

Abstract
Drug attrition rates have increased in past years, resulting in growing costs for the pharmaceutical industry and consumers. The reasons for this include the lack of in vitro models that correlate with clinical results, and poor preclinical toxicity screening assays. The in vitro production of human cardiac progenitor cells and cardiomyocytes from human pluripotent stem cells provides an amenable source of cells for applications in drug discovery, disease modeling, regenerative medicine, and cardiotoxicity screening. In addition, the ability to derive human induced pluripotent stem cells from somatic tissues, combined with current high-throughput screening and pharmacogenomics, may help realize the use of these cells to fulfill the potential of personalized medicine. In this review, we discuss the use of pluripotent stem cell-derived cardiomyocytes for drug discovery and cardiotoxicity screening, as well as current hurdles that must be overcome for wider clinical applications of this promising approach.
Keywords: pluripotent stem cells, cardiovascular disease, toxicity screening, drug discovery
INTRODUCTION
Heart disease is the most significant cause of morbidity and mortality in the industrialized world, accounting for nearly 33% of all deaths in 2008 within the United States alone [1]. Over 82 million Americans suffer from some form of cardiovascular disease (CVD), accounting for nearly 27% of the entire population [2]. Billions of dollars are spent on drug therapy in the U.S. every year, and drugs for CVD are some of the most widely used on the market. However, despite the latest advances in research, much remains to be learned about pharmacological and cellular treatments for cardiovascular disease. Recently, the isolation and propagation of pluripotent stem cells such as human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs) has provided new opportunities for the development of novel approaches to treat heart disease [3-6]. While the heart does have some regenerative ability due to the presence of proposed cardiac stem cells, human adult cardiomyocytes (CMs) have only a limited regenerative capacity (with an estimated renewal rate of approximately 1% per year during its peak) and lose their ability to divide postpartum [7-10]. The therapeutic potential of engraftment of human pluripotent stem cell-derived CMs to repair cardiac damage is highly promising, but many technological challenges remain for this approach to advance from the bench to the bedside [11-14]. In addition, cell-based therapeutic strategies for cardiac repair must satisfy a considerable number of criteria that can critically affect safety and potential risks to patients such as cell biodistribution, tumorigenic potential, and immunogenicity [15, 16].
In contrast to potential therapeutic engraftment studies, the use of human pluripotent stem cell-derived CMs for drug discovery and screening has already proven promising, as hiPSC-CMs have been shown to respond to cardioactive drugs in a similar way as hESCs, comparable to empirical results seen in a clinical setting [17-19]. There is much need for the replacement of current in vitro and in vivo cardiotoxicity and arrhythmogenesis models that rely on animal- or tumor-derived cell lines, lines immortalized by genetic modifications, and isolated tissues such as perfused animal hearts [20-22]. These functional assays are used at various stages of drug development, including target identification and validation, library screening for early hits and leads, and pharmacological analysis of lead optimization and potential drug candidate selection. In these assays, pharmacologically targeted receptors and proteins of interest are transfected into cell lines or expressed in animal models to mimic a functional human system. However, many new chemical entities (NCEs) in early preclinical studies have failed because targets validated in both in vitro assays and in vivo animal models often prove to be unreliable and non-predictive when translated to humans. In addition to drug discovery, the use of pluripotent stem cells as tools for modeling cardiac development and disease is another important application. This goal often relies on the use of microarrays and other genomic approaches for the phenotyping of novel cardiac-associated genes during pluripotent stem cell differentiation [23, 24]. With the number of drugs approved by the U.S. Food and Drug Administration (FDA) decreasing every year, these models could also eventually spur the discovery of novel drug pathways to target using pharmacological therapy, ultimately with the potential to lead to new classes of drugs.
Developmental biology of the heart
The human heart is the first organ to be formed and function in utero, and all subsequent life events are dependent on its capacity to meet the demands for oxygenating the developing organism. The heart is derived from the mesodermal germ layer that is established by a signaling cascade starting with NODAL [25, 26]. The high levels of NODAL expression in the proximal epiblast at E5.0 maintain bone morphogenetic protein-4 (BMP4) expression in the extraembryonic ectoderm adjacent to the epiblast [27-29]. BMP4 acts by inducing WNT3 expression in the proximal epiblast [30]. It is at this pre-gastrulation stage that the expression of mesoendodermal markers such as T, MIXL1, EOMES and GSC is detected [31-34]. Unpatterned mesoderm expresses kinase insert domain receptor (KDR, FLK1/VEGFR2) and platelet-derived growth factor receptor alpha (PDGFRA) [35, 36]. Lateral plate mesoderm, which forms the cardiac lineage, subsequently down-regulates PDGFRA, whereas paraxial mesoderm down-regulates KDR [37]. The lateral mesoderm moves in an anterior direction to populate positions on either side of the midline, creating the cardiac crescent [38]. This epithelial crescent initially functions as a contractile cardiac tube of myocardium, which then undergoes a rightward looping and expansion of the myocardium, culminating in a segmented structure and the formation of cardiac chambers [39].
Recent analyses have revealed that two distinct mesodermal heart fields contribute to the developing heart. The first heart field is responsible for the formation of the primitive heart tube, generating endocardial and myocardial cell populations therein. Cells of the second heart field migrate onto a scaffold generated for it by the primary heart field in order to establish the cardiac chambers. Both heart fields contribute to most cardiac structures; however, recent molecular analyses have discerned possible differences in their innate developmental potential. This becomes important when one is considering what might be the most competent cell type to engraft and repair a failing heart. Unlike the genesis of skeletal muscle, for which a single transcription factor, myogenic differentiation 1 (MYOD1), is sufficient, cardiogenesis appears to rely on a complex web of interacting factors. Myocyte-specific enhancer factor 2C (MEF2C) coordinates the activation of myofibrillar protein encoding genes, whereas the serum response factor (SRF) acts in concert with other factors like NKX2-5, GATA4, and MYOCD to modulate the expression of structural cardiac proteins such as troponins, actins and myosins (e.g., TNNT2, TNNI3, ACTN2, MYH6, and MYH7) [40-43]. The expression of NKX2-5 and GATA4 are heavily dependent on the activating signals of the BMPs. Furthermore, ventricular genes are specifically activated by the IRX4 while simultaneously suppressing expression of atrial genes within the ventricular chambers [44]. These observations are now further complicated by reports of the existence of two populations of cardiac progenitors, one for each of the first and second heart fields [45-47]. Importantly, these populations share significant pathway components in their regulation, such as a common dependence on NKX2-5 in orchestrating their differentiation [39]. However, their differences also underscore a potentially critical divergence in their biological roles; most notably, the secondary heart field is proposed to contain a population of cardiac progenitor stem cells. This secondary heart field multipotent cardiovascular progenitor appears to be characterized by its expression of the LIM homeodomain transcription factor ISL1, in combination with expression of other cardiac genes like NKX2-5 and KDR.
Differentiation of CM from human pluripotent stem cells
The recent discovery of murine iPSCs (miPSCs) in 2006 and hiPSCs in 2007 as an alternative, non-embryonic derived source of pluripotent stem cells has resulted in significant advances in the field of stem cell research over the past five years [3, 4, 48, 49]. This groundbreaking work has opened the door for a new area of stem cell research, as it can avoid many of the ethical and political concerns associated with hESCs. Unlike hESCs, hiPSCs can be generated from somatic cells such as fibroblasts via delivery and ectopic expression of the Yamanaka factors (Oct4, Sox2, Klf4, and c-Myc), also known as OKSM. Recent studies have also demonstrated the successful derivation of iPSCs from other sources, including amniotic fluid, human blood and keratinocytes [50-52]. Multiple methods to deliver these critical factors have been published, such as the use of integrating approaches (lentiviruses and retroviruses), as well as non-integrating approaches such as adenoviruses, Sendai virus, polycistronic minicircle vectors, and self-replicating selectable episomes [53-55]. Once pluripotency is established, hiPSCs can be differentiated into CMs by various methods [56]. Traditionally, human pluripotent stem cells are differentiated to the cardiac lineage in culture by allowing colonies to form three-dimensional aggregates in suspension known as “human embryoid bodies” (hEBs). It is thought that these three-dimensional structures recapitulate some of the growth factor gradients and cell-cell interactions that normally exist in the human embryo. In general, there are currently three well-described techniques for cardiac differentiation of hESC. The first is an EB-based technique that uses a defined proprietary medium, in addition to ascorbic acid, hypoxic conditions, and a rigorously defined growth factor regiment [57]. In the second method, hESCs are cultured directly on stromal cells such as murine OP9 or visceral endoderm-like cells [58, 59]. The third approach is a monolayer-based method that uses a medium consisting of RPMI 1640, the supplement B-27, and growth factors activin A and BMP4 with high-density hESC cultured on Matrigel [60, 61]. In this last technique, the medium is not changed for 4 days (between days 1 and 5), which allows for a buildup of secreted factors to occur. In all three systems, contracting cells are witnessed after 8-12 days. Similar methods can also be used for the differentiation of hiPSCs into CMs.
The use of hiPSC-CM for drug discovery and development
Novel drug discovery, development, and safety testing make up a long, arduous and expensive process that is plagued by the lack of economical and reliable methods that can accurately mimic the human physiological response. High drug attrition rates continue to bedevil the research and development process, including the more than 40% of NCEs that enter phase III clinical trials which fail due to ineffectiveness and/or unforeseen toxicities [62]. The pharmaceutical industry currently invests ~ $1.5 billion to take a candidate drug from primary screen to market, a process lasting an average of 10-15 years, and many drugs are withdrawn due to side effects associated with off- and on-target electrophysiological alterations and biochemical cardiotoxicity [63]. For example, in an eight-year span from 2000-2007, drugs such as the non-steroidal anti-inflammatory drug (NSAID) Vioxx (rofecoxib), the gastrointestinal prokinetic drug Propulsid (cisapride), and the 5- hydroxytryptamine receptor 4 (5HT4) agonist Zelnorm (tegaserod) were all withdrawn from the U.S. market following unforeseen clinical cardiotoxic and/or arrhythmogenic toxicities. Off-target cardiac toxicity is the most common cause of regulatory delay in approval and market withdrawal of newly developed pharmaceuticals [20, 64]. In addition, members of several non-cardiovascular drug classes, including the anthracyclines, non-steroidal anti-inflammatory drugs (NSAIDs) and certain first- and second-generation antipsychotics, carry cardiovascular-related black box warnings (Table 1). Decreasing the failure rate of novel therapeutics could result in a significant reduction in both the length of time of drug development as well as pharmaceutical company and consumer costs.
Table 1.
Approved drugs with cardiovascular-related black box warnings
| Trade name (generic name) | Indication(s) | Black box warning and associated cardiotoxicity |
|---|---|---|
| Anthracyclines including Adriamycin (doxorubicin), Cerubidine (daunorubicin), and Ellence (epirubicin) | Anticancer | Black box warnings (cardiotoxicity) |
| Avandia (rosiglitazone) | Type 2 diabetes | Black box warning (congestive heart failure and myocardial infarction) |
| Betapace (sotalol) | Antiarrhythmic | Black box warning (proarrhythmic effects) |
| Clozaril (clozapine) | Antipsychotic | Black box warning (myocarditis) |
| Cordarone, Pacerone (amiodarone) | Antiarrhythmic | Black box warning (proarrhythmic effects) |
| Herceptin (trastuzumab) | Breast cancer | Black box warning (cardiomyopathy) |
| Inapsine (droperidol) | Antiemetic and antipsychotic | Black box warning (QT prolongation and TdP) |
| Mellaril (thioridazine) | Antipsychotic | Black box warning (QT prolongation and TdP) |
| Novantrone (mitoxantrone) | Anticancer and multiple sclerosis | Black box warning (cardiotoxicity) |
| NSAIDs including Anaprox (naproxen), Cataflam (diclofenac), Celebrex (celecoxib), Orudis (ketoprofen) and Motrin (ibuprofen) | Pain and inflammation | Black box warnings (increased cardiovascular risk) |
| Sporanox (itraconzaole) | Antifungal | Black box warning (congestive heart failure) |
| Tambocor (flecainide) | Antiarrhythmic | Black box warning (ventricular arrhythmias) |
| Tasigna (nilotinib) | Leukemia | Black box warning (QT prolongation and TdP) |
| Tikosyn (dofetilide) | Antiarrhythmic | Black box warning (proarrhythmic effects) |
| Tyrosine kinase inhibitors including Gleevec (imatinib) and Sprycel (dasatinib) | Anticancer | No black box warnings (reports of cardiotoxicity) |
High drug attrition rates during early drug development are often related to suboptimal screening assays that do not accurately translate to in vivo results [65]. Some of the current barriers to improving the efficiency of novel drug discovery and development include the use of nonhuman animal models for the assessment of off-target toxicities and the lack of translation to potential human toxicities, the practice of performing early compound safety screening studies when only small quantities of the compound exist prior to scale-up for expensive animal model experiments, and the fact that small-scale early human clinical trials (usually around 20-50 patients) do not include rare but potentially relevant genetic backgrounds. CMs from animals may not translate to results observed in humans, and the utilization of primary human CMs is further limited by donor cell availability, problematic isolation procedures, and poor viability and proliferation capacity. The use of hiPSC-derived cardiac progenitor cells (CPCs) and CMs provide the potential to overcome these barriers by reducing the burden of each of these factors and therefore decreasing the time and cost of bringing new drugs to market. Importantly, hiPSC-CMs display many of the characteristics of normal in vivo CM, including molecular, structural and functional properties such as ion channel, transporter, and receptor expression, as well as similar electrophysiological properties and biochemical responses [66]. Other desirable properties of hiPSC-CMs include their ability to survive under cell culture conditions for extended periods of time, and the fact that they can be grown in controllable environmental conditions. In addition, a recent comparison of CMs derived from both hESCs and hiPSCs showed no observable differences in the time course for the development of contracting cells between these two types of pluripotent stem cells [67]. Large-scale generation of CMs from pluripotent stem cells, or disease-specific hiPSC lines such as patients with heart disease, dilated cardiomyopathy, LEOPARD syndrome, long QT syndrome, and Timothy syndrome hold the potential to serve as a high-throughput human-based model for both drug development and cardiotoxicity screening. All of these desirable properties favor the use of pluripotent stem cells for drug testing and toxicology screening [68-72]. This model could provide the pharmaceutical industry with a valuable tool for the pre-clinical screening of candidate anti-arrhythmic and anti-heart failure pharmacological agents, as well as other classes of medicines for the evaluation of secondary off-target cardiac toxicities, such as various classes of chemotherapeutic agents (Figure 1) [64, 73, 74].
Figure 1.
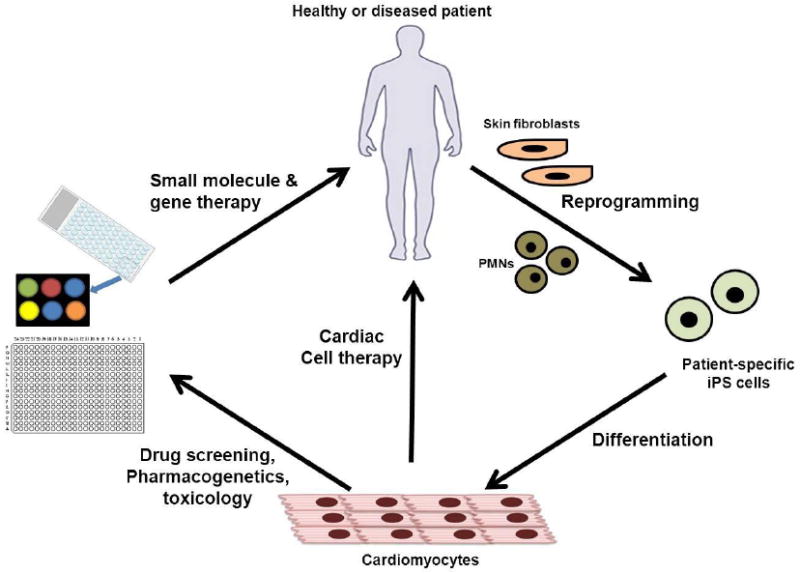
The use of human pluripotent stem cells for novel drug discovery, screening, and toxicity testing.
Evaluation of drug toxicity using hiPSC-CM
Action potentials are the rhythmic electrical oscillations of CM membrane potential that underlie the heartbeat as well as basic cardiac function. The action potential waveform results from ions crossing the plasma membrane through a variety of ion channels, the dysfunction of which can lead to an alteration of cardiac rhythm. Cardiac ion channel block and dysfunction arises from genetic mutations and/or interactions with drug compounds. Different species express different types of cardiac ion channels as well as differing relative levels of each channel type. Therefore, different toxicity assessments could be drawn from applying the same compound to CMs from different species. Dysfunction resulting in action potential prolongation is of particular concern as it can lead to ventricular arrhythmias and sudden death. The most common cause of cardiac action potential prolongation is drug-induced blockade of the human Ether-à-go-go-Related Gene (hERG) channel, a rapid component of the delayed rectifier potassium current (IKr) that controls action potential repolarization. Inhibition of hERG causes one of the most common negative cardiovascular drug effects, long QT syndrome (LQTS). LQTS is associated with an increased risk of ventricular arrhythmias such as Torsade de Pointes (TdP). Drug-induced TdP is often caused by blockade of IKr channels, resulting in activation of L-type calcium channels, which are normally inactive during the repolarization phase. This can lead to an early after-depolarization caused by inward calcium currents, resulting in TdP and potentially life-threatening ventricular fibrillation. Drug-induced TdP is the most common reason for the withdrawal or restricted use of many cardiovascular and non-cardiovascular drugs [75].
The clinical evaluation of effect of a new drug for potential delayed repolarization is currently performed using a hERG-binding assay involving radiolabeled drugs and immortalized cell lines expressing single recombinant hERG potassium channels. However, this assay has multiple limitations. Specifically, it is expensive and does not accurately predict the occurrence of organ toxicity or lethal side effects secondary to pharmacological agents. Also, since the hERG channel only accounts for phases 2 and 3 of the action potential, it cannot completely predict drug-induced LQTS associated with the blockade of other channels Figure 2) [76]. Due to these drawbacks, the use of hiPSC-CMs would provide a unique and more predictive model for pharmaceutical companies to study the potential in vitro cardiotoxic effects of novel pharmacologicals in early drug development [77].
Figure 2.
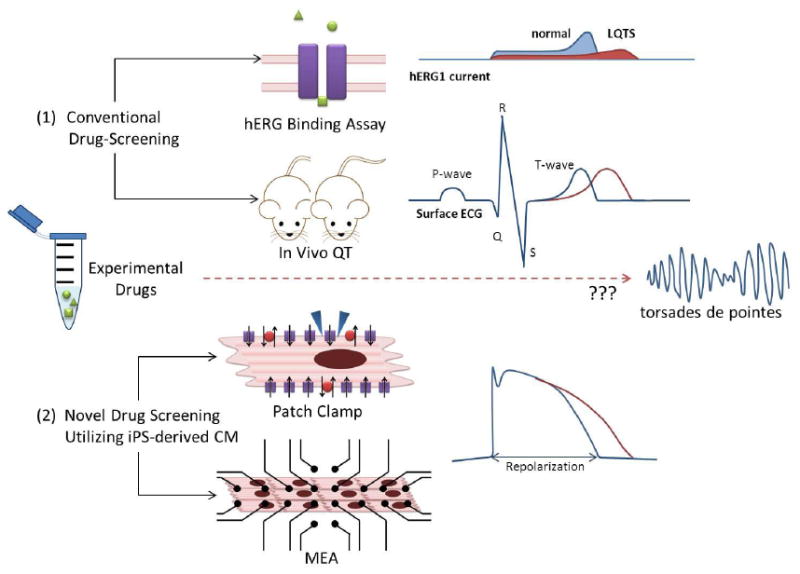
Cells expressing recombinant hERG potassium channels cannot completely predict drug-induced LQTS associated with the blockade of other channels.
The effects of drugs on pluripotent stem cell-derived CMs can be assessed using simple “number of beating hEBs” assays or more detailed studies such as the use of current laser capture microscopy and bioenergetics assays [78-80]. Patch-clamp assays and microelectrode arrays (MEAs) are often used to measure action potential duration (APD), field potential duration (FPD), as well as other measurements which can analyze the rhythm, excitation, repolarization, and conduction properties of beating CM clusters derived from both hESCs and hiPSCs after exposure to cardioactive drugs [81, 82]. Small-scale preliminary studies using MEAs on hiPSC-CMs have been performed, yielding promising results [83]. Both automated robotic high-throughput patch-clamp systems and MEA systems enabling high throughput screening of 96 or even 384 compounds simultaneously are already commercially available. These systems enable the measurement of the surface electrogenic activities of cell clusters and allow stable, long-duration recordings that are necessary to evaluate the relationship between dose-dependency and the induction of side effects by novel therapeutics. The washing out of drugs leads to the recovery of the field potential waveform almost to the baseline observed before the drug application, which means that this system can be used to test the reproducibility of pharmacological effects, and several drugs at varying concentrations may be tested using the same sample. In addition to high-throughput patch-clamp and MEA systems, recent studies have also demonstrated the ability to perform microfluidic-based single-cell, high-throughput, real-time polymerase chain reaction (RT-PCR) for the analysis of iPSCs and differentiated derivatives [84]. This technology also allows the ability to thoroughly investigate gene expression patterns in difficult to analyze heterogeneous cell populations from both healthy patients and patients with various disease states, and helps to overcome limitations associated with small sample sizes. The combination of these high-throughput assays with current in silico approaches provides a time-sensitive and cost-effective option for the evaluation of drug-induced cardiotoxicity in pluripotent stem cell-derived CMs [85]. The use of hiPSC-CMs also allows the possibility for drug testing on a panel of cell lines that might more closely reflect the genetic diversity of a population, including that seen in murine ESCs [86]. Genetic conditions that affect the heart may also be studied, including familial cardiomyopathy, familial lethal arrhythmias, and congenital heart diseases [87]. In addition, because hiPSC-CMs can be derived from both male and female patients, the use of these cells in drug screening may prove beneficial to evaluate gender differences in response to treatment. For example, almost two-thirds of TdP cases occur in women, who are more prone to drug-induced TdP [88, 89].
Publication standards for the characterization of hiPSC-CM
Despite the increasing popularity and use of iPSC-CM for drug discovery and toxicity screening, there are no published consensus guidelines for the characterization of these cells. These steps are of the utmost importance for the successful interpretation and reproducibility of subsequent experiments (Table 2). While beating cells will be observed at different time points depending on the specific differentiation method used, steps should be taken to assure that these contracting cells form striated sarcomeres, and express typical terminal cardiac markers and structural proteins such as α-actinin (ACTN2), cardiac troponin I (TNNI3), cardiac troponin T (cTNT), connexin43 (Cx43), myosin heavy chain 6 (MYH6), myosin light chain 2a (MLC2a), and troponin T type 2 (TNNT2). In addition, a detailed electrophysiological characterization should be done, including MEA recordings and patch-clamp techniques to record field potentials and action potentials, determine the percentage of atrial-, nodal-, and ventricular-like cells, as well as measure ion channel currents with and without the presence of specific agonists and antagonists. Calcium imaging and atomic force microscopy may also be useful to further characterize these cells, especially in cellular disease models where calcium transients and contraction forces play a significant role, such as dilated cardiomyopathy.
Table 2.
Characterization of hiPSC-CM
| Criteria | Assay(s) | Results |
|---|---|---|
| Electrophysiological properties | Multielectrode Array | Field potentials of beating cells including beat frequency, field potential duration (FPD), maximum positive amplitude (MPA), maximum negative amplitude (MNA), and interspike interval (ISI) |
| Patch Clamp | Action potential, classification of spontaneous action potentials (ventricular-, atrial-, or nodal-like) | |
| Expression of cardiac-specific genes and proteins | Immunohistochemistry and/or RT-PCR | Positive for cardiac markers and proteins such as ACTN2, cTNT, Cx43, MLC2a, MYH6, TNNI3, and TNNT2 |
| Structural properties and organization | Electron microscopy | Typical cardiomyocyte structural properties such as glycogen granules, mitochondria, myofibrils, sarcoplasmic reticulum striated sarcomeres, A-bands, I-bands, and Z-lines |
| Calcium signaling | Calcium imaging | Calcium transient waveforms, intracellular calcium voltage micromapping |
Current limitations
One of the major limitations preventing the large-scale use of these cells in drug discovery and screening is inefficient differentiation methodologies, which currently are incapable of producing CPCs or CMs at clinically or commercially relevant scales, nor to current good manufacturing practice (cGMP) standards. Current differentiation methods are designed around small-scale research paradigms [90]. These systems, although substantially improved over prior methods, are still vastly inefficient and provide mixed populations of terminal cells with a wide variability in cells derived from patients of different ages and sex [91]. For the scale-up to production levels, improvements in current differentiation techniques will need to be made, with a focus on large-scale pluripotent cell culture or automated culture [92, 93]. Also, hiPSC-CM are immature and do not express many markers of fully developed adult hearts. In addition, improvements will also need to be made in methodologies involving the purification of differentiated populations. Traditional methods for the purification of CMs, which can involve density gradient centrifugation or genetic modification for GFP or drug resistance expression, are unsuitable for large-scale or clinical practice, and routinely result in only a 5- to 10-fold enrichment in CM populations [94-96]. Also, producing hiPSC lines to cGMP standards in defined, feeder-free, and xeno-free conditions, as well as the exclusion of the use of retroviruses and oncogenic factors, represent other major challenges to stem cell biology. Recently, it has been shown that hiPSC lines can be derived in xeno-free conditions, and cardiac differentiation has been performed in serum-free conditions, albeit from cells cultured on mouse embryonic fibroblasts (MEFs) or animal tumor cell line-derived Matrigel [97, 98]. The further development of feeder-, serum-, and xeno-free conditions for the in vitro culture of pluripotent stem cells will be vital if they are to be used clinically in humans. Lastly, caution must be exercised when comparing results using hESCs versus hiPSCs. While it has been demonstrated that these two groups of stem cells share many properties, recent studies have also suggested that hiPSCs may not occupy the same pluripotent state as hESCs [99].
Conclusion
While the use of human pluripotent stem cell-derived CMs in regenerative medicine is a long-term goal, a growing body of studies already have shown promising results in the ability to use these cells in the field of drug discovery, development, and toxicity screening. The aim should not be to replace current methods of using animal-derived primary cells or immortalized cell lines, but to supplement their use with the combination of iPSC-CMs and high-throughput screening assays. This technology may one day provide a long sought after solution for multiple problems arising from the lengthy drug discovery process, including expensive preclinical studies that do not accurately translate to clinical results, high drug attrition rates, and market withdrawals.
Acknowledgments
We would like to acknowledge funding support from NIH R01 HL113006, Fondation Leducq 11CVD02, CIRM RB3-05129 (JCW) and AHA Postdoctoral Fellowship (PWB).
Footnotes
Disclosures
None
References
- 1.Gu Q, Dillon CF, Burt VL. Prescription drug use continues to increase: U.S. prescription drug data for 2007-2008. NCHS Data Brief. 2010;(42):1–8. [PubMed] [Google Scholar]
- 2.Roger VL, et al. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 4.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 5.Thomson JA, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–7. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 6.Park IH, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451(7175):141–6. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 7.Ellison GM et al. Myocyte death and renewal: modern concepts of cardiac cellular homeostasis. Nat Clin Pract Cardiovasc Med. 2007;4(Suppl 1):S52–9. doi: 10.1038/ncpcardio0773. [DOI] [PubMed] [Google Scholar]
- 8.MacLellan WR, Schneider MD. Genetic dissection of cardiac growth control pathways. Annu Rev Physiol. 2000;62:289–319. doi: 10.1146/annurev.physiol.62.1.289. [DOI] [PubMed] [Google Scholar]
- 9.Nadal-Ginard B, et al. A matter of life and death: cardiac myocyte apoptosis and regeneration. J Clin Invest. 2003;111(10):1457–9. doi: 10.1172/JCI18611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torella D, et al. Resident human cardiac stem cells: role in cardiac cellular homeostasis and potential for myocardial regeneration. Nat Clin Pract Cardiovasc Med. 2006;3(Suppl 1):S8–13. doi: 10.1038/ncpcardio0409. [DOI] [PubMed] [Google Scholar]
- 11.Boudoulas KD, Hatzopoulos AK. Cardiac repair and regeneration: the Rubik's cube of cell therapy for heart disease. Dis Model Mech. 2009;2(7-8):344–58. doi: 10.1242/dmm.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Habib M, Caspi O, Gepstein L. Human embryonic stem cells for cardiomyogenesis. J Mol Cell Cardiol. 2008;45(4):462–74. doi: 10.1016/j.yjmcc.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Laflamme MA, Murry CE. Regenerating the heart. Nat Biotechnol. 2005;23(7):845–56. doi: 10.1038/nbt1117. [DOI] [PubMed] [Google Scholar]
- 14.Murry CE, Field LJ, Menasche P. Cell-based cardiac repair: reflections at the 10-year point. Circulation. 2005;112(20):3174–83. doi: 10.1161/CIRCULATIONAHA.105.546218. [DOI] [PubMed] [Google Scholar]
- 15.Hyun I, et al. New ISSCR guidelines underscore major principles for responsible translational stem cell research. Cell Stem Cell. 2008;3(6):607–9. doi: 10.1016/j.stem.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 16.Goldring CE, et al. Assessing the safety of stem cell therapeutics. Cell Stem Cell. 2011;8(6):618–28. doi: 10.1016/j.stem.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 17.Lian Q, et al. Future perspective of induced pluripotent stem cells for diagnosis, drug screening and treatment of human diseases. Thromb Haemost. 2010;104(1):39–44. doi: 10.1160/TH10-05-0269. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka T, et al. In vitro pharmacologic testing using human induced pluripotent stem cell-derived cardiomyocytes. Biochem Biophys Res Commun. 2009;385(4):497–502. doi: 10.1016/j.bbrc.2009.05.073. [DOI] [PubMed] [Google Scholar]
- 19.Yokoo N, et al. The effects of cardioactive drugs on cardiomyocytes derived from human induced pluripotent stem cells. Biochem Biophys Res Commun. 2009;387(3):482–8. doi: 10.1016/j.bbrc.2009.07.052. [DOI] [PubMed] [Google Scholar]
- 20.Kannankeril PJ, Roden DM. Drug-induced long QT and torsade de pointes: recent advances. Curr Opin Cardiol. 2007;22(1):39–43. doi: 10.1097/HCO.0b013e32801129eb. [DOI] [PubMed] [Google Scholar]
- 21.Carlsson L. In vitro and in vivo models for testing arrhythmogenesis in drugs. J Intern Med. 2006;259(1):70–80. doi: 10.1111/j.1365-2796.2005.01590.x. [DOI] [PubMed] [Google Scholar]
- 22.Thomsen MB, et al. Assessing the proarrhythmic potential of drugs: current status of models and surrogate parameters of torsades de pointes arrhythmias. Pharmacol Ther. 2006;112(1):150–70. doi: 10.1016/j.pharmthera.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 23.Miller RA, et al. Efficient array-based identification of novel cardiac genes through differentiation of mouse ESCs. PLoS One. 2008;3(5):e2176. doi: 10.1371/journal.pone.0002176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao F, et al. Transcriptional and functional profiling of human embryonic stem cell-derived cardiomyocytes. PLoS One. 2008;3(10):e3474. doi: 10.1371/journal.pone.0003474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varlet I, Collignon J, Robertson EJ. nodal expression in the primitive endoderm is required for specification of the anterior axis during mouse gastrulation. Development. 1997;124(5):1033–44. doi: 10.1242/dev.124.5.1033. [DOI] [PubMed] [Google Scholar]
- 26.Conlon FL, et al. A primary requirement for nodal in the formation and maintenance of the primitive streak in the mouse. Development. 1994;120(7):1919–28. doi: 10.1242/dev.120.7.1919. [DOI] [PubMed] [Google Scholar]
- 27.Ben-Haim N, et al. The nodal precursor acting via activin receptors induces mesoderm by maintaining a source of its convertases and BMP4. Dev Cell. 2006;11(3):313–23. doi: 10.1016/j.devcel.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Brennan J, et al. Nodal signalling in the epiblast patterns the early mouse embryo. Nature. 2001;411(6840):965–9. doi: 10.1038/35082103. [DOI] [PubMed] [Google Scholar]
- 29.Winnier G, et al. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995;9(17):2105–16. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- 30.Liu P, et al. Requirement for Wnt3 in vertebrate axis formation. Nat Genet. 1999;22(4):361–5. doi: 10.1038/11932. [DOI] [PubMed] [Google Scholar]
- 31.Pearce JJ, Evans MJ. Mml a mouse Mix-like gene expressed in the primitive streak. Mech Dev. 1999;87(1-2):189–92. doi: 10.1016/s0925-4773(99)00135-5. [DOI] [PubMed] [Google Scholar]
- 32.Wilkinson DG, Bhatt S, Herrmann BG. Expression pattern of the mouse T gene and its role in mesoderm formation. Nature. 1990;343(6259):657–9. doi: 10.1038/343657a0. [DOI] [PubMed] [Google Scholar]
- 33.Blum M, et al. Gastrulation in the mouse: the role of the homeobox gene goosecoid. Cell. 1992;69(7):1097–106. doi: 10.1016/0092-8674(92)90632-m. [DOI] [PubMed] [Google Scholar]
- 34.Ciruna BG, Rossant J. Expression of the T-box gene Eomesodermin during early mouse development. Mech Dev. 1999;81(1-2):199–203. doi: 10.1016/s0925-4773(98)00243-3. [DOI] [PubMed] [Google Scholar]
- 35.Ema M, Takahashi S, Rossant J. Deletion of the selection cassette, but not cis-acting elements, in targeted Flk1-lacZ allele reveals Flk1 expression in multipotent mesodermal progenitors. Blood. 2006;107(1):111–7. doi: 10.1182/blood-2005-05-1970. [DOI] [PubMed] [Google Scholar]
- 36.Takakura N, et al. PDGFR alpha expression during mouse embryogenesis: immunolocalization analyzed by whole-mount immunohistostaining using the monoclonal anti-mouse PDGFR alpha antibody APA5. J Histochem Cytochem. 1997;45(6):883–93. doi: 10.1177/002215549704500613. [DOI] [PubMed] [Google Scholar]
- 37.Sakurai H, et al. In vitro modeling of paraxial and lateral mesoderm differentiation reveals early reversibility. Stem Cells. 2006;24(3):575–86. doi: 10.1634/stemcells.2005-0256. [DOI] [PubMed] [Google Scholar]
- 38.Tam PP, Behringer RR. Mouse gastrulation: the formation of a mammalian body plan. Mech Dev. 1997;68(1-2):3–25. doi: 10.1016/s0925-4773(97)00123-8. [DOI] [PubMed] [Google Scholar]
- 39.Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. 2005;6(11):826–35. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- 40.McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002;27(1):40–7. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- 41.Antonini G, et al. Natural history of cardiac involvement in myotonic dystrophy: correlation with CTG repeats. Neurology. 2000;55(8):1207–9. doi: 10.1212/wnl.55.8.1207. [DOI] [PubMed] [Google Scholar]
- 42.Belaguli NS, et al. Cardiac tissue enriched factors serum response factor and GATA-4 are mutual coregulators. Mol Cell Biol. 2000;20(20):7550–8. doi: 10.1128/mcb.20.20.7550-7558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Q, et al. Comparative studies on the expression patterns of three troponin T genes during mouse development. Anat Rec. 2001;263(1):72–84. doi: 10.1002/ar.1078. [DOI] [PubMed] [Google Scholar]
- 44.Hescheler J, et al. Embryonic stem cells: a model to study structural and functional properties in cardiomyogenesis. Cardiovasc Res. 1997;36(2):149–62. doi: 10.1016/s0008-6363(97)00193-4. [DOI] [PubMed] [Google Scholar]
- 45.Keller G. Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Genes Dev. 2005;19(10):1129–55. doi: 10.1101/gad.1303605. [DOI] [PubMed] [Google Scholar]
- 46.Boheler KR, et al. Cardiomyocytes derived from embryonic stem cells. Methods Mol Med. 2005;108:417–35. doi: 10.1385/1-59259-850-1:417. [DOI] [PubMed] [Google Scholar]
- 47.Boheler KR, et al. Differentiation of pluripotent embryonic stem cells into cardiomyocytes. Circ Res. 2002;91(3):189–201. doi: 10.1161/01.res.0000027865.61704.32. [DOI] [PubMed] [Google Scholar]
- 48.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 49.Wernig M, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448(7151):318–24. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- 50.Novak A, et al. Enhanced reprogramming and cardiac differentiation of human keratinocytes derived from plucked hair follicles, using a single excisable lentivirus. Cell Reprogram. 2010;12(6):665–78. doi: 10.1089/cell.2010.0027. [DOI] [PubMed] [Google Scholar]
- 51.Haase A, et al. Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell. 2009;5(4):434–41. doi: 10.1016/j.stem.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 52.Li C, et al. Pluripotency can be rapidly and efficiently induced in human amniotic fluid-derived cells. Hum Mol Genet. 2009;18(22):4340–9. doi: 10.1093/hmg/ddp386. [DOI] [PubMed] [Google Scholar]
- 53.Jia F, et al. A nonviral minicircle vector for deriving human iPS cells. Nat Methods. 2010;7(3):197–9. doi: 10.1038/nmeth.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fusaki N, et al. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85(8):348–62. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu J, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324(5928):797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burridge PW, et al. Production of de novo cardiomyocytes: human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell. 2012;10(1):16–28. doi: 10.1016/j.stem.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keller GM. In vitro differentiation of embryonic stem cells. Curr Opin Cell Biol. 1995;7(6):862–9. doi: 10.1016/0955-0674(95)80071-9. [DOI] [PubMed] [Google Scholar]
- 58.Mummery C, et al. Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation. 2003;107(21):2733–40. doi: 10.1161/01.CIR.0000068356.38592.68. [DOI] [PubMed] [Google Scholar]
- 59.Nakano T, Kodama H, Honjo T. Generation of lymphohematopoietic cells from embryonic stem cells in culture. Science. 1994;265(5175):1098–101. doi: 10.1126/science.8066449. [DOI] [PubMed] [Google Scholar]
- 60.Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132(4):661–80. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 61.Nishikawa SI, et al. Progressive lineage analysis by cell sorting and culture identifies FLK1+VE-cadherin+ cells at a diverging point of endothelial and hemopoietic lineages. Development. 1998;125(9):1747–57. doi: 10.1242/dev.125.9.1747. [DOI] [PubMed] [Google Scholar]
- 62.The bitterest pill. Nature. 2006;444(7119):532–3. doi: 10.1038/444532a. [DOI] [PubMed] [Google Scholar]
- 63.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3(8):711–5. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 64.Force T, Kolaja KL. Cardiotoxicity of kinase inhibitors: the prediction and translation of preclinical models to clinical outcomes. Nat Rev Drug Discov. 2011;10(2):111–26. doi: 10.1038/nrd3252. [DOI] [PubMed] [Google Scholar]
- 65.Lawrence CL, et al. In vitro models of proarrhythmia. Br J Pharmacol. 2008;154(7):1516–22. doi: 10.1038/bjp.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ma J, et al. High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am J Physiol Heart Circ Physiol. 2011;301(5):H2006–17. doi: 10.1152/ajpheart.00694.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang J, et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104(4):e30–41. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Braam SR, et al. Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem Cell Res. 2010;4(2):107–16. doi: 10.1016/j.scr.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 69.Chapin RE, Stedman DB. Endless possibilities: stem cells and the vision for toxicology testing in the 21st century. Toxicol Sci. 2009;112(1):17–22. doi: 10.1093/toxsci/kfp202. [DOI] [PubMed] [Google Scholar]
- 70.Davila JC, et al. Use and application of stem cells in toxicology. Toxicol Sci. 2004;79(2):214–23. doi: 10.1093/toxsci/kfh100. [DOI] [PubMed] [Google Scholar]
- 71.Ebert AD, Liang P, Wu JC. Induced Pluripotent Stem Cells as a Disease Modeling and Drug Screening Platform. J Cardiovasc Pharmacol. 2012 doi: 10.1097/FJC.0b013e318247f642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dick E, et al. Evaluating the utility of cardiomyocytes from human pluripotent stem cells for drug screening. Biochem Soc Trans. 2010;38(4):1037–45. doi: 10.1042/BST0381037. [DOI] [PubMed] [Google Scholar]
- 73.Caspi O, et al. In vitro electrophysiological drug testing using human embryonic stem cell derived cardiomyocytes. Stem Cells Dev. 2009;18(1):161–72. doi: 10.1089/scd.2007.0280. [DOI] [PubMed] [Google Scholar]
- 74.McNeish J. Embryonic stem cells in drug discovery. Nat Rev Drug Discov. 2004;3(1):70–80. doi: 10.1038/nrd1281. [DOI] [PubMed] [Google Scholar]
- 75.Pollard CE, Valentin JP, Hammond TG. Strategies to reduce the risk of drug- induced QT interval prolongation: a pharmaceutical company perspective. Br J Pharmacol. 2008;154(7):1538–43. doi: 10.1038/bjp.2008.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brimecombe JC, Kirsch GE, Brown AM. Test article concentrations in the hERG assay: losses through the perfusion, solubility and stability. J Pharmacol Toxicol Methods. 2009;59(1):29–34. doi: 10.1016/j.vascn.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 77.Lahti AL, et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Model Mech. 2012;5(2):220–30. doi: 10.1242/dmm.008409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chaudhary KW, et al. Embryonic stem cells in predictive cardiotoxicity: laser capture microscopy enables assay development. Toxicol Sci. 2006;90(1):149–58. doi: 10.1093/toxsci/kfj078. [DOI] [PubMed] [Google Scholar]
- 79.Mohr JC, et al. The microwell control of embryoid body size in order to regulate cardiac differentiation of human embryonic stem cells. Biomaterials. 2010;31(7):1885–93. doi: 10.1016/j.biomaterials.2009.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rana P, et al. Characterization of Human Induced Pluripotent Stem Cell Derived Cardiomyocytes: Bioenergetics and Utilization in Safety Screening. Toxicol Sci. 2012 doi: 10.1093/toxsci/kfs233. [DOI] [PubMed] [Google Scholar]
- 81.Takei S, et al. Bone morphogenetic protein-4 promotes induction of cardiomyocytes from human embryonic stem cells in serum-based embryoid body development. Am J Physiol Heart Circ Physiol. 2009;296(6):H1793–803. doi: 10.1152/ajpheart.01288.2008. [DOI] [PubMed] [Google Scholar]
- 82.Reppel M, et al. The electrocardiogram of human embryonic stem cell-derived cardiomyocytes. J Electrocardiol. 2005;38(4 Suppl):166–70. doi: 10.1016/j.jelectrocard.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 83.Harmer AR, et al. Optimisation and validation of a medium-throughput electrophysiology-based hNav1.5 assay using IonWorks. J Pharmacol Toxicol Methods. 2008;57(1):30–41. doi: 10.1016/j.vascn.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 84.Sanchez-Freire V, et al. Microfluidic single-cell real-time PCR for comparative analysis of gene expression patterns. Nat Protoc. 2012;7(5):829–38. doi: 10.1038/nprot.2012.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Muzikant AL, Penland RC. Models for profiling the potential QT prolongation risk of drugs. Curr Opin Drug Discov Devel. 2002;5(1):127–35. [PubMed] [Google Scholar]
- 86.Meyer T, et al. QT-screen: high-throughput cardiac safety pharmacology by extracellular electrophysiology on primary cardiac myocytes. Assay Drug Dev Technol. 2004;2(5):507–14. doi: 10.1089/adt.2004.2.507. [DOI] [PubMed] [Google Scholar]
- 87.Sun N, et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. 2012;4(130):130ra47. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cubeddu LX. QT prolongation and fatal arrhythmias: a review of clinical implications and effects of drugs. Am J Ther. 2003;10(6):452–7. doi: 10.1097/00045391-200311000-00013. [DOI] [PubMed] [Google Scholar]
- 89.Drici MD, Clement N. Is gender a risk factor for adverse drug reactions? The example of drug-induced long QT syndrome. Drug Saf. 2001;24(8):575–85. doi: 10.2165/00002018-200124080-00002. [DOI] [PubMed] [Google Scholar]
- 90.Yang L, et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008;453(7194):524–8. doi: 10.1038/nature06894. [DOI] [PubMed] [Google Scholar]
- 91.Kiskinis E, Eggan K. Progress toward the clinical application of patient-specific pluripotent stem cells. J Clin Invest. 2010;120(1):51–9. doi: 10.1172/JCI40553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schulz TC, et al. A large-scale proteomic analysis of human embryonic stem cells. BMC Genomics. 2007;8:478. doi: 10.1186/1471-2164-8-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thomas RJ, et al. Automated, scalable culture of human embryonic stem cells in feeder-free conditions. Biotechnol Bioeng. 2009;102(6):1636–44. doi: 10.1002/bit.22187. [DOI] [PubMed] [Google Scholar]
- 94.Anderson D, et al. Transgenic enrichment of cardiomyocytes from human embryonic stem cells. Mol Ther. 2007;15(11):2027–36. doi: 10.1038/sj.mt.6300303. [DOI] [PubMed] [Google Scholar]
- 95.Huber I, et al. Identification and selection of cardiomyocytes during human embryonic stem cell differentiation. FASEB J. 2007;21(10):2551–63. doi: 10.1096/fj.05-5711com. [DOI] [PubMed] [Google Scholar]
- 96.Xu XQ, et al. Highly enriched cardiomyocytes from human embryonic stem cells. Cytotherapy. 2008;10(4):376–89. doi: 10.1080/14653240802105307. [DOI] [PubMed] [Google Scholar]
- 97.Rajala K, et al. A defined and xeno-free culture method enabling the establishment of clinical-grade human embryonic, induced pluripotent and adipose stem cells. PLoS One. 2010;5(4):e10246. doi: 10.1371/journal.pone.0010246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rodriguez-Piza I, et al. Reprogramming of human fibroblasts to induced pluripotent stem cells under xeno-free conditions. Stem Cells. 2010;28(1):36–44. doi: 10.1002/stem.248. [DOI] [PubMed] [Google Scholar]
- 99.Narsinh KH, et al. Single cell transcriptional profiling reveals heterogeneity of human induced pluripotent stem cells. J Clin Invest. 2011;121(3):1217–21. doi: 10.1172/JCI44635. [DOI] [PMC free article] [PubMed] [Google Scholar]