quality. Operationally, basal measurements greater than 600pmol/min/5μg protein were associated with proton leakage and low quality mitochondria. The Y955C transgene and AZT each reduced basal respiration (Figure 5A). Interestingly, both parameters acted to reduce basal respiration further than individual effects. AZT reduced basal respiration by 20% whereas Y955C reduced it by 50%. Together Y955C and AZT reduced basal respiration by over 60% compared to wild-type controls. These results suggest that Y955C and AZT may not be working via an identical mechanism (i.e. mtDNA depletion by reduced pol γ function) to yield the observed reduction in mitochondrial function. Measurements of state III respiration showed no change among the experimental sets (Figure 5B), indicating that mitochondria from all treatment groups demonstrate the same capacity to produce ATP when provided adequate substrates. Y955C mice treated with AZT exhibited a 55% reduction in state IVo respiration compared to untreated controls (Figure 5C). While both AZT and Y955C reduced state IVo, AZT alone did not diminish the Y955C-induced state IVo respiratory reduction. These latter results suggest AZT and Y955C both promote mitochondrial coupling, with Y955C achieving near maximal coupling with little additional coupling achieved when combined with AZT treatment. Finally, analysis of the respiratory control ratio (RCR) revealed that both Y955C and AZT promote tighter coupling of the mitochondrial membrane (Figure 5D). A higher RCR is associated with increased capability of producing ATP and decreased proton leakage across the mitochondrial membrane. Data from these experiments suggest that the Y955C transgene and AZT may promote more efficient ATP production in the face of reduced ETC capacity and that some subcellular compensatory mechanism to energetic dysfunction may be operating.
Figure 5. Mitochondrial Function.
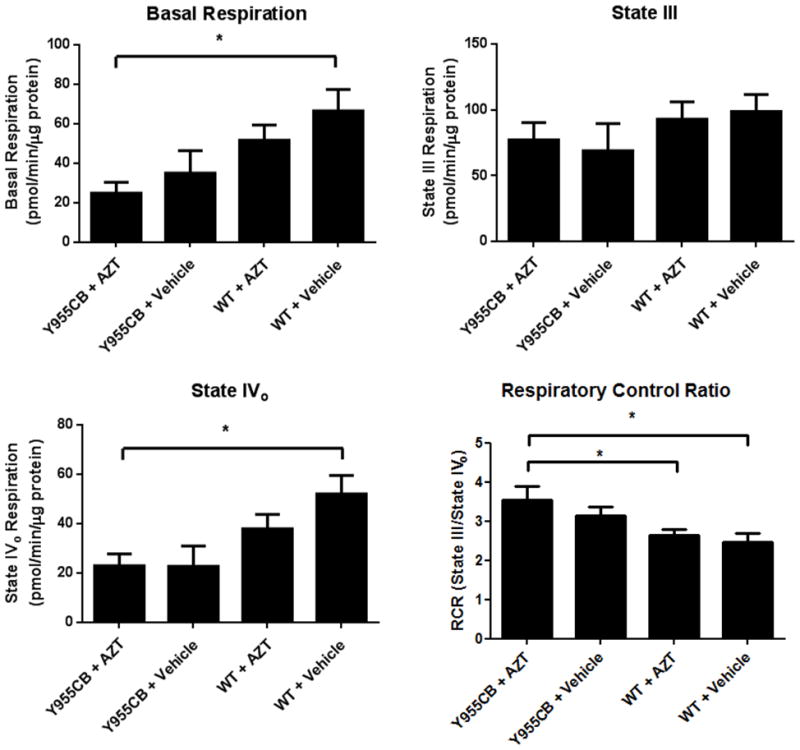
A) Basal respiration of the mitochondria (in the presence of substrates but absence of excess ADP) was suppressed in all treatment groups, with the Y955C transgene and AZT working cumulatively. B) State III respiration was unchanged in any experimental group, suggesting similar capabilities of ATP production in each group. C) State IVo respiration was reduced in each treatment group, with Y955C-AZT mice displaying a 55% reduction in proton leakage compared to vehicle-treated wild type mice. D) Respiratory control ratios (RCR) demonstrate enhanced coupling in the Y955C and AZT treatment groups, with Y955C and AZT cumulatively promoting increased RCR.
DISCUSSION
The Y955C mutation reduces the processivity of pol γ, resulting in increased replication time and decreased mitochondrial DNA abundance in human patients (13, 24, 25). Similarly, AZT has been demonstrated to reduce pol γ function in humans and mice, providing both a genetic and chemical method of measuring reduced pol γ function (6, 7). Previous experiments with cardiac-specific Y955C transgene expression on the FVB/N background demonstrated decreased mtDNA abundance and reduced cardiac contractility (2, 3). Here, we congenically backcrossed these mice to the ubiquitously-used C57Bl/6 mouse background, and pyhysiological changes were determined by echocardiography.
Findings here characterize the contribution of mitochondrial dysfunction to defective cardiac performance and the resultant development of cardiomyopathy from mtDNA depletion and ETC changes. Generation of a stable Y955C transgene on the C57Bl/6 background demonstrated no strain-specific changes, a point that bears emphasis. The Y955C transgene diminished LV function and caused the development of cardiomyopathy in C57Bl/6 mice as shown in FVB/N mice previously (2, 3). Additionally, Y955C transgenic mice display a 50% decrease in mtDNA abundance in both backgrounds. These results implicate pol γ as crucial in maintenance of cardiac function. It may be possible in future studies to approach congenic expression of the Y955C mutant in other inbred strains, or in genetic recombinant strains, but such studies are beyond the nature of the present one.
Our study utilized two independent approaches to determine cardiac fibrosis. The first morphometrically quantitates fibrosis using histological stains and computerized examination of slides. The second method uses quantitation of hydroxyproline in extracted cardiac samples (16). Importantly, both robust methods yielded similar results that corroborated one another. The Y955C transgenic mice display a significant increase in cardiac fibrosis without AZT treatment (Figures 2 and 3) that is consistent with other models of heart failure. Interestingly, AZT treatment was found to reduce cardiac fibrosis in the Y955C transgenic mice, though levels of fibrosis remained elevated compared to controls. These results may suggest that AZT can attenuate the generation of fibrosis, but the subcellular mechanism requires further study. It may be possible to consider that nucleotide pool levels and TK1/TK2 competition and/or inhibition have been demonstrated as important additional considerations in AZT metabolism and utilization. Similarly, further studies may be useful in determining these effects with the Y955C transgenic mouse to enable clarification of these mechanisms of fibrosis (9, 26, 27).
The analysis of mitochondrial function in Y955C transgenic and AZT-treated mice provides novel insight into the effects of mtDNA changes on mitochondrial function. Utilizing oximetric analysis on isolated mitochondria following 35 week AZT or vehicle treatment, we noted a decrease in the basal respiration of the mitochondria from Y955C and AZT treatments. This decrease in basal respiration implicated abnormal mitochondrial function in both Y955C transgenic mitochondria and AZT exposure. However, both the Y955C transgene and AZT decreased the basal respiration, again suggesting a secondary effect of AZT that promotes mitochondrial dysfunction. We found no change in state III respiration, demonstrating that the mitochondria from both Y955C transgenic and AZT-treated mice have a similar capacity to generate ATP under ADP-stimulated conditions (Figure 5B). The change in mitochondrial function was determined to be a decrease in state IVo respiration (Figure 5C). These data suggest that the isolated mitochondria from the Y955C mice and AZT-treated mice may be more tightly coupled than the wild-type untreated mitochondria and exhibit a lower proton leak. This novel finding may be explained by the following hypotheses. First, the Y955C transgene or AZT-treated cardiomyocytes are only retaining mitochondria that have the highest energy production and eliminating damaged mitochondria. Such an effect would lead to a population that is more tightly coupled to maintain ATP production to meet cardiomyocyte energetic demands and reduce proton leakage across the mitochondria membrane. The second hypothesis suggests that enhanced coupling of the mitochondria by an adaptive process in the cardiomyocytes increases energy production efficiency in light of reduced mtDNA abundance. The mechanism of such a process is unknown, though oxidative stress from the Y955C transgene or AZT treatment may be involved (3, 28). Finally, the increase in coupled mitochondria may be the result of the mitochondrial isolation process per se. Our methods for producing an enriched mitochondrial preparation select the best mitochondria from the Y955C or AZT-treated mice. Although it cannot be ruled out, enhanced state III respiration would also be expected to be observed in better prepared mitochondria. The RCR can be used to normalize modulation of state III and state IVo and substantiate observed changes observed in state IVo respiration.
These studies provide insights into the effects of reduced mtDNA abundance by pol γ inhibition. Results suggest the Y955C mutation promotes mtDNA depletion and increases mitochondrial coupling, while AZT elicits an increase in mitochondrial coupling and increases mtDNA abundance possibly in a compensatory way. This mitochondrial dysfunction, via increased oxidative stress or altered mitochondrial functionality, promotes the development of cardiac fibrosis, with the fibrosis observed been intimately linked to the mitochondria due to the models utilized. These studies support a theory of “mito-centric” heart failure with energy starvation at its core (1). They implicate mitochondrial dysfunction in the progression of cardiomyopathy and other cardiovascular diseases.
Supplementary Material
Acknowledgments
These studies were supported by DHHS/NIH/NIDA DA030996 to WL.
Abbreviations
- DCM
dilated cardiomyopathy
- ETC
electron transport chain
- LV
left ventricle
- LVEDD
left ventricle end diastolic dimension
- mtDNA
mitochondrial DNA
- NRTI
nucleoside reverse transcriptase inhibitors
- TG
transgenic mouse
References
- 1.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007 Mar 15;356(11):1140–51. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 2.Kohler JJ, Hosseini SH, Green E, Hoying-Brandt A, Cucoranu I, Haase CP, et al. Cardiac-targeted transgenic mutant mitochondrial enzymes: mtDNA defects, antiretroviral toxicity and cardiomyopathy. Cardiovasc Toxicol. 2008 Summer;8(2):57–69. doi: 10.1007/s12012-008-9015-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis W, Day BJ, Kohler JJ, Hosseini SH, Chan SS, Green EC, et al. Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase gamma. Lab Invest. 2007 Apr;87(4):326–35. doi: 10.1038/labinvest.3700523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kohler JJ, Hosseini SH, Lewis W. Mitochondrial DNA impairment in nucleoside reverse transcriptase inhibitor-associated cardiomyopathy. Chem Res Toxicol. 2008 May;21(5):990–6. doi: 10.1021/tx8000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. 2010 May 14;106(9):1541–8. doi: 10.1161/CIRCRESAHA.109.212753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis W, Kohler JJ, Hosseini SH, Haase CP, Copeland WC, Bienstock RJ, et al. Antiretroviral nucleosides, deoxynucleotide carrier and mitochondrial DNA: evidence supporting the DNA pol gamma hypothesis. AIDS. 2006 Mar 21;20(5):675–84. doi: 10.1097/01.aids.0000216367.23325.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis W, Day BJ, Copeland WC. Mitochondrial toxicity of NRTI antiviral drugs: an integrated cellular perspective. Nat Rev Drug Discov. 2003 Oct;2(10):812–22. doi: 10.1038/nrd1201. [DOI] [PubMed] [Google Scholar]
- 8.Koczor CA, Torres RA, Lewis W. The role of transporters in the toxicity of nucleoside and nucleotide analogs. Expert Opin Drug Metab Toxicol. 2012 Apr 18; doi: 10.1517/17425255.2012.680885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kohler JJ, Hosseini SH, Cucoranu I, Hoying-Brandt A, Green E, Johnson D, et al. Murine cardiac mtDNA: effects of transgenic manipulation of nucleoside phosphorylation. Lab Invest. 2009 Feb;89(2):122–30. doi: 10.1038/labinvest.2008.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Purevjav E, Nelson DP, Varela JJ, Jimenez S, Kearney DL, Sanchez XV, et al. Myocardial Fas ligand expression increases susceptibility to AZT-induced cardiomyopathy. Cardiovasc Toxicol. 2007;7(4):255–63. doi: 10.1007/s12012-007-9004-9. [DOI] [PubMed] [Google Scholar]
- 11.Friis-Moller N, Thiebaut R, Reiss P, Weber R, Monforte AD, De Wit S, et al. Predicting the risk of cardiovascular disease in HIV-infected patients: the data collection on adverse effects of anti-HIV drugs study. Eur J Cardiovasc Prev Rehabil. 2010 Oct;17(5):491–501. doi: 10.1097/HJR.0b013e328336a150. [DOI] [PubMed] [Google Scholar]
- 12.Lewis W, Haase CP, Raidel SM, Russ RB, Sutliff RL, Hoit BD, et al. Combined antiretroviral therapy causes cardiomyopathy and elevates plasma lactate in transgenic AIDS mice. Lab Invest. 2001 Nov;81(11):1527–36. doi: 10.1038/labinvest.3780366. [DOI] [PubMed] [Google Scholar]
- 13.Estep PA, Johnson KA. Effect of the Y955C mutation on mitochondrial DNA polymerase nucleotide incorporation efficiency and fidelity. Biochemistry. 2011 Jul 26;50(29):6376–86. doi: 10.1021/bi200280r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graziewicz MA, Bienstock RJ, Copeland WC. The DNA polymerase gamma Y955C disease variant associated with PEO and parkinsonism mediates the incorporation and translesion synthesis opposite 7,8-dihydro-8-oxo-2′-deoxyguanosine. Hum Mol Genet. 2007 Nov 15;16(22):2729–39. doi: 10.1093/hmg/ddm227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Copeland WC, Ponamarev MV, Nguyen D, Kunkel TA, Longley MJ. Mutations in DNA polymerase gamma cause error prone DNA synthesis in human mitochondrial disorders. Acta Biochim Pol. 2003;50(1):155–67. [PubMed] [Google Scholar]
- 16.Woessner JF., Jr The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys. 1961 May;93:440–7. doi: 10.1016/0003-9861(61)90291-0. [DOI] [PubMed] [Google Scholar]
- 17.Starcher B. A ninhydrin-based assay to quantitate the total protein content of tissue samples. Anal Biochem. 2001 May 1;292(1):125–9. doi: 10.1006/abio.2001.5050. [DOI] [PubMed] [Google Scholar]
- 18.Faulx MD, Ernsberger P, Vatner D, Hoffman RD, Lewis W, Strachan R, et al. Strain-dependent beta-adrenergic receptor function influences myocardial responses to isoproterenol stimulation in mice. Am J Physiol Heart Circ Physiol. 2005 Jul;289(1):H30–6. doi: 10.1152/ajpheart.00636.2004. [DOI] [PubMed] [Google Scholar]
- 19.Kania G, Blyszczuk P, Eriksson U. Mechanisms of cardiac fibrosis in inflammatory heart disease. Trends Cardiovasc Med. 2009 Nov;19(8):247–52. doi: 10.1016/j.tcm.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Zannad F, Dousset B, Alla F. Treatment of congestive heart failure: interfering the aldosterone-cardiac extracellular matrix relationship. Hypertension. 2001 Nov;38(5):1227–32. doi: 10.1161/hy1101.099484. [DOI] [PubMed] [Google Scholar]
- 21.Dai DF, Chen T, Szeto H, Nieves-Cintron M, Kutyavin V, Santana LF, et al. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011 Jun 28;58(1):73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narula N, Zaragoza MV, Sengupta PP, Li P, Haider N, Verjans J, et al. Adenine nucleotide translocase 1 deficiency results in dilated cardiomyopathy with defects in myocardial mechanics, histopathological alterations, and activation of apoptosis. JACC Cardiovasc Imaging. 2011 Jan;4(1):1–10. doi: 10.1016/j.jcmg.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012 Apr 13;110(8):1109–24. doi: 10.1161/CIRCRESAHA.111.246140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atanassova N, Fuste JM, Wanrooij S, Macao B, Goffart S, Backstrom S, et al. Sequence-specific stalling of DNA polymerase gamma and the effects of mutations causing progressive ophthalmoplegia. Hum Mol Genet. 2011 Mar 15;20(6):1212–23. doi: 10.1093/hmg/ddq565. [DOI] [PubMed] [Google Scholar]
- 25.Graziewicz MA, Longley MJ, Bienstock RJ, Zeviani M, Copeland WC. Structure-function defects of human mitochondrial DNA polymerase in autosomal dominant progressive external ophthalmoplegia. Nat Struct Mol Biol. 2004 Aug;11(8):770–6. doi: 10.1038/nsmb805. [DOI] [PubMed] [Google Scholar]
- 26.Hosseini SH, Kohler JJ, Haase CP, Tioleco N, Stuart T, Keebaugh E, et al. Targeted transgenic overexpression of mitochondrial thymidine kinase (TK2) alters mitochondrial DNA (mtDNA) and mitochondrial polypeptide abundance: transgenic TK2, mtDNA, and antiretrovirals. Am J Pathol. 2007 Mar;170(3):865–74. doi: 10.2353/ajpath.2007.060655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koczor CA, Lewis W. Nucleoside reverse transcriptase inhibitor toxicity and mitochondrial DNA. Expert Opin Drug Metab Toxicol. 2010 Dec;6(12):1493–504. doi: 10.1517/17425255.2010.526602. [DOI] [PubMed] [Google Scholar]
- 28.Kohler JJ, Cucoranu I, Fields E, Green E, He S, Hoying A, et al. Transgenic mitochondrial superoxide dismutase and mitochondrially targeted catalase prevent antiretroviral-induced oxidative stress and cardiomyopathy. Lab Invest. 2009 Jul;89(7):782–90. doi: 10.1038/labinvest.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.