

Abstract
Cyclic addition of a hydrazine nitrogen to unactivated alkynes catalyzed by non-metals. Starting from readily accessible γ-silyl allenyl esters, β-alkynyl hydrazines are prepared in one step and subsequently undergo unprecedented cyclization reactions in the presence of ammonium and phosphonium catalysts leading to dehydro-azaproline products. These heterocycles are also produced in high enantiomeric excesses using chiral ammonium phase transfer catalysts via a kinetic resolution pathway.
Keywords: non-metal alkyne-activation, organo-catalyst, beta-alkynyl hydrazines, dehydro-azaproline, kinetic resolution
Heteroatom additions to unactivated alkynes are thought to be best mediated by transition metal catalysts such as palladium[1] and gold.[2] Recent reports by Hammond[3] and others[4] demonstrate that cyclizations of alkynes with nitrogen and oxygen nucleophiles are possible using stoichiometric tetrabutylammonium fluoride. However, these several examples appear to be limited to aryl or α,α-difluoro-alkynes. As described below, we have discovered by serendipity a non-metal catalyzed addition of amine to unactivated alkynes in a cyclic context to yield azaproline derivatives (scheme 1). Although the nature of this catalysis reaction is not clear, these reactions proceed in excellent yields and lead to enantio-enriched azaprolines under kinetic resolution conditions using ammonium phase transfer catalysts.
Scheme 1.

Catalytic generation of azaprolines via cyclization of β-alkynyl hydrazines.
We decided to further investigate this cyclization reaction partly out of mechanistic curiosity but also mindful that azaprolines have taken on an increasingly important role in bioorganic[5] and medicinal chemistry investigations.[6] Indeed, there has been a great deal of interest in the synthesis of azaproline derivatives over the last decade. The earliest approach pioneered by Carreira involved diastereoselective [3+2] additions of diazoalkanes with α,β-unsaturated chiral esters.[7] More recently this method has been expanded to include other dipolar diazo reactions[8] including those catalyzed by chiral magnesium bisoxazole[9] and titanium BINOL-ate[10] and Lewis acids. The pyrazolidine ring of azaprolines has also been prepared via palladium catalyzed cyclizations of optically active allenylic hydrazines[11] or hydrazine adducts produced from racemic allenyl phosphine oxides.[12] However, in the present communication, we report related cyclization reactions catalyzed by non-metal cations which have allowed for an exciting class of chiral ammonium phase transfer catalysts to be brought to bear to produce non-racemic azaproline derivatives (scheme 1).
Based on our previous experience with γ-silyl allenyl esters,[13] we hypothesized that base catalyzed addition of dinitrogen electrophiles such as azidodicarboxylates should lead to β-alkynyl hydrazine intermediates. Indeed, with substrates 1 and 2 (EWG = CO2But and CO2Et) DBU catalyzed this transformation but the reactions were sluggish especially with larger α-substituents (table 1). Following a procedure similar to one we had previously reported,[14] γ-silyl allenyl and propargyl thioesters (EWG = COSBut) were prepared and, as expected, these substrates underwent rapid addition to azidodicarboxylates with DBU even at −20 °C. Less basic amines failed to give hydrazine products even with these thioesters except with DMF as the reaction solvent. This solvent gave no enantioselectivity when chiral amine catalysts such as quinine were used to catalyze the addition reaction to give 4.
Table 1.
DBU catalyzed additions of allenyl esters to azidodicarboxylates.

| |||||||
|---|---|---|---|---|---|---|---|
| entry | EWG | R1 | R3Si | R2 | T (°C) | time (h) | %yield 4a |
| 1 | CO2But | Ph | TES | iPr | rt | 12 | 83 |
| 2 | CO2Et | H | TES | iPr | 0 | 4 | 87 |
| 3 | CO2Et | H | TES | Bn | 0 | 4 | 67 |
| 4 | COSBut | Ph | TES | iPr | −20 | 1 | 69 |
| 5 | COSBut | Ph | TMS | iPr | −20 | 1 | 67 |
| 6 | COSBut | Ph | TIPS | iPr | −20 | 1 | 75 |
| 7 | COSBut | Ph | TES | tBu | −20 | 1 | 78 |
| 8 | COSBut | vinyl | TES | iPr | −20 | 1 | 71 |
| 9 | COSBut | 2-naph | TES | iPr | −20 | 1 | 76 |
| 10 | COSBut | o-tol | TES | iPr | −20 | 1 | 79 |
| 11 | COSBut | p-Cl-Ph | TES | iPr | −20 | 1 | 68 |
Isolated yields. With thioester substrates <5% of γ-addition hydrazine products (allenes) were also observed.
For improved characterization, we thought it useful to remove the silyl group from product 4. However, we were surprised to discover that the desilylation of 4 using tetrabutylammonium fluoride (TBAF) led to dehydro-azaproline 5 in nearly quantitative yields. Our subsequent studies of this system revealed that the ammonium salt acts catalytically to form the heterocycle and that the reaction appears to tolerate a variety of substituents at the quaternary carbon center (table 2). We note that the TBAF used was a commercially available THF solution containing up to 5% dissolved water, likely the stoichiometric agent responsible for silyl group removal. While the present TBAF catalyzed cyclization step is unprecedented, a literature report details the cyclization of aryl alkynes with nitrogen and oxygen nucleophiles mediated by stoichiometric TBAF.[4] In addition, Hammond and co-workers describe a cyclization with a nitrogen nucleophile with an alkyne flanked with an α,α-difluoro methylene requiring two equivalents of TBAF.[3]
Table 2.
TBAF catalyzed cyclization of β-alkynyl hydrazines.

| |||||
|---|---|---|---|---|---|
| entry | EWG | R1 | R2 | R3Si | %yield 5a |
| 1 | CO2But | Ph | iPr | TES | 99 |
| 2 | COSBut | Ph | iPr | TES | 97b |
| 3 | COSBut | Ph | iPr | TMS | 98 |
| 4 | COSBut | Ph | iPr | TIPS | 99 |
| 5 | COSBut | Ph | tBu | TES | 94 |
| 6 | CO2Et | H | iPr | TES | 99 |
| 7 | COSBut | vinyl | iPr | TES | 99 |
| 8 | COSBut | p-ClPh | iPr | TES | 99 |
| 9 | COSBut | o-tol | iPr | TES | 99 |
| 10 | COSBut | 2-naph | iPr | TES | 99 |
| 11 | CO2Et | H | Bn | TES | 99 |
| 12c | CONR2 | vinyl | iPr | TES | 97 |
Isolated yields.
Starting with non-racemic 4 leads to product with the same ee.
Amide of pyrrolidine
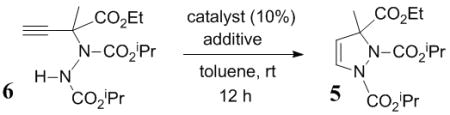
To better understand the role of the non-metal catalyst in the present reaction, several factors were examined. To separate issues of desilylation versus cyclization, we prepared silyl-free substrate 6 using stoichiometric TBAF in isopropanol (alcohol solvents do not lead to cyclization products). Now, using TBAF as the catalyst (0.1 eq), the cyclization reaction of 6 gives excellent yields in THF, DCM, and toluene. The use of tetrabutylammonium bromide (TBAB) as a catalyst gave no cyclization product except with the addition of CsF (table 3, entry 3). However, the anion need not be fluoride as added carbonate leads to the same result with TBAB (entry 6). Interestingly, tetrabutylphosphonium bromide (TBPB) also effectively catalyzes the cyclization[15] with either fluoride or carbonate additives. Clearly, neither ammonium nor fluoride are strictly required to catalyze this cyclization reaction.
Table 3.
Role of cation and anion in catalyzed cyclization of 4.
| |||
|---|---|---|---|
| entry | cat | additive (eq) | %yield 5a |
| 1 | TBAF | - | 99 |
| 2 | TBABa | - | NR |
| 3 | TBAB | CsF (0.5) | 97 |
| 4 | TBPBb | - | NR |
| 5 | TBPB | CsF (0.5) | 98 |
| 6 | TBAB | K2CO3 (0.5) | 95 |
| 7 | TBPB | K2CO3 (0.5) | 94 |
tetratbutylammonium bromide.
tetrabutylphosphonium bromide
The discovery that the present cyclization reaction is mediated by catalytic non-metal cations suggested that kinetic resolution might be possible. Our initial experiments with α-benzyl ester and chiral ammonium bromides 7 and 8 (table 4, entries 1–2) gave no reaction. However, less steric bulk at the quaternary carbon leads to a successful reactions (entries 4–7) which proceed fairly rapidly to approximately 50% conversion with catalysts 7 and 8. Importantly, this reaction leads to products (and remaining starting materials) of high enantioselectivities. Catalysts containing the bromide counter ion required added CsF to achieve product. This cyclization reaction is particularly sensitive to steric crowding. Thus with allyl substitution at α-position, catalyst 7 proved ineffective whereas the much smaller 8 leads to product with good resolution (table 4, entries 6–7). As expected, recovered starting material from a kinetic resolution reaction (entry 7) led to cyclization product 5 using catalytic TBAF in high enantiomeric excess (entry 8).
Table 4.
Non-racemic azaproline products via kinetic resolution.

| ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | EWG | R1 | R2 | cat | additive | %yield | time (h) | %ee |
| 1 | CO2Et | Ph | iPr | 7-Br | CsF | NR | 12 | - |
| 2 | CO2Et | Ph | iPr | 8-Br | CsF | NR | 12 | - |
| 4 | CO2Et | H | Bn | 7-Br | CsF | 47 | 48 | 93a |
| 5 | CO2Et | H | Bn | 7-F | - | 48 | 48 | 93a |
| 6 | COSBut | vinyl | iPr | 8-Br | CsF | 54 | 12 | 76 |
| 7 | COSBut | vinyl | iPr | 8-F | - | 51 | 12 | 81 |
| 8 | COSBut | vinyl | iPr | TBAF | - | 100 | 1 | 99b |
| 9c | CONR2 | vinyl | iPr | 8-F | - | 73d | 8 | 4 |
Based on chiral HPLC analysis of remaining starting material.
Recovered starting material from previous reaction (entry 7) was utilized.
Amide of pyrrolidine.
Recovered 25% starting material.
It appears that a modestly basic counteranion (F− or CO32−) is required for this cyclization reaction presumably to deprotonate the attacking carbamate nitrogen. Hiroya and Sakamoto have proposed an ammonium cation activation of alkynes in cycloaddition reactions involving aryl alkynes.[4a] While additional theoretical and physical investigations are needed to substantiate these claims, we remain intrigued by the possibility that the carbamate nitrogen nucleophile in our case attacks the alkyne group similarly activated by an ammonium or phosphonium catalyst. In this manner, the catalyzing ability of a chiral organic cation is expected to be influenced by the configuration of the quaternary center immediately adjacent to the alkyne perhaps explaining our successful kinetic resolution in this cyclization reaction.
The main thrust of this project was to develop a kinetic resolution technique to take advantage of an unprecedented non-metal catalyzed cyclic heteroatom addition mentioned above. Nevertheless we briefly explored asymmetric phase-transfer catalyzed additions of azidodicarboxylates to our γ-silyl allenyl ester system following a recent precedent by Maruoka involving carbon electrophiles.[16] While aqueous-organic biphasic systems resulted in no reaction with allenyl oxy-esters, we observed that CsOH•H2O in toluene led to their successful hydrazination. However, this solid-liquid phase reaction led to entirely racemic product when a variety of commercially available phase transfer catalysts (PTC) we employed.[17] We returned to the organic-aqueous systems with thioester substrates and observed addition products in good yields and moderate enantioselectivities (eq. 1). For this study we employed only several commercially available PTCs including cinchona-based catalysts and Maruoka catalyst 7. These results suggest that a suitable catalyst can be engineered for higher selectivities.
 |
(1) |
In conclusion, a robust α-selective hydrazination of allenyl esters has been developed. This reaction was also expanded to include asymmetric catalysis conditions using chiral phase transfer catalysts leading to promising results. Importantly, these hydrazino adducts underwent ammonium and phosphonium catalyzed cyclization to afford dehydro-azaproline derivatives. This amine addition to unactivated alkynes is uniquely mediated by non-metal cation catalysts. While the precise mechanism of this cyclization reaction is not yet understood, highly enantio-enriched azaproline derivatives have been prepared using readily available chiral ammonium salts as catalysts. Further mechanistic studies and exploration of the reactivity of the dehydro-azaproline system are currently underway.
Experimental Section
Neutral base catalyzed α-amination: Allene/alkyne mixture (1 mmol) was added to a round bottom flask which was then charged with solvent (5 mL). The reaction was cooled to the required temperature followed by addition of base (20 mol%). After 5 min of stirring, azidodicarboxylate (1.2 equiv) was slowly added and the reaction was allowed to stir. Reaction was monitored by thin layer chromatography and, upon completion, the reaction was quenched with aqueous HCl (1 M). The organic layer was removed and the aqueous layer was extracted twice with ether. All organic layers were combined, concentrated in vacuo, and purified by silica gel flash chromatography.
TBAF catalyzed cyclization: The substrate (0.5 mmol) was dissolved in THF (2 mL), followed by addition of TBAF (1 M THF, 20mol %). The reaction was stirred for 30 min, quenched with aqueous HCl (1 M) and extracted with ether. The organic layer was removed and the aqueous layer was extracted twice with ether. All organic layers were combined, concentrated in vacuo, and purified by silica gel flash chromatography to give nearly quantitative yield of product.
General procedure for kinetic resolution with ammonium fluoride catalysts: substrate (0.5 mmol) was added to a round bottom flask which was then charged with toluene (5 mL). Catalyst (10 mol%) was then added followed by stirring for the required time. The reaction was quenched with aqueous HCl (1 M) and extracted with ether. All organic layers were combined, concentrated in vacuo, and purified by silica gel flash chromatography. The separated products and starting materials were then analyzed by chiral HPLC for enantiomeric excess.
For spectroscopic data on all products see Supporting Information.
Supplementary Material
Footnotes
The work was supported by the National Institutes of of Mental Health (087932-01) and NSF (0311369).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Alonso F, Beletskaya IP, Yus M. Chem Rev. 2004;104:3079. doi: 10.1021/cr0201068. [DOI] [PubMed] [Google Scholar]
- 2.Shen HC. Tetrahedron. 2008;64:3885. [Google Scholar]
- 3.Fustero S, Fernandez B, Bello P, del Pozo C, Arimitsu A, Hammond GB. Org Lett. 2007;9:4251. doi: 10.1021/ol701811z. [DOI] [PubMed] [Google Scholar]
- 4.a) Hiroya K, Jouka R, Kameda M, Yasuhara A, Sakamoto T. Tetrahedron. 2001;57:9697. [Google Scholar]; b) Hiroya K, Suzuki N, Yasuhara A, Egawa Y, Kasano A, Sakamoto T. J Chem Soc, Perkin Trans 1. 2000:4339. [Google Scholar]; c) Yasuhara A, Kanamori Y, Kaneko M, Numata A, Kondo Y, Sakamoto T. J Chem Soc, Perkin Trans 1. 1999:529. [Google Scholar]
- 5.Liu B, Brandt JD, Moeller KD. Tetrahedron. 2003;59:8515. [Google Scholar]
- 6.Lange UEW, Baucke D, Hornberger W, Mack H, Seitz W, Hoffken HW. Bioorg Med Chem Lett. 2006;16:2648. doi: 10.1016/j.bmcl.2006.01.046. [DOI] [PubMed] [Google Scholar]
- 7.Mish MR, Guerra M, Carreira EM. J Am Chem Soc. 1997;119:8379. [Google Scholar]
- 8.a) Sibi MP, Stanley LM, Soeta T. Org Lett. 2007;9:1553. doi: 10.1021/ol070364x. [DOI] [PubMed] [Google Scholar]; b) Jung ME, Min SJ, Houk KN, Ess D. J Org Chem. 2004;69:9085. doi: 10.1021/jo048741w. [DOI] [PubMed] [Google Scholar]; c) Di M, Rein KS. Tet Lett. 2004;45:4703. [Google Scholar]
- 9.Sibi MP, Stanley LM, Jasperse CP. J Am Chem Soc. 2005;127:8276. doi: 10.1021/ja051650b. [DOI] [PubMed] [Google Scholar]
- 10.Kano T, Hashimoto T, Maruoka K. J Am Chem Soc. 2006;128:2174. doi: 10.1021/ja056851u. [DOI] [PubMed] [Google Scholar]
- 11.Yang Q, Jiang X, Ma S. Chem Eur J. 2007;13:9310. doi: 10.1002/chem.200700620. [DOI] [PubMed] [Google Scholar]
- 12.Santos JM, Lopez Y, Aparicio D, Palacios F. J Org Chem. 2008;73:550. doi: 10.1021/jo702050t. [DOI] [PubMed] [Google Scholar]
- 13.Maity P, Lepore SD. J Am Chem Soc. 2009;131:4196. doi: 10.1021/ja810136m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maity P, Lepore SD. J Org Chem. 2009;74:158. doi: 10.1021/jo801563x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.One phosphonium catalyzed carbocyclization was reported recently: Hu J, Wu LY, Wang XC, Hu YY, Niu YN, Liu XY, Yang S, Liang YM. Adv Synth Catal. 2010;352:351.
- 16.Hashimoto T, Sakata K, Maruoka K. Angew Chem Int Ed. 2009;48:5014. doi: 10.1002/anie.200901977. [DOI] [PubMed] [Google Scholar]
- 17.This suggests that the addition reactions may be occurring at the toluene/KOH interface without the intermediacy of the chiral PTC.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.