

Abstract
Background
Glioblastoma multiforme, the most common type of primary brain tumor in adults, is driven by cells with neural stem (NS) cell characteristics. Using derivation methods developed for NS cells, it is possible to expand tumorigenic stem cells continuously in vitro. Although these glioblastoma-derived neural stem (GNS) cells are highly similar to normal NS cells, they harbor mutations typical of gliomas and initiate authentic tumors following orthotopic xenotransplantation. Here, we analyzed GNS and NS cell transcriptomes to identify gene expression alterations underlying the disease phenotype.
Methods
Sensitive measurements of gene expression were obtained by high-throughput sequencing of transcript tags (Tag-seq) on adherent GNS cell lines from three glioblastoma cases and two normal NS cell lines. Validation by quantitative real-time PCR was performed on 82 differentially expressed genes across a panel of 16 GNS and 6 NS cell lines. The molecular basis and prognostic relevance of expression differences were investigated by genetic characterization of GNS cells and comparison with public data for 867 glioma biopsies.
Results
Transcriptome analysis revealed major differences correlated with glioma histological grade, and identified misregulated genes of known significance in glioblastoma as well as novel candidates, including genes associated with other malignancies or glioma-related pathways. This analysis further detected several long non-coding RNAs with expression profiles similar to neighboring genes implicated in cancer. Quantitative PCR validation showed excellent agreement with Tag-seq data (median Pearson r = 0.91) and discerned a gene set robustly distinguishing GNS from NS cells across the 22 lines. These expression alterations include oncogene and tumor suppressor changes not detected by microarray profiling of tumor tissue samples, and facilitated the identification of a GNS expression signature strongly associated with patient survival (P = 1e-6, Cox model).
Conclusions
These results support the utility of GNS cell cultures as a model system for studying the molecular processes driving glioblastoma and the use of NS cells as reference controls. The association between a GNS expression signature and survival is consistent with the hypothesis that a cancer stem cell component drives tumor growth. We anticipate that analysis of normal and malignant stem cells will be an important complement to large-scale profiling of primary tumors.
Background
Glioblastoma (grade IV astrocytoma) is the most common and severe type of primary brain tumor in adults. The prognosis is poor, with a median survival time of 15 months despite aggressive treatment [1]. Glioblastomas display extensive cellular heterogeneity and contain a population of cells with properties characteristic of neural stem (NS) cells [2]. It has been proposed that such corrupted stem cell populations are responsible for maintaining cancers, and give rise to differentiated progeny that contribute to the cellular diversity apparent in many neoplasias. Data supporting this hypothesis have been obtained for several types of malignancies, including a variety of brain cancers [2]. Importantly, a recent study using a mouse model of glioblastoma demonstrated that tumor recurrence after chemotherapy originates from a malignant cell population with NS cell features [3]. Characterizing human glioblastoma cancer stem cells to understand how they differ from normal tissue stem cell counterparts may therefore provide key insights toward the identification of new therapeutic opportunities.
Fetal and adult NS cells can be isolated and maintained as untransformed adherent cell lines in serum-free medium supplemented with growth factors [4,5]. Using similar protocols, it is possible to expand NS cells from gliomas [6]. These glioma-derived NS (GNS) cells are very similar in morphology to normal NS cells, propagate continuously in culture and share expression of many stem and progenitor cell markers, such as SOX2 and Nestin. Like normal progenitor cells of the central nervous system, they can also differentiate into neurons, astrocytes and oligodendrocytes to varying degrees [5,6]. In contrast to NS cells, however, GNS cells harbor extensive genetic abnormalities characteristic of the disease and form tumors that recapitulate human gliomas when injected into mouse brain regions corresponding to sites of occurrence in patients.
In this study, we compare gene expression patterns of GNS and NS cells to discover transcriptional anomalies that may underlie tumorigenesis. To obtain sensitive and genome-wide measurements of RNA levels, we conducted high-throughput sequencing of transcript tags (Tag-seq) on GNS cell lines from three glioblastoma cases and on two normal NS cell lines, followed by quantitative reverse transcription PCR (qRT-PCR) validation in a large panel of GNS and NS cell lines. Tag-seq is an adaptation of serial analysis of gene expression (SAGE) to high-throughput sequencing and has considerable sensitivity and reproducibility advantages over microarrays [7,8]. Compared to transcriptome shotgun sequencing (RNA-seq), Tag-seq does not reveal full transcript sequences, but has the advantages of being strand-specific and unbiased with respect to transcript length.
A large body of microarray expression data for glioblastoma biopsies has been generated through multiple studies [9-13]. These data have been extensively analyzed to detect gene expression differences among samples, with the aim to identify outliers indicative of aberrant expression [11,14,15], discover associations between gene expression and prognosis [12,16] or classify samples into clinically relevant molecular subtypes [9,10,13,17]. However, expression profiling of tumor specimens is limited by the inherent cellular heterogeneity of malignant tissue and a lack of reference samples with similar compositions of corresponding normal cell types. GNS cells represent a tractable alternative for such analyses, as they constitute a homogeneous and self-renewing cell population that can be studied in a wide range of experimental contexts and contrasted with genetically normal NS cells. By combining the sensitive Tag-seq method with the GNS/NS model system we obtain a highly robust partitioning of malignant and normal cell populations, and identify candidate oncogenes and tumor suppressors not previously associated with glioma.
Materials and methods
Cell culture and sample preparation
GNS and NS cells were cultured in N2B27 serum-free medium [18], a 1:1 mixture of DMEM/F-12 and Neurobasal media (Invitrogen, Paisley, UK) augmented with N2 (Stem Cell Sciences, Cambridge, UK) and B27 (Gibco, Paisley, UK) supplements. Self-renewal was supported by the addition of 10 ng/ml epidermal growth factor and 20 ng/ml fibroblast growth factor 2 to the complete medium. Cells were plated at 20,000/cm2 in laminin-coated vessels (10 μg/ml laminin-1 (Sigma, Dorset, UK) in phosphate-buffered saline for 6 to 12 h), passaged near confluence using Accutase dissociation reagent (Sigma) and were typically split at 1:3 for NS cells and 1:3 to 1:6 for GNS cells. For expression analysis, cells were dissociated with Accutase and RNA was extracted using RNeasy (Qiagen, West Sussex, UK), including a DNase digestion step. RNA quality was assessed on the 2100 Bioanalyzer (Agilent, Berkshire, UK).
Transcriptome tag sequencing
Tag-seq entails the capture of polyadenylated RNA followed by extraction of a 17-nucleotide (nt) sequence immediately downstream of the 3′-most NlaIII site in each transcript. These 17 nt 'tags' are sequenced in a high-throughput manner and the number of occurrences of each unique tag is counted, resulting in digital gene expression profiles where tag counts reflect expression levels of corresponding transcripts [8].
Tag-seq libraries were prepared using the Illumina NlaIII DGE protocol. Briefly, polyadenylated RNA was isolated from 2 µg total RNA using Sera-Mag oligo(dT) beads (Thermo Scientific, Leicestershire, UK). First-strand cDNA was synthesized with SuperScript II reverse transcriptase (Invitrogen) for 1 h at 42°C, followed by second-strand synthesis by DNA polymerase I for 2.5 h at 16°C in the presence of RNase H. cDNA products were digested with NlaIII for 1 h at 37°C and purified to retain only the 3′-most fragments bound to the oligo(dT) beads. Double-stranded GEX adapter 1 oligonucleotides, containing an MmeI restriction site, were ligated to NlaIII digestion products with T4 DNA ligase for 2 h at 20°C. Ligation products were then digested with MmeI at the adapter-cDNA junction site, thereby creating 17 bp tags free in solution. GEX adapter 2 oligos were ligated to the MmeI cleavage site by T4 DNA ligase for 2 h at 20°C, and the resulting library constructs were PCR-amplified for 15 cycles with Phusion DNA polymerase (Finnzymes, Essex, UK).
Libraries were sequenced at Canada's Michael Smith Genome Sciences Centre, Vancouver BC on the Illumina platform. Transcript tags were extracted as the first 17 nt of each sequencing read and raw counts obtained by summing the number of reads for each observed tag. To correct for potential sequencing errors, we used the Recount program [19], setting the Hamming distance parameter to 1. Recount uses an expectation maximization algorithm to estimate true tag counts (that is, counts in the absence of error) based on observed tag counts and base-calling quality scores. Tags matching adapters or primers used in library construction and sequencing were identified and excluded using TagDust [20] with a target false discovery rate (FDR) of 1%. Tags derived from mitochondrial or ribosomal RNA were identified and excluded by running the bowtie short-read aligner [21] against a database consisting of all ribosomal RNA genes from Ensembl [22], all ribosomal repeats in the UCSC Genome Browser RepeatMasker track for genome assembly GRCh37 [23], and the mitochondrial DNA sequence; only perfect matches to the extended 21 nt tag sequence (consisting of the NlaIII site CATG followed by the observed 17 nt tag) were accepted. Remaining tags were assigned to genes using a hierarchical strategy based on the expectation that tags are most likely to originate from the 3'-most NlaIII site in known transcripts (Additional files 1 and 2). To this end, expected tag sequences (virtual tags) were extracted from the SAGE Genie database [24] and Ensembl transcript sequences. In addition, bowtie was applied to determine unique, perfect matches for sequenced tags to the reference genome.
The Bioconductor package DESeq [25] was used to normalize tag counts, call differentially expressed genes and obtain variance-stabilized expression values for correlation calculations. Tests for enrichment of Gene Ontology and InterPro terms were performed in R, using Gene Ontology annotation from the core Bioconductor package org.Hs.eg and InterPro annotation from Ensembl. Each term associated with a gene detected by Tag-seq was tested. Signaling pathway impact analysis was carried out using the Bioconductor package SPIA [26]. To identify major differences common to the GNS cell lines investigated, we filtered the set of genes called differentially expressed at 1% FDR, further requiring (i) two-fold or greater change in each GNS cell line compared to each NS cell line, with the direction of change being consistent among them; and (ii) expression above 30 tags per million in each GNS cell line (if upregulated in GNS cells) or each NS cell line (if downregulated in GNS cells). Sequencing data and derived gene expression profiles are available from ArrayExpress [27] under accession E-MTAB-971.
Quantitative RT-PCR validation
Custom-designed TaqMan low-density array microfluidic cards (Applied Biosystems, Paisley, UK) were used to measure the expression of 93 genes in 22 cell lines by qRT-PCR. This gene set comprises 82 validation targets from Tag-seq analysis, eight glioma and developmental markers, and three endogenous control genes (18S ribosomal RNA, TUBB and NDUFB10). The 93 genes were interrogated using 96 different TaqMan assays (three of the validation targets required two different primer and probe sets to cover all known transcript isoforms matching differentially expressed tags). A full assay list with raw and normalized threshold cycle (Ct) values is provided in Additional file 3. To capture biological variability within cell lines, we measured up to four independent RNA samples per line. cDNA was generated using SuperScript III (Invitrogen) and real-time PCR carried out using TaqMan fast universal PCR master mix. Ct values were normalized to the average of the three control genes using the Bioconductor package HTqPCR [28]. Differentially expressed genes were identified by the Wilcoxon rank sum test after averaging replicates.
Tumor gene expression analysis
Public microarray data, survival information and other associated metadata were obtained from The Cancer Genome Atlas (TCGA) and four independent studies (Table 1). All tumor microarray data were from samples obtained upon initial histologic diagnosis. We used processed (level 3) data from TCGA, consisting of one expression value per gene and sample (Additional file 4). For the other data sets, we processed the raw microarray data with the RMA method in the Bioconductor package affy [29] and retrieved probe-gene mappings from Ensembl 68 [22]. For genes represented by multiple probesets, expression values were averaged across probesets for randomization tests, heatmap visualization and GNS signature score calculation. Differential expression was computed using limma [30]. Randomization tests were conducted with the limma function geneSetTest, comparing log2 fold-change for core up- or downregulated genes against the distribution of log2 fold-change for randomly sampled gene sets of the same size.
Table 1.
Public gene expression data sets used in this study
| Number of cases | |||||||
|---|---|---|---|---|---|---|---|
| Citation | Citation | Microarray platform (Affymetrix) | Glioblastoma | Grade III astrocytoma | Other grade III glioma | Grade I-II glioma | Non-neoplastic brain |
| The Cancer Genome Atlas (TCGA) [11,46] | NA | Exon 1.0 ST | 397 | 0 | 0 | 0 | 10 |
| Gravendeel et al. [13] | GSE16011 | U133 Plus 2.0 | 141 | 16 | 66 | 27 | 0 |
| Murat et al. [12] | GSE7696 | U133 Plus 2.0 | 70 | 0 | 0 | 0 | 0 |
| Phillips et al. [9] | GSE4271 | U133A and U133B | 55 | 21 | 0 | 0 | 0 |
| Freije et al. [10] | GSE4412 | U133A and U133B | 50 | 8 | 16 | 0 | 0 |
Survival analysis was carried out with the R library survival. To combine expression values of multiple genes for survival prediction, we took an approach inspired by Colman et al. [16]. The normalized expression values xij, where i represents the gene and j the sample, were first standardized to be comparable between genes by subtracting the mean across samples and dividing by the standard deviation, thus creating a matrix of z-scores:
Using a set U of nU genes upregulated in GNS cell lines and a set D of nD genes downregulated in these cells, we then computed a GNS signature score sj for each sample j by subtracting the mean expression of the downregulated genes from the mean expression of the upregulated genes:
IDH1 mutation calls for TCGA samples were obtained from Firehose data run version 2012-07-07 [31] and data files from the study by Verhaak et al. updated 2011-11-28 [32].
Array comparative genomic hybridization
We re-analyzed the array comparative genomic hybridization (CGH) data described by Pollard et al. [6]. CGH was performed with Human Genome CGH Microarray 4x44K arrays (Agilent), using genomic DNA from each cell line hybridized in duplicate (dye swap) and normal human female DNA as reference (Promega, Southampton, UK). Log2 ratios were computed from processed Cy3 and Cy5 intensities reported by the software CGH Analytics (Agilent). We corrected for effects related to GC content and restriction fragment size using a modified version of the waves array CGH correction algorithm [33]. Briefly, log2 ratios were adjusted by sequential loess normalization on three factors: fragment GC content, fragment size, and probe GC content. These were selected after investigating dependence of log ratio on multiple factors, including GC content in windows of up to 500 kb centered around each probe. The Bioconductor package CGHnormaliter [34] was then used to correct for intensity dependence and log2 ratios scaled to be comparable between arrays using the 'scale' method in the package limma [35]. Replicate arrays were averaged and the genome (GRCh37) segmented into regions with different copy number using the circular binary segmentation algorithm in the Bioconductor package DNAcopy [36], with the option undo.SD set to 1. Aberrations were called using the package CGHcall [37] with the option nclass set to 4. CGH data are available from ArrayExpress [27] under accession E-MTAB-972.
Results
Transcriptome analysis highlights pathways affected in glioma
We applied Tag-seq to four GNS cell lines (G144, G144ED, G166 and G179) and two human fetal NS cell lines (CB541 and CB660), all previously described [5,6]. G144 and G144ED were independently established from the same parental tumor in different laboratories. Tag-seq gene expression values were strongly correlated between these two lines (Pearson r = 0.94), demonstrating that the experimental procedure, including cell line establishment, library construction and sequencing, is highly reproducible. The two NS cell transcriptome profiles were also well correlated (r = 0.87), but there were greater differences among G144, G166 and G179 (r ranging from 0.78 to 0.82). This is expected, as G144, G166 and G179 originate from different and histologically distinct glioblastoma cases.
We used the Tag-seq data to identify differences in gene expression between the three GNS cell lines G144, G166 and G179 and the two normal NS cell lines CB541 and CB660. At a FDR of 10%, this analysis revealed 485 genes to be expressed at a higher average level in GNS cells (upregulated) and 254 genes to be downregulated (Additional file 5). GNS cells display transcriptional alterations common in glioblastoma, including upregulation of the epidermal growth factor receptor (EGFR) gene and downregulation of the tumor suppressor PTEN [11]. Enrichment analysis using Gene Ontology and the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway database confirmed the set of 739 differentially expressed genes to be enriched for pathways related to brain development, glioma and cancer (Tables 2 and 3). We also observed enrichment of regulatory and inflammatory genes, such as signal transduction components, cytokines, growth factors and DNA-binding factors. Several genes related to antigen presentation on MHC class I and II molecules were upregulated in GNS cells, consistent with the documented expression of their corresponding proteins in glioma tumors and cell lines [38,39]. In addition, we detected 25 differentially expressed long non-coding RNAs (Additional file 6). Several of these display an expression pattern similar to a neighboring protein-coding gene, including cancer-associated genes DKK1 and CTSC [40,41] (Figure 1) and developmental regulators IRX2, SIX3 and ZNF536 [42], suggesting that they may be functional RNAs regulating nearby genes [43] or represent transcription from active enhancers [44].
Table 2.
Selected Gene Ontology terms and InterPro domains enriched among differentially expressed genes
| Differentially expressed (729 genes) | Upregulated (485 genes) | Downregulated (254 genes) | ||||
|---|---|---|---|---|---|---|
| Genes | P | Genes | P | Genes | P | |
| Biological process Gene Ontology terms | ||||||
| Immune response | 70 | 2.4 × 10-12 | 61 | 3.0 × 10-16 | 9 | NS |
| Nervous system development | 106 | 1.9 × 10-10 | 62 | 0.0055 | 44 | 2.3 × 10-5 |
| Cell adhesion | 74 | 9.8 × 10-8 | 56 | 1.9 × 10-7 | 18 | NS |
| Antigen processing and presentation | 17 | 4.3 × 10-7 | 17 | 5.4 × 10-10 | 0 | NS |
| Cell differentiation | 128 | 7.4 × 10-7 | 74 | NS | 54 | 1.8 × 10-4 |
| Cell migration | 44 | 3.0 × 10-4 | 30 | 0.0262 | 14 | NS |
| Cell proliferation | 86 | 3.4 × 10-4 | 59 | 0.0136 | 27 | NS |
| Cellular ion homeostasis | 36 | 0.0138 | 33 | 1.7 × 10-5 | 3 | NS |
| Molecular function Gene Ontology terms | ||||||
| Cytokine activity | 27 | 2.3 × 10-8 | 25 | 7.1 × 10-11 | 2 | NS |
| Signal transducer activity | 111 | 2.8 × 10-7 | 66 | 0.0584 | 45 | 0.0017 |
| Receptor activity | 83 | 8.0 × 10-7 | 48 | NS | 35 | 0.0016 |
| Sequence-specific DNA binding | 52 | 2.7 × 10-4 | 34 | 0.0526 | 18 | NS |
| MHC class II receptor activity | 5 | 0.0077 | 5 | 9.2 × 10-4 | 0 | NS |
| Growth factor activity | 20 | 0.0109 | 17 | 0.0019 | 3 | NS |
| InterPro domains | ||||||
| Immunoglobulin-like | 45 | 3.1 × 10-8 | 32 | 6.0 × 10-6 | 13 | NS |
| MHC classes I/II-like antigen recognition protein | 14 | 1.1 × 10-7 | 14 | 3.2 × 10-10 | 0 | NS |
| Homeobox | 28 | 8.5 × 10-6 | 18 | 0.0124 | 10 | NS |
Number of genes differentially expressed at 10% FDR and annotated with the indicated Gene Ontology and InterPro terms. P-values indicating the statistical significance of enrichment of these terms were computed with Fisher's exact test and corrected for multiple testing using the Bonferroni method. NS, not significant (P > 0.1).
Table 3.
Representative KEGG pathways from signaling pathway impact analysis of gene expression differences between GNS and NS cell lines
| Pathway | Genes | P | Predicted status in GNS cells |
|---|---|---|---|
| Cytokine-cytokine receptor interaction | 29 | 4.4 × 10-12 | Activated |
| Chemokine signaling pathway | 15 | 5.3 × 10-6 | Activated |
| Neuroactive ligand-receptor interaction | 21 | 2.2 × 10-4 | Inhibited |
| Antigen processing and presentation | 11 | 6.8 × 10-4 | Activated |
| MAPK signaling pathway | 24 | 0.0106 | Activated |
| Glioma | 10 | 0.0131 | Activated |
| ECM-receptor interaction | 10 | 0.0405 | Inhibited |
| Calcium signaling pathway | 15 | 0.0405 | Activated |
The number of genes found to be differentially expressed at 10% FDR and belonging to the selected pathways are indicated. P-values and status predictions were obtained by signaling pathway impact analysis [26], taking fold-change estimates and pathway topology into account. P-values were FDR-corrected for multiple testing. ECM, extracellular matrix; MAPK, mitogen-activated protein kinase.
Figure 1.

Correlated expression of CTSC and a nearby non-coding RNA. (a) CTSC (cathepsin C) is located in a gene desert harboring an uncharacterized non-coding gene transcribed in the opposite orientation [GenBank:BC038205]. (b) Both CTSC and the non-coding RNA have strongly elevated expression in GNS relative to NS cell lines, with highest levels in G179.
To visualize gene expression differences in a pathway context, we compiled an integrated pathway map that includes the pathways most commonly affected in glioblastoma, as well as pathways related to antigen processing and presentation, apoptosis, angiogenesis and invasion (Additional file 1). The map contains 182 genes, of which 66 were differentially expressed between GNS and NS cells at 10% FDR (Additional file 7). Figure 2 depicts a condensed version focused on the pathways most frequently affected in glioblastoma. This approach allowed us to identify differentially expressed genes that participate in glioma-related pathways, but have not been directly implicated in glioma. These include several genes associated with other neoplasms (Table 4). Our comparison between GNS and NS cells thus highlights genes and pathways that are known to be affected in glioma as well as novel candidates, and suggests the GNS/NS comparison is a compelling model for investigating the molecular attributes of glioma.
Figure 2.

Expression changes in pathways most commonly affected in glioma. Genes are represented by circles and colored according to fold-change between GNS and NS cells measured by Tag-seq (see color key), or grey unless statistically significant (10% FDR). Gene complexes and families (hexagons), small molecules (chevrons) and affected cellular processes (squares) are included. Edges indicate activation (green), inhibition (red), contains (black with circular tip), becomes (black with half arrow) and other interactions (grey).
Table 4.
Novel candidate glioma genes identified by differential expression and pathway analysis
| Genea | Log2 fold-changeb | Prior association with glioma | Implication in other neoplasms | Reference |
|---|---|---|---|---|
| CACNA1A | 7.1 | None | Prostate cancer (mouse model) | [80] |
| CACNA1C | -8.2 | None | Liver cancer | [81] |
| CACNG7 | -2.6 | None | None | -- |
| CACNG8 | -4.8 | None | None | -- |
| CAMK1D | -2.4 | None | Breast cancer | [82] |
| CPLX2 | 6.4 | None | None | -- |
| DDIT3 (CHOP, GADD153) | 4.4 | Limited | General (cellular stress response) | [83-86] |
| DUSP16 | 4.2 | None | Burkitt's lymphoma | [87] |
| FGF19 | - | None | Liver, lung and colon cancer | [88] |
| ITGA4 (CD49D) | 3.0 | Limited | Chronic lymphocytic leukemia, breast cancer and others | [89-91] |
| ITGBL1 | + | None | None | -- |
| MAP3K5 (ASK1) | 5.1 | Limited | Gastric cancer and histiocytoma | [92-94] |
| NFATC2 (NFAT1) | + | Limited | Breast cancer | [95-98] |
| NFKBIZ | 5.1 | None | Liposarcoma | [99] |
| NR0B1 (DAX1) | + | None | Lung adenocarcinoma and Ewing's sarcoma | [100,101] |
| NR1D1 | 2.9 | None | Breast cancer | [102] |
| PARP3, PARP12 | 4.1, 2.9 | By homologyc | The PARP gene family is involved in DNA repair and several other processes related to tumorigenesis | [103,104] |
| PERP | 3.8 | None | Lung and skin cancer | [105,106] |
| PPEF1 | 4.4 | Limited | None | [107] |
| SNAP25 | 3.3 | None | Lung cancer | [108] |
| SYT1 | -2.5 | None | None | -- |
| TNFRSF14 | 4.0 | None | Follicular lymphoma | [109] |
| TNFSF4 (OX40L) | 4.0 | None | Generally implicated in immune response to tumors | [110] |
aAliases are listed in parentheses. bGene expression log2 fold-change between GNS and NS cell lines compared by Tag-seq. Some genes were detected exclusively in GNS or NS cells (indicated in column 2 by + or -, respectively). cThe homolog PARP1 has been implicated in glioma. PARP, poly(ADP-ribose) polymerase.
Core expression changes in GNS lines are mirrored in glioma tumors and correlate with histological grade
To capture major gene expression changes common to G144, G166 and G179, we set strict criteria on fold changes and tag counts (see Materials and methods). This approach yielded 32 upregulated and 60 downregulated genes, in the following referred to as 'core' differentially expressed genes (Additional file 8). This set includes genes with established roles in glioblastoma (for example, PTEN [11] and CEBPB [45]), as well as others not previously implicated in the disease (see Discussion). To investigate whether these core differentially expressed genes have similar expression patterns in GNS cells and primary tumors, we made use of public microarray data (Table 1). Perfect agreement between tissue- and cell-based results would not be expected, as tissues comprise a heterogeneous mixture of cell types. Nevertheless, analysis of microarray expression data from TCGA [11,46] for 397 glioblastoma cases (Additional file 4) revealed a clear trend for core upregulated GNS genes to be more highly expressed in glioblastoma tumors than in non-neoplastic brain tissue (P = 0.02, randomization test; Figure 3a) and an opposite trend for core downregulated genes (P = 3 × 10-5; Figure 3c).
Figure 3.

Core gene expression changes in GNS lines are mirrored in glioblastoma tumors. (a-d) Expression in tumors for genes that are strongly upregulated (a,b) or downregulated (c,d) in GNS cells. The gene sets were identified by comparison of Tag-seq expression profiles for GNS and NS cell lines (see main text). Bars depict average fold-change between glioblastoma and non-neoplastic brain tissue (a,c) (TCGA data set) and between glioblastoma and grade III astrocytoma (b,d) (Phillips and Freije data sets combined). Black bars indicate genes with significant differential expression in the microarray data (P < 0.01). Heatmaps show expression in individual samples relative to the average in non-neoplastic brain (a,c) or grade III astrocytoma (b,d). One gene (CHCHD10) not quantified in the TCGA data set is omitted from (a).
We hypothesized that the expression of these genes might also differ between glioblastoma and less severe astrocytomas. We therefore examined their expression patterns in microarray data from the studies of Phillips et al. [9] and Freije et al. [10], which both profiled grade III astrocytoma cases in addition to glioblastomas (Table 1). The result was similar to the comparison with non-neoplastic brain tissue above; there was a propensity for core upregulated genes to be more highly expressed in glioblastoma than in the lower-grade tumor class (P = 10-6; Figure 3b), while core downregulated genes showed the opposite pattern (P = 10-4; Figure 3d). The set of core differentially expressed genes identified by Tag-seq thus defines an expression signature characteristic of glioblastoma and related to astrocytoma histological grade.
Large-scale qRT-PCR validates Tag-seq results and identifies a robust gene set distinguishing GNS from NS cells
To assess the accuracy of Tag-seq expression level estimates and investigate gene activity in a larger panel of cell lines, we assayed 82 core differentially expressed genes in 16 GNS cell lines (derived from independent patient tumors) and six normal NS cell lines by qRT-PCR using custom-designed TaqMan microfluidic arrays. The 82 validation targets (Additional file 3) were selected from the 92 core differentially expressed genes based on the availability of TaqMan probes and considering prior knowledge of gene functions. For the cell lines assayed by both Tag-seq and qRT-PCR, measurements agreed remarkably well between the two technologies: the median Pearson correlation for expression profiles of individual genes was 0.91 and the differential expression calls were corroborated for all 82 genes (Figure 4a). Across the entire panel of cell lines, 29 of the 82 genes showed statistically significant differences between GNS and NS cells at an FDR of 5% (Figure 4b,c). This set of 29 genes generally distinguishes GNS cells from normal NS cell counterparts, and may therefore have broad relevance for elucidating properties specific to tumor-initiating cells.
Figure 4.

qRT-PCR validates Tag-seq results and identifies a robust gene set distinguishing GNS from NS cells. (a) Fold-change estimates (indicating expression level in GNS relative to NS cell lines) from Tag-seq and qRT-PCR for each of the 82 genes measured by qRT-PCR. Greater than two-fold difference in expression (dashed lines at y = ±1) was confirmed for all genes. (b) Heatmap of 29 genes differentially expressed between 16 GNS and six NS cell lines. Colors indicate qRT-PCR ΔΔCt values, that is, normalized expression on a log2 scale, where zero corresponds to the average expression between the two groups (GNS and NS cells). (c) Expression levels of the top six genes (by Wilcoxon test P-value) distinguishing GNS from NS cell lines, measured by qRT-PCR and presented as percentage of NS geometric mean.
A GNS cell expression signature is associated with patient survival
To further explore the relevance in glioma for these recurrent differences between GNS and NS cell transcriptomes, we integrated clinical information with tumor expression data. We first tested for associations between gene expression and survival time using the TCGA data set consisting of 397 glioblastoma cases (Table 1). For each gene, we fitted a Cox proportional hazards model with gene expression as a continuous explanatory variable and computed a P-value by the score test (Table 5). The set of 29 genes found to distinguish GNS from NS cells across the 22 cell lines assayed by qRT-PCR was enriched for low P-values compared to the complete set of 18,632 genes quantified in the TCGA data set (P = 0.02, one-sided Kolmogorov-Smirnov test), demonstrating that expression analysis of GNS and NS cell lines had enriched for genes associated with patient survival. Seven of the 29 genes had a P-value below 0.05 and, for six of these, the direction of the survival trend was concordant with the expression in GNS cells, such that greater similarity to the GNS cell expression pattern indicated poor survival. Specifically, DDIT3, HOXD10, PDE1C and PLS3 were upregulated in GNS cells and expressed at higher levels in glioblastomas with poor prognosis, while PTEN and TUSC3 were downregulated in GNS cells and expressed at lower levels in gliomas with poor prognosis.
Table 5.
Survival tests for 29 genes distinguishing GNS from NS lines
| TCGA data set | Gravendeel data set (glioblastoma cases) | |||||
|---|---|---|---|---|---|---|
| Gene | Category | Coefficienta | P | Probesetb | Coefficienta | P |
| ADD2 | Upregulated | -0.13 | 0.2858 | 237336_at | -0.17 | 0.1420 |
| CD9 | Upregulated | 0.18 | 0.0731 | 201005_at | 0.17 | 0.0689 |
| CEBPB | Upregulated | 0.19 | 0.1028 | 212501_at | 0.17 | 0.0651 |
| DDIT3 | Upregulated | 0.17 | 0.0128 | 209383_at | 0.09 | 0.2777 |
| FOXG1 | Upregulated | 0.13 | 0.0861 | 206018_at | 0.11 | 0.0380 |
| HMGA2 | Downregulated | 0.13 | 0.1456 | 1561633_at | -0.84 | 0.2459 |
| HOXD10 | Upregulated | 0.12 | 0.0108 | 229400_at | 0.15 | 0.0021 |
| IRX2 | Downregulated | -0.19 | 0.2346 | 228462_at | -0.20 | 4.4 × 10-4 |
| LMO4 | Upregulated | 0.24 | 0.1046 | 209205_s_at | 0.20 | 0.1435 |
| LYST | Upregulated | 0.05 | 0.5590 | 203518_at | 0.10 | 0.4151 |
| MAF | Downregulated | 0.10 | 0.5873 | 209348_s_at | 0.38 | 0.0074 |
| MAP6 | Downregulated | 0.16 | 0.3063 | 235672_at | -0.30 | 0.0087 |
| MT2A | Upregulated | 0.16 | 0.1554 | 212185_x_at | 0.27 | 0.0127 |
| MYL9 | Downregulated | 0.08 | 0.3764 | 201058_s_at | 0.15 | 0.0252 |
| NDN | Downregulated | -0.04 | 0.4874 | 209550_at | -0.22 | 6.0 × 10-5 |
| NELL2 | Downregulated | 0.08 | 0.1021 | 203413_at | 0.14 | 0.0215 |
| PDE1C | Upregulated | 0.20 | 0.0105 | 236344_at | 0.21 | 0.0134 |
| PLA2G4A | Upregulated | -0.06 | 0.3198 | 210145_at | 0.30 | 2.9 × 10-4 |
| PLCH1 | Downregulated | 0.10 | 0.3165 | 214745_at | 0.45 | 0.0094 |
| PLS3 | Upregulated | 0.13 | 0.0381 | 201215_at | 0.30 | 0.0069 |
| PRSS12 | Upregulated | -0.11 | 0.1865 | 213802_at | 0.20 | 0.0296 |
| PTEN | Downregulated | -0.53 | 0.0047 | 228006_at | -0.40 | 0.0062 |
| SDC2 | Downregulated | 0.22 | 0.0044 | 212158_at | 0.28 | 5.8 × 10-4 |
| ST6GALNAC5 | Downregulated | 0.01 | 0.9116 | 220979_s_at | 0.08 | 0.2416 |
| SULF2 | Upregulated | -0.11 | 0.1525 | 233555_s_at | -0.15 | 0.0930 |
| SYNM | Downregulated | -0.06 | 0.5620 | 212730_at | 0.08 | 0.2613 |
| TAGLN | Downregulated | 0.03 | 0.5947 | 205547_s_at | 0.17 | 0.0030 |
| TES | Downregulated | -0.05 | 0.5759 | 202720_at | 0.07 | 0.5499 |
| TUSC3 | Downregulated | -0.14 | 0.0079 | 209227_at | -0.18 | 0.0060 |
aFitted coefficient from Cox model; a positive coefficient indicates that higher expression is associated with poor survival and a negative coefficient indicates the opposite. bFor the Gravendeel data set, the result for the most significant probeset interrogating the gene is shown.
We reasoned that, if a cancer stem cell subpopulation in glioblastoma tumors underlies these survival trends, it may be possible to obtain a stronger and more robust association with survival by integrating expression information for multiple genes up- or downregulated in GNS cells. We therefore combined the expression values for the genes identified above (DDIT3, HOXD10, PDE1C, PLS3, PTEN and TUSC3) into a single value per tumor sample, termed 'GNS signature score' (see Materials and methods). This score was more strongly associated with survival (P = 10-6) than were the expression levels of any of the six individual genes (P ranging from 0.005 to 0.04; Table 5).
To test whether these findings generalize to independent clinical sample groups, we examined the glioblastoma data sets described by Gravendeel et al. [13] and Murat et al. [12], consisting of 141 and 70 cases, respectively (Table 1). The GNS signature score was correlated with patient survival in both of these data sets (P = 3 × 10-5 and 0.006, respectively; Figure 5a; Additional file 9). At the level of individual GNS signature genes, five were significantly associated with survival (P < 0.05) in both of the two largest glioblastoma data sets we investigated (TCGA and Gravendeel): HOXD10, PDE1C, PLS3, PTEN and TUSC3 (Table 5). In addition to glioblastoma (grade IV) tumors, Gravendeel et al. also characterized 109 grade I to III glioma cases (Table 1). Inclusion of these data in survival analyses made the association with the GNS signature even more apparent (Figure 5b). This is consistent with the above observation that core transcriptional alterations in GNS cells correlate with histological grade of primary tumors. Analysis of data from the studies of Phillips et al. [9] and Freije et al. [10], which profiled both grade III and IV gliomas (Table 1), further confirmed the correlation between GNS signature and survival (Figure 5b). In summary, the association between GNS signature and patient survival was reproducible in five independent data sets comprising 867 glioma cases in total (Table 1).
Figure 5.
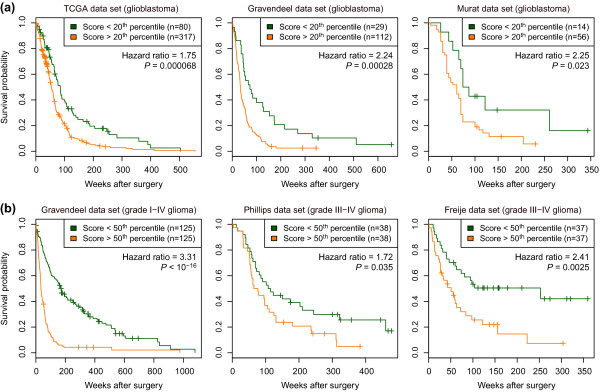
Association between GNS signature score and patient survival. (a,b) Kaplan-Meier plots illustrate the association between signature score and survival for three independent glioblastoma data sets (a) and three data sets that include gliomas of lower grade (b) (Table 1). Higher scores indicate greater similarity to the GNS cell expression profile. Hazard ratios and log-rank P-values were computed by fitting a Cox proportional hazards model to the data. Percentile thresholds were chosen for illustration; the association with survival is statistically significant across a wide range of thresholds (Additional file 9) and the P-values given in the text and Table 6 were computed without thresholding, using the score as a continuous variable.
We controlled for a range of potential confounding factors; these did not explain the survival trends observed (Additional file 10). Investigating a relationship to known predictors of survival in glioma, we noted that the GNS signature score correlates with patient age at diagnosis, suggesting that the GNS cell-related expression changes are associated with the more severe form of the disease observed in older patients (Figure 6a). Of the genes contributing to the GNS signature, HOXD10, PLS3, PTEN and TUSC3 correlated with age both in the TCGA and Gravendeel data sets (Additional file 11).
Figure 6.
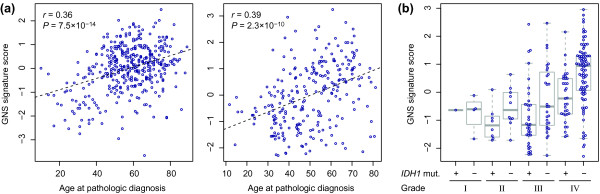
Association between GNS signature and other survival predictors. (a) Scatter plots demonstrate the correlation between GNS signature score and age at diagnosis for the TCGA (left) and Gravendeel (right) data sets. The regression line, Pearson correlation coefficient (r) and P-value indicating statistical significance of the correlation are shown. (b) GNS signature score for samples in the Gravendeel data set, stratified by IDH1 mutation status and histological grade. Blue circles represent individual samples (independent cases) and grey boxplots summarize their distribution. Only cases with known IDH1 status are shown (127 mutated, 77 wild type).
Most grade III astrocytomas and a minority of glioblastomas carry a mutation affecting codon 132 of the IDH1 gene resulting in an amino acid change (R132H, R132S, R132C, R132G, or R132L). The presence of this mutation is associated with lower age at disease onset and better prognosis [47,48]. All 16 GNS cell lines profiled in this study were derived from glioblastoma tumors, and the IDH1 locus was sequenced in each cell line (data not shown); none of them harbor the mutation. We therefore investigated whether the GNS signature is characteristic of IDH1 wild-type glioblastomas. IDH1 status has been determined for most cases in the TCGA and Gravendeel data sets (Table 6) [11,13,17]. As expected, we found that gliomas with the IDH1 mutation tend to have lower GNS signature scores than IDH1 wild-type gliomas of the same histological grade (Figure 6b). However, we also found the GNS signature to have a stronger survival association than IDH1 status (Table 6). The signature remained a significant predictor of patient survival when controlling for IDH1 status (Table 6), demonstrating that it contributes independent information to the survival model and does not simply represent a transcriptional state of IDH1 wild-type tumors. This was evident in glioblastomas as well as grade I to III gliomas; the effect is thus not limited to grade IV tumors.
Table 6.
Significance of survival association for GNS signature and IDH1 status
| Single covariate | Two covariates | ||||
|---|---|---|---|---|---|
| Data set | Number of cases | GNS signature | IDH1 status | GNS signature | IDH1 status |
| TCGA | 270 | 5.3 × 10-5 | 0.0015 | 0.0091 | 0.1489 |
| Gravendeel, glioblastoma cases | 118 | 2.7 × 10-5 | 0.0031 | 9.2 × 10-4 | 0.0840 |
| Gravendeel, grade I to III glioma cases | 86 | 6.5 × 10-4 | 0.5776 | 6.3 × 10-4 | 0.5408 |
Wald test P-values, indicating association with survival, for each covariate in a Cox proportional hazards model with one or two covariates (GNS signature, IDH1 status or both). Cases with unknown IDH1 mutation status were excluded.
To investigate whether the correlation between GNS signature and age could be explained by the higher proportion of cases with IDH1 mutation among younger patients, we repeated the correlation analysis described above (Figure 6a), limiting the data to glioblastoma cases without IDH1 mutation. For the TCGA data set, the correlation was decreased somewhat (Pearson r = 0.25 compared to 0.36 for the full data set) but still highly significant (P = 6 × 10-5), demonstrating that the correlation with age is only partially explained by IDH1 status. This result was confirmed in the Gravendeel data set, where the effect of controlling for IDH1 status and grade was negligible (r = 0.38 compared to 0.39 for the full data set including grade I to III samples). Among the individual signature genes, both HOXD10 and TUSC3 remained correlated with age in both data sets when limiting the analysis to IDH1 wild-type glioblastoma cases (Additional file 11).
Influence of copy number alterations on the GNS transcriptome
Previous analysis of chromosomal aberrations in G144, G166 and G179 by spectral karyotyping and array CGH detected genetic variants characteristic of glioblastoma [6]. To assess the influence of copy-number changes on the GNS transcriptome, we compared CGH profiles (Figure 7) with Tag-seq data. On a global level, there was an apparent correlation between chromosomal aberrations and gene expression levels (Figure 8a,b), demonstrating that copy-number changes are a significant cause of the observed expression differences. Among the 29 genes differentially expressed between GNS and NS cells in the larger panel assayed by qRT-PCR, there was a tendency for downregulated genes to be lost: 10 out of 15 downregulated genes were in regions of lower than average copy number in one or more GNS cell lines, compared to 4 out of 14 upregulated genes (P = 0.046, one-sided Fisher's exact test).
Figure 7.
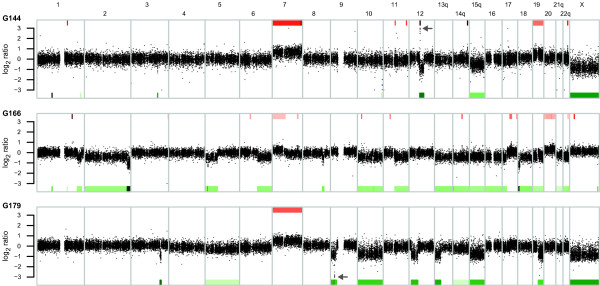
CGH profiles for GNS lines. Dots indicate log2 ratios for array CGH probes along the genome, comparing each GNS cell line to normal female DNA. Colored segments indicate gain (red) and loss (green) calls, with color intensity proportional to mean log2 ratio over the segment. Aberrations known to be common in glioblastoma [11,79] were identified, including gain of chromosome 7 and losses of large parts of chromosomes 10, 13, 14 and 19 in more than one GNS cell line, as well as focal gain of CDK4 in G144 (arrow, chromosome 12) and focal loss of the CDKN2A-CDKN2B locus in G179 (arrow, chromosome 9). The X chromosome was called as lost in G144 and G179 because these two cell lines are from male patients; sex-linked genes were excluded from downstream analyses of aberration calls.
Figure 8.
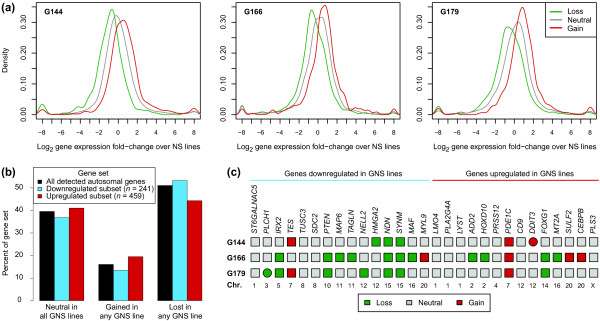
Copy number changes in GNS cell lines correlate with gene expression levels. (a) Curves show distributions of expression level differences between GNS and NS cells, stratified by aberration calls. The distributions for genes in segments without aberrations (neutral) peak near the 0 mark, corresponding to an equal expression level in GNS and NS cells. Conversely, genes in lost and gained regions tend to be expressed at lower and higher levels, respectively. In each plot, log2 fold changes were computed between the indicated GNS cell line and the mean of the two NS cell lines, and capped at (-8, 8) for visualization purposes. To obtain robust fold-change distributions, genes with low expression (<25 tags per million) in both cancer and normal cell types were excluded; consequently, between 6,014 and 6,133 genes underlie each plot. (b) For each of the three gene sets listed in the legend (inset), bars represent the percentage of genes with the indicated copy number status. (c) Aberration calls for the 29 genes broadly distinguishing GNS from NS cells by qRT-PCR. Circles indicate focal (<10 Mb) aberrations; boxes indicate larger chromosomal segments.
Despite the global correlation between gene expression and copy number, many individual expression changes could not be explained by structural alterations. For example, only a minority of upregulated genes (21%) were located in regions of increased copy number, including whole-chromosome gains (Figure 8b), the survival-associated genes HOXD10, PLS3, and TUSC3 lacked copy-number aberrations consistent with their expression changes, and the survival-associated gene DDIT3 was only genetically gained in G144, although highly expressed in all three GNS cell lines (Figure 8c). In general, the 29 genes that robustly distinguish GNS from NS cells did not show a consistent pattern of aberrations: only three genes (PDE1C, NDN and SYNM) were located in regions similarly affected by genetic lesions in all lines. Thus, in addition to copy-number alterations, other factors are important in shaping the GNS transcriptome, and regulatory mechanisms may differ among GNS cell lines yet produce similar changes in gene expression.
Discussion
To reveal transcriptional changes that underlie glioblastoma, we performed an in-depth analysis of gene expression in malignant stem cells derived from patient tumors in relation to untransformed, karyotypically normal NS cells. These cell types are closely related and it has been hypothesized that gliomas arise by mutations in NS cells or in glial cells that have reacquired stem cell features [2]. We measured gene expression by high-throughput RNA tag sequencing (Tag-seq), a method that features high sensitivity and reproducibility compared to microarrays [7]. qRT-PCR validation further demonstrates that Tag-seq expression values are highly accurate. Other cancer samples and cell lines have recently been profiled with the same method [8,47] and it should be feasible to directly compare those results to the data presented here.
Through Tag-seq expression profiling of normal and cancer stem cells followed by qRT-PCR validation in a wider panel of 22 cell lines, we identified 29 genes strongly discriminating GNS from NS cells. Some of these genes have previously been implicated in glioma, including four with a role in adhesion and/or migration, CD9, ST6GALNAC5, SYNM and TES [49-52], and two transcriptional regulators, FOXG1 and CEBPB. FOXG1, which has been proposed to act as an oncogene in glioblastoma by suppressing growth-inhibitory effects of transforming growth factor β [53], showed remarkably strong expression in all 16 GNS cell lines assayed by qRT-PCR. CEBPB was recently identified as a master regulator of a mesenchymal gene expression signature associated with poor prognosis in glioblastoma [45]. Studies in hepatoma and pheochromocytoma cell lines have shown that the transcription factor encoded by CEBPB (C/EBPβ) promotes expression of DDIT3 [54], another transcriptional regulator that we found to be upregulated in GNS cells. DDIT3 encodes the protein CHOP, which in turn can inhibit C/EBPβ by dimerizing with it and acting as a dominant negative [54]. This interplay between CEBPB and DDIT3 may be relevant for glioma therapy development, as DDIT3 induction in response to a range of compounds sensitizes glioma cells to apoptosis (see, for example, [55]).
Our results also corroborate a role in glioma for several other genes with limited prior links to the disease. This list includes PLA2G4A, HMGA2, TAGLN and TUSC3, all of which have been implicated in other neoplasias (Additional file 12). PLA2G4A encodes a phospholipase that functions in the production of lipid signaling molecules with mitogenic and pro-inflammatory effects. In a subcutaneous xenograft model of glioblastoma, expression of PLA2G4A by the host mice was required for tumor growth [56]. For HMGA2, a transcriptional regulator downregulated in most GNS cell lines, low or absent protein expression has been observed in glioblastoma compared to low-grade gliomas [57], and HMGA2 polymorphisms have been associated with survival time in glioblastoma [58]. The set of 29 genes found to generally distinguish GNS from NS cells also includes multiple genes implicated in other neoplasias, but without direct links to glioma (Additional file 12). Of these, the transcriptional regulator LMO4, may be of particular interest, as it is well studied as an oncogene in breast cancer and regulated through the phosphoinositide 3-kinase pathway [59], which is commonly affected in glioblastoma [11].
Five of these 29 genes have not been directly implicated in cancer. This list comprises one gene downregulated in GNS cells (PLCH1) and four upregulated (ADD2, LYST, PDE1C and PRSS12). PLCH1 is involved in phosphoinositol signaling [60], like the frequently mutated phosphoinositide 3-kinase complex [11]. ADD2 encodes a cytoskeletal protein that interacts with FYN, a tyrosine kinase promoting cancer cell migration [61,62]. For PDE1C, a cyclic nucleotide phosphodiesterase gene, we found higher expression to correlate with shorter survival after surgery. Upregulation of PDE1C has been associated with proliferation in other cell types through hydrolysis of cAMP and cGMP [63,64]. PRSS12 encodes a protease that can activate tissue plasminogen activator (tPA) [65], an enzyme that is highly expressed by glioma cells and has been suggested to promote invasion [66].
By considering expression changes in a pathway context, we identified additional candidate glioblastoma genes, such as the putative cell adhesion gene ITGBL1 [67], the orphan nuclear receptor NR0B1, which is strongly upregulated in G179 and is known to be upregulated and mediate tumor growth in Ewing's sarcoma [68], and the genes PARP3 and PARP12, which belong to the poly(ADP-ribose) polymerase (PARP) family of ADP-ribosyl transferase genes involved in DNA repair (Table 4). The upregulation of these PARP genes in GNS cells may have therapeutic relevance, as inhibitors of their homolog PARP1 are in clinical trials for brain tumors [69].
Transcriptome analysis thus identified multiple genes of known significance in glioma pathology as well as several novel candidate genes and pathways. These results are further corroborated by survival analysis, which revealed a GNS expression signature associated with patient survival time in five independent data sets. This finding is compatible with the notion that gliomas contain a GNS component of relevance for prognosis. Five individual GNS signature genes were significantly associated with survival of glioblastoma patients in both of the two largest data sets: PLS3, HOXD10, TUSC3, PDE1C and the well-studied tumor suppressor PTEN. PLS3 (T-plastin) regulates actin organization and its overexpression in the CV-1 cell line resulted in partial loss of adherence [70]. Elevated PLS3 expression in GNS cells may thus be relevant for the invasive phenotype. The association between transcriptional upregulation of HOXD10 and poor survival is surprising, because HOXD10 protein levels are suppressed by a microRNA (miR-10b) highly expressed in gliomas, and it has been suggested that HOXD10 suppression by miR-10b promotes invasion [71]. Notably, the HOXD10 mRNA upregulation we observe in GNS cells also occurs in glioblastoma tumors, as shown by comparison with grade III astrocytoma (Figure 3b). Similarly, miR-10b is present at higher levels in glioblastoma compared to gliomas of lower grade [71]. It is conceivable that HOXD10 transcriptional upregulation and post-transcriptional suppression is indicative of a regulatory program associated with poor prognosis in glioma.
Tumors from older patients featured an expression pattern more similar to the GNS signature. One of the genes contributing to this trend, TUSC3, is known to be silenced by promoter methylation in glioblastoma, particularly in patients aged over 40 years [72]. Loss or downregulation of TUSC3 has been found in other cancers, such as of the colon, where its promoter becomes increasingly methylated with age in the healthy mucosa [73]. Taken together, these data suggest that transcriptional changes in healthy aging tissue, such as TUSC3 silencing, may contribute to the more severe form of glioma in older patients. Thus, the molecular mechanisms underlying the expression changes described here are likely to be complex and varied. To capture these effects and elucidate their causes, transcriptome analysis of cancer samples will benefit from integration of diverse genomic data, including structural and nucleotide-level genetic alterations, as well as DNA methylation and other chromatin modifications.
To identify expression alterations common to most glioblastoma cases, other studies have profiled tumor resections in relation to non-neoplastic brain tissue [47,74,75]. While such comparisons have been revealing, their power is constrained by discrepancies between reference and tumor samples - for instance, the higher neuronal content of normal brain tissue compared to tumors. Gene expression profiling of tumor tissue further suffers from mixed signal due to a stromal cell component and heterogeneous populations of cancer cells, only some of which contribute to tumor progression and maintenance [2]. Part of a recent study bearing a closer relationship to our analysis examined gene expression in another panel of glioma-derived and normal NS cells [76], but included neurosphere cultures, which often contain a heterogeneous mixture of self-renewing and differentiating cells.
Here, we have circumvented these issues by profiling uniform cultures of primary malignant stem cell lines that can reconstitute the tumor in vivo [6], in direct comparison to normal counterparts of the same fundamental cell type [4,5]. While the resulting expression patterns largely agree with those obtained from glioblastoma tissues, there are notable differences. For example, we found the breast cancer oncogene LMO4 (discussed above) to be upregulated in most GNS cell lines, although its average expression in glioblastoma tumors is low relative to normal brain tissue (Figure 3a). Similarly, TAGLN and TES were absent or low in most GNS cell lines, but displayed the opposite trend in glioblastoma tissue compared to normal brain (Figure 3c) or grade III astrocytoma (Figure 3d). Importantly, both TAGLN and TES have been characterized as tumor suppressors in malignancies outside the brain and the latter is often silenced by promoter hypermethylation in glioblastoma [77,78].
Conclusions
Our results support the use of GNS cells as a relevant model for investigating the molecular basis of glioblastoma, and the use of NS cell lines as controls in this setting. Transcriptome sequencing revealed aberrant gene expression patterns in GNS cells and defined a molecular signature of the proliferating cell population that drives malignant brain cancers. These transcriptional alterations correlate with several prognostic indicators and are strongly associated with patient survival in both glioblastoma and lower-grade gliomas, suggesting that a greater GNS cell component contributes to poorer prognosis. Several genes observed to be consistently altered in GNS cells have not previously been implicated in glioma, but are known to play a role in other neoplasias or in cellular processes related to malignancy. Such alterations include changes in oncogene and tumor suppressor expression not detectable by microarray profiling of post-surgical glioma biopsies. These findings demonstrate the utility of cancer stem cell models for advancing the molecular understanding of tumorigenesis.
Abbreviations
CGH, comparative genomic hybridization; DMEM, Dulbecco's modified Eagle's medium; FDR, false discovery rate; GNS, glioma neural stem; MHC, major histocompatibility complex; NS, neural stem; nt, nucleotide; PARP, poly(ADP-ribose) polymerase; qRT-PCR, quantitative reverse-transcription polymerase chain reaction; RNA-seq, high-throughput shotgun sequencing of RNA transcripts; SAGE, serial analysis of gene expression; Tag-seq, high-throughput sequencing of transcript tags; TCGA, The Cancer Genome Atlas.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PGE analyzed data, designed experiments and drafted the manuscript. DT analyzed data and helped draft the manuscript. SHS and CE performed experiments. PB and SMP conceived and supervised the study and participated in writing the manuscript. All authors read and approved the final content.
Supplementary Material
Supplemental methods. Detailed method descriptions for (1) assignment of tags to genes, (2) differential expression analysis of Tag-seq data, and (3) construction of the integrated glioma pathway map. Format: PDF.
Classification of sequenced tags. Table listing the number of sequenced tags in each sample and the proportion of tags assigned to different categories by the Tag-seq data processing pipeline. Format: XLS.
qRT-PCR data. Table with raw and normalized Ct values from TaqMan qRT-PCR assays. Format: XLS.
TCGA sample IDs. Table listing sample identifiers for the TCGA expression and survival data used. Format: XLS.
Differentially expressed genes at 10% FDR. Table with expression values, fold-changes and P-values for the genes found to be expressed at a higher or lower average level in GNS cells compared to NS cells by Tag-seq (10% FDR). Format: XLS.
Differentially expressed non-coding RNAs. Table of non-coding RNAs found to be differentially expressed between GNS and NS cells by Tag-seq. Format: XLS.
Integrated pathway map. Network diagram of the integrated glioma pathway, with differentially expressed genes colored according to fold-change between GNS and NS cells. Format: PDF.
Core differentially expressed genes. Table of genes with large expression changes common to the GNS cell lines G144, G166 and G179, relative to the normal NS cell lines CB541 and CB660. Format: XLS.
Kaplan-Meier plots for multiple GNS signature score thresholds. Survival curves illustrating the association between GNS signature and patient survival for three independent glioblastoma data sets and a range of percentile thresholds on GNS signature score. Format: PDF.
Controls for survival tests on GNS expression signature. Text and table detailing how confounding factors were controlled for when testing for an association between the GNS expression signature and patient survival. Format: XLS.
Correlation between age at diagnosis and GNS signature gene expression. Scatter plots demonstrating the correlation between age at diagnosis and expression of GNS signature genes. Format: PDF.
Disease association of GNS cell-specific genes. Literature survey for the set of 29 genes found to distinguish GNS from NS cells across a panel of 22 different lines. The table details whether each gene has previously been implicated in glioma or other neoplasias, and includes references to relevant publications. Format: XLS.
Contributor Information
Pär G Engström, Email: engstrom@ebi.ac.uk.
Diva Tommei, Email: diva@ebi.ac.uk.
Stefan H Stricker, Email: s.stricker@ucl.ac.uk.
Christine Ender, Email: c.ender@ucl.ac.uk.
Steven M Pollard, Email: steven.pollard@ucl.ac.uk.
Paul Bertone, Email: bertone@ebi.ac.uk.
Acknowledgements
We thank Marco Marra for access to sequencing facilities, Tina Wong and Yongjun Zhao for technical assistance, Simon Anders for helpful discussions and Colin Watts for providing primary cultures and cell lines. This work was funded by EMBL, Cancer Research UK (grant C25858/A9160) and the Brain Tumour Charity. CE is supported by a Marie Curie Intra-European Fellowship. SMP holds an Alex Bolt Research Fellowship.
References
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff R-O. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- Park DM, Rich JN. Biology of glioma cancer stem cells. Mol Cells. 2009;28:7–12. doi: 10.1007/s10059-009-0111-2. [DOI] [PubMed] [Google Scholar]
- Chen J, Li Y, Yu T-S, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti L, Pollard SM, Gorba T, Reitano E, Toselli M, Biella G, Sun Y, Sanzone S, Ying Q-L, Cattaneo E, Smith A. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. 2005;3:e283. doi: 10.1371/journal.pbio.0030283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Pollard S, Conti L, Toselli M, Biella G, Parkin G, Willatt L, Falk A, Cattaneo E, Smith A. Long-term tripotent differentiation capacity of human neural stem (NS) cells in adherent culture. Mol Cell Neurosci. 2008;38:245–258. doi: 10.1016/j.mcn.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M, Squire JA, Smith A, Dirks P. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4:568–580. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 't Hoen PAC, Ariyurek Y, Thygesen HH, Vreugdenhil E, Vossen RHAM, de Menezes RX, Boer JM, van Ommen G-JB, den Dunnen JT. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008;36:e141. doi: 10.1093/nar/gkn705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissy AS, Morin RD, Delaney A, Zeng T, McDonald H, Jones S, Zhao Y, Hirst M, Marra MA. Next-generation tag sequencing for cancer gene expression profiling. Genome Res. 2009;19:1825–1835. doi: 10.1101/gr.094482.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Freije WA, Castro-Vargas FE, Fang Z, Horvath S, Cloughesy T, Liau LM, Mischel PS, Nelson SF. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64:6503–6510. doi: 10.1158/0008-5472.CAN-04-0452. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou M-F, de Tribolet N, Regli L, Wick W, Kouwenhoven MCM, Hainfellner JA, Heppner FL, Dietrich P-Y, Zimmer Y, Cairncross JG, Janzer R-C, Domany E, Delorenzi M, Stupp R, Hegi ME. Stem cell-related "self-renewal" signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol. 2008;26:3015–3024. doi: 10.1200/JCO.2007.15.7164. [DOI] [PubMed] [Google Scholar]
- Gravendeel LAM, Kouwenhoven MCM, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE, Daemen A, Bleeker FE, Bralten LBC, Kloosterhof NK, De Moor B, Eilers PHC, van der Spek PJ, Kros JM, Sillevis Smitt PAE, van den Bent MJ, French PJ. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69:9065–9072. doi: 10.1158/0008-5472.CAN-09-2307. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masica DL, Karchin R. Correlation of somatic mutation and expression identifies genes important in human glioblastoma progression and survival. Cancer Res. 2011;71:4550–4561. doi: 10.1158/0008-5472.CAN-11-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman H, Zhang L, Sulman EP, McDonald JM, Shooshtari NL, Rivera A, Popoff S, Nutt CL, Louis DN, Cairncross JG, Gilbert MR, Phillips HS, Mehta MP, Chakravarti A, Pelloski CE, Bhat K, Feuerstein BG, Jenkins RB, Aldape K. A multigene predictor of outcome in glioblastoma. Neuro Oncol. 2010;12:49–57. doi: 10.1093/neuonc/nop007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O'Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Q-L, Smith AG. Defined conditions for neural commitment and differentiation. Methods Enzymol. 2003;365:327–341. doi: 10.1016/s0076-6879(03)65023-8. [DOI] [PubMed] [Google Scholar]
- Wijaya E, Frith MC, Suzuki Y, Horton P. Recount: expectation maximization based error correction tool for next generation sequencing data. Genome Inform. 2009;23:189–201. [PubMed] [Google Scholar]
- Lassmann T, Hayashizaki Y, Daub CO. TagDust--a program to eliminate artifacts from next generation sequencing data. Bioinformatics. 2009;25:2839–2840. doi: 10.1093/bioinformatics/btp527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg S. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, Gil L, Gordon L, Hendrix M, Hourlier T, Johnson N, Kähäri AK, Keefe D, Keenan S, Kinsella R, Komorowska M, Koscielny G, Kulesha E, Larsson P, Longden I, McLaren W, Muffato M, Overduin B, Pignatelli M, Pritchard B, Riat HS. et al. Ensembl 2012. Nucleic Acids Res. 2012;40:D84–90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, Diekhans M, Dreszer TR, Giardine BM, Harte RA, Hillman-Jackson J, Hsu F, Kirkup V, Kuhn RM, Learned K, Li CH, Meyer LR, Pohl A, Raney BJ, Rosenbloom KR, Smith KE, Haussler D, Kent WJ. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39:D876–882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon K, Osorio EC, Greenhut SF, Schaefer CF, Shoemaker J, Polyak K, Morin PJ, Buetow KH, Strausberg RL, De Souza SJ, Riggins GJ. An anatomy of normal and malignant gene expression. Proc Natl Acad Sci USA. 2002;99:11287–11292. doi: 10.1073/pnas.152324199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim J-S, Kim CJ, Kusanovic JP, Romero R. A novel signaling pathway impact analysis. Bioinformatics. 2009;25:75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ArrayExpress. http://www.ebi.ac.uk/arrayexpress
- Dvinge H, Bertone P. HTqPCR: high-throughput analysis and visualization of quantitative real-time PCR data in R. Bioinformatics. 2009;25:3325–3326. doi: 10.1093/bioinformatics/btp578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, Irizarry RA. affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Broad GDAC Firehose. https://confluence.broadinstitute.org/display/GDAC/Home
- TCGA Data Portal: An integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. https://tcga-data.nci.nih.gov/docs/publications/gbm_exp/ [DOI] [PMC free article] [PubMed]
- Leprêtre F, Villenet C, Quief S, Nibourel O, Jacquemin C, Troussard X, Jardin F, Gibson F, Kerckaert JP, Roumier C, Figeac M. Waved aCGH: to smooth or not to smooth. Nucleic Acids Res. 2010;38:e94. doi: 10.1093/nar/gkp1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Houte BPP, Binsl TW, Hettling H, Heringa J. CGHnormaliter: a Bioconductor package for normalization of array CGH data with many CNAs. Bioinformatics. 2010;26:1366–1367. doi: 10.1093/bioinformatics/btq155. [DOI] [PubMed] [Google Scholar]
- Smyth GK, Speed T. Normalization of cDNA microarray data. Methods. 2003;31:265–273. doi: 10.1016/S1046-2023(03)00155-5. [DOI] [PubMed] [Google Scholar]
- Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23:657–663. doi: 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- van de Wiel MA, Kim KI, Vosse SJ, van Wieringen WN, Wilting SM, Ylstra B. CGHcall: calling aberrations for array CGH tumor profiles. Bioinformatics. 2007;23:892–894. doi: 10.1093/bioinformatics/btm030. [DOI] [PubMed] [Google Scholar]
- Tran CT, Wolz P, Egensperger R, Kösel S, Imai Y, Bise K, Kohsaka S, Mehraein P, Graeber MB. Differential expression of MHC class II molecules by microglia and neoplastic astroglia: relevance for the escape of astrocytoma cells from immune surveillance. Neuropathol Appl Neurobiol. 1998;24:293–301. doi: 10.1046/j.1365-2990.1998.00120.x. [DOI] [PubMed] [Google Scholar]
- Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, Mortini P, Ferrone S, Doglioni C, Marincola FM, Galli R, Parmiani G, Maccalli C. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res. 2010;16:800–813. doi: 10.1158/1078-0432.CCR-09-2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smadja DM, d' Audigier C, Weiswald L-B, Badoual C, Dangles-Marie V, Mauge L, Evrard S, Laurendeau I, Lallemand F, Germain S, Grelac F, Dizier B, Vidaud M, Bièche I, Gaussem P. The Wnt antagonist Dickkopf-1 increases endothelial progenitor cell angiogenic potential. Arterioscler Thromb Vasc Biol. 2010;30:2544–2552. doi: 10.1161/ATVBAHA.110.213751. [DOI] [PubMed] [Google Scholar]
- Dang C, Gottschling M, Manning K, O'Currain E, Schneider S, Sterry W, Stockfleth E, Nindl I. Identification of dysregulated genes in cutaneous squamous cell carcinoma. Oncol Rep. 2006;16:513–519. [PubMed] [Google Scholar]
- Qin Z, Ren F, Xu X, Ren Y, Li H, Wang Y, Zhai Y, Chang Z. ZNF536, a novel zinc finger protein specifically expressed in the brain, negatively regulates neuron differentiation by repressing retinoic acid-induced gene transcription. Mol Cell Biol. 2009;29:3633–3643. doi: 10.1128/MCB.00362-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R, Shiekhattar R. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T-K, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, Lasorella A, Aldape K, Califano A, Iavarone A. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas. http://cancergenome.nih.gov
- Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu I-M, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Hartigan J, Smith DR, Strausberg RL, Marie SKN, Shinjo SMO, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G. et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Deimling A, Korshunov A, Hartmann C. The next generation of glioma biomarkers: MGMT methylation, BRAF fusions and IDH1 mutations. Brain Pathol. 2011;21:74–87. doi: 10.1111/j.1750-3639.2010.00454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boëda B, Briggs DC, Higgins T, Garvalov BK, Fadden AJ, McDonald NQ, Way M. Tes, a specific Mena interacting partner, breaks the rules for EVH1 binding. Mol Cell. 2007;28:1071–1082. doi: 10.1016/j.molcel.2007.10.033. [DOI] [PubMed] [Google Scholar]
- Kolesnikova TV, Kazarov AR, Lemieux ME, Lafleur MA, Kesari S, Kung AL, Hemler ME. Glioblastoma inhibition by cell surface immunoglobulin protein EWI-2, in vitro and in vivo. Neoplasia. 2009;11:77–86. doi: 10.1593/neo.81180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroes RA, He H, Emmett MR, Nilsson CL, Leach FE 3rd, Amster IJ, Marshall AG, Moskal JR. Overexpression of ST6GalNAcV, a ganglioside-specific alpha2,6-sialyltransferase, inhibits glioma growth in vivo. Proc Natl Acad Sci USA. 2010;107:12646–12651. doi: 10.1073/pnas.0909862107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Huiatt TW, Paulin D, Li Z, Robson RM. Synemin interacts with the LIM domain protein zyxin and is essential for cell adhesion and migration. Exp Cell Res. 2010;316:491–505. doi: 10.1016/j.yexcr.2009.10.015. [DOI] [PubMed] [Google Scholar]
- Seoane J, Le H-V, Shen L, Anderson SA, Massagué J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- Fawcett TW, Eastman HB, Martindale JL, Holbrook NJ. Physical and functional association between GADD153 and CCAAT/enhancer-binding protein beta during cellular stress. J Biol Chem. 1996;271:14285–14289. doi: 10.1074/jbc.271.24.14285. [DOI] [PubMed] [Google Scholar]
- Kang YJ, Kim IY, Kim EH, Yoon MJ, Kim SU, Kwon TK, Choi KS. Paxilline enhances TRAIL-mediated apoptosis of glioma cells via modulation of c-FLIP, survivin and DR5. Exp Mol Med. 2011;43:24–34. doi: 10.3858/emm.2011.43.1.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkous AG, Yazlovitskaya EM, Hallahan DE. Cytosolic phospholipase A2 and lysophospholipids in tumor angiogenesis. J Natl Cancer Inst. 2010;102:1398–1412. doi: 10.1093/jnci/djq290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akai T, Ueda Y, Sasagawa Y, Hamada T, Date T, Katsuda S, Iizuka H, Okada Y, Chada K. High mobility group I-C protein in astrocytoma and glioblastoma. Pathol Res Pract. 2004;200:619–624. doi: 10.1016/j.prp.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Liu Y, Shete S, Etzel CJ, Scheurer M, Alexiou G, Armstrong G, Tsavachidis S, Liang F-W, Gilbert M, Aldape K, Armstrong T, Houlston R, Hosking F, Robertson L, Xiao Y, Wiencke J, Wrensch M, Andersson U, Melin BS, Bondy M. Polymorphisms of LIG4, BTBD2, HMGA2, and RTEL1 genes involved in the double-strand break repair pathway predict glioblastoma survival. J Clin Oncol. 2010;28:2467–2474. doi: 10.1200/JCO.2009.26.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montañez-Wiscovich ME, Seachrist DD, Landis MD, Visvader J, Andersen B, Keri RA. LMO4 is an essential mediator of ErbB2/HER2/Neu-induced breast cancer cell cycle progression. Oncogene. 2009;28:3608–3618. doi: 10.1038/onc.2009.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Choi JW, Lim S, Kwon O, Seo JK, Ryu SH, Suh P-G. Phospholipase C-η1 is activated by intracellular Ca(2+) mobilization and enhances GPCRs/PLC/Ca(2+) signaling. Cell Signal. 2011;23:1022–1029. doi: 10.1016/j.cellsig.2011.01.017. [DOI] [PubMed] [Google Scholar]
- Shima T, Okumura N, Takao T, Satomi Y, Yagi T, Okada M, Nagai K. Interaction of the SH2 domain of Fyn with a cytoskeletal protein, beta-adducin. J Biol Chem. 2001;276:42233–42240. doi: 10.1074/jbc.M102699200. [DOI] [PubMed] [Google Scholar]
- Yadav V, Denning MF. Fyn is induced by Ras/PI3K/Akt signaling and is required for enhanced invasion/migration. Mol Carcinog. 2011;50:346–352. doi: 10.1002/mc.20716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolci S, Belmonte A, Santone R, Giorgi M, Pellegrini M, Carosa E, Piccione E, Lenzi A, Jannini EA. Subcellular localization and regulation of type-1C and type-5 phosphodiesterases. Biochem Biophys Res Commun. 2006;341:837–846. doi: 10.1016/j.bbrc.2006.01.035. [DOI] [PubMed] [Google Scholar]
- Rybalkin SD, Yan C, Bornfeldt KE, Beavo JA. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ Res. 2003;93:280–291. doi: 10.1161/01.RES.0000087541.15600.2B. [DOI] [PubMed] [Google Scholar]
- Mitsui S, Yamaguchi N, Osako Y, Yuri K. Enzymatic properties and localization of motopsin (PRSS12), a protease whose absence causes mental retardation. Brain Res. 2007;1136:1–12. doi: 10.1016/j.brainres.2006.11.094. [DOI] [PubMed] [Google Scholar]
- Goh KYC, Poon WS, Chan DTM, Ip CP. Tissue plasminogen activator expression in meningiomas and glioblastomas. Clin Neurol Neurosurg. 2005;107:296–300. doi: 10.1016/j.clineuro.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Berg RW, Leung E, Gough S, Morris C, Yao WP, Wang SX, Ni J, Krissansen GW. Cloning and characterization of a novel beta integrin-related cDNA coding for the protein TIED ("ten beta integrin EGF-like repeat domains") that maps to chromosome band 13q33: A divergent stand-alone integrin stalk structure. Genomics. 1999;56:169–178. doi: 10.1006/geno.1998.5707. [DOI] [PubMed] [Google Scholar]
- García-Aragoncillo E, Carrillo J, Lalli E, Agra N, Gómez-López G, Pestaña A, Alonso J. DAX1, a direct target of EWS/FLI1 oncoprotein, is a principal regulator of cell-cycle progression in Ewing's tumor cells. Oncogene. 2008;27:6034–6043. doi: 10.1038/onc.2008.203. [DOI] [PubMed] [Google Scholar]
- Leonetti C, Biroccio A, Graziani G, Tentori L. Targeted therapy for brain tumours: role of PARP inhibitors. Curr Cancer Drug Targets. 2012;12:218–236. doi: 10.2174/156800912799277403. [DOI] [PubMed] [Google Scholar]
- Arpin M, Friederich E, Algrain M, Vernel F, Louvard D. Functional differences between L- and T-plastin isoforms. J Cell Biol. 1994;127:1995–2008. doi: 10.1083/jcb.127.6.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Yan W, Wang Y, Sun G, Luo H, Zhang J, Wang X, You Y, Yang Z, Liu N. MicroRNA-10b induces glioma cell invasion by modulating MMP-14 and uPAR expression via HOXD10. Brain Res. 2011;1389:9–18. doi: 10.1016/j.brainres.2011.03.013. [DOI] [PubMed] [Google Scholar]
- Li Q, Jedlicka A, Ahuja N, Gibbons MC, Baylin SB, Burger PC, Issa JP. Concordant methylation of the ER and N33 genes in glioblastoma multiforme. Oncogene. 1998;16:3197–3202. doi: 10.1038/sj.onc.1201831. [DOI] [PubMed] [Google Scholar]
- Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998;58:5489–5494. [PubMed] [Google Scholar]
- Tso C-L, Shintaku P, Chen J, Liu Q, Liu J, Chen Z, Yoshimoto K, Mischel PS, Cloughesy TF, Liau LM, Nelson SF. Primary glioblastomas express mesenchymal stem-like properties. Mol Cancer Res. 2006;4:607–619. doi: 10.1158/1541-7786.MCR-06-0005. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Yeh R-F, Ray A, Wang NJ, Smirnov I, Yu M, Hariono S, Silber J, Feiler HS, Gray JW, Spellman PT, Vandenberg SR, Berger MS, James CD. Comparative analyses of gene copy number and mRNA expression in glioblastoma multiforme tumors and xenografts. Neuro-oncology. 2009;11:477–487. doi: 10.1215/15228517-2008-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lottaz C, Beier D, Meyer K, Kumar P, Hermann A, Schwarz J, Junker M, Oefner PJ, Bogdahn U, Wischhusen J, Spang R, Storch A, Beier CP. Transcriptional profiles of CD133+ and CD133- glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Res. 2010;70:2030–2040. doi: 10.1158/0008-5472.CAN-09-1707. [DOI] [PubMed] [Google Scholar]
- Assinder SJ, Stanton J-AL, Prasad PD. Transgelin: an actin-binding protein and tumour suppressor. Int J Biochem Cell Biol. 2009;41:482–486. doi: 10.1016/j.biocel.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Mueller W, Nutt CL, Ehrich M, Riemenschneider MJ, von Deimling A, van den Boom D, Louis DN. Downregulation of RUNX3 and TES by hypermethylation in glioblastoma. Oncogene. 2007;26:583–593. doi: 10.1038/sj.onc.1209805. [DOI] [PubMed] [Google Scholar]
- Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, Vivanco I, Lee JC, Huang JH, Alexander S, Du J, Kau T, Thomas RK, Shah K, Soto H, Perner S, Prensner J, Debiasi RM, Demichelis F, Hatton C, Rubin MA, Garraway LA, Nelson SF, Liau L, Mischel PS, Cloughesy TF, Meyerson M, Golub TA, Lander ES, Mellinghoff IK. et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc Natl Acad Sci USA. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morey Kinney SR, Smiraglia DJ, James SR, Moser MT, Foster BA, Karpf AR. Stage-specific alterations of DNA methyltransferase expression, DNA hypermethylation, and DNA hypomethylation during prostate cancer progression in the transgenic adenocarcinoma of mouse prostate model. Mol Cancer Res. 2008;6:1365–1374. doi: 10.1158/1541-7786.MCR-08-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer KJ, Mas VR, David K, Maluf DG, Bornstein K, Fisher RA. Identifying genes for establishing a multigenic test for hepatocellular carcinoma surveillance in hepatitis C virus-positive cirrhotic patients. Cancer Epidemiol Biomarkers Prev. 2009;18:2929–2932. doi: 10.1158/1055-9965.EPI-09-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamaschi A, Kim YH, Kwei KA, La Choi Y, Bocanegra M, Langerød A, Han W, Noh D-Y, Huntsman DG, Jeffrey SS, Børresen-Dale A-L, Pollack JR. CAMK1D amplification implicated in epithelial-mesenchymal transition in basal-like breast cancer. Mol Oncol. 2008;2:327–339. doi: 10.1016/j.molonc.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragel BT, Couldwell WT, Gillespie DL, Jensen RL. Identification of hypoxia-induced genes in a malignant glioma cell line (U-251) by cDNA microarray analysis. Neurosurg Rev. 2007;30:181–187; discussion 187. doi: 10.1007/s10143-007-0070-z. [DOI] [PubMed] [Google Scholar]
- Meng X, Leyva ML, Jenny M, Gross I, Benosman S, Fricker B, Harlepp S, Hébraud P, Boos A, Wlosik P, Bischoff P, Sirlin C, Pfeffer M, Loeffler J-P, Gaiddon C. A ruthenium-containing organometallic compound reduces tumor growth through induction of the endoplasmic reticulum stress gene CHOP. Cancer Res. 2009;69:5458–5466. doi: 10.1158/0008-5472.CAN-08-4408. [DOI] [PubMed] [Google Scholar]
- Kaul A, Maltese WA. Killing of cancer cells by the photoactivatable protein kinase C inhibitor, calphostin C, involves induction of endoplasmic reticulum stress. Neoplasia. 2009;11:823–834. doi: 10.1593/neo.09388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IY, Kang YJ, Yoon MJ, Kim EH, Kim SU, Kwon TK, Kim IA, Choi KS. Amiodarone sensitizes human glioma cells but not astrocytes to TRAIL-induced apoptosis via CHOP-mediated DR5 upregulation. Neuro-oncology. 2011;13:267–279. doi: 10.1093/neuonc/noq195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Syed N, Taylor J, Smith P, Griffin B, Baens M, Bai M, Bourantas K, Stebbing J, Naresh K, Nelson M, Tuthill M, Bower M, Hatzimichael E, Crook T. DUSP16 is an epigenetically regulated determinant of JNK signalling in Burkitt's lymphoma. Br J Cancer. 2010;103:265–274. doi: 10.1038/sj.bjc.6605711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnoyers LR, Pai R, Ferrando RE, Hötzel K, Le T, Ross J, Carano R, D'Souza A, Qing J, Mohtashemi I, Ashkenazi A, French DM. Targeting FGF19 inhibits tumor growth in colon cancer xenograft and FGF19 transgenic hepatocellular carcinoma models. Oncogene. 2008;27:85–97. doi: 10.1038/sj.onc.1210623. [DOI] [PubMed] [Google Scholar]
- Korshunov A, Sycheva R, Golanov A. Genetically distinct and clinically relevant subtypes of glioblastoma defined by array-based comparative genomic hybridization (array-CGH). Acta Neuropathol. 2006;111:465–474. doi: 10.1007/s00401-006-0057-9. [DOI] [PubMed] [Google Scholar]
- Majid A, Lin TT, Best G, Fishlock K, Hewamana S, Pratt G, Yallop D, Buggins AGS, Wagner S, Kennedy BJ, Miall F, Hills R, Devereux S, Oscier DG, Dyer MJS, Fegan C, Pepper C. CD49d is an independent prognostic marker that is associated with CXCR4 expression in CLL. Leuk Res. 2011;35:750–756. doi: 10.1016/j.leukres.2010.10.022. [DOI] [PubMed] [Google Scholar]
- Lee K-M, Han W, Kim JB, Shin I, Ko E, Park IA, Lee DS, Oh K, Noh D-Y. The CD49d+/high subpopulation from isolated human breast sarcoma spheres possesses tumor-initiating ability. Int J Oncol. 2012;40:665–672. doi: 10.3892/ijo.2011.1289. [DOI] [PubMed] [Google Scholar]
- van den Boom J, Wolter M, Kuick R, Misek DE, Youkilis AS, Wechsler DS, Sommer C, Reifenberger G, Hanash SM. Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. Am J Pathol. 2003;163:1033–1043. doi: 10.1016/S0002-9440(10)63463-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibon F, Mariani O, Derré J, Mairal A, Coindre J-M, Guillou L, Sastre X, Pédeutour F, Aurias A. ASK1 (MAP3K5) as a potential therapeutic target in malignant fibrous histiocytomas with 12q14-q15 and 6q23 amplifications. Genes Chromosomes Cancer. 2004;40:32–37. doi: 10.1002/gcc.20012. [DOI] [PubMed] [Google Scholar]
- Hayakawa Y, Hirata Y, Nakagawa H, Sakamoto K, Hikiba Y, Kinoshita H, Nakata W, Takahashi R, Tateishi K, Tada M, Akanuma M, Yoshida H, Takeda K, Ichijo H, Omata M, Maeda S, Koike K. Apoptosis signal-regulating kinase 1 and cyclin D1 compose a positive feedback loop contributing to tumor growth in gastric cancer. Proc Natl Acad Sci USA. 2011;108:780–785. doi: 10.1073/pnas.1011418108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosieniak G, Pyrzynska B, Kaminska B. Nuclear factor of activated T cells (NFAT) as a new component of the signal transduction pathway in glioma cells. J Neurochem. 1998;71:134–141. doi: 10.1046/j.1471-4159.1998.71010134.x. [DOI] [PubMed] [Google Scholar]
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Mancini M, Toker A. NFAT proteins: emerging roles in cancer progression. Nat Rev Cancer. 2009;9:810–820. doi: 10.1038/nrc2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chigurupati S, Venkataraman R, Barrera D, Naganathan A, Madan M, Paul L, Pattisapu JV, Kyriazis GA, Sugaya K, Bushnev S, Lathia JD, Rich JN, Chan SL. Receptor channel TRPC6 is a key mediator of Notch-driven glioblastoma growth and invasiveness. Cancer Res. 2010;70:418–427. doi: 10.1158/0008-5472.CAN-09-2654. [DOI] [PubMed] [Google Scholar]
- Göransson M, Andersson MK, Forni C, Ståhlberg A, Andersson C, Olofsson A, Mantovani R, Aman P. The myxoid liposarcoma FUS-DDIT3 fusion oncoprotein deregulates NF-kappaB target genes by interaction with NFKBIZ. Oncogene. 2009;28:270–278. doi: 10.1038/onc.2008.378. [DOI] [PubMed] [Google Scholar]
- Oda T, Tian T, Inoue M, Ikeda J-I, Qiu Y, Okumura M, Aozasa K, Morii E. Tumorigenic role of orphan nuclear receptor NR0B1 in lung adenocarcinoma. Am J Pathol. 2009;175:1235–1245. doi: 10.2353/ajpath.2009.090010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey M, Smith R, Lessnick SL. NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing's sarcoma. Mol Cancer Res. 2006;4:851–859. doi: 10.1158/1541-7786.MCR-06-0090. [DOI] [PubMed] [Google Scholar]
- Kourtidis A, Jain R, Carkner RD, Eifert C, Brosnan MJ, Conklin DS. An RNA interference screen identifies metabolic regulators NR1D1 and PBP as novel survival factors for breast cancer cells with the ERBB2 signature. Cancer Res. 2010;70:1783–1792. doi: 10.1158/0008-5472.CAN-09-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangerich A, Bürkle A. How to kill tumor cells with inhibitors of poly(ADP-ribosyl)ation. Int J Cancer. 2011;128:251–265. doi: 10.1002/ijc.25683. [DOI] [PubMed] [Google Scholar]
- Smith SJ, Long A, Barrow JH, Macarthur DC, Coyle B, Grundy RG. Pediatric high-grade glioma: identification of poly(ADP-ribose) polymerase as a potential therapeutic target. Neuro-oncology. 2011;13:1171–1177. doi: 10.1093/neuonc/nor115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Luo Z, Li Z, Liu Y, Zhao Q. PERP gene therapy attenuates lung cancer xenograft via inducing apoptosis and suppressing VEGF. Cancer Biol Ther. 2011;12:[Epub ahead of print]. doi: 10.4161/cbt.12.12.18435. [DOI] [PubMed] [Google Scholar]
- Marques MR, Horner JS, Ihrie RA, Bronson RT, Attardi LD. Mice lacking the p53/p63 target gene Perp are resistant to papilloma development. Cancer Res. 2005;65:6551–6556. doi: 10.1158/0008-5472.CAN-05-0366. [DOI] [PubMed] [Google Scholar]
- Liu Q, Nguyen DH, Dong Q, Shitaku P, Chung K, Liu OY, Tso JL, Liu JY, Konkankit V, Cloughesy TF, Mischel PS, Lane TF, Liau LM, Nelson SF, Tso C-L. Molecular properties of CD133+ glioblastoma stem cells derived from treatment-refractory recurrent brain tumors. J Neurooncol. 2009;94:1–19. doi: 10.1007/s11060-009-9919-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff L, Castrop F, Bauer M, Höfler H, Gratzl M. Expression of vesicular monoamine transporters, synaptosomal-associated protein 25 and syntaxin1: a signature of human small cell lung carcinoma. Cancer Res. 2001;61:2138–2144. [PubMed] [Google Scholar]
- Cheung K-JJ, Johnson NA, Affleck JG, Severson T, Steidl C, Ben-Neriah S, Schein J, Morin RD, Moore R, Shah SP, Qian H, Paul JE, Telenius A, Relander T, Lam W, Savage K, Connors JM, Brown C, Marra MA, Gascoyne RD, Horsman DE. Acquired TNFRSF14 mutations in follicular lymphoma are associated with worse prognosis. Cancer Res. 2010;70:9166–9174. doi: 10.1158/0008-5472.CAN-10-2460. [DOI] [PubMed] [Google Scholar]
- Redmond WL, Ruby CE, Weinberg AD. The role of OX40-mediated co-stimulation in T-cell activation and survival. Crit Rev Immunol. 2009;29:187–201. doi: 10.1615/CritRevImmunol.v29.i3.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods. Detailed method descriptions for (1) assignment of tags to genes, (2) differential expression analysis of Tag-seq data, and (3) construction of the integrated glioma pathway map. Format: PDF.
Classification of sequenced tags. Table listing the number of sequenced tags in each sample and the proportion of tags assigned to different categories by the Tag-seq data processing pipeline. Format: XLS.
qRT-PCR data. Table with raw and normalized Ct values from TaqMan qRT-PCR assays. Format: XLS.
TCGA sample IDs. Table listing sample identifiers for the TCGA expression and survival data used. Format: XLS.
Differentially expressed genes at 10% FDR. Table with expression values, fold-changes and P-values for the genes found to be expressed at a higher or lower average level in GNS cells compared to NS cells by Tag-seq (10% FDR). Format: XLS.
Differentially expressed non-coding RNAs. Table of non-coding RNAs found to be differentially expressed between GNS and NS cells by Tag-seq. Format: XLS.
Integrated pathway map. Network diagram of the integrated glioma pathway, with differentially expressed genes colored according to fold-change between GNS and NS cells. Format: PDF.
Core differentially expressed genes. Table of genes with large expression changes common to the GNS cell lines G144, G166 and G179, relative to the normal NS cell lines CB541 and CB660. Format: XLS.
Kaplan-Meier plots for multiple GNS signature score thresholds. Survival curves illustrating the association between GNS signature and patient survival for three independent glioblastoma data sets and a range of percentile thresholds on GNS signature score. Format: PDF.
Controls for survival tests on GNS expression signature. Text and table detailing how confounding factors were controlled for when testing for an association between the GNS expression signature and patient survival. Format: XLS.
Correlation between age at diagnosis and GNS signature gene expression. Scatter plots demonstrating the correlation between age at diagnosis and expression of GNS signature genes. Format: PDF.
Disease association of GNS cell-specific genes. Literature survey for the set of 29 genes found to distinguish GNS from NS cells across a panel of 22 different lines. The table details whether each gene has previously been implicated in glioma or other neoplasias, and includes references to relevant publications. Format: XLS.