

Abstract
Pathogenic viral infections have exerted selection pressure on their hosts to evolve cellular antiviral inhibitors referred to as restriction factors. Examples of such molecules are APOBEC3G, APOBEC3F and TRIM5α. APOBEC3G and APOBEC3F are cytidine deaminases that are able to strongly inhibit retroviral replication by at least two mechanisms. They are counteracted by the lentiviral Vif protein. TRIM5α binds to sensitive, incoming retroviruses via its C-terminal PRY/SPRY domain and rapidly recruits them to the proteasome before significant viral DNA synthesis can occur. Both of these proteins robustly block retroviral replication in a species-specific way. It remains an open but important question as to whether innate restriction factors such as these can be harnessed to inhibit HIV-1 replication in humans.
APOBEC3G and TRIM5α are potent antiviral restriction factors
Retroviruses and reverse-transcribing transposable elements have been invading their host’s genomes for millions of years. Consequently, host organisms have been obliged to evolve ways to protect themselves from infection by pathogens that can cause a variety of diseases. Over the past few years it has become apparent that there is a system of antiviral activities mediated by intracellular proteins referred to as restriction factors. These proteins can robustly block infection of sensitive viruses after they have entered the host cell. They are species specific in that each species can restrict a subset of viruses with exquisite sensitivity. With the failure of recent HIV vaccine trials [1] there is an increased interest in this type of immunity and the question of whether such inhibitors can be harnessed to restrict HIV-1 infection in humans. In this review we focus on recent developments considering the intracellular innate immune mediators, or restriction factors, APOBEC3 and TRIM5α.
APOBEC3G-mediated virus restriction and DNA editing
APOBEC3G (apolipoprotein B mRNA-editing enzyme catalytic polypeptide 3G or A3G, initially known as CEM-15) was first identified as a cellular factor that renders human cells nonpermissive for infection by HIV-1 viruses lacking a Vif gene [2]. A3G is a member of a family of cytidine deaminases that catalyse the hydrolysis of cytidines (C) to uridines (U). A3G targets cytidines in the negative sense single-stranded DNA that is generated during HIV-1 reverse transcription, a process also referred to as DNA editing or hypermutation [3]. The C to U editing leads to guanine (G) to adenine (A) substitutions in the positive sense DNA strand as reverse transcription is completed and these mutations might inactivate viral gene products or regulatory genetic elements. A3G is incorporated into virus particles budding from the infected cell and editing occurs during reverse transcription upon infection of the next target cell. The HIV-1-encoded Vif protein (viral infectivity factor) counteracts A3G by targeting it for ubi-quitylation and subsequent degradation by the proteasome, such that intracellular levels of A3G in the producer cell are reduced as is its packaging into progeny virions [4,5]. In addition to A3G, several other members of the APOBEC3 protein family (A3) have been shown to act against a wide range of viruses and transposable elements [6]. Of these, human A3F also inhibits HIV-1 in a Vif-dependent manner [7-10]. The mode of A3G/F action is summarized in Figure 1.
Figure 1.

Model of APOBEC antiviral function. (a) Effect of hA3G on the life cycle of wild-type HIV-1. The integrated HIV-1 provirus (blue) is transcribed and translated like a normal cellular gene. The viral Vif protein (purple) acts as an adaptor protein, connecting hA3G (green) to an E3 ubiquitin ligase complex comprising ElonginB, ElonginC, Cullin5 and RING-box-1 (rbx1), thereby inducing the polyubiquitylation and proteasomal degradation of hA3G. Viral proteins (turquoise) and genomic RNA (red) assemble and bud through the plasma membrane, releasing viral particles. Virions then enter target cells and, after reverse transcription, uncoating and nuclear entry have taken place, the viral DNA is integrated into the host genome. Formation of this provirus thus completes the replication cycle. (b) Effect of hA3G on the life cycle of HIV-1/Δvif. The HIV-1 provirus is transcribed and translated as before, but no Vif is synthesized. Cellular hA3G is therefore not degraded but instead is packaged into nascent budding virions. On infection of target cells, the presence of hA3G leads to a block in infection by any one or more of the following mechanisms. (1) hA3G might impede the formation of reverse transcripts through an unknown mechanism. (2) Cytidine deamination of nascent reverse transcripts by hA3G could be sufficient to hinder ensuing rounds of infectious virion production owing to inactivating mutations in viral genes and/or proteins. (3) Edited reverse transcripts might be recognized by cellular DNA repair pathways, leading to the degradation of these transcripts. (4) It is also possible that hA3G induces the degradation of HIV-1 reverse transcripts through an editing-independent mechanism, perhaps by recruiting a cellular endonuclease. (Reproduced with permission from [6]).
The APOBEC3G structure
A recent report of the NMR structure of a truncated A3G containing the cytidine deaminase catalytic centre has shed some light on the mechanism of DNA editing [11]. This structure revealed that A3G adopts a fold that is very similar to other cytidine deaminases and is characterised by a five-stranded mixed β-sheet and five α helices of which predominantly α1, β3 and α2 define the zinc-coordination motif that constitutes the catalytic centre (Figure 2a). The core structure of the catalytic centre is conserved in cytidine deaminases ranging from bacteria to vertebrates. There are, however, major differences with regard to the substrate specificity because other cytidine deaminases act either on free nucleotides or RNA, whereas A3G acts exclusively on single-stranded DNA. Chen et al. have begun to address the substrate specificity of A3G by monitoring changes in chemical shift upon titration of single-stranded DNA by NMR. Subsequently, residues that showed differential shift were mutated and mutant A3G editing properties assayed. In this manner, Chen et al. were able to construct a model for DNA binding, which indicates that contacts with the DNA substrate are predominantly made by arginines in proximity to the catalytic centre (Figure 2b). Ofthese, R215 and R313 are conserved throughout the APOBEC protein family, but they are not present in other cytidine deaminases. A3G preferentially edits the second cytidine in a CpC dinucleotide context of the DNA substrate as opposed to A3F, which preferentially edits in a TpC context [3]. The current model suggests that the conserved R313 is in close proximity of the first base in the target sequence, which leaves open the question of how the difference in target site specificity between A3F and A3G is determined.
Figure 2.
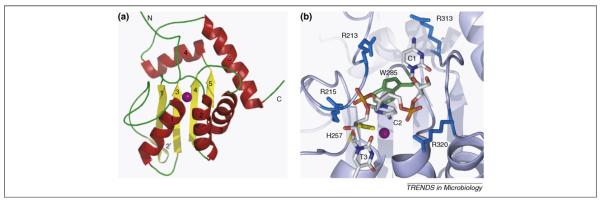
NMR structure of APOBEC3G and a model for substrate DNA binding. (a) Ribbon representation of the NMR structure of A3G [11]. Beta strands are shown in yellow and are numbered 1–5 (2′ indicates a short disrupted β strand); α helices are shown in red and are numbered 1–5. A purple sphere indicates the position of the zinc ion. (b) Model of the interaction of APOBEC3G with a trinucleotide 5′-C1-C2-T3–3′ DNA substrate. C2 represents the nucleotide that is being edited and is flipped out from the phosphodiester backbone for positioning towards the catalytic zinc ion (purple) and H257 (yellow). W285 (green) can facilitate positioning of C2 through stacking interactions. Arginines are shown in blue, and several conserved arginines of APOBEC3G have the potential to interact with the DNA. R213, R215 and R320 are shown in close proximity to the DNA backbone phosphates, whereas R313 could interact with C1.
The contribution of hypermutation to virus restriction
Although much attention was initially directed at hypermutation as the means by which A3G restricts HIV replication, there is now a large body of evidence to indicate that A3G additionally inhibits HIV by an editing-independent mechanism. This was first demonstrated using catalytically inactive mutants of A3G, which maintain a significant level of antiviral activity in the absence of detectable editing [12]. It has since been shown that editing is dispensable for inhibition of the hepatitis B virus and several transposable elements [6,13-16]. In addition, editing-defective mutants of A3F maintain a robust antiviral phenotype [17]. It was recognized before the discovery of the APOBEC proteins that Vif-deleted HIV-1 viruses show reduced accumulation of reverse transcription products [18]. There is supporting evidence that A3G interferes with the process of reverse transcription, but the precise mechanism remains to be elucidated [19,20].
Further support for an editing-independent mode of HIV inhibition by A3G comes from a study by Chiu et al. [21]. These authors have shown that A3G can also act as an inhibitor of incoming HIV in resting CD4+ T cells. Upon activation, CD4+ T cells become permissive to HIV infection and this coincides with assembly of A3G into high-molecular-weight RNP complexes that harbour an RNA component that is inhibitory to A3G action [22]. In the resting cell, the uncomplexed A3G interferes with the accumulation of reverse transcription products. Importantly, this post-entry restriction posed by A3G leads to hypermutation of a minority of the incoming viral genomes. The relative contributions of editing-dependent and editing-independent mechanisms to virus inhibition remain a hotly debated topic in this field of research. Recent studies have demonstrated that the antiviral properties of editing-defective A3G appear more compromised when assayed in a spreading infection rather than a single-cycle infectivity assay [23,24]. Thus, despite the intensive efforts to elucidate the mechanism by which APOBEC proteins mediate virus and retro-element restriction, there is to date no consensus opinion.
APOBEC3G and virus evolution
Although the antiviral properties of the APOBEC proteins are not yet fully understood, it has often been speculated that the editing activity of A3 proteins could in fact contribute to viral diversity. Hypermutated viral genomes are present in some HIV-infected individuals [25-27] and Vif proteins from different HIV-1 isolates show marked differences in their ability to downregulate A3F or A3G [28], suggesting that these proteins could on occasion gain access to the viral genome during the natural course of infection. It has also been noted that some common drug-resistance mutations in the HIV genome have all the hallmarks of A3-mediated mutation, which involves a change from G to A at a preferred A3 target sequence [29]. A recent report has substantiated the claim that A3 proteins can contribute to the emergence of drug-resistance mutations [30]. Mulder et al. propagated HIV-1 viruses with defective Vif alleles in human peripheral blood mononuclear cells (PBMCs) and determined both the fitness and mutational load of progeny viruses. The extent of hypermutation was shown to correlate inversely with the A3G-neutralising activity of the Vif protein, with partially defective alleles leading to fewer mutations than alleles with a more severe defect. In the absence of a drug, the emergence of the M184I mutation that confers resistance to 3TC was observed in the viral genomes, whereas the mutations M184 V and M184T that cannot be generated by G to A substitutions were not observed. Viral populations with the M184I mutation had compromised viral fitness as a consequence of the high mutational load in the virus genome. However, upon co-infection with a wild-type HIV-1 strain the dormant viral quasi-species facilitated the emergence of a replication competent and 3TC-resistant virus, presumably through template switching-mediated recombination.
Despite the evidence that A3 proteins can contribute to the emergence of drug resistance in vitro, it is not obvious how these proteins have contributed to genome evolution of HIV during its natural history. The genome of HIV is particularly A-rich, which might indicate past exposure to C to U editing enzymes. However, two recent surveys have concluded that a case for A3-driven HIV evolution is confounded by additional factors, such as the fidelity of the RT enzyme and imbalances in the dNTP pool of infected cells [31,32]. Indeed, G to A substitutions occur only marginally more frequently than A to G substitutions in drug-resistance mutations and the latter cannot be induced by A3 proteins [32]. The prospect that therapeutic strategies targeted at A3F/G would fail due to their contribution to viral diversity might thus not be as bleak as has been suggested [29].
Restriction of retroviruses by TRIM5α
Tripartite motif protein 5α (TRIM5α) was identified as a cellular inhibitor of HIV-1 replication in rhesus monkey cells in 2004 [33]. Before this time it was referred to as Ref1 in humans [34] and Lv1 in monkeys [35]. It is a member of the tripartite motif family encoded by about 70 genes in humans [36]. The function of most TRIMs remains poorly characterised, although several have been implicated in innate immune functions. The tripartite motif, or RBCC, comprises a RING domain, 1 or 2 B-boxes and a coiled coil with the characteristic ordering and spacing that defines the TRIM family. Conserved order and spacing suggests that the RBCC acts as a single domain rather than each component having an independent activity. TRIM5’s RING has E3 ubiquitin ligase activity [37], the B-box2 has unknown but essential function [38] and the coiled coil is responsible for multimerisation into homotrimers, as well as heterotrimers with closely related TRIM molecules [39]. Like many TRIMs, TRIM5α also has a C-terminal PRY/SPRY, or B30.2 domain, which includes the viral recruitment motifs [40]. The PRY/SPRY can be perceived as a pattern-recognition domain that recognizes the shape of sensitive incoming retroviral capsids. It recruits the TRIM5 enzyme (RING-Bbox) to the incoming virions leading to inhibition of viral infectivity. TRIM proteins commonly have multiple isoforms generated by alternate splicing [33]. A variety of TRIM5-spliced isoforms exist but only the longest, the α form, includes a PRY/SPRY and consequently only the α form has antiviral activity. The shorter isoforms have dominant negative activity and rescue restricted infection when overexpressed [33,41].
Antiviral TRIM5 orthologues have been found in diverse species including primates, rabbits and cattle [42-44]. The PRY/SPRY domain shows evidence of strong positive selection throughout primate evolution [45]. Positive selection in TRIM5 suggests counterevolution between the host and the virus, an outcome referred to as the Red Queen effect (Box 1) [45]. Presumably, infection by pathogenic retroviruses selected for change, leading to the species-specific inhibition of infection that we see today. For example, the rhesus macaque TRIM5α strongly inhibits HIV-1 infectivity whereas the human protein only inhibits HIV-1 weakly, if at all [33]. The human protein, however, inhibits other viruses including equine infectious anaemia virus and a particular murine leukaemia virus (MLV-N) [46]. There is also evidence for selection within a species with polymorphism in rhesus macaques leading to allele-specific antiviral specificity against Old World lineage lentiviruses [47,48].
Box 1. TRIM5, CypA and the Red Queen.
Restriction factors put viruses under selective pressure to change by mutation. In turn, pathogenic infection puts selective pressure on the host. This leads to counterevolution between viruses and host restriction factors and both the virus and the host are under constant selective pressure to gain the advantage. This molecular arms race is referred to as the Red Queen effect, with reference to Lewis Caroll’s Through the Looking Glass in which the Red Queen says to Alice, “It takes all the running you can do to stay in the same place.” The running refers to evolutionary change, and the counterevolution between TRIM5 and primate lentiviruses is a great example. For instance, we see evidence for strong selection in the PRY/SPRY of TRIM5, particularly in variable regions thought to interact directly with the viral capsid. This is even true within a particular host, in the case of rhesus macaques. However, the viral capsid sequence is also variable, particularly in the external loop that recruits cyclophilin A to lentiviral capsids, which probably also binds TRIM5. CypA’s ability to isomerise a prolyl peptide bond in the HIV-1 CA appears to impact viral sensitivity to TRIM5α, and it has been proposed that it is acting as an uncloaking device to sensitize HIV-1 to TRIM5α in Old World monkeys. One would think that the relative speed of virus evolution gives the virus an overwhelming advantage, but the larger size and complexity of the host genome and the increased flexibility that this gives appear to ensure the host survives, at least in some cases. For example, on two independent occasions, primate hosts have simply replaced the virus-binding domain of TRIM5α for an entirely different and unrelated domain, cyclophilin A. The CypA domain is then also subject to change with a point mutation that leads to rhesus TRIMCyp’s ability to restrict HIV-2 rather than HIV-1. It is unlikely that HIV-1 and HIV-2 provided these selective pressures, but related viruses almost certainly did. As we understand these complex relationships better, we might be able to predict which viruses could be the most dangerous by virtue of their ability to escape this type of innate defence.
TRIMCyp, a novel form of TRIM5
In the New World owl monkey the TRIM5 locus has been modified by insertion of a cDNA encoding the enzyme cyclophilin A (CypA) into the last intron by L1-mediated retrotransposition [49,50]. This leads to the expression of a protein in which the PRY/SPRY has been replaced by a complete CypA protein. The factor is referred to as TRIMCyp and it restricts viruses whose incoming capsids bind the CypA domain including HIV-1, feline immunodeficiency virus, and certain simian immunodeficiency viruses (SIV) [38,49,51]. The CypA recruits the TRIM5 enzyme to the virus in the same way that the PRY/SPRY does for TRIM5α. All owl monkeys tested are homozygous for TRIMCyp suggesting that it gave a strong selective advantage to them at some point in their evolutionary history, leading to its fixation. Remarkably, a similar TRIMCyp has evolved independently in Old World monkeys [52-56]. Old World monkeys (from Africa and Asia) are genetically distinct from New World monkeys (from South America). In the case of the Old World rhesus macaque TRIMCyp, a downstream CypA insertion combined with a splice-site mutation leads to expression of a TRIMCyp fusion protein. The different location of the Old and New World CypA insertions indicates independent evolution of TRIMCyp in the Old and New World lineages of primates. Such convergent evolution signifies the advantage that encoding such a factor gives. The Old World TRIM5 allele encoding TRIMCyp exists amongst at least six further TRIM5 alleles in rhesus macaques that encode PRY/SPRY bearing TRIM5 s. Intriguingly, rhesus macaque TRIMCyp has geographical differences in allelic frequency, suggesting geographical differences in selection pressure [52]. The features of the TRIM5 RBCC that make it such an attractive partner for CypA remain unclear, especially given that fusion of CypA to several TRIM5-related and TRIM5-unrelated molecules makes efficient restriction factors, at least in vitro [57,58]. It is certainly possible that TRIM5α’s ability to recruit restricted virus to the proteasome is the key.
Cyclophilin A is itself an enzyme that is recruited to a variety of lentiviruses where, at least in the case of HIV-1, it catalyses isomerisation of a conserved prolyl peptide bond in the capsid between cis and trans [59]. CypA acts as a positive factor for HIV-1 replication in human cells but has an inhibitory effect on HIV-1 in simian cells [60]. Why this might be is not entirely clear but it appears that CypA sensitizes HIV-1 to simian TRIM5α, perhaps by altering the conformation of the TRIM5α recognition site on the capsid [61]. CypA does not impact HIV-1’s insensitivity to human TRIM5α and its activity as a cofactor, specifically in human cells, suggesting that it might help HIV-1 escape some other, as yet undefined, inhibitory activity [61].
The antiviral mechanism of TRIM5
The details of the mechanism of TRIM5α activity remain incompletely solved. Although there is good agreement that the TRIM5α PRY/SPRY is required for targeting to viral capsid structures [62], the mode of virus inactivation is less well understood. Upon chemical inhibition of the proteasome, TRIM5α-restricted viruses can complete reverse transcription but still fail to infect the cell [63]. This suggests that the block to reverse transcription is due to rapid recruitment of the TRIM5α-associated virus to the proteasome. Protection of the complex, by inhibition of the proteasome, therefore allows reverse transcription to occur. It is not clear how TRIM5α inhibits infection in the absence of an active proteasome. Some believe that TRIM5α directly uncoats the capsid protein from incoming virions, leading to accelerated uncoating [33], but others believe that this ‘uncoating’ is the activity of the proteasome [64]. Assaying uncoating by measuring pelletable capsids, in the presence of a proteasome inhibitor, is difficult to interpret because inhibiting the proteasome rescues a lot of virions that are otherwise degraded in a TRIM5-independent way [38]. Regardless, the TRIM5α enzyme (RING-Bbox2) is required for maximal inhibition of infectivity and it is likely that recruitment to the proteasome has an important role in the mechanism. Real-time microscopy of TRIM5α in action has been demonstrated recently [64]. In these experiments, TRIM5α is recruited to the incoming HIV-1 virions, which are then recruited to the TRIM5α containing cytoplasmic bodies. These events are exaggerated by inhibition of the proteasome, again supporting the notion that normally the proteasome degrades the TRIM5/virus complex. TRIM5α antiviral activity is summarized in Figure 3.
Figure 3.

Summary of the antiviral activity of TRIM5α. During unrestricted, productive infection, the retrovirus enters the cell, synthesises DNA by reverse transcription and enters the nucleus (N) where it integrates into a host chromosome (1). TRIM5α is found in dynamic structure in the cytoplasm called cytoplasmic bodies (CB) [64]. If sensitive incoming virions encounter TRIM5α they are rapidly recruited to the proteasome and degraded (2). If the proteasome is inhibited by MG132, then the virus does not get degraded but the TRIM5α-associated virus remains uninfectious (3) [63].
A role for interferon in restriction
TRIM5α is strongly induced by type 1 interferon, suggesting that during spreading infection in vivo TRIM5α levels might be high [65]. This observation encourages the notion that in certain cases restriction of virus might depend on high expression levels of TRIM5α. Examples include restriction of HIV-2 by high levels of human TRIM5α or SIVmac by rhesus TRIM5α [66]. Such activities might represent the situation in vivo during localized infection when interferon levels are high. Controversially, rhesus TRIM5α has been described recently as being able to block HIV-1 particle formation by degrading nascent gag molecules [67]. This study has been criticized for requiring high levels of TRIM5α expression [68], but again high levels might not represent an artefact in vivo. Further evidence for an ability of TRIMs to block retroviral replication in the late stages of the life cycle comes from a study of TRIM22, which appears to account for some of the interferon’s antiviral activity against HIV-1 budding [69]. Furthermore, in a screen of the TRIM family for antiviral activity, several proteins were shown to weakly inhibit retroviral budding [70]. These observations all merit closer investigation.
Concluding remarks and future perspectives
TRIM5 and APOBEC3G/F clearly present strong species-specific blocks to zoonotic transmission of sensitive viruses. For example, HIV-1 does not replicate in rhesus cells but can replicate, at least in vitro, if mutated to become insensitive to rhesus TRIM5α and APOBEC3G/F [71,72]. The degree to which these factors impact on pathogenesis in vivo remains incompletely solved (see Box 2 for further unanswered questions regarding TRIM5/APOBEC). As mentioned above, APOBEC3 could influence pathogenesis of HIV-1 in humans in the context of the emergence of drug resistance. Whether TRIM5α can impact HIV-1 pathogenesis in humans remains to be established. A recent study has suggested an association between a TRIM5α polymorphism (H43Y) that inactivates antiviral activity and accelerates AIDS disease progression [73]. Unfortunately, the frequency of this mutation in the studied cohorts was low, which makes it difficult to use these polymorphisms to address the role of TRIM5α in HIV-1 pathogenesis in humans. Regardless, it is of great interest to consider whether the TRIM5/APOBEC3 class of innate immune mediators can be stimulated to inhibit HIV-1 replication in humans. One can imagine a situation where high levels of TRIM5α are induced leading to a stronger, broader, less specific antiviral response, during which distinctive antiviral mechanisms come into play. Certainly, robust protective vaccination by live attenuated SIV in monkeys is not explained by classical adaptive immune responses in some studies [74,75] and might represent localized, interferon-induced upregulation of antiviral factors such as APOBEC3G/F and TRIM5α as well as other less well-defined restriction factors, such as the recently identified interferon-inducible tetherin [76]. The observation that the degree of attenuation is inversely correlated with the degree of protection supports the notion that continued attenuated replication is necessary in a specific subset of target cells [77]. Recent data also demonstrate that live attenuated vaccination can protect against vigorous uncloned challenge virus [78].
Box 2. Unanswered questions.
What is the mechanism by which APOBEC3G interferes with reverse transcription?
How is the DNA-editing target site specificity of different APOBEC proteins determined?
To what extent do APOBEC proteins contribute to viral diversity during the natural course of infection?
How exactly does TRIM5α block infectivity and can it block virus on the way out by interacting with newly formed gag molecules?
How does Cyclophilin A influence sensitivity to TRIM5α?
Does TRIM5α block any viruses other than retroviruses?
Does TRIM5α impact HIV-1/HIV-2 pathogenesis in humans?
How important for restriction are TRIM5α/APOBEC3G expression levels and interferon inducibility?
Does TRIM5/APOBEC3G have cellular functions unrelated to infection?
Can TRIM5/APOBEC3G specificity be enhanced or altered using drugs?
Nonstructural, or accessory, proteins from diverse viruses have been described as being able to subvert innate immune host defences [79]. We suspect that HIV-1’s ability to do similar tricks with its own nonstructural proteins Vif, Vpr, Vpu, Tat, and Rev might have been underestimated. Certainly Vif and Vpu have this ability, and we propose that the continued study of primate lentiviral accessory proteins will reveal much about host defences and their activity against retroviral infection. Indeed, it is often suggested that a better understanding of innate immunity against retroviruses might lead to better treatment for viral infection. What form these advances might take is not clear at present, although there are several possibilities. Gene therapy technologies could theoretically be used to modify human stem cells to be resistant to HIV-1 infection. The cells could be modified by expression of humanised TRIMCyp or mutants of human TRIM5α that restrict HIV-1. It is also possible that a more detailed understanding of interferon’s activity in stimulating expression of antiviral proteins might allow the design of more specific agonists that could stimulate antiviral activity more subtly than treatment with interferon itself, which has limited utility. It might even be possible to use interventions that stimulate innate immunity as adjuvants to improve more classical vaccination protocols by synergising with adaptive responses. We therefore hope that the study of innate immunity and restriction factors will eventually lead us to techniques of manipulating innate antivirals, and thereby the development of broadly effective therapeutics that rely on millions of years of host evolution and are therefore harder to escape by viral mutation.
Acknowledgements
We thank Flavia Autore for help with generating figures, Reuben Harris and Hiroshi Matsuo for providing the PDB file of A3G bound to DNA, Chad Swanson for critical reading of the manuscript, Torsten Schaller for critical reading of the manuscript and provision of Figure 2, and Neil Berry for helpful discussion of unpublished data.
References
- 1.Kaiser J. AIDS research: review of vaccine failure prompts a return to basics. Science. 2008;320:30–31. doi: 10.1126/science.320.5872.30. [DOI] [PubMed] [Google Scholar]
- 2.Sheehy AM, et al. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 3.Harris RS, et al. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 4.Sheehy AM, et al. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 5.Yu X, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 6.Holmes RK, et al. APOBEC-mediated viral restriction: not simply editing? Trends Biochem. Sci. 2007;32:118–128. doi: 10.1016/j.tibs.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Bishop KN, et al. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 2004;14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 8.Liddament MT, et al. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 2004;14:1385–1391. doi: 10.1016/j.cub.2004.06.050. [DOI] [PubMed] [Google Scholar]
- 9.Wiegand HL, et al. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 2004;23:2451–2458. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng YH, et al. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J. Virol. 2004;78:6073–6076. doi: 10.1128/JVI.78.11.6073-6076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen KM, et al. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature. 2008;452:116–119. doi: 10.1038/nature06638. [DOI] [PubMed] [Google Scholar]
- 12.Newman EN, et al. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 2005;15:166–170. doi: 10.1016/j.cub.2004.12.068. [DOI] [PubMed] [Google Scholar]
- 13.Turelli P, et al. Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004;303:1829. doi: 10.1126/science.1092066. [DOI] [PubMed] [Google Scholar]
- 14.Esnault C, et al. Dual inhibitory effects of APOBEC family proteins on retrotransposition of mammalian endogenous retroviruses. Nucleic Acids Res. 2006;34:1522–1531. doi: 10.1093/nar/gkl054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hulme AE, et al. Selective inhibition of Alu retrotransposition by APOBEC3G. Gene. 2007;390:199–205. doi: 10.1016/j.gene.2006.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiu YL, et al. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc. Natl. Acad. Sci. U. S. A. 2006;103:15588–15593. doi: 10.1073/pnas.0604524103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holmes RK, et al. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 2007;282:2587–2595. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- 18.von Schwedler U, et al. Vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J. Virol. 1993;67:4945–4955. doi: 10.1128/jvi.67.8.4945-4955.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwatani Y, et al. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007;35:7096–7108. doi: 10.1093/nar/gkm750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson JL, Hope TJ. APOBEC3G restricts early HIV-1 replication in the cytoplasm of target cells. Virology. 2008;375:1–12. doi: 10.1016/j.virol.2008.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiu YL, et al. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 2005;435:108–114. doi: 10.1038/nature03493. [DOI] [PubMed] [Google Scholar]
- 22.Soros VB, et al. Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog. 2007;3:e15. doi: 10.1371/journal.ppat.0030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyagi E, et al. Enzymatically active APOBEC3G is required for efficient inhibition of human immunodeficiency virus type 1. J. Virol. 2007;81:13346–13353. doi: 10.1128/JVI.01361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schumacher AJ, et al. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J. Virol. 2008;82:2652–2660. doi: 10.1128/JVI.02391-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janini M, et al. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4(+) T cells. J. Virol. 2001;75:7973–7986. doi: 10.1128/JVI.75.17.7973-7986.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kieffer TL, et al. G→A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. J. Virol. 2005;79:1975–1980. doi: 10.1128/JVI.79.3.1975-1980.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vartanian JP, et al. G→A hypermutation of the human immunodeficiency virus type 1 genome: evidence for dCTP pool imbalance during reverse transcription. Proc. Natl. Acad. Sci. U. S. A. 1994;91:3092–3096. doi: 10.1073/pnas.91.8.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon V, et al. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog. 2005;1:e6. doi: 10.1371/journal.ppat.0010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pillai SK, et al. Turning up the volume on mutational pressure: is more of a good thing always better? (A case study of HIV-1 Vif and APOBEC3) Retrovirology. 2008;5:26. doi: 10.1186/1742-4690-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mulder LC, et al. Cytidine deamination induced HIV-1 drug resistance. Proc. Natl. Acad. Sci. U. S. A. 2008;105:5501–5506. doi: 10.1073/pnas.0710190105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deforche K, et al. Estimating the relative contribution of dNTP pool imbalance and APOBEC3G/3F editing to HIV evolution in vivo. J. Comput. Biol. 2007;14:1105–1114. doi: 10.1089/cmb.2007.0073. [DOI] [PubMed] [Google Scholar]
- 32.Berkhout B, de Ronde A. APOBEC3G versus reverse transcriptase in the generation of HIV-1 drug-resistance mutations. AIDS. 2004;18:1861–1863. doi: 10.1097/00002030-200409030-00022. [DOI] [PubMed] [Google Scholar]
- 33.Stremlau M, et al. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 34.Towers G, et al. A conserved mechanism of retrovirus restriction in mammals. Proc. Natl. Acad. Sci. U. S. A. 2000;97:12295–12299. doi: 10.1073/pnas.200286297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cowan S, et al. Cellular inhibitors with Fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11914–11919. doi: 10.1073/pnas.162299499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nisole S, et al. TRIM family proteins: retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 2005;3:799–808. doi: 10.1038/nrmicro1248. [DOI] [PubMed] [Google Scholar]
- 37.Yamauchi K, et al. Ubiquitination of E3 ubiquitin ligase TRIM5alpha and its potential role. FEBS J. 2008;275:1540–1555. doi: 10.1111/j.1742-4658.2008.06313.x. [DOI] [PubMed] [Google Scholar]
- 38.Diaz-Griffero F, et al. Comparative requirements for the restriction of retrovirus infection by TRIM5alpha and TRIMCyp. Virology. 2007;369:400–410. doi: 10.1016/j.virol.2007.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mische CC, et al. Retroviral restriction factor TRIM5alpha is a trimer. J. Virol. 2005;79:14446–14450. doi: 10.1128/JVI.79.22.14446-14450.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yap MW, et al. A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr. Biol. 2005;15:73–78. doi: 10.1016/j.cub.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 41.Passerini LD, et al. Retroviral restriction factors Fv1 and TRIM5{alpha} act independently and can compete for incoming virus before reverse transcription. J. Virol. 2006;80:2100–2105. doi: 10.1128/JVI.80.5.2100-2105.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Si Z, et al. Evolution of a cytoplasmic tripartite motif (TRIM) protein in cows that restricts retroviral infection. Proc. Natl. Acad. Sci. U. S. A. 2006;103:7454–7459. doi: 10.1073/pnas.0600771103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ylinen LM, et al. Isolation of an active Lv1 gene from cattle indicates that tripartite motif protein-mediated innate immunity to retroviral infection is widespread among mammals. J. Virol. 2006;80:7332–7338. doi: 10.1128/JVI.00516-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schaller T, et al. An active TRIM5 in rabbits indicates an antiviral common ancestor for mammalian TRIM5 proteins. J. Virol. 2007;81:11713–11721. doi: 10.1128/JVI.01468-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sawyer SL, et al. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc. Natl. Acad. Sci. U. S. A. 2005;102:2832–2837. doi: 10.1073/pnas.0409853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keckesova Z, et al. The human and African green monkey TRIM5alpha genes encode Ref1 and Lv1 retroviral restriction factor activities. Proc. Natl. Acad. Sci. U. S. A. 2004;101:10780–10785. doi: 10.1073/pnas.0402474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newman RM, et al. Balancing selection and the evolution of functional polymorphism in Old World monkey TRIM5alpha. Proc. Natl. Acad. Sci. U. S. A. 2006;103:19134–19139. doi: 10.1073/pnas.0605838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson SJ, et al. Rhesus macaque TRIM5 alleles have divergent antiretroviral specificities. J. Virol. 2008;82:7243–7247. doi: 10.1128/JVI.00307-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sayah DM, et al. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature. 2004;430:569–573. doi: 10.1038/nature02777. [DOI] [PubMed] [Google Scholar]
- 50.Nisole S, et al. A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc. Natl. Acad. Sci. U. S. A. 2004;101:13324–13328. doi: 10.1073/pnas.0404640101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang F, et al. Antiretroviral potential of human tripartite motif-5 and related proteins. Virology. 2006;353:396–409. doi: 10.1016/j.virol.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 52.Wilson SJ, et al. Independent evolution of an antiviral TRIMCyp in rhesus macaques. Proc. Natl. Acad. Sci. U. S. A. 2008;105:3557–3562. doi: 10.1073/pnas.0709003105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Virgen CA, et al. Independent genesis of chimeric TRIM5-cyclophilin proteins in two primate species. Proc. Natl. Acad. Sci. U. S. A. 2008;105:3563–3568. doi: 10.1073/pnas.0709258105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brennan G, et al. TRIMCyp expression in Old World primates Macaca nemestrina and Macaca fascicularis. Proc. Natl. Acad. Sci. U. S. A. 2008;105:3569–3574. doi: 10.1073/pnas.0709511105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Newman RM, et al. Evolution of a TRIM5-CypA splice isoform in old world monkeys. PLoS Pathog. 2008;4:e1000003. doi: 10.1371/journal.ppat.1000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liao CH, et al. A novel fusion gene, TRIM5-Cyclophilin A in the pig-tailed macaque determines its susceptibility to HIV-1 infection. AIDS. 2007;21(Suppl 8):S19–S26. doi: 10.1097/01.aids.0000304692.09143.1b. [DOI] [PubMed] [Google Scholar]
- 57.Yap MW, et al. Trim-cyclophilin A fusion proteins can restrict human immunodeficiency virus type 1 infection at two distinct phases in the viral life cycle. J. Virol. 2006;80:4061–4067. doi: 10.1128/JVI.80.8.4061-4067.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schaller T, et al. Fusion of cyclophilin a to fv1 enables cyclosporine-sensitive restriction of human and feline immunodeficiency viruses. J. Virol. 2007;81:10055–10063. doi: 10.1128/JVI.00616-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bosco DA, et al. Catalysis of cis/trans isomerization in native HIV-1 capsid by human cyclophilin A. Proc. Natl. Acad. Sci. U. S. A. 2002;99:5247–5252. doi: 10.1073/pnas.082100499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berthoux L, et al. Cyclophilin A is required for TRIM5{alpha}-mediated resistance to HIV-1 in Old World monkey cells. Proc. Natl. Acad. Sci. U. S. A. 2005;102:14849–14853. doi: 10.1073/pnas.0505659102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Keckesova Z, et al. Cyclophilin A renders HIV-1 sensitive to old world monkey but not human TRIM5a antiviral activity. J. Virol. 2006;80:4683–4690. doi: 10.1128/JVI.80.10.4683-4690.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stremlau M, et al. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. U. S. A. 2006;103:5514–5519. doi: 10.1073/pnas.0509996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu X, et al. Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc. Natl. Acad. Sci. U. S. A. 2006;103:7465–7470. doi: 10.1073/pnas.0510483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Campbell EM, et al. Visualization of a proteasome-independent intermediate during restriction of HIV-1 by rhesus TRIM5alpha. J. Cell Biol. 2008;180:549–561. doi: 10.1083/jcb.200706154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sakuma R, et al. Alpha interferon enhances TRIM5{alpha}-mediated antiviral activities in human and rhesus monkey cells. J. Virol. 2007;81:10201–10206. doi: 10.1128/JVI.00419-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ylinen LM, et al. Differential restriction of HIV-2 and SIVmac by TRIM5alpha alleles. J. Virol. 2005;79:11580–11587. doi: 10.1128/JVI.79.18.11580-11587.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sakuma R, et al. Rhesus monkey TRIM5alpha restricts HIV-1 production through rapid degradation of viral Gag polyproteins. Nat.Med. 2007;13:631–635. doi: 10.1038/nm1562. [DOI] [PubMed] [Google Scholar]
- 68.Zhang F, et al. No effect of endogenous TRIM5alpha on HIV-1 production. Nat. Med. 2008;14:235–236. doi: 10.1038/nm0308-235. [DOI] [PubMed] [Google Scholar]
- 69.Barr SD, et al. The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog. 2008;4:e1000007. doi: 10.1371/journal.ppat.1000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Uchil PD, et al. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 2008;4:e16. doi: 10.1371/journal.ppat.0040016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hatziioannou T, et al. Generation of simian-tropic HIV-1 by restriction factor evasion. Science. 2006;314:95. doi: 10.1126/science.1130994. [DOI] [PubMed] [Google Scholar]
- 72.Kamada K, et al. Generation of HIV-1 derivatives that productively infect macaque monkey lymphoid cells. Proc. Natl. Acad. Sci. U. S. A. 2006;103:16959–16964. doi: 10.1073/pnas.0608289103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Manen D, et al. The effect of Trim5 polymorphisms on the clinical course of HIV-1 infection. PLoS Pathog. 2008;4:e18. doi: 10.1371/journal.ppat.0040018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Almond N, et al. Mechanisms of protection induced by attenuated simian immunodeficiency virus. I. Protection cannot be transferred with immune serum. J. Gen. Virol. 1997;78:1919–1922. doi: 10.1099/0022-1317-78-8-1919. [DOI] [PubMed] [Google Scholar]
- 75.Stebbings R, et al. CD8+ lymphocytes do not mediate protection against acute superinfection 20 days after vaccination with a live attenuated simian immunodeficiency virus. J. Virol. 2005;79:12264–12272. doi: 10.1128/JVI.79.19.12264-12272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Neil SJ, et al. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 77.Johnson RP, et al. Highly attenuated vaccine strains of simian immunodeficiency virus protect against vaginal challenge: inverse relationship of degree of protection with level of attenuation. J. Virol. 1999;73:4952–4961. doi: 10.1128/jvi.73.6.4952-4961.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Berry N, et al. Resistance to superinfection of a vigorously replicating uncloned stock of Simian Immunodifciency virus (SIVmac251) stimulates replication of a live attenuated virus vaccine (SIVmacC8) J. Gen. Virol. doi: 10.1099/vir.0.2008/001693-0. (in press) [DOI] [PubMed] [Google Scholar]
- 79.Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008;89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]