

Abstract
Background
The role of fibroblast growth factor and receptor (FGF/FGFR) signaling in bone development is well studied, partly because mutations in FGFRs cause human diseases of achondroplasia and FGFR-related craniosynostosis syndromes including Crouzon syndrome. The FGFR2c C342Y mutation is a frequent cause of Crouzon syndrome, characterized by premature cranial vault suture closure, midfacial deficiency and neurocranial dysmorphology. Here, using newborn Fgfr2cC342Y/+ Crouzon syndrome mice, we tested whether the phenotypic effects of this mutation go beyond the skeletal tissues of the skull, altering the development of other non-skeletal head tissues including the brain, the eyes, the nasopharynx and the inner ears.
Results
Quantitative analysis of 3D multimodal imaging (high resolution micro computed tomography and magnetic resonance microscopic images) revealed local differences in skull morphology and coronal suture patency between Fgfr2cC342Y/+ mice and unaffected littermates, as well as changes in brain shape but not brain size, significant reductions in nasopharyngeal and eye volumes, and no difference in inner ear volume in Fgfr2cC342Y/+ mice.
Conclusion
These findings provide an expanded catalogue of clinical phenotypes in Crouzon syndrome caused by aberrant FGF/FGFR signaling and evidence of the broad role for FGF/FGFR signaling in development and evolution of the vertebrate head.
Keywords: FGFR2, Crouzon syndrome, Pfeiffer syndrome, craniosynostosis, skull, brain, nasopharynx, eye, inner ear
INTRODUCTION
Signaling among fibroblast growth factor receptors (FGFRs) and their ligands (FGFs) is one of the few cell-cell signaling pathways conserved among all metazoans (Itoh and Ornitz, 2004). A growing body of evidence suggests that FGF/FGFR signaling is fundamental in the development of modern vertebrates and has played a critical role in the advent and evolution of many vertebrate characteristics including those which contribute to the vertebrate head (Bertrand et al., 2011). Mutations in FGFR2 are commonly associated with human craniosynostosis syndromes (Ornitz and Marie, 2002), common genetic disorders (~1:60,000) typified by premature closure of cranial vault sutures (especially the coronal suture), but in fact comprising a wide spectrum of anomalies (Wilkie, 1997; Cohen and MacLean, 2000; Ornitz and Marie, 2002). The role of FGF/FGFR signaling in bone development has been well established (Ornitz and Marie, 2002) and dysmorphology of the skull is well studied in craniosynostosis syndromes, due at least in part to the invariant occurrence of premature closure of cranial vault sutures and ensuing reconstructive surgery in human patients. However, FGF/FGFR signaling is critical to basic cellular processes of development (i.e., patterning, proliferation, differentiation, and migration) in tissues other than those with an osteogenic fate (Thisse and Thisse, 2005; Hebert, 2011). Using high resolution multimodal imaging we explore additional potential targets of FGF/FGFR signaling in head development by assessing the phenotypic effects of altered signaling in various skeletal and non-skeletal tissues of the head, including the skull, the brain, the nasopharynx, the eye, and the inner ear of mutant and unaffected littermates of a Crouzon syndrome mouse model.
The Fgfr2cC342Y/+ Crouzon syndrome mouse model carries a cysteine to tyrosine replacement at amino acid position 342 (Cys342Tyr; C342Y) in Fgfr2 (Eswarakumar et al., 2004) equivalent to the FGFR2 mutation most commonly associated with Crouzon syndrome [OMIM #123500], but also causative for Pfeiffer syndrome [OMIM # 101600]. The presence of digital and other organ abnormalities normally distinguish Crouzon from Pfeiffer syndrome patients, as differences in craniofacial phenotypes are subtle and there is marked overlap in the craniofacial features of the two syndromes (Rutland et al., 1995). In humans, Crouzon syndrome craniofacial phenotypes are variable but typically include premature closure of the coronal suture (either unilateral or bilateral) associated with a turribrachycephalic or brachycephalic cranial vault, generalized neurocranial dysmorphology, midfacial deficiency, shallow orbits, and ocular proptosis (Cohen and MacLean, 2000). Children with Crouzon syndrome can show additional symptoms affecting other head structures, such as cleft palate (Peterson and Pruzansky, 1974; Riley et al., 2007), upper airway obstruction (Moore, 1993; Sirotnak et al., 1995; Perkins et al., 1997; Scheid et al., 2002; Mitsukawa et al., 2004; Mitsukawa and Satoh, 2010; Randhawa et al., 2011), dysmorphology of the nasopharynx and contiguous structures that impair velopharyngeal function and nasal respiratory physiology (Peterson-Falzone et al., 1981; Johnson and Wilkie, 2011), and mild or moderate hearing loss (conductive, sensorineural, or mixed) often caused by recurrent otitis media effusion, ossicular chain fixation and external auditory canal atresia (Cremers, 1981; Vallino-Napoli, 1996; Orvidas et al., 1999; Cohen and MacLean, 2000; de Jong et al., 2011; Huh et al., 2012). Brain anomalies do not occur frequently in Crouzon syndrome but ventriculomegaly is fairly common (Proudman et al., 1995). Correspondence between the Crouzon mouse models and human patients with Crouzon syndrome has been demonstrated at the morphological, histological and molecular levels (Eswarakumar et al., 2004; Perlyn et al., 2006; Snyder-Warwick et al., 2010). Still, a significant knowledge gap exists between identification of causative genetic mutations and the development of strategies to prevent or treat these associated abnormalities. Consequently, most therapies are symptomatic and reconstructive-based and do not address the etiological origins of craniosynostosis phenotypes.
In Crouzon syndrome, the FGFR2 C342Y mutation is a gain-of-function mutation in the mesenchymal Fgfr2c variant. The distinct ligand-binding specificity and tissue-specific expression properties of alternatively spliced mRNA variants of FGFRs (b variants are epithelial-specific, c variants are mesenchymal-specific) may underlie the diverse phenotypic effects of craniosynostosis syndromes. The IIIc isoform of FGFR2 is preferentially expressed in mesenchymal tissues (Orr-Urtreger et al., 1993) and is required by the osteoblast lineage for normal skeletogenesis. However, as FGFR isoform production enables regulatory interplay between epithelial and mesenchymal layers during development in response to FGFs (Eswarakumar et al., 2005; Degnin et al., 2010), and mutations such as FGFR2 C342Y in the IIIc isoform result in loss of ligand specificity and constitutive activation of FGFR2 triggering abnormal signaling without the presence of ligand (Neilson and Friesel, 1995), the effects of a mutation in the IIIc isoform could affect cell lineages other than the those destined to become bone.
To provide a broad characterization of the potential concurrent phenotypic effects of mutations in the FGFR2c isoform on skeletal and non-skeletal phenotypes of the murine head we conduct an extensive quantitative analysis of heads of newborn Fgfr2cC342Y/+ Crouzon syndrome mice and their unaffected littermates using multimodal imaging. We test the hypothesis that a mutation in the Fgfr2 IIIc splice variant causes numerous and varied changes in quantitative effects on several skeletal and non-skeletal head tissues of varied embryonic origin. Head phenotypes of newborn Fgfr2cC342Y/+ Crouzon mice and unaffected littermates are quantified using landmark and volumetric data collected from high-resolution micro-computed tomography (µCT) and magnetic resonance microscopy (MRM) images. Under the null hypothesis we expect that effects of a mutation of the Fgfr2 IIIc variant will primarily affect bone tissue and thus significant differences between mutant and unaffected mice will be limited to the skull. The null hypothesis is rejected if we determine statistically significant contrasts between mutant and unaffected littermates in non-skeletal tissues (i.e., brain, nasopharynx, eye, inner ear), indicating that the Fgfr2 IIIc variant affects morphogenesis of several tissues in addition to those formed from mesenchyme. The alternative hypothesis proposes multiple tissue effects of FGF/FGFR signaling in head development, and if supported provides novel data pertaining to the broad effects of FGFR mutations on the development of head tissues and evidence for the importance of this signaling pathway in the integration and coordination of changes that occurred with the evolution of the vertebrate head (Bertrand et al., 2011; Martínez-Abadías et al., 2011).
RESULTS
We compared the phenotypic variation of skeletal and non-skeletal structures of P0 heads of Fgfr2cC342Y/+ Crouzon mice and their unaffected littermates (Table 1) using an array of morphometric and statistical methods (see Experimental procedures for more details) that allow us to characterize overall variation and pinpoint highly localized shape and size differences among Fgfr2cC342Y/+ and unaffected littermates.
Table 1.
Sample sizes (N) of Fgfr2cC342Y/+ mutant mice and unaffected littermates used in analyses.
| Genotype | µCTa | MRMb | MRM | MRM |
|---|---|---|---|---|
| Skull | Ear and Nasopharynx |
Brain | Eye (vitreous humor) |
|
| N | N | N | N | |
| Fgfr2cC342Y/+ | 28 | 8 | 12 | 10 |
| Fgfr2c+/+ | 31 | 11 | 12 | 10 |
| Total | 59 | 19 | 24 | 20 |
Analysis of skull morphology was accomplished using data from micro computed tomography (µCT).
Analysis of soft tissue structures was accomplished using data from magnetic resonance microscopy (MRM). MRM samples represent subsets of the same individuals that underwent µCT imaging. These samples vary in size due to quality of images for segmentation of various structures.
Skull phenotypes of Fgfr2cC342Y/+ newborn mice show global and localized differences relative to unaffected littermates
Skull shape was captured by collecting the 3D coordinates of 39 anatomical landmarks on µCT images of P0 heads (Table 2). Generalized Procrustes analysis was used to superimpose the coordinate data and to extract shape information (Rohlf and Slice, 1990a; Dryden and Mardia, 1998). This procedure minimizes the influence of size and adopts a single orientation for all specimens by shifting the landmark configurations to a common position, scaling them to a standard size and rotating them until a best fit of corresponding landmarks is achieved. For the configuration of landmarks representing each animal, a measure of size is computed as the centroid size, the square root of the summed distances between each landmark coordinate and the centroid of the landmark configuration. Shape variation as represented by Procrustes coordinates was analyzed using Principal Component Analysis (PCA), a data exploration technique that performs an orthogonal decomposition of the data and transforms the resulting Procrustes coordinates into a smaller number of uncorrelated variables called principal components (PCs).
Table 2.
Anatomical definitions of skull landmarks collected from µCT images and shown on Fig 1. Skull regions: G- global skull; F- facial skeleton; B- cranial base; V- cranial vault; P- palate. These landmarks may be viewed at: http://www.getahead.psu.edu/LandmarkNewVersion/P0mouseskull_updated_applet.html
| Landmark (Left, Right) |
Anatomical definition | Skull region |
|---|---|---|
| 1, 2 | Most antero-medial point of the nasal bone | G, F |
| 3, 4 | Most postero-medial point of the nasal bone | G, F |
| 5, 6 | Intersection of frontal process of maxilla with frontal and lacrimal bones | G, F |
| 7, 8 | Intersection of malar with zygomatic process of maxilla, taken on maxilla | F |
| 9, 10 | Intersection of malar with zygomatic process of maxilla, taken on malar | F |
| 11, 12 | Intersection of zygoma with zygomatic process of temporal, taken on zygoma | G, F |
| 13, 14 | Most anterior point of the anterior palatine foramen | G, F |
| 15, 16 | Most posterior point of the anterior palatine foramen | G, F |
| 17, 18 | Most infero-lateral point of the premaxillary-maxillary suture, taken on premaxilla | G, F |
| 19 | Midline point on the premaxilla between the incisor and the nasal cavity just anterior of the incisive foramen, right side only | G, F |
| 20 | Most antero-superior point of the vomer | G, F |
| 21 | Most posterior point of the vomer | G, B |
| 22 | Most anterior point of the indentation in the center of the presphenoid | G, B |
| 23, 24 | Most anterior point on the anterior projection on the presphenoid | G, B |
| 25, 26 | Postero-medial point of the inferior portion of the left alisphenoid | G, B |
| 27, 28 | Most antero-lateral indentation at the posterior edge of the horizontal plate of the palatine bone | G, F |
| 29, 30 | Most posterior tip of the medial pterygoid process | G, B |
| 31, 32 | Most antero-lateral point on corner of the basioccipital at the basi occipital synchondrosis | G, B |
| 33, 34 | Most infero-lateral point on the squamous occipital | G, V |
| 35 | Mid-point on the posterior margin of the foramen magnum, taken on basioccipital | G, B |
| 36 | Mid-point on the anterior margin of the foramen magnum, taken on squamosal occipital | G, B |
| 37, 38 | Superior posterior point on the ectocranial surface of occipital lateralis on the foramen magnum | G, B |
| 39, 40 | Most posterior point on the posterior extension of the forming squamosal | G, V |
| 41, 42 | Most lateral intersection of the frontal and parietal bones, taken on the frontal | V |
| 43, 44 | Most lateral intersection of the frontal and parietal bones, taken on the parietal | V |
| 49, 50 | Most postero-medial point on the parietal | G, V |
| 51 | Most antero-medial point on the body of the sphenoid | G, B |
| P1, P2 | Most anterolateral point on the posterior palatine plate | P |
| P3, P4 | Most anteriomedial point on the posterior palatine plate | P |
| P5, P6 | Most posteromedial point on the posterior palatine plate | P |
| P7, P8 | Most posterolateral point on the posterior palatine plate | P |
The PCA based on the Procrustes coordinates of the 39 cranial landmarks that define the global skull configuration (Table 2; Fig 1A, 1B) showed clear separation between Crouzon syndrome Fgfr2cC342Y/+ mutant mice and unaffected littermates along PC1, which explained 24.36% of total morphological variation (Fig. 1A). Shape changes associated with positive values of PC1 and corresponding to Fgfr2cC342Y/+ mutant mice are located on the parietal and temporal regions of the cranial vault, as well as the cranial base and the face. PC2 accounted for 12.49% of total shape variation but did not separate mutant and unaffected littermates, indicating skull shape variation along this axis is shared by the two groups (Fig. 1A). The multivariate regression of global skull shape on size showed that allometry (size-related differences in shape) was not a significant factor affecting newborn skull shape variation (i.e., Fgfr2cC342Y/+ mutant and unaffected littermates were similar in size), with size predicting only 2.18% of shape variation (p = 0.21).
Figure 1.

Combined results of PCA of skull based on Procrustes coordinates and of EDMA of landmark coordinates. A) Scatter plots of individual scores based on PCA from global skull morphology of Fgfr2cC342Y/+ mutant mice and unaffected littermates along first and second Principal Components axes (PC1 and PC2). B) Lateral (top) and inferior (bottom) views of 3D isosurfaces of µCT reconstruction of unaffected P0 mouse skull with mandible removed displaying the 39 landmarks used in global skull shape analysis. Facial landmarks are shown in red, cranial base landmarks in green, cranial vault landmarks in blue. C-F) Results of PCA analyses based on Procrustes coordinates of regional configurations of skull landmarks on left and EDMA analysis of regional configuration of points on right. Scatter plots of individual morphologies of the facial skeleton (C), cranial base (D), cranial vault (E) and palate (F) along first two principal axes (PC1 and PC2). Since size-related differences in shape (allometry) were significant (P ≤0.05) in the analysis of the face and palate, Figs 1C and 1F represent shape variation after mathematically adjusting for correlations among shape variables due to allometric effects following (Drake and Klingenberg, 2008). Allometry did not have a significant effect on cranial base or cranial vault. EDMA results show linear distances within each regional configuration of points that show at least a 5% difference in length and are significantly different between groups by confidence intervals. Blue lines are significantly larger in mutant mice relative to unaffected littermates; fuchsia lines are significantly smaller in mutant mice.
To explore regional shape variation, we summarized four distinct configurations of 3D landmark coordinates representing the facial skeleton, the cranial base, the cranial vault and the palate (Table 2, Fig 1B) and analyzed them separately using PCA of Procrustes-based coordinates. To avoid problems associated with mathematical invariance that make the Procrustes parameters used for inference non-identifiable (Lele and McCulloch, 2002; Richtsmeier et al., 2002), we also used Euclidean Distance Matrix analysis (EDMA) (Lele and Richtsmeier, 2001), a coordinate system invariant approach to statistically test for localized differences in shape between Fgfr2cC342Y/+ mutant mice and unaffected littermates (see Experimental procedures). Separation of mutant and unaffected mice is apparent in the results of the PCA of the facial skeleton (Fig 1C) and cranial base (Fig 1D) but is less clear for cranial vault (Fig 1E) and especially palatal shapes (Fig 1F). The hypothesis of similarity in facial skeletal shape tested with EDMA was rejected (p = 0.03) and revealed statistically significant differences in facial shape of the two groups of mice. The facial skeleton of Fgfr2cC342Y/+ mutant mice is statistically reduced relative to unaffected littermates for measures on the nasal bones and premaxilla (derivatives of the medial nasal prominence) and the rostrocaudal length of the maxillary palatal shelves (formed in the maxillary prominences) (Fig 1C, Fig 2A,B; Video S1). Statistical results indicate a reduction in midline elements of the facial skeleton and expansion in height and width of the more caudal aspect of the face in mice carrying the Fgfr2cC342Y/+ mutation. Cranial base shapes of mutant and unaffected mice are statistically different (p = 0.001) and a distinct demarcation between the effects of the C342Y mutation on rostral and caudal cranial base elements is apparent (Fig 1D, Fig 2C,D; Video S2) with the line of demarcation of these opposite effects localized to the basisphenoid bone. Dimensions measured from the midsphenoidal body to rostral aspects of the cranial base are relatively reduced in mutant mice while dimensions of the caudal aspects of the cranial base are relatively enlarged in mutant mice (Fig 2D). EDMA demonstrated that the anterior cranial vault of Fgfr2cC342Y/+ mutant mice is reduced rostrocaudally local to the parietal and squamous temporal bones, but posteriorly it is increased along rostrocaudal and mediolateral axes (interparietal, squamous occipital) relative to unaffected littermates, with an overall statistically different shape (p = 0.001) (Fig 2E). Finally, EDMA also confirmed statistically different palatal shapes in the two groups of mice (p = 0.001) (Fig 2F). Analysis of palatal landmarks revealed increased rostrocaudal dimensions of the horizontal plates of the palatine bones but reduction along the mediolateral axis with increased distance between these plates at the inter-palatine suture in Fgfr2cC342Y/+ P0 mice relative to unaffected littermates, though no mice in our sample had overt cleft palate.
Figure 2.

Linear distances (measured between 3D landmarks) that are significantly different in Fgfr2cC342Y/+ Crouzon syndrome mutant mice relative to unaffected littermates when landmarks from global skull are analyzed by EDMA (Lele and Richtsmeier, 2001). Blue lines are significantly larger in mutant mice relative to unaffected littermates. Fuchsia lines are significantly smaller in mutant mice. Significant differences are shown on facial skeleton and palate (lateral (A), and superior (B) views) and on cranial vault and cranial base (lateral (C) and superior view (D)). Fuller 3D views are available as videos (Videos S1 and S2).
Suture patency was visualized and qualitatively scored in Fgfr2cC342Y/+ Crouzon mice and unaffected littermates from the 3D µCT isosurfaces (Fig 3). Over 95% of Fgfr2cC342Y/+ mutant mice showed partial to complete coronal suture fusion bilaterally at P0 (Table 3, Fig 3C). Coronal sutures were completely patent in most unaffected littermates. Fgfr2cC342Y/+ mutant mice show a higher tendency towards patency of several facial sutures relative to unaffected littermates (inter-premaxillary, inter-palatine, maxillary-palatine), with the exception of fusion of zygomatic-maxillary suture. Sutures connecting the anterior neurocranium to the facial skeleton (fronto-maxillary, fronto-premaxillary and fronto-nasal sutures) are patent in both mutant and unaffected littermates (Table 3, Fig 3).
Figure 3.

Patterns of suture patency in Fgfr2cC342Y/+ Crouzon syndrome mice and unaffected littermates at P0. Sutures are shown on µCT reconstructions of an unaffected neonatal mouse skull showing lateral view of complete skull (A) and inferior view of palate (B) with incisors at top: 1-frontal-nasal; 2-frontal-premaxilla; 3-frontal-maxilla; 4-coronal; 5-premaxilla-maxilla; 6-zygomatic-maxilla; 7-inter-premaxillary; 8: maxillary-palatine; 9: inter-palatine. C) Patterns of suture patency in Fgfr2cC342Y/+ Crouzon syndrome mice and unaffected littermates at P0. a a sutural state where only the most anterior section remains patent (see Table 3). b a sutural state where none of the suture appears patent (see Table 3). See Table 3 for supporting quantitative information relating to sample size and patency patterns for individual sutures.
Table 3.
Counts (and corresponding percentages) for suture patency in Fgfr2cC342Y/+ Crouzon syndrome mice and unaffected littermates at P0.
| SUTUREa | CORONAL | ZYGOMATIC- MAXILLARY |
PREMAXILLARY- MAXILLARY |
INTER- PREMAXILLARYb |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Open | Partial | Fused | TOTAL | Open | Fused | TOTAL | Open | TOTAL | 233 | 333 | TOTAL | |
| Fgfr2c+/+ | 20 (65%) | 10 (32%) | 1c (3%) | 31 | 31 (100%) | 0 (0%) | 31 | 31 (100%) | 31 | 9 (23%) | 30 (77%) | 39 |
| Fgfr2cC342Y/+ | 1 (4%) | 4 (14%) | 23 (82%) | 28 | 26 (93%) | 2 (7%) | 28 | 28 (100%) | 28 | 16 (47%) | 18 (53%) | 34 |
| Totald | 21 | 14 | 24 | 59 | 57 | 2 | 59 | 59 | 59 | 14 | 48 | 73 |
| SUTURE | INTER-PALATINE | MAXILLARY-PALATINE | FRONTO-MAXILLARY |
FRONTO- PREMAXILLARY |
FRONTO-NASAL | |||||||
| Open | Partial | TOTAL | Open | Fused | TOTAL | Open | TOTAL | Open | TOTAL | Open | TOTAL | |
| Fgfr2c+/+ | 30 (77%) | 9 (23%) | 39 | 26 (67%) | 13 (33%) | 39 | 39 (100%) | 39 | 39 (100%) | 39 | 39 (100%) | 39 |
| Fgfr2cC342Y/+ | 30 (88%) | 4 (12%) | 34 | 28 (82%) | 6 (18%) | 34 | 34 (100%) | 34 | 34 (100%) | 34 | 34 (100%) | 34 |
| Totald | 60 | 13 | 73 | 54 | 19 | 73 | 73 | 73 | 73 | 73 | 73 | 73 |
For each group, qualitative scores on suture patency (open, partial, fused) as visualized on µCT images are provided. Open: No closure or overlap visualized at either side of the suture (both sides scored as open). Partial: Partial fusion or “bridging” apparent. Fused: almost no open areas visualized along sutural margins. For a more complete definition of patency states see Materials and Methods.
The inter-premaxilla suture was divided into thirds: the most anterior third, medial third, and most posterior third. Each section was scored on the degree of fusion: a score of 1 indicated the suture was completely patent; 2 indicated partial fusion; 3 indicated complete fusion. The sutures were scored from anterior to posterior. A score of 123 indicates the anterior third is open, the medial third is partially fused, and posterior third is fused; 233 refers to inter-premaxillary sutures that remained patent anteriorly but were otherwise not patent; 333 refers to inter-premaxillary sutures that showed no visible patency by inspection. See Supplementary methods for details.
This Fgfr2c+/+ mouse displayed complete fusion of the left coronal suture and partial fusion of the right coronal suture.
Totals vary for suture counts in cases where a mouse was damaged during processing and specific suture(s) could not be scored for patency reliably.
Brain phenotypes of Fgfr2cC342Y/+ mutant mice at birth show localized shape differences relative to unaffected littermates
Global brain shape was defined by the 3D coordinates of 15 anatomical landmarks (Table 4, Fig 4D,E,F). The PCA analysis based on the Procrustes coordinates of brain landmarks did not show a distinct separation between Crouzon syndrome Fgfr2cC342Y/+ mutant mice and unaffected littermates along PC1 (27.47% of total morphological variation) or PC2 (17.30% of total shape variation), indicating that the majority of brain shape variation is shared by the two groups (Fig 4A). Brain volume, estimated as the geometric mean of all linear distances measured among a set of 15 neural landmarks (Darroch and Mosimann, 1985) (Table 4) was not significantly different in Fgfr2cC342Y/+ mutant mice and unaffected littermates (p = 0.862; Mann-Whitney U test; IBM SPSS 20.0; IBM, New York). Multivariate regression of global brain shape on size showed that allometry was not a significant factor affecting newborn brain shape variation, with size predicting only 4.65% of shape variation (p = 0.36).
Table 4.
Brain landmarks used in analysis. These can be visualized on 3D MRM reconstructions in Fig 4 and at our website: http://getahead.psu.edu.
| Landmark (Left, Right) |
Anatomical definition | Midline (M) Bilateral (B) |
Surface (S) Subsurface (ss) |
|---|---|---|---|
| 1, 2 | Most superolateral point of intersection of olfactory bulb with anterior frontal lobe surface | B | S |
| 3, 4 | Most caudolateral point on occipital lobe surface | B | S |
| 5 | Most caudal point on cerebellar surface | M | S |
| 6, 7 | Most lateral point on cerebellar surface | B | S |
| 8, 9 | Centroid of head of caudate nucleus | B | ss |
| 10, 11 | Origin of middle cerebral artery from Circle of Willis on ventral cerebral surface | B | S |
| 12 | Genu of corpus callosum | M | ss |
| 13 | Anterior commissure | M | ss |
| 14 | Splenium of corpus callosum | M | ss |
| 15 | Intersection of pons with most caudal aspect of the ventral cerebral surface | M | S |
Figure 4.

Results of PCA of brain based on Procrustes coordinates. Scatter plots of individual brain morphologies along PC1 and PC2 of PCA of whole brain (A), surface brain landmarks (B), and subsurface brain landmarks (C). Size related differences in shape (allometry) did not have a significant effect on shape difference of the whole brain or surface landmarks, but there was a significant allometric effect of size (P ≤ 0.05) in the analysis of the subsurface landmarks. Brain landmarks used in analysis visualized on 3D MRM reconstructions of unaffected mouse brain. Views are: D: superior; E: lateral; F : sagittal section. Landmark numbering corresponds with Table 4. For additional views and definitions of these landmarks see: http://www.getahead.psu.edu/LandmarkNewVersion/Mousebrain.html
To explore regional shape variation, we divided the 3D landmark coordinates into two distinct configurations: landmarks located on the brain surface (K=10) and those located deep to the surface (K=5) (Table 4; Fig 4D, E, F) and analyzed them separately using PCA of Procrustes-based coordinates and EDMA as described for skull analyses. PCA demonstrates no distinct separation between mutant and unaffected brain shapes for the surface shapes (Fig 4B), but the subsurface landmarks reveal a tendency towards separation suggesting that the Fgfr2c mutation affects surface and subsurface regions of the brain differently (Fig 4C). Size related differences in shape (allometry) did not have a significant effect on shape difference of the whole brain or surface landmarks, but there was a significant allometric effect of size (p ≤0.05) in the analysis of the subsurface landmarks, indicating a size difference in the distribution of subsurface features in the brains of mutant and unaffected mice.
EDMA non-parametric hypothesis testing for overall shape difference between mutant and unaffected littermates for brain surface and subsurface landmark subsets did not show statistical differences between groups (p = 0.297, p = 0.565, respectively), but non-parametric confidence interval testing revealed significant localized differences in the neonatal brains of Fgfr2cC342Y/+ mice and unaffected littermates (Fig 5). Specifically, dimensions describing the height of the cerebrum of Fgfr2cC342Y/+ mutant mice are significantly increased (Fig 5B,C), while the mediolateral breadth of the rostral cerebrum is significantly decreased in Fgfr2cC342Y/+ mice relative to unaffected littermates (Fig 5A). The caudolateral surface of the brain is located farther from the midline in mutant mice as compared to unaffected mice (Fig 5A).
Figure 5.

Significant differences between Fgfr2cC342Y/+ Crouzon syndrome mutant mice relative to unaffected littermates as analyzed by EDMA shown on 3D reconstruction of MRM images (A superior view, B lateral view) and a sagittal slice (C). Blue lines represent neural dimensions that are significantly larger in mutant mice relative to unaffected littermates. Fuschia lines are significantly smaller in mutant mice. Landmarks are identified in Table 4 and Fig 4.
Nasopharyngeal and vitreous humor volume, but not inner ear volume differs in Fgfr2cC342Y/+ mutant mice relative to unaffected littermates
To analyze and compare the effects of the Fgfr2 IIIc mutation on other non-skeletal head tissues, we obtained volumetric quantitative measures of the nasopharynx, eye ball and inner ear from MRM images. Volume of the nasopharynx was estimated for each mouse by combining standard segmentation techniques with brain landmarks (Aldridge et al., 2010) that were used to define a plane determining the caudal end of the nasopharynx (see methods). Fgfr2cC342Y/+ mutant mice had significantly restricted nasopharyngeal volumes (2.81± 0.17 mm3) compared to unaffected littermates (3.28 ± 0.13 mm3; p = 0.012, Mann-Whitney U) (Fig. 6). Neither the length of the rostral aspect of the nasopharynx (estimated as the distance from most rostro-superior point of the nasopharynx to the most caudo-superior point), nor the length of the caudal aspect of the nasopharynx (estimated as the distance between the most caudo-superior point to the most infero-caudal point on the nasopharynx) are significantly different in mutant as compared to unaffected littermates. 3D visualizations of the virtual endocasts of the nasopharynges (Fig. 6) suggest differences in the complex shape of these negative spaces in mutant and unaffected littermates and reduction in the cross section of the nasal passages.
Figure 6.

Nasopharynx, vestibular canal and cochlea of Fgfr2cC342Y/+ Crouzon mice (A) and unaffected littermates (B) segmented from MRM images showing relative locations and magnified reconstructions of soft tissue structures (C, D). Nasopharynx volumes were significantly restricted in Fgfr2cC342Y/+ Crouzon mice (C) compared to unaffected littermates (D), while vitreous volumes of the eye were significantly larger in Fgfr2cC342Y/+ Crouzon mice. No significant differences between Fgfr2cC342Y/+ Crouzon mice and unaffected littermates were detected for inner ear volume.
Vitreous humor, an avascular mass of transparent, gelled, intercellular substance occupying a chamber posterior to the lens and making up a large proportion of the eye volume, was segmented from MRM images and used as a volumetric measure for globe of the eye (Fig. 6). There was no significant difference in vitreous humor volume estimated for the left and right globes of the eyes of Fgfr2cC342Y/+ mutant mice or unaffected littermates, indicating no asymmetry for eye volume in either sample. Therefore left and right eye volumes were averaged for each animal and used in statistical analysis. The mean vitreous humor volume of the eyes of Fgfr2cC342Y/+ mutant mice (0.60 ± 0.04 mm3) is significantly larger as compared to that of unaffected littermates (0.36 ± 0.03 mm3) (p < 0.001, Mann-Whitney U).
Virtual endocasts of the fluid filled spaces of the cochlea and vestibular canals were segmented using MRM images of each mouse using standard segmentation techniques (Fig. 6; see Experimental procedures). Total inner ear volumes were estimated (cochlea and vestibular canals) from these segmentations. No difference in inner ear volume was found between newborn mutant (0.97 ± 0.04 mm3) and unaffected (1.02 ± 0.03 mm3) littermates (p = 0.248, Mann-Whitney U).
DISCUSSION
In agreement with what has been previously described for adult Fgfr2cC342Y/+ mice (Eswarakumar et al., 2004; Snyder-Warwick et al., 2010), the dysmorphology of skulls of newborn Fgfr2cC342Y/+ mice parallels skull phenotypes of human infants with Crouzon syndrome, even though most of the bones of the murine neonatal neurocranium and cranial base are only partially formed at P0. Skulls of Fgfr2cC342Y/+ mice and unaffected littermates are similar in size at P0, but different in shape (Figs 1, 2). These differences are accompanied by premature closure of the coronal sutures and variation in patency of facial sutures (Fig 3, Table 3). Similar to previous studies (Eswarakumar et al., 2004; Snyder-Warwick et al., 2010) the incidence of cleft palate is extremely low in mice heterozygous for this mutation. Our analysis is the first to estimate additional effects of the Fgfr2 IIIc mutation on shape and size of several non-skeletal tissues of the head in newborn Fgfr2cC342Y/+ mice. We catalogue additional targets of the Fgfr2 C342Y mutation and reveal specific significant changes in brain shape (but not brain size) (Figs 4, 5), significant reductions of nasopharyngeal volume and significantly increased eye volume (Fig 6), supporting the alternative hypothesis that a mutation in the Fgfr2 IIIc variant affects development of various tissues of the embryonic head.
Direct and indirect effects of the Fgfr2 IIIc mutation
Genetic variants causative for the FGFR-related craniosynostosis syndromes are many, and variation in midfacial deficiency and neurocranial dysmorphology are characteristic of these syndromes (Wilkie, 2005). These traits are usually attributed to dysmorphogenesis of the skull, a composite and complex osseous structure, and our Fgfr2cC342Y/+ mice show many, and some of the most severe malformations seen in the skulls of humans carrying this mutation. Though it is true that mineralized skeletal tissue provides a structural framework for overall head morphology (Abzhanov et al., 2007), mineralization of intramembranous and endochondral bones of the skull occurs late in development relative to the appearance of other soft tissue structures and spaces (e.g., brain, naso- and oropharynx, eyes). Moreover, the skull is not a product of the activities of isolated osteogenic precursor cells, but takes its form from the interaction of multiple cell types that send and respond to signals from many sources (Noden and Trainor, 2005), all combining in a highly coordinated 4-dimensional space-time environment to differentiate into the varying tissues that together form the head.
Historically, the functional matrix (FM) hypothesis proposed that the presence, size and shape, growth, and position in space of all skeletal tissues are secondary, compensatory and mechanically obligatory responses to the temporally prior demands of related soft tissue functional matrices (Moss, 1962). The proposed mechanism (e.g., biomechanical, biophysical) was put forward prior to current knowledge of the genetic basis of development and failed to provide an explanation for cause and effect in development, though the generation of signaling via mechanical forces through cell-cell contact between tissues continues to be a productive area of research (e.g.,(Yu et al., 2001)). Specific to craniosynostosis, the FM hypothesis predicted that changes in the orientation of fiber tracts of the dura mater changed tensile forces that affected the position of cranial bone anlagen and suture development resulting in premature suture fusion (Moss, 1959). More recently, data pertaining to localized expression of FGF/FGFR pathways, ligand specificity, the interaction of gene products, and cellular events happening local to the suture have been gathered with the knowledge that FGFs are integral to the growth of normal bone, and FGFR mutations are perhaps the direct and critical players in the process of premature suture fusion. Work presented here and previously (Martínez-Abadías et al., 2010) demonstrate that coronal suture closure is neither the primary nor sole locus of skull dysmorphology in mouse models for craniosynostosis syndromes.
Determining to what extent size and shape changes quantified in various tissues result from a direct influence of the mutation on each tissue, and/or are the result of indirect effects of an alternate malforming tissue (those we analyzed or alternatives) on the morphogenesis of another tissue requires further analysis. We provide a preliminary analysis of the interdependence of development among traits of the head by estimating the correlation of size of skeletal and non-skeletal structures of the head (Table 5). The correlation analysis reveals similar patterns of relationships among these tissues in Fgfr2cC342Y/+ mice and their unaffected littermates. In mutant and unaffected mice the correlation among skull regions is strongest and the correlation among non-skeletal traits is low to moderate. In both groups, the correlation between sizes of developing skeletal and non-skeletal cranial traits is very low or absent, a finding that is surprising given the apparently precise coordination among multiple developing tissues during head morphogenesis. Our preliminary analyses suggest that this detailed coordination occurs with little or no discernible statistical association among skeletal and non-skeletal trait size at P0. However, change in size is simply one aspect of development. Study of early postnatal growth (from P0 to P2) in the Fgfr2+/P253R mouse model for Apert syndrome indicates that size and shape change differently in the skull and brain, suggesting some level of independence in the pattern of growth for the two tissues (Hill et al., 2012). Future correlation analyses with larger sample sizes and additional data (i.e., quantitative measures of shape) will provide further insights into the interdependence of these developing tissues. We propose that variation in localized shape change of complex head traits contribute to the obvious differences in relative position and overall spatial association of traits in Fgfr2cC342Y/+ mice relative to unaffected littermates (Fig 7).
Table 5.
Correlations of size of skeletal and non-skeletal structures of the head in Fgfr2cC342Y/+ Crouzon syndrome mice (below the diagonal) and unaffected littermates (above the diagonal).
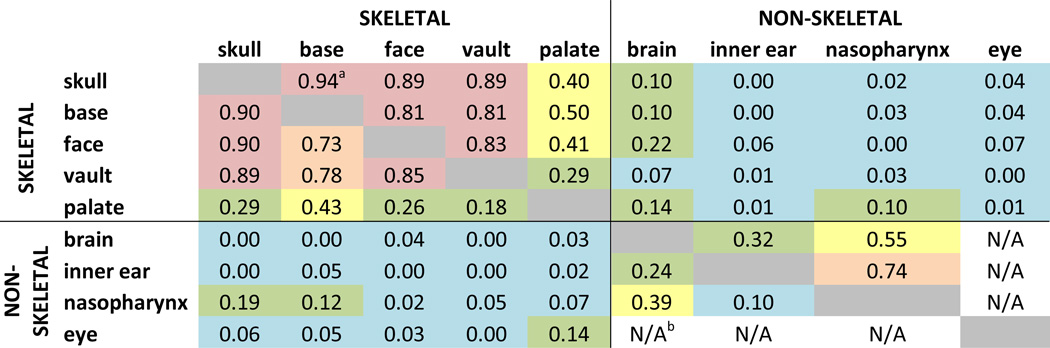 |
Squared correlation coefficient between measures of size for each structure. Color code represents a subjective scale for these correlation coefficients: red, high correlation (>0.80); orange, moderate to high correlation (≥0.6<0.8); yellow, low to moderate correlation (≥0.3 <0.6); green, low correlation (≥0.1<0.3); blue: no correlation (<0.1).
N/A: not available because of small sample size (N<3). Note that some values involving non-skeletal regions are based on small sample sizes (see Table 1).
Figure 7.

Relative position of brain, globe of the eye, nasopharynx, skull, and inner ear in Fgfr2cC342Y/+ mutant mouse (A) and unaffected littermate (B). In each panel the top view shows the superimposition of skull as visualized by µCT and soft tissues segmented from MRM; the bottom view represents MRM data only. Scales are internally consistent for each imaging modality. Skull=yellow, brain=purple, vitreous humor=green, nasopharynx=blue, cochea and vestibular canals=blue. Additional soft tissues visualized on MRM shown in blue/green.
Complete bicoronal craniosynostosis was observed in 75% of Fgfr2cC342Y/+ newborn skulls (Table 3), in agreement with previous observations of bilateral coronal suture closure in 80% of adult mice (Perlyn et al., 2006), and confirming that coronal suture fusion occurs prenatally when this mutation is present. Fusion of the presphenoid-basisphenoid synchondrosis observed in adult Fgfr2cC342Y/+ mice (Perlyn et al., 2006) was not apparent in our newborn sample where mineralized bones of the cranial base are small and clearly separated from each other (Fig 2 and Videos S1, S2). Consequently premature fusion of mineralized cranial base elements cannot be the cause of the significant changes in cranial base, facial or cranial vault morphology quantified in Fgfr2cC342Y/+ skulls at P0. Our shape analysis reveals a boundary of cranial base effects at the midpoint of the cranial base where rostral cranial base elements and tissues that occupy the non-mineralized spaces between them are reduced in size while those caudal to the basisphenoid are elongated (Figs 1 and 2). Whether or not this boundary of effects corresponds with the division between neural crest-derived (presphenoid, basisphenoid) and mesoderm-derived (basioccipital) elements of the cranial base (McBratney-Owen et al., 2008) cannot be determined directly by image data. Variation in cranial vault suture patency and changes in bone and brain shape contribute to neurocranial dysmorphology in Fgfr2cC342Y/+ mice.
Midfacial deficiency in Fgfr2cC342Y/+ mice was evidenced by a reduction in bones of the face and palate (Figs 1, 2), variation in facial suture patency (Fig 3), and significantly reduced nasopharyngeal volume (Fig 6). Though midfacial hypoplasia, a characteristic feature of craniosynostosis phenotypes, is often associated with maldevelopment of facial bones, we have no evidence to limit the primary target of Fgfr2 mutations to osteoprogenitor cells. The primitive pharynx is composed of endoderm internally, that is Fgf-dependent for morphogenesis and it has been shown in zebrafish that pharyngeal endoderm plays a role in promoting the survival of local skeletogenic neural crest cells and patterning of bones and cartilages of the pharyngeal arches (Crump et al., 2004). Consequently, pharyngeal endoderm may also be a primary target of aberrant FGF/FGFR signaling that directly affects nasopharyngeal shape and indirectly affects the size and shape of facial bones developing within the pharyngeal arches. There is no doubt of the role of this Fgfr2 mutation in osteogenesis and maldevelopment of skeletal components, as this is well studied. Our analyses reveal additional possible primary effects of the Fgfr2 IIIc variant in non-skeletal tissues of the head that might contribute to dysmorphogenesis of skull bones.
FGF/FGFR signaling and the role of head development in vertebrate evolution
Our analysis establishes parallels in human and mouse head phenotypes resulting from the C342Y mutation on FGFR2, revealing strong conservation of the evolved developmental genetic program that underlies mammalian head development and further validates the use of this mouse model in the analysis of the development of Crouzon syndrome phenotypes. These data provide new factors for future molecular and cellular investigations targeting the effects of this mutation on tissue specific cell signaling that contributes to craniofacial dysmorphogenesis and the complex etiology of Crouzon syndrome head phenotypes. At the same time, our data provide a basis for thinking more broadly about organismal response to mutations as developmental reactions that are constrained to follow established evolutionary patterns.
FGFs are small proteins characterized by a conserved functional domain that act by binding to FGFRs causing them to homodimerize, and eventually activate and phosphorylate multiple cytoplasmic signaling cascades (Eswarakumar et al., 2005). FGFRs are key regulators of numerous cellular processes (Turner and Grose, 2010). The large number of genes coding for FGFs (at least 22 in vertebrates), along with four coding for their FGFRs appear to have been generated from two rounds of whole genome duplication with many functions added over evolutionary time through the accumulation of genes and of splice variants with different ligand binding specifications (Itoh and Ornitz, 2004; Popovici et al., 2005). The expression of FGFs in neural crest cell derivatives, cranial placodes and specific regions of the vertebrate brain, and the conservation of distinct ligand-binding properties of alternatively spliced mRNA variants of FGFRs increases their functional diversity and renders FGF/FGFR signaling pervasive in development of the head (Yeh et al., 2003; Itoh and Ornitz, 2004; Eswarakumar et al., 2005).
A growing body of evidence suggests that FGF/FGFR signaling is fundamental to diverse processes in normal vertebrate development (Ornitz and Itoh, 2001; Thisse and Thisse, 2005; Wilkie, 2005; Szabo-Rogers et al., 2008; Hatch, 2010; Hebert, 2011). Evidence presented here of the multiple phenotypic effects of a mutation in FGFR2 demonstrates the influence of FGF/FGFR signaling on the development of multiple cell and tissue types. Importantly, this evidence also suggests a role for FGF/FGFR signaling in the accommodation of changing tissue associations that occur during head morphogenesis; a role that would require compliant cell to cell signaling mechanisms in which cells of various tissues are cooperative partners in the development of the overall assembly. This type of system would not arise de novo with the introduction of a mutation in a modern vertebrate, but instead is a product of the evolution of developmental systems.
In a previous study of morphological integration of various parts of the skull in two Fgfr2 mutant mouse models for Apert syndrome (Martínez-Abadías et al., 2011), we suggested that FGF/FGFR signaling is a covariance-generating mechanism established early in vertebrate evolution that acts as a global factor modulating coordinated development of various skull components. We further suggested that this mechanism could constrain clinically relevant shape changes of the skull induced by Fgfr2 mutations (Martínez-Abadías et al., 2011). While skull morphological integration was assessed in that study by statistical analysis of covariation among the shapes of skull components, we show here that this covariance generating system is not restricted to skeletal tissues.
Covariation generated by FGF/FGFR signaling occurs at the level of the cell. FGF/FGFR signaling may both induce specific patterns of gene expression and confer specific responses to naïve cells providing dynamic accommodation of shape change as the head assembles from simple homogenous cell populations into precisely coordinated traits of mixed cell origin. Cells respond to the FGF/FGFR signal by changing size or shape, by differentiating, by dying, by dividing, by sending an appropriate signal, by enabling a cell to receive and respond appropriately to a signal, or by changing the genes the receiving cell uses to respond. Importantly, the tissue-specific and global responses of head tissues to FGFR mutations are constrained by cell-to-cell signaling patterns produced over millions of years of evolution. Knowledge of the evolution of these systems may ultimately bring us closer to harnessing their dynamics for therapeutic purposes.
CONCLUSIONS
In this work, we present a detailed analysis of the global and local phenotypic effects of the C342Y mutation on Fgfr2c on various skeletal and non-skeletal tissues of the head. The approach that we present is directly applicable to the study of other animal models of human disease. Our findings contribute to the growing evidence that FGF/FGFR signaling is part of a multifaceted set of interactions among genes and regulatory networks that drive communication among cells in the development of the head. The nature of the signals vary by cell type, location, and developmental timing so that the challenge becomes one of understanding the complexity of cell-cell signaling and phenotypic assembly at many levels across time (evolutionary and developmental) and space. In addition to the highly successful foundation of developmental biology built largely on molecular analyses of cells and tissues at specific developmental time points, the knowledge that complex craniofacial traits vary together by exploiting patterns of signaling, association, and covariation established over hundreds of millions of years of evolution should be used in the design of new tools for decoding the complex effects of specific mutations on head morphogenesis.
EXPERIMENTAL PROCEDURES
Mice generation
The generation and analysis of Crouzon syndrome Fgfr2cC342Y/+ mouse model are described elsewhere (Eswarakumar et al., 2004; Snyder-Warwick et al., 2010). Fgfr2cC342Y/+ mice were maintained on a CD1 genetic background. Our sample (Table 1) was generated and imaged after death in compliance with animal welfare guidelines approved by the Mount Sinai School of Medicine and the Pennsylvania State University Animal Care and Use Committees. Newborn mice (P0, gestation time was 19.0 ± 0.5 days) were euthanized by inhalation anesthetics and fixed in 4% paraformaldehyde. The genotypes were determined by PCR analysis of tail DNA.
µCT and MRM imaging protocols at P0
High resolution micro computed tomography (µCT) images with pixel size and slice thickness ranging from 0.014 to 0.016 mm were acquired by the Center for Quantitative X-Ray Imaging at the Pennsylvania State University (www.cqi.psu.edu) using the HD-600 OMNI-X high-resolution X-ray computed tomography system (Bio-Imaging Research Inc, Lincolnshire, IL). Image data were reconstructed on a 1024 × 1024 pixel grid as 16 bit TIFFs but reduced to 8 bit for image analysis. High resolution micro magnetic resonance microscopy (MRM) images were acquired of the same specimens for which we have µCT images by the High Field MRI Facility at the Pennsylvania State University (www.imaging.psu.edu/facilities/high-field). The fixed animals were immersed in 2% Magnevist (Bayer Health Care, Wayne, NJ) phosphor-buffered solution for 10 days to reduce the T1 and T2 relaxation time. All experiments were conducted on a vertical 14.1 Tesla Varian (Varian Inc., Palo Alto, CA) imaging system with direct drive technology. To prevent drying and to minimize magnetic susceptibility artifacts during scanning, specimens were immersed in a flourinert liquid, FC-43 (3M, St. Paul, MN). A standard imaging experiment with an isotropic resolution of 80 µm comprised a field of view of 15.4 × 14 × 11 mm3 and a matrix size of 192 × 132 (75% partial Fourier: 176) × 137. With eight averages and a repetition time of 75 ms (echo time 25 ms) the total scan time was three hours. Matlab (The MathWorks, Inc., Natick, MA) was used for image postprocessing. By zero-filling all directions by a factor of two, the pixel resolution of a standard imaging experiment was 40 (µm)3.
Landmark data collection
Three-dimensional (3D) coordinates of 59 skull landmarks (Table 2, Fig 1) were recorded on the 3D µCT isosurfaces reconstructed using AVIZO 6.2 (Visualization Sciences Group, VSG). Landmarks are locations on biological forms that hold biological significance and that can be precisely defined and located with an acceptable degree of measurement error (Richtsmeier et al., 2002). 3D coordinates of landmark locations were digitized twice on each specimen by the same observer and measurement error was minimized by averaging the coordinates of the two trials (Richtsmeier et al., 1995; Aldridge et al., 2005). To ascertain the accuracy and reproducibility of landmark placement, intraobserver error (i.e., absolute difference between the two trials) was checked for every landmark. If landmark placement differed by more than 0.05 mm, the position of the landmark was remeasured. In addition to skull shape defined by a selection of 39 landmarks that cover the whole skull, subsets of 3D landmarks were defined to represent the shape of the three major regions of the mammalian skull: facial skeleton (N=22), cranial vault (N=10), cranial base (N=15), and palate (N=8) (Table 2, Fig 1).
For analysis of the brain, non-neural tissue was removed from the MRM images using Amira 5.2 (Visualization Sciences Group, VSG). Three-dimensional coordinates of 15 brain landmarks (Table 4, Fig 4) were collected from the 3D reconstructions of the brain using etdips (http://www.cc.nih.gov/cip/software/etdips/). 3D coordinates of landmark locations were digitized twice by the same observer and averaged to minimize measurement error. Coordinate data were checked for gross errors, and the absolute difference between the two trials was compared; if landmark placement differed by more than 0.08 mm (i.e., two pixels), the position of the landmark was remeasured. Brain shape defined by all landmarks was analyzed, as were two subsets of landmarks defined to represent the shape of the surface of the brain and the shape of the brain deep to the surface (Table 4, Fig 4). As the dominant feature of Crouzon syndrome is craniosynostosis and skull dysmorphology, we hypothesized that the effects of the Fgfr2c C342Y mutation would have differential effects on brain structures located adjacent to the cranium (i.e., the surface) as compared to structures located farther from the cranium (i.e., deeper, internal structures).
Shape analysis
To extract shape information for each dataset/subset of skull and brain landmarks we performed separate General Procrustes Analysis (GPA) to superimpose the coordinate data (Rohlf and Slice, 1990b; Dryden and Mardia, 1998) using the MorphoJ software package (Klingenberg, 2008). This procedure minimizes the influence of size and adopts a single orientation for all specimens by shifting the landmark configurations to a common position, scaling them to a standard size and rotating them until a best fit of corresponding landmarks is achieved. GPA removes the effects of scale, translation and rotation (Rohlf and Slice, 1990b), but does not eliminate the allometric shape variation that is related to size. In order to test how allometry affected our samples, we computed a regression of shape (represented by Procrustes coordinates) on centroid size (Drake and Klingenberg, 2008).
To statistically evaluate shape differences between Fgfr2cC342Y/+ mice and unaffected littermates, we used Euclidean Distance Matrix Analysis (EDMA; (Lele and Richtsmeier, 2001)). EDMA is a 3D morphometric technique that is invariant to the group of transformations consisting of translation, rotation, and reflection (Lele and McCulloch, 2002) (Richtsmeier et al., 2002). The original 3D coordinates of landmark locations collected from the forms were re-written and analyzed as a matrix of all unique linear distances among landmarks called the form matrix, FM. The original landmark coordinate data are used to estimate a mean FM for each sample being considered following Lele and Richtsmeier (Lele and Richtsmeier, 2001). Form difference between samples is evaluated by calculating ratios of like-linear distances using the mean FMs of the two samples. The matrix of ratios, the form difference matrix (FDM) is used to estimate a statistic against which information from boot strapped samples are evaluated. The null hypothesis of similarity in shape is rejected if estimates from the data fall outside of the 95% bootstrapped confidence intervals. Further examination of the FDM identifies patterns of localized differences in form between the two samples. To statistically test for localized differences in form, an alternate nonparametric bootstrap procedure calculates the confidence interval for each linear distance (α = 0.10). If this interval contains the value 1.0, the null hypothesis of similarity for that linear distance is accepted. Confidence interval testing revealed the localized effects of the Fgfr2c C342Y mutation on the facial skeleton, the cranial base, the neurocranium and the palate. EDMA analyses were performed using WinEDMA v. 1.0.1 (Cole, 2002).
Evaluation of cranial suture fusion
All sutures were scored using isosurfaces segmented for bone using hydroxyapatite phantoms imaged with the specimens as described above. In the case of the appearance of fusion, we examined individual slice images for that portion of the suture to verify fusion. All sutures were scored bilaterally, excepting the inter-premaxillary and inter-palatine sutures that are midline sutures. For each specimen, sutures were scored qualitatively as open when more than 75% of the length of the suture was completely patent; partial when more than 25% but less than 75% of the length of the suture was patent; and fused when less than 25% of the length of the suture was patent. Given the discrete nature of patency of the right and left zygomatic-maxillary and premaxilla-maxillary facial sutures, we assigned a discrete score of either open or fused. Because the inter-premaxillary suture showed highly localized variation in patency, we attempted to determine whether or not this variation was patterned by dividing the suture into thirds for scoring. The divisions are: the most anterior third, medial third, and most posterior third (see Table 3).
Segmentation of upper airway, vitreous humor, and inner ear volumes
The nasopharynx, eyes and inner ear volumes of a subset of specimens were analyzed (Table 1). The irregular and relatively smooth shapes of these biological structures prevented the acquisition of repeatable landmark data. Consequently, volumetric analyses were conducted. To meet requirements for segmentation, the nasopharynx of the individual had to be visibly perfused with Magnevist solution (a contrast medium used in MRM; see Imaging protocols). Structures were segmented from MRM images of each mouse twice using label fields and the segmentation editor of Avizo version 6.3 (VSG). Quantification of volume was performed using the MaterialStatistics module of Avizo, computed as the number of voxels multiplied by the size of a single voxel. Mean volumes from the trials were used in analyses and statistical analyses were conducted in SPSS version 20 (IBM SPSS Statistics, SPSS Inc., v. 20). Threshold values (min/max range = 89–255) were set for each specimen depending upon the individual MRM scan. As the inner ears and vitreous body are closed structures, boundaries of these structures were defined by the change in density of tissues that defined the outer limits of these regions. In essence, virtual endocasts of the fluid filled inner ear structures and the vitreous humor-filled vitreous body were segmented and their volumes were estimated. Since the nasopharynx is a structure that continues to the oropharynx and trachea, an inferior endpoint was needed in order to limit the segmentation. To accomplish this, 3D coordinates of 4 neural landmarks (denoting the most dorsolateral point of intersection of the olfactory bulb with rostral frontal lobe surface (left and right) and the origin of middle of cerebral artery from the circle of Willis on ventral cerebral surface (left and right)) (Aldridge et al., 2010) were located on the MRM of the brain using Avizo. These landmarks were used to create an oblique plane through the specimen that defined the caudal endpoint of the nasopharynx as measured in this study. The nasopharynx of each specimen was segmented and the volume estimated twice by the same observer.
Supplementary Material
Take-Home Points.
The most common Fgfr2 Crouzon syndrome mutation affects development of cranial tissues of varying embryological origin, as revealed by precise morphometric analysis of multi-modal images of heads of Fgfr2cC342Y/+ Crouzon syndrome mouse model.
Expanded catalogue of clinical cranial phenotypes in Crouzon syndrome caused by aberrant FGF/FGFR signaling includes skull, brain, nasopharynx, and eye dysmorphologies.
Effects of the Fgfr2c C342Y mutation on various head tissues points to primary effects of this mutation on cell-cell signaling required in the development of each of these tissues and their integration.
Findings contribute to the growing evidence that FGF/FGFR signaling is part of a multifaceted set of interactions among genes and regulatory networks that drive communication among cells and tissues in the development of the head that has been highly conserved in vertebrate evolution.
Multimodal imaging and 3D morphometric methods used to evaluate differences in tissue and organ development in Fgfr2cC342Y/+ Crouzon syndrome mice can be applied to other models of human disease.
ACKNOWLEDGEMENTS
We thank Timothy Ryan, Timothy Stecko and Thomas Neuberger for excellent technical work in acquiring µCT and MRM images. Ken Weiss, Anne Buchanan, Chris Percival, and Yann Heuzé offered helpful suggestions throughout the development of this study. David Ornitz generously provided the Fgfr2cC342Y/+ Crouzon syndrome mice for this study. This research was supported in part by NIH/NIDCR grants R01DE018500, 3R01DE018500-02S1 3R01DE018500-02S2 and R01 1R01DE022988.
Footnotes
Conflict of interest statement: All authors declare that there are no conflicts of interest
REFERENCES CITED
- Abzhanov A, Rodda SJ, McMahon AP, Tabin CJ. Regulation of skeletogenic differentiation in cranial dermal bone. Development. 2007;134:3133–3144. doi: 10.1242/dev.002709. [DOI] [PubMed] [Google Scholar]
- Aldridge K, Boyadjiew S, Capone G, DeLeon V, Richtsmeier JT. Precision and error of three-dimensional phenotypic measures acquired from 3dMD photogrammetric images. Amer J Med Genet. 2005;138:247–253. doi: 10.1002/ajmg.a.30959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge K, Hill CA, Austin JR, Percival C, Martinez-Abadias N, Neuberger T, Wang Y, Jabs EW, Richtsmeier JT. Brain phenotypes in two FGFR2 mouse models for Apert syndrome. Dev Dyn. 2010;239:987–997. doi: 10.1002/dvdy.22218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand S, Camasses A, Somorjai I, Belgacem MR, Chabrol O, Escande ML, Pontarotti P, Escriva H. Amphioxus FGF signaling predicts the acquisition of vertebrate morphological traits. Proc Natl Acad Sci U S A. 2011;108:9160–9165. doi: 10.1073/pnas.1014235108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MM Jr, MacLean RE, editors. Craniosynostosis: Diagnosis, Evaluation, and Management. New York: Oxford University Press; 2000. pp. 361–365. [Google Scholar]
- Cole T., III WinEDMA. Windows-Based Software for Euclidean Distance Matrix Analysis. 2002 In. pp. [Google Scholar]
- Cremers CW. Hearing loss in Pfeiffer's syndrome. Int J Pediatr Otorhinolaryngol. 1981;3:343–353. doi: 10.1016/0165-5876(81)90059-8. [DOI] [PubMed] [Google Scholar]
- Crump JG, Maves L, Lawson ND, Weinstein BM, Kimmel CB. An essential role for Fgfs in endodermal pouch formation influences later craniofacial skeletal patterning. Development. 2004;131:5703–5716. doi: 10.1242/dev.01444. [DOI] [PubMed] [Google Scholar]
- Darroch J, Mosimann J. Canonical and principal components of shape. Biometrika. 1985;72:241–252. [Google Scholar]
- de Jong T, Toll MS, de Gier HH, Mathijssen IM. Audiological profile of children and young adults with syndromic and complex craniosynostosis. Arch Otolaryngol Head Neck Surg. 2011;137:775–778. doi: 10.1001/archoto.2011.115. [DOI] [PubMed] [Google Scholar]
- Degnin CR, Laederich MB, Horton WA. FGFs in endochondral skeletal development. J Cell Biochem. 2010;110:1046–1057. doi: 10.1002/jcb.22629. [DOI] [PubMed] [Google Scholar]
- Drake AG, Klingenberg CP. The pace of morphological change: historical transformation of skull shape in St Bernard dogs. Proc Biol Sci. 2008;275:71–76. doi: 10.1098/rspb.2007.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryden I, Mardia K. Statistical shape analysis. Chichester: John Wiley & Sons; 1998. [Google Scholar]
- Eswarakumar VP, Horowitz MC, Locklin R, Morriss-Kay GM, Lonai P. A gain-of-function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis. Proc Natl Acad Sci U S A. 2004;101:12555–12560. doi: 10.1073/pnas.0405031101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Hatch NE. FGF signaling in craniofacial biological control and pathological craniofacial development. Crit Rev Eukaryot Gene Expr. 2010;20:295–311. doi: 10.1615/critreveukargeneexpr.v20.i4.20. [DOI] [PubMed] [Google Scholar]
- Hebert JM. FGFs: Neurodevelopment's Jack-of-all-Trades - How Do They Do it? Front Neurosci. 2011;5:133. doi: 10.3389/fnins.2011.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill C, Martínez-Abadías N, Motch S, Austin J, Wang Y, Jabs E, Richtsmeier J, Aldridge K. Growth of the skull and brain differ postnatally in a mouse odel for Apert syndrome. Amer J Med Genet. 2012 doi: 10.1002/ajmg.a.35805. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh SH, Jones J, Warchol ME, Ornitz DM. Differentiation of the lateral compartment of the cochlea requires a temporally restricted FGF20 signal. PLoS Biol. 2012;10:e1001231. doi: 10.1371/journal.pbio.1001231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Johnson D, Wilkie AO. Craniosynostosis. Eur J Hum Genet. 2011;19:369–376. doi: 10.1038/ejhg.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg C. Faculy of Life Sciences. UK: University of Manchester; 2008. MophoJ. http://www.flywings.org.uk/MorphoJ_page.htm. [Google Scholar]
- Lele S, McCulloch C. Invariance, identifiability and morphometrics. JASA. 2002;971:796–806. [Google Scholar]
- Lele S, Richtsmeier JT. An invariant approach to the statistical analysis of shapes. London: Chapman and Hall/CRC Press; 2001. 308 pp. [Google Scholar]
- Martínez-Abadías N, Heuze Y, Wang Y, Jabs EW, Aldridge K, Richtsmeier JT. FGF/FGFR signaling coordinates skull development by modulating magnitude of morphological integration: evidence from Apert syndrome mouse models. PLoS One. 2011;6:e26425. doi: 10.1371/journal.pone.0026425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Abadías N, Percival C, Aldridge K, Hill C, Ryan T, Sirivunnabood S, Wang Y, Jabs E, Richtsmeier J. Beyond the closed suture in Apert syndrome mouse models: evidence of primary effects of FGFR2 signaling on facial shape at birth. Dev Dyn. 2010;239:3058–3071. doi: 10.1002/dvdy.22414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBratney-Owen B, Iseki S, Bamforth SD, Olsen BR, Morriss-Kay GM. Development and tissue origins of the mammalian cranial base. Dev Biol. 2008;322:121–132. doi: 10.1016/j.ydbio.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsukawa N, Satoh K. Midfacial distraction using a transfacial pinning technique for syndromic craniosynostosis with obstructive respiratory disorders. J Plast Reconstr Aesthet Surg. 2010;63:1990–1994. doi: 10.1016/j.bjps.2010.01.029. [DOI] [PubMed] [Google Scholar]
- Mitsukawa N, Satoh K, Hayashi T, Furukawa Y, Uemura T, Hosaka Y. A reflectable case of obstructive sleep apnea in an infant with Crouzon syndrome. J Craniofac Surg. 2004;15:874–878. doi: 10.1097/00001665-200409000-00035. discussion 878–879. [DOI] [PubMed] [Google Scholar]
- Moore MH. Upper airway obstruction in the syndromal craniosynostoses. Br J Plast Surg. 1993;46:355–362. doi: 10.1016/0007-1226(93)90039-e. [DOI] [PubMed] [Google Scholar]
- Moss M. The pathogenesis of premature cranial synostosis in man. Acta Anat. 1959;37:351–370. doi: 10.1159/000141479. [DOI] [PubMed] [Google Scholar]
- Moss M. The functional matrix. In: Kraus B, Riedel R, editors. Vistas in orthodontics. Chicago: Lea & Febiger; 1962. pp. 85–98. [Google Scholar]
- Neilson KM, Friesel RE. Constitutive activation of fibroblast growth factor receptor-2 by a point mutation associated with Crouzon syndrome. J Biol Chem. 1995;270:26037–26040. doi: 10.1074/jbc.270.44.26037. [DOI] [PubMed] [Google Scholar]
- Noden DM, Trainor PA. Relations and interactions between cranial mesoderm and neural crest populations. J Anat. 2005;207:575–601. doi: 10.1111/j.1469-7580.2005.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-3-reviews3005. REVIEWS3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446–1465. doi: 10.1101/gad.990702. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Bedford MT, Burakova T, Arman E, Zimmer Y, Yayon A, Givol D, Lonai P. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2) Dev Biol. 1993;158:475–486. doi: 10.1006/dbio.1993.1205. [DOI] [PubMed] [Google Scholar]
- Orvidas LJ, Fabry LB, Diacova S, McDonald TJ. Hearing and otopathology in Crouzon syndrome. Laryngoscope. 1999;109:1372–1375. doi: 10.1097/00005537-199909000-00002. [DOI] [PubMed] [Google Scholar]
- Perkins JA, Sie KC, Milczuk H, Richardson MA. Airway management in children with craniofacial anomalies. Cleft Palate Craniofac J. 1997;34:135–140. doi: 10.1597/1545-1569_1997_034_0135_amicwc_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Perlyn CA, DeLeon VB, Babbs C, Govier D, Burell L, Darvann T, Kreiborg S, Morriss-Kay G. The craniofacial phenotype of the Crouzon mouse: analysis of a model for syndromic craniosynostosis using three-dimensional MicroCT. Cleft Palate-Craniofacial Journal. 2006;43:740–748. doi: 10.1597/05-212. [DOI] [PubMed] [Google Scholar]
- Peterson-Falzone SJ, Pruzansky S, Parris PJ, Laffer JL. Nasopharyngeal dysmorphology in the syndromes of Apert and Crouzon. Cleft Palate Journal. 1981;18:237–250. [PubMed] [Google Scholar]
- Peterson SJ, Pruzansky S. Palatal anomalies in the syndromes of Apert and Crouzon. Cleft Palate Journal. 1974;11:394–403. [PubMed] [Google Scholar]
- Popovici C, Roubin R, Coulier F, Birnbaum D. An evolutionary history of the FGF superfamily. Bioessays. 2005;27:849–857. doi: 10.1002/bies.20261. [DOI] [PubMed] [Google Scholar]
- Proudman TW, Clark BE, Moore MH, Abbott AH, David DJ. Central nervous system imaging in Crouzon's syndrome. J Craniofac Surg. 1995;6:401–405. doi: 10.1097/00001665-199509000-00016. [DOI] [PubMed] [Google Scholar]
- Randhawa PS, Ahmed J, Nouraei SR, Wyatt ME. Impact of long-term nasopharyngeal airway on health-related quality of life of children with obstructive sleep apnea caused by syndromic craniosynostosis. J Craniofac Surg. 2011;22:125–128. doi: 10.1097/SCS.0b013e3181f6f82c. [DOI] [PubMed] [Google Scholar]
- Richtsmeier J, Paik C, Elfert P, Cole TI, Dahlman H. Precision, repeatability, and validation of the localization of cranial landmarks using computed tomography scans. Cleft Palate Craniofac J. 1995;32:217–227. doi: 10.1597/1545-1569_1995_032_0217_pravot_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Richtsmeier JT, Deleon VB, Lele SR. The promise of geometric morphometrics. Yearbook of Physical Anthropology, Vol 45. 2002;45:63–91. doi: 10.1002/ajpa.10174. [DOI] [PubMed] [Google Scholar]
- Riley BM, Mansilla MA, Ma J, Daack-Hirsch S, Maher BS, Raffensperger LM, Russo ET, Vieira AR, Dode C, Mohammadi M, Marazita ML, Murray JC. Impaired FGF signaling contributes to cleft lip and palate. Proc Natl Acad Sci U S A. 2007;104:4512–4517. doi: 10.1073/pnas.0607956104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlf FJ, Slice D. Extensions of the Procrustes method for the optimal superimposition of landmarks. Syst. Zool. 1990a;39:40–59. [Google Scholar]
- Rohlf FJ, Slice D. Extensions of the Procrustes method for the optimal superimposition of landmarks. Syst Zool. 1990b;39:40–59. [Google Scholar]
- Rutland P, Pulleyn LJ, Reardon W, Baraitser M, Hayward R, Jones B, Malcolm S, Winter RM, Oldridge M, Slaney SF, Poole MD, Wilkie AOM. Identical Mutations in the Fgfr2 Gene Cause Both Pfeiffer and Crouzon Syndrome Phenotypes. Nature Genetics. 1995;9:173–176. doi: 10.1038/ng0295-173. [DOI] [PubMed] [Google Scholar]
- Scheid SC, Spector AR, Luft JD. Tracheal cartilaginous sleeve in Crouzon syndrome. Int J Pediatr Otorhinolaryngol. 2002;65:147–152. doi: 10.1016/s0165-5876(02)00132-5. [DOI] [PubMed] [Google Scholar]
- Sirotnak J, Brodsky L, Pizzuto M. Airway obstruction in the Crouzon syndrome: case report and review of the literature. Int J Pediatr Otorhinolaryngol. 1995;31:235–246. doi: 10.1016/0165-5876(94)01090-k. [DOI] [PubMed] [Google Scholar]
- Snyder-Warwick AK, Perlyn CA, Pan J, Yu K, Zhang L, Ornitz DM. Analysis of a gain-of-function FGFR2 Crouzon mutation provides evidence of loss of function activity in the etiology of cleft palate. Proc Natl Acad Sci U S A. 2010;107:2515–2520. doi: 10.1073/pnas.0913985107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo-Rogers HL, Geetha-Loganathan P, Nimmagadda S, Fu KK, Richman JM. FGF signals from the nasal pit are necessary for normal facial morphogenesis. Dev Biol. 2008;318:289–302. doi: 10.1016/j.ydbio.2008.03.027. [DOI] [PubMed] [Google Scholar]
- Thisse B, Thisse C. Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev Biol. 2005;287:390–402. doi: 10.1016/j.ydbio.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nature Reviews Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- Vallino-Napoli LD. Audiologic and otologic characteristics of Pfeiffer syndrome. Cleft Palate Craniofac J. 1996;33:524–529. doi: 10.1597/1545-1569_1996_033_0524_aaocop_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Wilkie AO. Craniosynostosis: genes and mechanisms. Hum Mol Genet. 1997;6:1647–1656. doi: 10.1093/hmg/6.10.1647. [DOI] [PubMed] [Google Scholar]
- Wilkie AO. Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev. 2005;16:187–203. doi: 10.1016/j.cytogfr.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Yeh BK, Igarashi M, Eliseenkova AV, Plotnikov AN, Sher I, Ron D, Aaronson SA, Mohammadi M. Structural basis by which alternative splicing confers specificity in fibroblast growth factor receptors. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2266–2271. doi: 10.1073/pnas.0436500100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JC, Lucas JH, Fryberg K, Borke JL. Extrinsic tension results in FGF-2 release, membrane permeability change, and intracellular Ca++ increase in immature cranial sutures. Journal of Craniofacial Surgery. 2001;12:391–398. doi: 10.1097/00001665-200107000-00018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.