

Abstract
The architecture of plant metabolism includes substantial duplication of metabolite pools and enzyme catalyzed reactions in different subcellular compartments. This poses challenges for understanding the regulation of metabolism particularly in primary metabolism and amino acid biosynthesis. To explore the extent to which amino acids are made in single compartments and to gain insight into the metabolic precursors from which they derive, we used steady state 13C labelling and analysed labelling in protein amino acids from plastid and cytosol. Ribulose 1,5-bisphosphate carboxylase/oxygenase (Rubisco) is a major component of green tissues and its large and small subunits are synthesized from different pools of amino acids in the plastid and cytosol, respectively. Developing Brassica napus embryos were cultured in the presence of [U-13C]-sucrose, [U-13C]-glucose, [U-13C]-glutamine or [U-13C]-alanine to generate proteins. The large subunits (LSU) and small subunits (SSU) of Rubisco were isolated and the labelling in their constituent amino acids was analysed by gas chromatography-mass spectrometry. Amino acids including alanine, glycine and serine exhibited different 13C enrichment in the LSU and SSU, demonstrating that these pools have different metabolic origins and are not isotopically equilibrated between the plastid and cytosol on the time scale of cellular growth. Potential extensions of this novel approach to other macromolecules, organelles and cell types of eukaryotes are discussed.
Keywords: compartmentation, isotopic labelling, metabolic flux analysis, primary metabolism
INTRODUCTION
Subcellular compartmentation is a defining characteristic of eukaryotic organisms. Organelles serve multiple purposes but foremost among these is the compartmentalization of metabolism. By segregating enzymes and metabolic fluxes into distinct compartments, more complex and sophisticated control over metabolism can be achieved (Winter, Robinson & Heldt 1993). In addition, anabolic and catabolic pathways can be separated to allow simultaneous synthesis and breakdown of metabolites. As one example, fatty acids can be synthesized in the plastids of plants while fatty acid degradation by beta-oxidation simultaneously takes place predominantly in the peroxisomes (Cooper & Beevers 1969; Germain et al. 2001; Baker et al. 2006; Khan, Adham & Zolman 2012) and possibly to a lesser extent in the mitochondria (Dieuaide et al. 1993; Masterson & Wood 2009). Separated anabolic and catabolic pathways also occur in multiple compartments for the metabolism of proteins (Vierstra 1993), purine bases (Zrenner et al. 2006) and carbohydrates (Zeeman et al. 2007a; Zeeman, Smith & Smith 2007b). The different organelle environments permit distinct metabolic fates for compounds based upon their location [e.g. compartmentation of glycolytic intermediates directed towards different products (Giege et al. 2003; Graham et al. 2007; Andriotis et al. 2010)].
A number of experimental and bioinformatic approaches are available to study the compartmentation of metabolism. Subcellular fractionation has been used to probe metabolism within individual compartments (Stocking 1959; Gerhardt & Heldt 1984; Winter et al. 1993). However, fractionation methods may alter metabolite concentrations and impact metabolic activity as small molecules can leak out of organelles during their isolation. Development of rapid isolation techniques with non-aqueous fractionation of organelles (Gerhardt & Heldt 1984; Gerhardt, Stitt & Heldt 1987; Stitt et al. 1989; Farre et al. 2001) has improved quantification of some of the more abundant and stable metabolites. Recent advances have included coupling this technology with metabolomics to provide new insights to metabolic function (Benkeblia, Shinano & Osaki 2007; Geigenberger, Tiessen & Meurer 2011). Nonetheless, the approach remains technically challenging and the extent to which metabolites may move between compartments during isolation remains uncertain despite the use of marker enzymes to identify organelles and determine the purity of separation.
Protein localization with fluorescent tags, immuno-tracking and sequence-based targeting predictions have provided information on subcellular localization of enzymes. However, many predictions are not unambiguous (see Supporting Information Table S1) and the presence of enzymes in a compartment determined from these methods does not quantitatively describe pathway activity. Overall, these methodological limitations make it challenging to evaluate many of the in vivo metabolic fluxes that occur within different subcellular compartments. This in turn limits our understanding of complex systems (Allen, Libourel & Shachar-Hill 2009a) and sometimes forces us to redefine descriptions of enzymatic roles (Schwender et al. 2004). These limitations impede the success of metabolic engineering strategies that require targeting of overexpressed enzyme activities to the appropriate subcellular location (Ma et al. 2000).
Many studies of metabolism utilize stable isotope labelling to infer fluxes. Incorporation of information based on subcellular synthesis of fatty acids, carbohydrates and other metabolites has resulted in models with increased sophistication for developing seeds of oilseed rape (Schwender, Ohlrogge & Shachar-Hill 2003; Schwender, Shachar-Hill & Ohlrogge 2006; Junker et al. 2007), sunflower (Alonso et al. 2007), maize (Alonso, Val & Shachar-Hill 2010, 2011), Arabidopsis seeds (Lonien & Schwender 2009), soybean (Sriram et al. 2004; Iyer et al. 2008; Allen, Ohlrogge & Shachar-Hill 2009b) and cells of Arabidopsis (Williams et al. 2008; Masakapalli et al. 2010). Information on compartmentation is also frequently deduced based on literature or sequence-based predictions, which can lead to errors, particularly in non-model species. Furthermore, direct labelling evidence is needed if compartmentalized fluxes are to be defined as literature information that establishes subcellular metabolic location does not establish values for fluxes but instead increases the number of parameters that must be modelled. Thus, the extent and accuracy of network structure and choice of the most informative labelled substrates are critical to model-derived conclusions about metabolism (Libourel & Shachar-Hill 2008; Masakapalli et al. 2010; Sweetlove et al. 2010).
In the past we have analysed and reported on several metabolites that have well-documented subcellular origins (Allen, Shachar-Hill & Ohlrogge 2007). For example, 13C isotopes of glucose that are incorporated into starch and cell wall monomers were used to assess the exchange of carbon in hexose phosphate pools between the plastid and cytosol (Allen et al. 2007); protein glycosylation provides another cytosolic reporter of hexose units (Sriram et al. 2007). Additionally, distinctive origins of cytosolic and plastid acetyl-CoA pools could be deduced from labelling of fatty acids because the biosynthesis up to 18 carbons in chain length takes place in the plastids of plants (Bao et al. 2000), whereas subsequent elongation to C20 and beyond occurs outside the plastid (Ohlrogge, Pollard & Stumpf 1978; Whitfield, Murphy & Hills 1993; Bao, Pollard & Ohlrogge 1998). Because acetyl-CoA is not transferred across membranes (Liedvogel & Stumpf 1982), different pools of acetyl-CoA for fatty acid biosynthesis were analysed through 13C labelling measurements (Schwender & Ohlrogge 2002; Allen et al. 2007).
Stable isotope labelling of proteins that are assembled from amino acid pools in different subcellular compartments is a previously untested source of compartmental information. For eukaryotes, most translation takes place in the cytosol using mRNA that originated in the cell nucleus. However, because plastids and mitochondria have maintained their own genomes throughout evolution, there are a number of proteins that are synthesized in these compartments (Unseld et al. 1997; Martin et al. 2002; Burger, Gray & Lang 2003). As examples, many photosynthetic proteins involved with light harvesting and ATP generation are produced in the chloroplast (Martin et al. 2002), while enzymes important to the electron transport chain such as cytochrome reductase/oxidase proteins and NADH dehydrogenases are assembled specifically in mitochondria (Wintz, Chen & Pillay 1989; Unseld et al. 1997). Biosynthesis of protein in a specific location will be based upon the amino acid pools in that location.
We hypothesized that examination of labelling of protein subunits synthesized in different locations might reveal variations in isotope enrichment of amino acids which in turn could provide insights into compartmentalized fluxes of subcellular metabolism. To assess this hypothesis, developing Brassica napus (rapeseed) embryos were labelled with [U-13C]-sucrose, [U-13C]-glucose, [U-13C]-glutamine or [U-13C]-alanine. Analysis of the large and small subunits of ribulose 1,5-bisphosphate carboxylase/oxygenase (Rubisco), translated in the plastid and cytosol, respectively, demonstrates that this approach can be informative about important intermediates of central metabolism as well as highlighting the simultaneous synthesis and turnover of amino acids in multiple compartments.
RESULTS
A major objective of this study was to explore if novel information about compartmentation of plant metabolism can be obtained by comparison of proteins synthesized in the cytosol to those synthesized in the plastid. As an initial test of this approach, we focused on Rubisco, the most abundant soluble protein of plant leaves, and the enzyme responsible for a majority of all carbon fixation (Dhingra, Portis & Daniell 2004). The Rubisco structure, a 480 000 to 590 000 kDa complex, consists of eight large subunits (LSU) and eight small subunits (SSU), approximately 50–55 and 12–15 kDA in size, respectively. The LSU and SSU genes are encoded by the plastid and nuclear genomes, respectively (Blair & Ellis 1973; Gray & Kekwick 1974; Baker et al. 1975). The genes encoding SSU and LSU are transcribed from each genome and their mRNA is translated to protein by distinct ribosomal assemblies localized in each compartment. Therefore, the SSU protein is derived from amino acid pools present in the cytosol, whereas LSU protein synthesis depends on plastid pools of amino acids. After protein synthesis, the SSU is imported into the plastid where assembly of the complex takes place. Comparison of other pairs of proteins assembled in the cytosol versus the plastid might also reveal similar information. However, a major advantage of analysis of Rubisco is that because the SSU and LSU are components of a multi-subunit enzyme complex, with fixed stoichiometry, the timing of SSU and LSU protein synthesis is synchronized and temporal differences will be minimized.
During development in siliques, B. napus embryos are green and express substantial levels of Rubisco (King, Badger & Furbank 1998; White et al. 2000; Ruuska et al. 2002; Ruuska, Schwender & Ohlrogge 2004). To analyse the SSU and LSU, we tested separation and isolation of subunits by gel electrophoresis with or without prior separation using molecular weight cut-off membranes. Although the subunits were well separated by polyacrylamide gel electrophoresis (PAGE), the recovery from crude extracts after electrophoresis resulted in LSU and SSU preparations contaminated with other proteins and in low yields. Therefore, we used antibody-coupled beads to isolate Rubisco from other proteins present in crude extracts (Fig. 1). LSU and SSU subunits of the affinity-purified Rubisco were then resolved by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE). Direct protein hydrolysis of gel excised bands resulted in low amino acid yields and contaminants presumed to be amides that are a by-product of the polyacrylamide gel matrix. Therefore, protein was transferred to polyvinylidene fluoride (PVDF) membranes, hydrolyzed, and tert-butyl-dimethylsilyl derivatives (TBDMS) of amino acids were prepared and analysed by gas chromatography-mass spectrometry (GC-MS) (Fig. 1).
Figure 1.
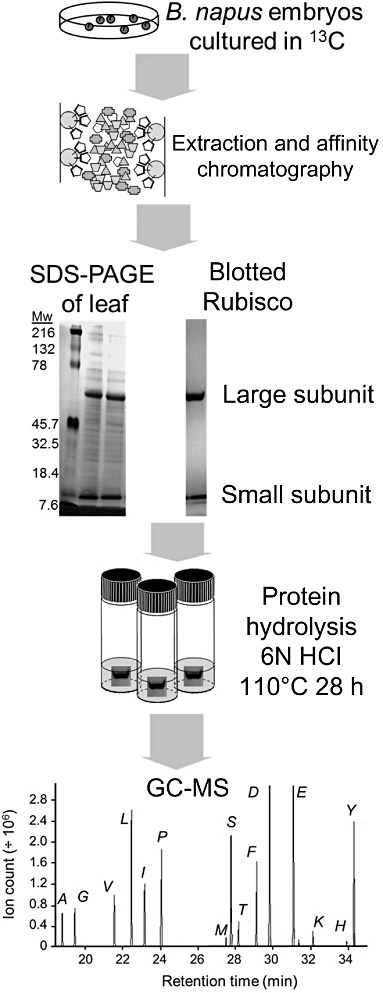
Schematic for analysis of ribulose 1,5-bisphosphate carboxylase/oxygenase (Rubisco) small and large subunits. Brassica napus embryos were cultured in 13C-labelled substrates, protein extracted and Rubisco was isolated by affinity chromatography. Sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate small and large subunits. Gels were electroblotted to polyvinylidene fluoride (PVDF) membrane, protein bands isolated and hydrolyzed with 6N HCl. Resulting amino acids were derivatized to the tert-butyl-dimethylsilyl products and analysed for isotopic enrichment by gas chromatography-mass spectrometry (GC-MS).
When developing B. napus, embryos are excised from siliques at an early stage of development and cultured with light (50 µE); embryo development continues until mature viable seeds are produced with protein and lipid compositions similar to embryos that develop in planta (Schwender & Ohlrogge 2002). To mimic the in planta environment, media contained sucrose, glucose, glutamine and alanine at concentrations similar to those of the endosperm that surrounds embryos (Schwender & Ohlrogge 2002). The growth rate observed in this study was 0.3–0.4 mg DW day−1 which is comparable with previously reported growth in culture and in planta (Pomeroy et al. 1991; Schwender & Ohlrogge 2002). Media were supplemented with: [U-13C]-glucose, [U-13C]-sucrose, [U-13C]-glutamine or [U-13C]-alanine; substrates that enter metabolism through different reactions and transporters and are consumed at different rates. Therefore, each labelling experiment will result in unique metabolite enrichments and provide complementary information. Substrates were replaced with 100% [U-13C] label except the predominant carbon source for embryos, sucrose that was supplied as 1:1 combination of [U-13C] labelled and unlabelled sucrose. [U-13C] label was supplied for 2 weeks during which isotopic steady state conditions are observed (Schwender et al. 2003, 2006). Under these conditions, greater than 95% of total biomass accumulates during labelling, equivalent to 4.5–5 doublings. Therefore, the biomass analysed represents the steady-state metabolic labelling of metabolism during embryo development under conditions very similar to in planta growth.
Pools of metabolic intermediates may exchange between cytosol and plastid via the triose, hexose, pentose, four carbon and other transporters (Linka & Weber 2010). If exchange is rapid resulting in complete isotopic equilibration of these intermediates and/or amino acid pools between the cytosol and plastid, identical labelling patterns would be expected for proteins synthesized in these two locations. Here, equilibration refers to the labelling patterns in a particular amino acid becoming equal in plastid and cytosol through interchange of pools. The same result would be expected if synthesis of a metabolite only occurs in one compartment. When B. napus embryos were cultured with 13C labelled substrates, several amino acids of the large and small subunits of Rubisco were enriched differently. Of these experiments, [U-13C]-glucose and [U-13C]-sucrose provided the most statistically significant differences in labelling of LSU and SSU.
A comparison of the labelling in amino acids of LSU and SSU is presented in Fig. 2 and in the Supporting Information Tables S2–S6. Figure 2b presents the differences in label between LSU and SSU subunits for all experiments, with shading to indicate significance (t-test, two-tailed, 99%). Of the 20 amino acids present in protein, 10 could be analysed with high reproducibility and reliability. Asparagine and glutamine are deamidated during hydrolysis to their acidic counterparts; therefore ‘ASX’ and ‘GLX’ represent combined aspartate/asparagine and glutamate/glutamine. Cysteine and tryptophan are lost oxidatively during acid hydrolysis, and valine could not be resolved from an unidentified contaminant. Finally, methionine, lysine, arginine, histidine and tyrosine levels were too low to reliably analyse reflecting their lower abundance in protein and more challenging fragmentation patterns for GC-MS. The mass enrichments for different GC-MS fragments of the same amino acid are not independent, therefore the average labelling per carbon was used to compare LSU and SSU labelling (defined in figure caption). Additional average enrichment per carbon data is provided in Supporting Information Table S2.
Figure 2.
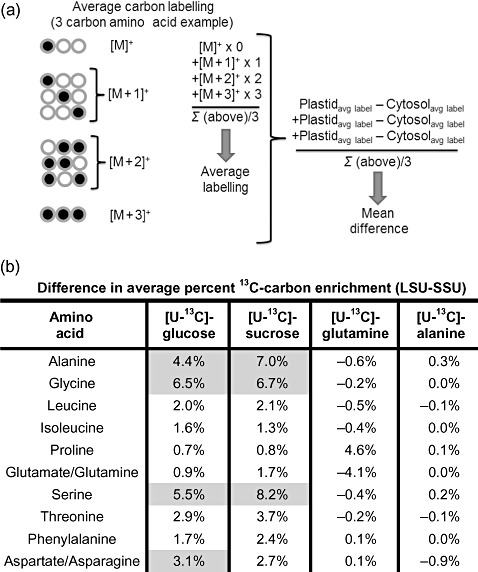
Differences in 13C enrichment of large subunit (LSU) and small subunit (SSU)-derived amino acids. Gas chromatography-mass spectrometry (GC-MS) results were subject to statistical comparison of the average carbon labelling for each amino acid. (a) The average labelling difference was calculated as shown by multiplying the fractional label of each mass weight by the number of labelled carbons for that mass weight, then summing these overall weights and dividing by the total number of carbons. The difference is defined as the average change in enrichment between the LSU and SSU fragments. (b) Differences in average carbon labelling of LSUs and SSUs for four labelling experiments. Significance was calculated using two-tailed Student's t-test with 99% levels shaded for minimum of n = 3 except for several amino acids within the alanine labelling experiment that were duplicates.
Alanine is simultaneously made in multiple compartments and is not isotopically equilibrated between the plastid and cytosol
As indicated in Figs 2 and 3, after labelling with [U-13C]-glucose the enrichment of alanine was significantly different (t-test, 99%) in LSU (plastidic synthesis) compared with the SSU (cytosolic synthesis) (n = 3). Average 13C enrichment per carbon of alanine in the LSU was 21% and in the SSU was 16%, a relative difference of 22% and absolute difference of 4.4% (Fig. 2). Figure 3 presents two GC-MS fragments for alanine from [U-13C]-glucose labelling. Data derived from both the MS fragment representing all three of the backbone carbons of alanine and a second fragment (2nd and 3rd carbons of alanine) were consistent within each Rubisco subunit. The observed differences in labelling when [U-13C]-glucose was supplied were confirmed by the results of sucrose labelling experiments. These data indicate that alanine within the LSU is derived from precursors that are more highly labelled than alanine from the SSU protein. This provides a clear in vivo demonstration that cytosolic and plastidic pools of alanine are not in isotopic equilibrium.
Figure 3.
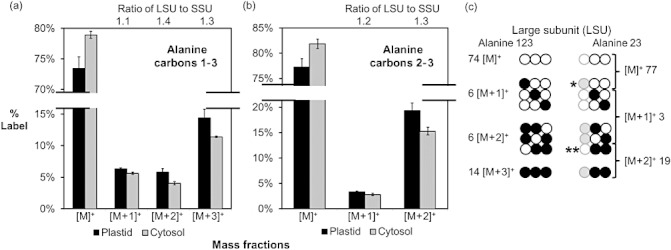
13C labelling differences in alanine of large subunits (LSUs) and small subunits (SSUs) of ribulose 1,5-bisphosphate carboxylase/oxygenase (Rubisco). Alanine enrichment varied between the cytosol and plastid pools as revealed by labelling with [U-13C]-glucose (n = 3, SD). Differences are reflected in isotopomers of both (a) three carbon and (b) two carbon gas chromatography-mass spectrometry (GC-MS) fragments. (c) Together these two fragments can be used to calculate the labelling of specific isotopomers that are indicated by (*) and (**). Alanine (LSU) labelled exclusively in the first carbon (*) represents 77 − 74 = 3% of all alanine isotopomers and is one half of the pool of singly labelled isotopomers. Enrichment of doubly labelled molecules in the second and third positions (**) is 19 − 14 = 5% out of a total pool of 6% and is the predominantly abundant isotopomer ([M+2]+ enrichment).
As unlabelled alanine is provided in the media, this could highlight the disequilibrium by reducing the 13C enrichment of alanine in the cytosol compared with the plastid. Thus, a greater incorporation of unlabelled exogenous alanine into the cytosolic pool is the most straightforward explanation for the lower levels of SSU alanine 13C enrichment.
Precursors for plastid and cytosolic alanine are distinct
Although the 13C distribution pattern in Fig. 3 was similar for LSU and SSU, statistically significant differences between the subunits were observed. If the differences between subunits (Fig. 2) were merely a result of dilution, then all labelled isotopomers for the SSU would be fractionally reduced to the same extent relative to the LSU. This is because the labelling profile of an unlabelled source of alanine will be composed of isotopomers exclusively of weight [M]+ (i.e. 100%) and 0% in [M+1]+ to [M+3]+ (after correction for natural abundance). Therefore, adding unlabelled alanine would increase the unlabelled [M]+ fraction and would uniformly decrease all other fractions. However, this was not observed. Instead, as indicated in Fig. 3, the ratio of LSU to SSU 13C enrichment varied in the different mass weights of alanine. The LSU to SSU enrichment for [M+1]+ was 1.1 whereas for [M+2]+ it was 1.4 (i.e. statistically significant fractional change of 30%), Fig. 3, and [M+3]+LSU/[M+3]+SSU is 1.3. These differences reveal that relative fluxes through the alternative metabolic pathways are different in plastids and cytosol.
Alanine labelling implies contributions of both glycolytic and pentose phosphate pathways
[U-13C]-hexose that is metabolized via glycolysis will retain equal 13C label in each carbon atom of the three carbon alanine backbone (reflected by the M+3 isotopomer of alanine). As indicated in Fig. 3, the [M+3]+ mass isomer represented only approximately 50% of the labelled [M+1]+, [M+2]+ and [M+3]+ mass isomers. The intermediate [M+1]+ and [M+2]+ mass isomers represent alanine molecules that contain both unlabelled and labelled carbons, which result from bond breaking of 13C backbone derived from the fully labelled hexose precursor. The analysis of fragment ions (Fig. 3) indicates that [M+2]+ isotopomers are predominantly labelled in the second and third positions of the alanine backbone. The breaking and reforming of bonds between carbons 1, 2, 3 and 4, 5, 6 of hexose occur in the pentose phosphate pathways (both oxidative and non-oxidative reactions) and through the Rubisco bypass (Schwender et al. 2004). The bypass incorporates carbon dioxide into 3-phosphoglyceric acid that can ultimately lead to alanine. These results imply substantial flux of hexose to pyruvate not only through glycolysis but also via the pentose phosphate or Rubisco bypass reactions. Thus, the enrichments are consistent with a significant Rubisco bypass contribution to phosphoglyceric acid production as previously observed (Schwender et al. 2004).
In vivo labelling supports leucine biosynthesis in the plastid
Leucine is derived from pyruvate and acetyl-CoA. Proteomic [Supporting Information Table S1 (Zybailov et al. 2008)] and GFP studies (Maloney et al. 2010) indicate that committed enzymes in leucine biosynthesis are in the plastid (Binder 2010). Given the differences in alanine enrichment (that represents pyruvate), we examined whether leucine labelling demonstrated similar variation. Leucine enrichment in the LSU and SSU revealed no significant differences for any labelling experiment (Fig. 2). Thus, either the precursors of leucine (i.e. pyruvate and acetyl-CoA) are equivalently labelled across locations in metabolism, or leucine is made in one location and transported to other compartments.
To assess these two alternatives, we first analysed the labelling of plastidic and cytosolic acetyl-CoA using the McLafferty fragments of fatty acids as a reporter (Schwender & Ohlrogge 2002; Allen et al. 2007; Lonien & Schwender 2009). Synthesis of fatty acids up to 18 carbon chain lengths are derived from plastid acetyl-CoA whereas elongation of fatty acids for chain lengths greater than 18 carbons utilize acetyl-CoA from the cytosol (Ohlrogge et al. 1978; Whitfield et al. 1993; Bao et al. 1998; Schwender & Ohlrogge 2002). Figure 4 presents the 13C enrichment of terminal acetyl groups of C18 and C22 fatty acids after labelling with 13C-glutamine. The much higher enrichment (i.e. 31% average carbon label) in the C22-derived McLafferty fragment (i.e. the cytosolic acetyl group) than labelling of the C18 fatty acids (3%) reflects the compartmentation of the acetyl-CoA pools. Leucine was labelled to a similar extent as the C18 fatty acids, indicating that the plastid acetyl-CoA pool and not the cytosolic pool is the precursor of leucine. For reference, alanine from LSU and SSU is also presented. The low enrichments for both alanine pools indicate that the carbon skeleton of glutamine contributes little to alanine biosynthesis in agreement with previous work (Schwender et al. 2006; Allen et al. 2009b).
Figure 4.
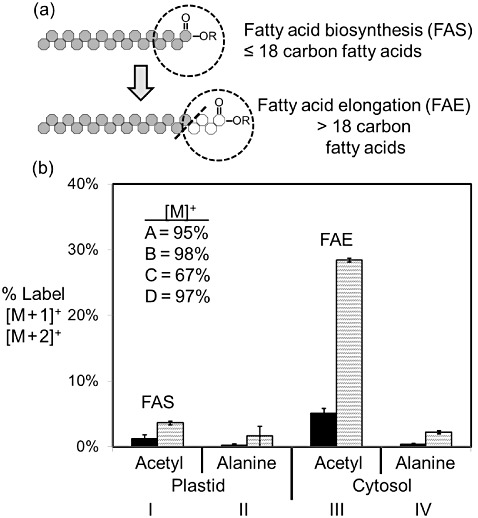
Labelling of fatty acid carbons from plastid and cytosolic reactions reflect distinct pools of acetyl-CoA. (a) The terminal acetate group of fatty acids synthesized in the plastid is enriched differently from fatty acids elongated in the cytosol (n = 3, SD). (b) In this experiment, embryo metabolism was inspected after labelling with [U-13C]-glutamine. The histogram presents the enrichment of four products of pyruvate including: acetyl-CoA from the plastid used in fatty acid biosynthesis (I), alanine from ribulose 1,5-bisphosphate carboxylase/oxygenase (Rubisco) large subunits (LSUs) (plastid synthesized) (II), acetyl-CoA from the cytosol used in fatty acid elongation (III) and alanine from Rubisco small subunits (SSU) (cytosol synthesized) (IV). The results indicate glutamine is not a significant source of carbon for fatty acid biosynthesis in the plastid (I) consistent with Schwender & Ohlrogge (2002) and Schwender et al. (2006) but provides carbon for fatty acid elongation steps (III). Glutamine is not a source of carbon for alanine biosynthesis in either location as indicated (II and IV).
We further estimated these differences by simulating the labelling profile that is expected from precursors (Fig. 5). Using alanine as a readout for pyruvate and the average labelling from acetyl-CoA, we calculated the expected abundances of mass isotopomers for leucine made from cytosol and plastid precursors. The simulated values are compared with the measured values in the figure. The enriched cytosolic acetyl-CoA results in a simulated profile with more abundant [M+1]+ and reduced [M]+ compared with measured leucine. The simulation confirms that cytosolic acetyl-CoA was not used in leucine biosynthesis. Furthermore, the profile from plastid precursors is consistent with the measured enrichments for leucine from the LSU. These results provide in vivo confirmation that leucine biosynthesis occurs predominantly in the plastid, consistent with supporting evidence (Singh 1999). In addition, the data reinforce previous findings in B. napus embryos (Schwender et al. 2006) that acetyl-CoA pools are not in equilibrium between plastids and cytosol.
Figure 5.
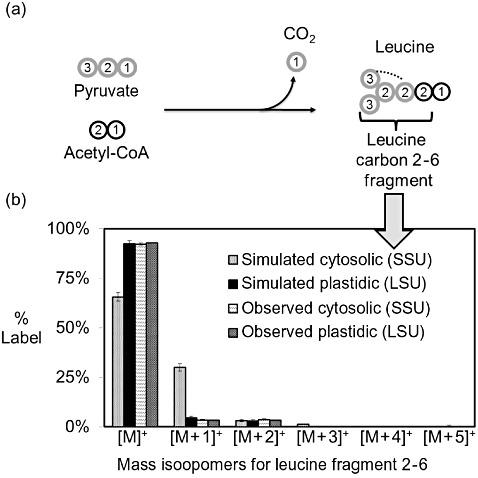
In vivo labelling confirms leucine biosynthesis in the plastid. The enrichment in leucine carbons 2–5 was determined from [U-13C]-glutamine labelling experiments (n = 3, SD). There were no statistical differences between the large subunit (LSU) and small subunit (SSU)-derived leucine, indicating that the cytosol and plastid pools are equivalent within the sensitivity of technique. (a) Schematic of leucine carbons derived from pyruvate and acetyl-CoA. (b) The expected enrichment for leucine biosynthesized in either plastid or cytosol was calculated from labelling in carbons derived from acetyl-CoA and pyruvate. The labelling in acetyl-CoA from fatty acid biosynthesis or elongation (see Fig. 4) was used to predict the 13C-label contribution to the first carbon of the leucine 2–5 fragment. Alanine carbons two and three (see Fig. 4) were used to represent 13C-labelling of pyruvate. The simulated labelling if carbons are derived from plastid pools matches the observed results. In contrast, labelling expected if synthesis occurs from cytosolic pools differs greatly from the observed results, particularly in the unlabelled ([M]+) and single labelled ([M+1]+) mass isotopomers. These results indicate that leucine is synthesized in the plastid and equilibrated across the plastid membrane to generate the cytosolic pool.
Serine and glycine LSU and SSU pools are not in isotopic equilibrium
Both serine and glycine are more highly 13C-enriched in LSU than in SSU (Fig. 2) and are labelled differently than phenylalanine. Phenylalanine is also made from triose (via phosphoenolpyruvate) but does not display differences between LSU and SSU (Fig. 6), which probably reflects a single site of synthesis. The differences in average 13C enrichment for glycine and serine large and small subunits are similar to alanine (Fig. 2). However, unlike alanine, the maximally labelled [M+3]+ mass isotopomer in serine and [M+2]+ isotopomer in glycine are less abundant relative to other mass fractions (Fig. 6a and Supporting Information Tables S2–S4). The increased abundance of isotopomers with one or two 13C atoms indicates multiple bond-breaking events in metabolism and is indicative of pentose phosphate pathways as previously discussed. However, there are additional pathways for glycine and serine biosynthesis that may contribute to the isotopomer labelling patterns. For example, glycine labelling could be altered by reversible reactions through glycine oxidase, glycine decarboxylase, glycine dehydrogenase, glycine aminotransferase and/or serine hydroxyl-methyltransferase. The pathways result in partially labelled glycine but the extent of their subcellular activity in seeds is unknown. Of these possibilities, photorespiratory metabolism is unlikely because developing embryos of B. napus have concentrations of CO2 approaching 2000 times atmospheric levels (Goffman et al. 2005). Instead, the high CO2 levels may enhance the reversibility of glycine decarboxylase and other enzymes that utilize a CO2 substrate. Furthermore, one carbon groups are necessary for synthesis of other amino acids (i.e. histidine and methionine) and impact serine hydroxymethyltransferase and glycine dehydrogenase activities.
Figure 6.
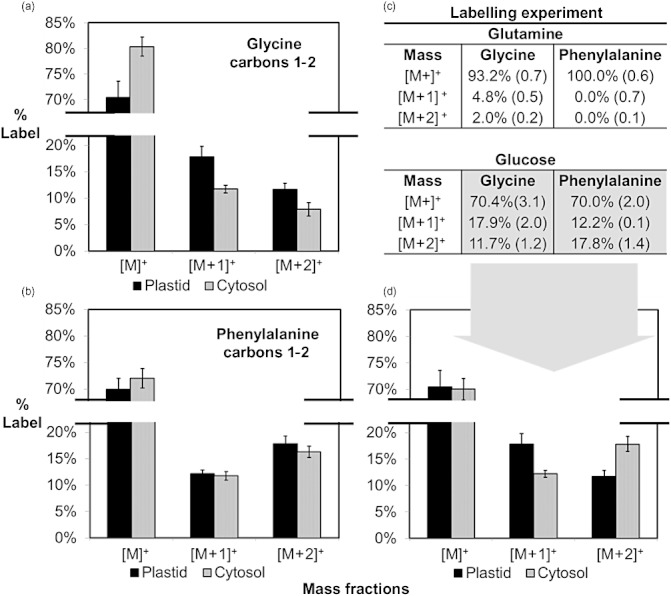
Glycine labelling differs from phenylalanine. Comparison of glycine and phenylalanine carbons from labelling experiments (n = 3, SD). Glycine and the first two carbons of phenylalanine are derived in part from three carbon intermediates including 3-phosphoglycerate and phosphoenolpyruvate. (a) Glycine shows different mass enrichments between large subunits (LSUs) and small subunits (SSUs), (b) while phenylalanine is enriched similarly in both. (c) Glycine is significantly labelled by [U-13C]-glutamine whereas phenylalanine is unlabelled. (d) Glucose labelling reveals differences between glycine and phenylalanine that are derived from different metabolic processes.
Interestingly, our results reveal that glycine also became labelled when cultures were provided with [U-13C]-glutamine (Fig. 6c). The absence of gluconeogenesis in filling oilseed embryos (Schwender et al. 2003; Allen et al. 2009b) implies that glycine label may come from threonine aldolase, utilizing threonine as a precursor. In comparison, the first two carbons of aromatic amino acids (i.e. that are derived from phosphoenolpyruvate) remain unlabelled in the glutamine labelling experiment (Fig. 6c).
LSU and SSU amino acids from aspartate and glutamate families are similarly labelled
The 13C-enrichment of four and five carbon amino acids is presented in Fig. 7 and in Supporting Information Fig. S1, respectively. The aspartate family (aspartate, asparagine, methionine and threonine) is derived from oxaloacetate. The glutamate family (glutamate, glutamine, proline and arginine) is synthesized from glutamine supplied to the seed or from 2-oxoglutarate. These amino acids are tied to central metabolites of the TCA cycle and their biosynthesis is linked to enzymes with established subcellular locations such as phosphoenolpyruvate carboxylase and succinate dehydrogenase. The results from 13C-glucose labelling reveal reduced differences in enrichment in isoleucine, aspartate/asparagine and threonine between LSU and SSU. Other labelling experiments presented in the figure reinforce these observations. The similar pool enrichments imply that either the amino acids or their precursors are isotopically equilibrated with regard to labelling across the plastid envelope.
Figure 7.
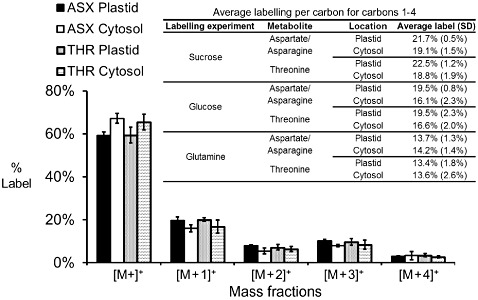
Labelling of four carbon products is similar across organelles. Comparison of aspartate/asparagine labelling with threonine from large subunits (LSUs) and small subunits (SSUs) (n = 3, SD). The labelling in four carbon amino acids is similar between subcellular compartments as indicated by label enrichment from [U-13C]-glucose. Labelling for other experiments is summarized in the inset table.
DISCUSSION
In this study, we investigated whether steady-state isotope labelling of proteins synthesized in different subcellular compartments would result in differential labelling of these proteins. We reasoned that if differences could be observed, these could provide a novel strategy to probe compartmentation of plant metabolism. Because Rubisco is a highly abundant protein with eight large and eight small subunits that are coordinately synthesized in the plastid and cytosol (Blair & Ellis 1973; Gray & Kekwick 1974; Baker et al. 1975), it was chosen as a test case for this study. Our results clearly indicate that some amino acids exhibit differences between the LSU and SSU in both the extent and the isotopomer patterns of 13C labelling. As described, these experiments have: (1) provided new information on the biosynthesis of alanine; (2) revealed that alanine, serine and glycine pools are not isotopically equilibrated between the cytosol and plastid; (3) confirmed in vivo that leucine synthesis is mostly confined to the plastid; and (4) provided evidence that amino acids from aspartate and glutamate families are nearly in isotopic equilibrium between the plastid and cytosol.
Other approaches have also provided in vivo information at the subcellular level. Compartment-specific labelling patterns for protein glycans have been established from hydroxyacetone and levulinic acid breakdown products using strong acid hydrolysis of protein (Sriram et al. 2007). Each of these labelled compounds reports specifically on cytosolic glycolysis. Starch that is synthesized in the plastid from ADP-glucose reflects the plastidic carbohydrate pool and has also been analysed by both GC-MS and nuclear magnetic resonance (NMR) (Allen et al. 2007). These methods have been used to resolve subcellular hexose phosphate pools. For example, Alonso et al. report a compartmentalized flux model of developing sunflower embryos based on the differences between starch and sucrose labelling (Alonso et al. 2007). Compared with starch, glycan and cell wall analysis that report on a limited number of precursor pools, amino acids provide more diverse information on primary metabolism.
When not overlooked, the successful engineering of metabolism at a compartmentalized level has provided major advantages. For example, the targeting of enzymes for the production of poly-hydroxybutyrate (PHB) to plastids of Arabidopsis resulted in a 100-fold increase in PHB accumulation compared with cytosolic targeting (Nawrath, Poirier & Somerville 1994). Nonetheless, such marked success stories are the exception, which may in part be because there are insufficient methods to assess metabolism at the subcellular level. This is one of the main challenges in producing validated flux models of plant seeds and other eukaryotic systems that include the compartmentation of metabolism (Kruger & Ratcliffe 2009; Allen et al. 2009a). Therefore, methods to assess the degree of compartmentation and biosynthetic location of metabolites, including amino acids, are important. In turn, these more accurate descriptions may help distinguish metabolic differences between wild type, mutants and transgenics that are of interest in basic biology and metabolic engineering. In combination with other compartmented analytes (such as the use of fatty acid labelling in this study) and flux analysis, the labelling in metabolic precursors in different compartments can be determined. Together, these approaches can be used to tackle other open questions about the compartmentation of amino acid metabolism in plants. For example, assessing the relative contributions of different compartments to the synthesis of the important amino acids cysteine (Hell & Wirtz 2011) and proline (Verslues & Sharma 2010) would be valuable and potentially accessible by modest modifications of the methods described here.
Limitations of the study
In this study, enrichments in amino acids and their patterns of isotopomer labelling report on the isotopic equilibration of subcellular pools, and in some cases on possible difference in pathways for the biosynthesis of amino acids (or precursors) in different compartments. The technique establishes the degree of similarity of pools from different organelles and indicates when multiple pools may be involved. We note that if pools equilibrate fully across the plastid envelope, information on differences in metabolism in the plastid and cytosol is not available. Partial isotopic equilibration between compartments will also reduce labelling differences and therefore changes in enrichment are not necessarily proportional to the relative contribution of synthesis of an amino acid in different compartments. However, such exchange cannot create or increase labelling inequalities. Therefore, observed differences in labelling provide a clear indication of multiple origins of those amino acids. The comparison of these results to benchmark data on metabolite or enzyme localization can provide a guide for further experimentation.
The concentrations of enriched amino acids, labelled substrate selection and sensitivity of instrumentation are important. In this study, several amino acids could not be accurately compared between the LSU and SSU because of low concentrations or contaminations and losses during hydrolysis. Future more extensive analysis will be enabled by improving sensitivities of MS instruments.
Outlook for and extension of methodology
Ribosomes and protein synthesis occur in mitochondria, cytosol and plastid in plants, and in mitochondria and cytosol of other eukaryotic organisms. Therefore, this method is extendable to essentially any eukaryote, where sufficient amounts of protein are available. In addition, the same concept can be applied to compare metabolism in specific cell types of multicellular organisms. For example, developing seeds are an organ with at least four cell types: seed coat, endosperm, embryo radical and embryo cotyledons. Mitochondrial multi-protein complexes with individual proteins produced in the cytosol or mitochondria would be present in all cell types and their examination would provide a strategy to examine whether subcellular metabolic fluxes are partitioned differently in these different cell types.
Even within a single tissue, cells are exposed to substantially different environments. For example, abaxial and adaxial cells of developing oilseed embryos differ by several-fold in ATP and oxygen levels, corresponding to light and oxygen availability (Vigeolas et al. 2003; Borisjuk et al. 2005; Rolletschek et al. 2005). Analysis of the Rubisco subunits from dissected sections of the seeds would provide further information on how these differences influence metabolism across a gradient of cells and within organelles.
MATERIALS AND METHODS
Chemicals
Methanol, hexane, isopropanol and acetonitrile, the labelled substrates: [U-13C]-glucose, [U-13C]-sucrose and [U-13C]-glutamine, and their unlabelled counterparts were all purchased from Sigma (Milwaukee, WI, USA). Hydrochloric acid was purchased from Mallinckrodt Baker (Phillipsburg, NJ, USA).
Plant material and embryo culturing
Greenhouse cultivation and embryo culturing of canola embryos with labelled organic substrates have been previously described (Schwender et al. 2006) and were used with slight modifications. B. napus plants were grown at 22 °C in 3 gallon pots that contained a 3:1 mixture of soil : vermiculite in a greenhouse irradiated with sunlight and supplemental lights at ∼150 µE to maintain at least 14 h of daylight conditions. Plants were watered three times daily and supplemented with Peters 20:20:20 nutrient solution. When embryos had developed on the plant to a torpedo stage, siliques were removed and surface sterilized in a 5% hypochlorite solution and rinsed with water. Seeds were removed from siliques and the seed coat was removed aseptically and embryos were transferred to Petri dishes for culture duration. The culture medium composition has been previously described (Schwender et al. 2003) and is composed of sucrose (80 mm), glucose (40 mm), glutamine (35 mm) and alanine (10 mm). For each labelling experiment, labelled substrates were substituted for 100% of their unlabelled counterpart, except for sucrose in which labelled sucrose replaced 50% of total sucrose. A complete list of macro- and micronutrients supplemented with cultures is given elsewhere (Schwender et al. 2003) and Gamborg's vitamins (Sigma) were also provided. Approximately 36 embryos were cultured in six Petri dishes for 14 d. Harvested embryos were rinsed in 1 L of water and either processed immediately or frozen in liquid nitrogen and stored at −80 °C.
Protein extraction, purification and hydrolysis
Embryos were placed in 1.5 mL tubes and extracted on a Retsch MM 301 Bead Beater using one steel ball and 0.6 mL of protein extraction buffer containing: 10 mm Tris-HCl, 150 mm NaCl, pH 7.4 and 60 µL of protease inhibitor stock (1 mini EDTA-free tablet per 500 µL water, Roche Indianapolis, IN, USA, #13821400). After 60 min at 30 Hz, the tubes were centrifuged at ∼13 200 g for 12 min at 4 °C and supernatant was directly loaded on a Seppro IgY-Rusbico Spin affinity purification column (GenWay Biotech Inc 28–288-23153-SC, San Diego, CA or Sigma). The extract and affinity beads were agitated overnight at 4 °C on an orbital shaker at low speed. Then the columns were washed of unbound proteins using extraction buffer and the intact Rubisco was recovered using a stripping buffer containing: 100 mm Glycine-HCl pH 2.5 and 10% v/v protease inhibitor stock. Affinity-purified Rubisco was subsequently processed through SDS-PAGE using 10.5–14% Tris-HCl Precast criterion gels (345-9949 Bio-Rad, Hercules, CA, USA) using a Bio-Rad Power Pac Universal Power Supply. Because the fractions of Rubisco from the column were dilute (∼0.3 µg µL−1), protein was first concentrated on the gel by loading 45 µL, running the gel for 40–60 s and reloading with more extract to focus a large amount of Rubisco within the stacking gel.
Using molecular weight markers and Rubisco harvested from leaf as standards, the large and small subunits of Rubisco were confirmed with Coomassie blue staining. Gels were electroblotted (Bio-Rad Criterion Cell) to membranes (Bio-Rad Immun-blot PVDF Membrane) overnight for ∼18 h at 15 V followed by 1 h at 100 V at 4 °C using a transfer buffer composed of 10 mm CAPS (pH 11), 10% methanol (v : v). Membranes were very briefly developed using 5% Coomassie blue stain contained in a 50% methanol, 10% acetic acid solution (Sigma). Next, bands were immediately and thoroughly destained by incubating in alternate solutions of water and methanol. Protein bands were excised and further washed. Protein amino acids were hydrolyzed from gel slices at 110 °C for 28 h in the presence of 6N HCl in an oxygen-deficient environment, dried by evaporation with nitrogen at 60 °C and stored at −20 °C until subsequent derivatization. Cation exchange was tested but did not further improve amino acid purity, reduced yields and was deemed unnecessary for processing by GC-MS.
Butyl amide reactions
Butyl amides were generated as has been previously described (Allen et al. 2007). Oil was extracted from Brassica embryos using a 2:1 (v : v) mixture of hexane : isopropanol. The extracted oil was reacted with 2 mL n-butylamine (Sigma) in the presence of 3 mL hexane. Reactions proceeded for 48 h at 75 °C and were quenched by the addition of concentrated HCl that resulted in phase partitioning of unreacted butylamine from hexane and butylamides. The non-polar hexane phase was concentrated by evaporation under nitrogen before GC-MS analysis. Specific details to GC-MS settings can be found elsewhere (Allen et al. 2007).
GC-MS of amino acids
For 13C labelling, protein amino acids were converted to their tert-butyl-dimethylsilyl derivatives using MTBSTFA (Sigma) and analysed with GC-MS (Das Neves & Vasconcelos 1987; Dauner, Bailey & Sauer 2001). A Thermo – Trace Ultra gas chromatogram linked to a DSQII mass spectrometer and a TriPlus autosampler was used for analysis. A DB5 (30 m × 0.2 mm × 0.33 µm) silica column was used for separations. The column utilized a helium carrier gas and split injections of 10:1 (injection temperature 250 °C, detector temperature 280 °C). The temperature profile consisted of a starting temperature of 40 °C for 1 min followed by a 50 °C per min ramp to 100 °C, a hold time of 1 min, then 5 °C per min to a temperature of 200 °C, followed by 10 °C per min to a temperature of 300 °C. The EI (70 eV) MS was tuned before runs using the m/z 69, 219, and 502 ions of perflurotributylamine as a calibrant. Total ion chromatograms were used to scan the weights from 50 to 500 m/z and establish retention times that were then used in coordination with selected ion monitoring to quantitate the relative labelling. Ions of interest and correction for natural abundance have been described elsewhere (Antoniewicz, Kelleher & Stephanopoulos 2007; Allen et al. 2009b).
Acknowledgments
The authors gratefully acknowledge: Dr Mike Pollard for his key role in suggesting this approach and idea development, along with Dr Dan Jones and Beverly Chamberlin (MSU Mass Spectrometry Facility), and Dr Joe Leykam (Macromolecular Structure, Sequencing and Synthesis Facility), for help with instrumentation; and Drs Jorg Schwender, Ana Alonso, Tim Durrett, Phil Bates and Igor Libourel for valuable discussions. Additionally, the authors thank the editor and anonymous referees for helpful comments and suggestions that have improved the manuscript. Any product or trademark mentioned here does not imply a warranty, guarantee or endorsement by the USDA over other suitable products.
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article:
Figure S1. Labelling in glutamate-derivedcompounds is equivalent across organelles. Glutamate-derivedproducts proline and glutamine/glutamate are equivalently labelledfor multiple labelling experiments (n = 3, SD). Arginine enrichments were qualitatively similar but not reported because of complex fragmentation patterns described elsewhere (Allen & Ratcliffe 2009).
Table S1. Subcellular biosynthetic origin ofamino acid pathway enzymes. Information on the subcellularlocalization in Arabidopsis of the indicated amino acid biosynthesis enzymes was derived from a number of literature sources and websites, including the examples given. Multiple locations reported for a number of enzymes indicate that the biosynthetic locations of amino acids are either not conclusive or suggest that amino acids can be synthesized in multiple subcellular compartments.
Table S2. Average carbon labelling for LSU and SSU. The average labelling per carbon was calculated as described in Fig. 2 of the main text and the differences were compared between the LSU and SSU that represent plastidic and cytosolic pools, respectively.
Table S3. Isotope fractional labelling in (a)plastidic and (b) cytosolic amino acids after provision of[U-13C]-glucose to filling B. napus embryos.
Table S4. Isotope fractional labelling in (a)plastidic and (b) cytosolic amino acids after provision of[U-13C]-sucrose to filling B. napus embryos.
Table S5. Isotope fractional labelling in (a)plastidic and (b) cytosolic amino acids after provision of[U-13C]-glutamine to filling B. napus embryos.
Table S6. Isotope fractional labelling in (a)plastidic and (b) cytosolic amino acids after provision of[U-13C]-alanine to filling B. napus embryos.
Appendix S1. Supplement references.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors.Any queries (other than missing material) should be directed to the corresponding author for the article.
REFERENCES
- Allen DK, Ratcliffe RG. Quantification of Isotope Label. In: Schwender J, editor. Plant Metabolic Networks. New York: Springer; 2009. pp. 105–149. [Google Scholar]
- Allen DK, Shachar-Hill Y, Ohlrogge JB. Compartment-specific labeling information in 13C metabolic flux analysis of plants. Phytochemistry. 2007;68:2197–2210. doi: 10.1016/j.phytochem.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Allen DK, Libourel IGL, Shachar-Hill Y. Metabolic flux analysis in plants: coping with complexity. Plant, Cell & Environment. 2009a;32:1241–1257. doi: 10.1111/j.1365-3040.2009.01992.x. [DOI] [PubMed] [Google Scholar]
- Allen DK, Ohlrogge JB, Shachar-Hill Y. The role of light in soybean seed filling metabolism. The Plant Journal. 2009b;58:220–234. doi: 10.1111/j.1365-313X.2008.03771.x. [DOI] [PubMed] [Google Scholar]
- Alonso AP, Goffman FD, Ohlrogge JB, Shachar-Hill Y. Carbon conversion efficiency and central metabolic fluxes in developing sunflower (Helianthus annuus L.) embryos. The Plant Journal. 2007;52:296–308. doi: 10.1111/j.1365-313X.2007.03235.x. [DOI] [PubMed] [Google Scholar]
- Alonso AP, Val DL, Shachar-Hill Y. Understanding fatty acid synthesis in developing maize embryos using metabolic flux analysis. Metabolic Engineering. 2010;12:488–497. doi: 10.1016/j.ymben.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Alonso AP, Val DL, Shachar-Hill Y. Central metabolic fluxes in the endosperm of developing maize seeds and their implications for metabolic engineering. Metabolic Engineering. 2011;13:96–107. doi: 10.1016/j.ymben.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Andriotis VME, Kruger NJ, Pike MJ, Smith AM. Plastidial glycolysis in developing Arabidopsis embryos. New Phytologist. 2010;185:649–662. doi: 10.1111/j.1469-8137.2009.03113.x. [DOI] [PubMed] [Google Scholar]
- Antoniewicz MR, Kelleher JK, Stephanopoulos G. Accurate assessment of amino acid mass isotopomer distributions for metabolic flux analysis. Analytical Chemistry. 2007;79:7554–7559. doi: 10.1021/ac0708893. [DOI] [PubMed] [Google Scholar]
- Baker A, Graham IA, Holdsworth M, Smith SM, Theodoulou FL. Chewing the fat: β-oxidation in signalling and development. Trends in Plant Science. 2006;11:124–132. doi: 10.1016/j.tplants.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Baker TS, Eisenberg D, Eiserling FA, Weissman L. The structure of form I crystals of D ribulose 1,5 diphosphate carboxylase. Journal of Molecular Biology. 1975;91:391–399. doi: 10.1016/0022-2836(75)90267-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao XM, Pollard M, Ohlrogge J. The biosynthesis of erucic acid in developing embryos of Brassica rapa. Plant Physiology. 1998;118:183–190. doi: 10.1104/pp.118.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao XM, Focke M, Pollard M, Ohlrogge J. Understanding in vivo carbon precursor supply for fatty acid synthesis in leaf tissue. The Plant Journal. 2000;22:39–50. doi: 10.1046/j.1365-313x.2000.00712.x. [DOI] [PubMed] [Google Scholar]
- Benkeblia N, Shinano T, Osaki M. Metabolite profiling and assessment of metabolome compartmentation of soybean leaves using non-aqueous fractionation and GC-MS analysis. Metabolomics. 2007;3:297–305. [Google Scholar]
- Binder S. Branched-chain amino acid metabolism in Arabidopsis thaliana. The Arabidopsis Book. 2010;8:e0137. doi: 10.1199/tab.0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair GE, Ellis RJ. Protein synthesis in chloroplasts. I. Light driven synthesis of the large subunit of fraction I protein by isolated pea chloroplasts. Biochimica et Biophysica Acta. 1973;319:223–234. doi: 10.1016/0005-2787(73)90013-0. [DOI] [PubMed] [Google Scholar]
- Borisjuk L, Nguyen TH, Neuberger T, et al. Gradients of lipid storage, photosynthesis and plastid differentiation in developing soybean seeds. New Phytologist. 2005;167:761–776. doi: 10.1111/j.1469-8137.2005.01474.x. [DOI] [PubMed] [Google Scholar]
- Burger G, Gray MW, Lang BF. Mitochondrial genomes: anything goes. Trends in Genetics. 2003;19:709–716. doi: 10.1016/j.tig.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Cooper TG, Beevers H. Beta oxidation in glyoxysomes from castor bean endosperm. Journal of Biological Chemistry. 1969;244:3514–3520. [PubMed] [Google Scholar]
- Das Neves HJC, Vasconcelos AMP. Capillary gas-chromatography of amino-acids, including asparagine and glutamine – sensitive gas-chromatographic mass-spectrometric and selected ion monitoring gas-chromatographic mass-spectrometric detection of the n,o(s)-tert-butyldimethylsilyl derivatives. Journal of Chromatography. 1987;392:249–258. doi: 10.1016/s0021-9673(01)94270-0. [DOI] [PubMed] [Google Scholar]
- Dauner M, Bailey JE, Sauer U. Metabolic flux analysis with a comprehensive isotopomer model in Bacillus subtilis. Biotechnology and Bioengineering. 2001;76:144–156. doi: 10.1002/bit.1154. [DOI] [PubMed] [Google Scholar]
- Dhingra A, Portis AR, Daniell H. Enhanced translation of a chloroplast-expressed RbcS gene restores small subunit levels and photosynthesis in nuclear RbcS antisense plants. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:6315–6320. doi: 10.1073/pnas.0400981101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieuaide M, Couee I, Pradet A, Raymond P. Effects of glucose starvation on the oxidation of fatty acids by maize root tip mitochondria and peroxisomes: evidence for mitochondrial fatty acid β-oxidation and acyl-CoA dehydrogenase activity in a higher plant. Biochemical Journal. 1993;296:199–207. doi: 10.1042/bj2960199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farre EM, Tiessen A, Roessner U, Geigenberger P, Trethewey RN, Willmitzer L. Analysis of the compartmentation of glycolytic intermediates, nucleotides, sugars, organic acids, amino acids, and sugar alcohols in potato tubers using a nonaqueous fractionation method. Plant Physiology. 2001;127:685–700. doi: 10.1104/pp.010280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geigenberger P, Tiessen A, Meurer J. Use of non-aqueous fractionation and metabolomics to study chloroplast function in Arabidopsis. Methods in Molecular Biology. 2011;775:135–160. doi: 10.1007/978-1-61779-237-3_8. [DOI] [PubMed] [Google Scholar]
- Gerhardt R, Heldt HW. Measurement of subcellular metabolite levels in leaves by fractionation of freeze-stopped material in nonaqueous media. Plant Physiology. 1984;75:542–547. doi: 10.1104/pp.75.3.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt R, Stitt M, Heldt HW. Subcellular metabolite levels in spinach leaves: regulation of sucrose synthesis during diurnal alterations in photosynthetic partitioning. Plant Physiology. 1987;83:399–407. doi: 10.1104/pp.83.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain V, Rylott EL, Larson TR, Sherson SM, Bechtold N, Carde JP, Bryce JH, Graham IA, Smith SM. Requirement for 3-ketoacyl-CoA thiolase-2 in peroxisome development, fatty acid β-oxidation and breakdown of triacylglycerol in lipid bodies of Arabidopsis seedlings. The Plant Journal. 2001;28:1–12. doi: 10.1046/j.1365-313x.2001.01095.x. [DOI] [PubMed] [Google Scholar]
- Giege P, Heazlewood JL, Roessner-Tunali U, Millar AH, Fernie AR, Leaver CJ, Sweetlove LJ. Enzymes of glycolysis are functionally associated with the mitochondrion in Arabidopsis cells. The Plant Cell. 2003;15:2140–2151. doi: 10.1105/tpc.012500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffman FD, Alonso AP, Schwender J, Shachar-Hill Y, Ohlrogge JB. Light enables a very high efficiency of carbon storage in developing embryos of rapeseed. Plant Physiology. 2005;138:2269–2279. doi: 10.1104/pp.105.063628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JWA, Williams TCR, Morgan M, Fernie AR, Ratcliffe RG, Sweetlove LJ. Glycolytic enzymes associate dynamically with mitochondria in response to respiratory demand and support substrate channeling. The Plant Cell. 2007;19:3723–3738. doi: 10.1105/tpc.107.053371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JC, Kekwick RGO. The synthesis of the small subunit of ribulose 1,5 bisphosphate carboxylase in the French bean Phaseolus vulgaris. European Journal of Biochemistry. 1974;44:491–500. doi: 10.1111/j.1432-1033.1974.tb03507.x. [DOI] [PubMed] [Google Scholar]
- Hell R, Wirtz M. Molecular biology, biochemistry and cellular physiology of cysteine metabolism in Arabidopsis thaliana. The Arabidopsis Book. 2011;9:e0154. doi: 10.1199/tab.0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer VV, Sriram G, Fulton DB, Zhou R, Westgate ME, Shanks JV. Metabolic flux maps comparing the effect of temperature on protein and oil biosynthesis in developing soybean cotyledons. Plant, Cell & Environment. 2008;31:506–517. doi: 10.1111/j.1365-3040.2008.01781.x. [DOI] [PubMed] [Google Scholar]
- Junker BH, Lonien J, Heady LE, Rogers A, Schwender J. Parallel determination of enzyme activities and in vivo fluxes in Brassica napus embryos grown on organic or inorganic nitrogen source. Phytochemistry. 2007;68:2232–2242. doi: 10.1016/j.phytochem.2007.03.032. [DOI] [PubMed] [Google Scholar]
- Khan BR, Adham AR, Zolman BK. Peroxisomal Acyl-CoA oxidase 4 activity differs between Arabidopsis accessions. Plant Molecular Biology. 2012;78:45–58. doi: 10.1007/s11103-011-9843-4. [DOI] [PubMed] [Google Scholar]
- King SP, Badger MR, Furbank RT. CO2 refixation characteristics of developing canola seeds and silique wall. Australian Journal of Plant Physiology. 1998;25:377–386. [Google Scholar]
- Kruger NJ, Ratcliffe RG. Insights into plant metabolic networks from steady-state metabolic flux analysis. Biochimie. 2009;91:697–702. doi: 10.1016/j.biochi.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Libourel IGL, Shachar-Hill Y. Metabolic flux analysis in plants: from intelligent design to rational engineering. Annual Review of Plant Biology. 2008;59:625–650. doi: 10.1146/annurev.arplant.58.032806.103822. [DOI] [PubMed] [Google Scholar]
- Liedvogel B, Stumpf PK. Origin of acetate in spinach leaf cell. Plant Physiology. 1982;69:897–903. doi: 10.1104/pp.69.4.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linka N, Weber APM. Intracellular metabolite transporters in plants. Molecular Plant. 2010;3:21–53. doi: 10.1093/mp/ssp108. [DOI] [PubMed] [Google Scholar]
- Lonien J, Schwender J. Analysis of metabolic flux phenotypes for two arabidopsis mutants with severe impairment in seed storage lipid synthesis. Plant Physiology. 2009;151:1617–1634. doi: 10.1104/pp.109.144121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Albert HH, Paull R, Moore PH. Metabolic engineering of invertase activities in different subcellular compartments affects sucrose accumulation in sugarcane cells. Australian Journal of Plant Physiology. 2000;27:1021–1030. [Google Scholar]
- Maloney GS, Kochevenko A, Tieman DM, Tohge T, Krieger U, Zamir D, Taylor MG, Fernie AR, Klee HJ. Characterization of the branched-chain amino acid aminotransferase enzyme family in tomato. Plant Physiology. 2010;153:925–936. doi: 10.1104/pp.110.154922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin W, Rujan T, Richly E, Hansen A, Cornelsen S, Lins T, Leister D, Stoebe B, Hasegawa M, Penny D. Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:12246–12251. doi: 10.1073/pnas.182432999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masakapalli SK, le Lay P, Huddleston JE, Pollock NL, Kruger NJ, George Ratcliffe R. Subcellular flux analysis of central metabolism in a heterotrophic Arabidopsis cell suspension using steady-state stable isotope labeling. Plant Physiology. 2010;152:602–619. doi: 10.1104/pp.109.151316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson C, Wood C. Influence of mitochondrial β-oxidation on early pea seedling development. New Phytologist. 2009;181:832–842. doi: 10.1111/j.1469-8137.2008.02717.x. [DOI] [PubMed] [Google Scholar]
- Nawrath C, Poirier Y, Somerville C. Targeting of the polyhydroxybutyrate biosynthetic pathway to the plastids of Arabidopsis thaliana results in high levels of polymer accumulation. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:12760–12764. doi: 10.1073/pnas.91.26.12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlrogge JB, Pollard MR, Stumpf PK. Studies on biosynthesis of waxes by developing jojoba seed tissue. Lipids. 1978;13:203–210. [Google Scholar]
- Pomeroy MK, Kramer JKG, Hunt DJ, Keller WA. Fatty acid changes during development of zygotic and microspore-derived embryos of Brassica napus. Physiologia Plantarum. 1991;81:447–454. [Google Scholar]
- Rolletschek H, Radchuk R, Klukas C, Schreiber F, Wobus U, Borisjuk L. Evidence of a key role for photosynthetic oxygen release in oil storage in developing soybean seeds. New Phytologist. 2005;167:777–786. doi: 10.1111/j.1469-8137.2005.01473.x. [DOI] [PubMed] [Google Scholar]
- Ruuska SA, Girke T, Benning C, Ohlrogge JB. Contrapuntal networks of gene expression during Arabidopsis seed filling. The Plant Cell. 2002;14:1191–1206. doi: 10.1105/tpc.000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruuska SA, Schwender J, Ohlrogge JB. The capacity of green oilseeds to utilize photosynthesis to drive biosynthetic processes. Plant Physiology. 2004;136:2700–2709. doi: 10.1104/pp.104.047977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwender J, Ohlrogge JB. Probing in vivo metabolism by stable isotope labeling of storage lipids and proteins in developing Brassica napus embryos. Plant Physiology. 2002;130:347–361. doi: 10.1104/pp.004275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwender J, Ohlrogge JB, Shachar-Hill Y. A flux model of glycolysis and the oxidative pentosephosphate pathway in developing Brassica napus embryos. Journal of Biological Chemistry. 2003;278:29442–29453. doi: 10.1074/jbc.M303432200. [DOI] [PubMed] [Google Scholar]
- Schwender J, Goffman FD, Ohlrogge JB, Shachar-Hill Y. Rubisco without the Calvin cycle improves the carbon efficiency of developing green seeds. Nature. 2004;432:779–782. doi: 10.1038/nature03145. [DOI] [PubMed] [Google Scholar]
- Schwender J, Shachar-Hill Y, Ohlrogge JB. Mitochondrial metabolism in developing embryos of Brassica napus. Journal of Biological Chemistry. 2006;281:34040–34047. doi: 10.1074/jbc.M606266200. [DOI] [PubMed] [Google Scholar]
- Singh B. Plant Amino Acids: Biochemistry and Biotechnology. New York: Marcel Dekker Inc; 1999. [Google Scholar]
- Sriram G, Fulton DB, Iyer VV, Peterson JM, Zhou RL, Westgate ME, Spalding MH, Shanks JV. Quantification of compartmented metabolic fluxes in developing soybean embryos by employing biosynthetically directed fractional C-13 labeling, [C-13, H-1] two-dimensional nuclear magnetic resonance, and comprehensive isotopomer balancing. Plant Physiology. 2004;136:3043–3057. doi: 10.1104/pp.104.050625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram G, Iyer VV, Fulton DB, Shanks JV. Identification of hexose hydrolysis products in metabolic flux analytes: a case study of levulinic acid in plant protein hydrolysate. Metabolic Engineering. 2007;9:442–451. doi: 10.1016/j.ymben.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Stitt M, Lilley RM, Gerhardt R, Heldt HW. Metabolite levels in specific cells and subcellular compartments of plant leaves. Methods in Enzymology. 1989;174:518–552. [Google Scholar]
- Stocking CR. Chloroplast isolation in nonaqueous media. Plant Physiology. 1959;34:56–61. doi: 10.1104/pp.34.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetlove LJ, Beard KFM, Nunes-Nesi A, Fernie AR, Ratcliffe RG. Not just a circle: flux modes in the plant TCA cycle. Trends in Plant Science. 2010;15:462–470. doi: 10.1016/j.tplants.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Unseld M, Marienfeld JR, Brandt P, Brennicke A. The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nature Genetics. 1997;15:57–61. doi: 10.1038/ng0197-57. [DOI] [PubMed] [Google Scholar]
- Verslues PE, Sharma S. Proline metabolism and its implications for plant-environment interaction. The Arabidopsis Book. 2010;8:e0140. doi: 10.1199/tab.0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierstra RD. Protein degradation in plants. Annual Review of Plant Physiology and Plant Molecular Biology. 1993;44:385–410. [Google Scholar]
- Vigeolas H, Van Dongen JT, Waldeck P, Hühn D, Geigenberger P. Lipid storage metabolism is limited by the prevailing low oxygen concentrations within developing seeds of oilseed rape. Plant Physiology. 2003;133:2048–2060. doi: 10.1104/pp.103.031963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JA, Todd J, Newman T, Focks N, Girke T, de Ilárduya OM, Jaworski JG, Ohlrogge JB, Benning C. A new set of arabidopsis expressed sequence tags from developing seeds. The metabolic pathway from carbohydrates to seed oil. Plant Physiology. 2000;124:1582–1594. doi: 10.1104/pp.124.4.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield HV, Murphy DJ, Hills MJ. Subcellular-localization of fatty-acid elongase in developing seeds of lunaria-annua and brassica-napus. Phytochemistry. 1993;32:255–258. [Google Scholar]
- Williams TCR, Miguet L, Masakapalli SK, Kruger NJ, Sweetlove LJ, Ratcliffe RG. Metabolic network fluxes in heterotrophic Arabidopsis cells: stability of the flux distribution under different oxygenation conditions. Plant Physiology. 2008;148:704–718. doi: 10.1104/pp.108.125195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter H, Robinson DG, Heldt HW. Subcellular volumes and metabolite concentrations in barley leaves. Planta. 1993;191:180–190. [Google Scholar]
- Wintz H, Chen HC, Pillay DTN. Partial characterization of the gene coding for subunit IV of soybean mitochondrial NADH dehydrogenase. Current Genetics. 1989;15:155–160. doi: 10.1007/BF00435463. [DOI] [PubMed] [Google Scholar]
- Zeeman SC, Delatte T, Messerli G, Umhang M, Stettler M, Mettler T, Streb S, Reinhold H, Kötting O. Starch breakdown: recent discoveries suggest distinct pathways and novel mechanisms. Functional Plant Biology. 2007a;34:465–473. doi: 10.1071/FP06313. [DOI] [PubMed] [Google Scholar]
- Zeeman SC, Smith SM, Smith AM. The diurnal metabolism of leaf starch. Biochemical Journal. 2007b;401:13–28. doi: 10.1042/BJ20061393. [DOI] [PubMed] [Google Scholar]
- Zrenner R, Stitt M, Sonnewald U, Boldt R. Pyrimidine and purine biosynthesis and degradation in plants. Annual Review of Plant Biology. 2006;57:805–836. doi: 10.1146/annurev.arplant.57.032905.105421. [DOI] [PubMed] [Google Scholar]
- Zybailov B, Rutschow H, Friso G, Rudella A, Emanuelsson O, Sun Q, van Wijk KJ. Sorting signals, N-terminal modifications and abundance of the chloroplast proteome. PLoS ONE. 2008;3:e1994. doi: 10.1371/journal.pone.0001994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.