

Measurement of residual dipolar couplings (RDCs) between pairs of nuclei has become a routine method for macromolecular structure determinations by solution NMR spectroscopy.1 Compared to the short-range restraints derived from inter-proton distances, torsion angles, and J couplings the RDCs can provide unique long-range orientation restraints of inter-nuclear vectors to the molecular alignment frame2, and thus are very useful in improving the accuracy and quality of protein NMR structure. This type of information is essential when the molecule under study is multidomain.3 RDCs can also provide important internal dynamic information of biomolecules.4 To measure the RDCs, a weak alignment of molecule in a magnetic field is required. As such, various technologies for creating the necessary partial alignment in solution have been developed, including direct magnetic alignment,5 alignment in different liquid crystals,6 or by dissolving the protein in a highly hydrated, anisotropically compressed polyacrylamide gel.7 Recently, new detergent-compatible liquid crystals based on DNA nanotubes8 and nucleic acid G-tetrad structures9 have been successfully exploited.
Although currently available alignment media are quite diverse, their application is still molecule specific. Most of them are suitable only to the application of water-soluble protein. Only a few are compatible with the detergents used in membrane protein study. In addition as the effective size gets larger alignment order will significantly increase and lead to extensive broadening of the NMR resonance signals due to the strong 1H-1H long-range dipolar couplings. The most effective way to reduce order is to scale down the alignment media concentration. Most media however are not stable at low concentration. Bicelles become unstable when the concentration is lower than 2.5% (w/v),10 polyethylene-glycol is easy to phase separate when the concentration is less than 3% (w/w),6d cetylpyridinium bromide-hexanol mixtures will lose 2H quadrupolar splitting at lower concentration, e.g., ~2.5% (w/w).11 Besides the DNA-based liquid crystals, the polyacrylamide gels is the only medium that can provide weak alignment in the presence of detergents.7 Potential drawbacks relate to the pore size of the gel matrices, which can make it difficult to dissolve larger protein–micelle complexes,12 and which can adversely affect the rotational correlation time.7b
Here we describe a new gel system that can weakly align a molecule for solution NMR spectroscopy. The aligned gels were obtained through the polymerization of ordered collagen in a magnetic field. Anisotropy in the collagen gel can be detected in the form of quadrupole splittings in the 2H NMR spectra. Experiments were carried out on uniformly 13C/15N enriched glutamine-binding protein (GlnBP), a 226 residues protein. The structure of GlnBP has been determined by X-ray crystallography.13 To date, there are around 28 different types of collagen described in literature.14 But only type I and III collagens isolated from tissues can polymerize in vitro.15 In this study, the rat tail tendon type I is selected for preparing the collagen gels. The pH value and temperature are two controlling factors for inducing the collagen polymerization. The polymerization is usually very weak at lower pH value or lower temperature but becomes significantly faster at pH range 6~8 and temperature range of 25~42°C. The collagen monomer was prepared as described in Saffarian et. al..16 It is kept dissolved in acetic acid at pH value ~3. We neutralized the collagen solution by adding 2M Tris/HCl, pH 8.0. The pH value was measured to make sure it is ~7.5 (It can be adjusted by adding NaOH). An appropriate amount of protein, D2O and NaCl were added into the collagen solution to make a final sample of 200 mM Tris/HCl, 200 mM NaCl, ~150 uM protein and 10% D2O. This was all carried out at 4°C. The mixture was put into an NMR tube and placed in a Bruker DMX 600 MHz magnet. To achieve initial alignment of the collagen monomer in the magnetic field the temperature was kept at 4°C for 3 hours. Subsequently it was increased to room temperature at a rate of 1°C/10 min. This was followed by elevating the temperature to 37°C at a rate of 1°C/15 min for polymerization. The quadrupolar splitting of the 2H NMR signal of the solvent could be observed when the temperature reached 32°C. This splitting stabilized in two hours after the temperature is increased to 37°C.
As expected, the quadrupolar splitting observed in the 2H NMR spectrum increased with increasing collagen concentration (Figure 1), indicating that ordering effect induced by collagen gel can be tuned by adjusting its concentration. After polymerization, the 2H splitting of the collagen gel becomes stable and totally temperature independent over the entire range tested (5-40°C). The polymerization is associated by the color change of the sample, from clear to slightly opaque white. The opaqueness is correlated to the concentration of the collagen (see the Supporting Information).
Figure 1.
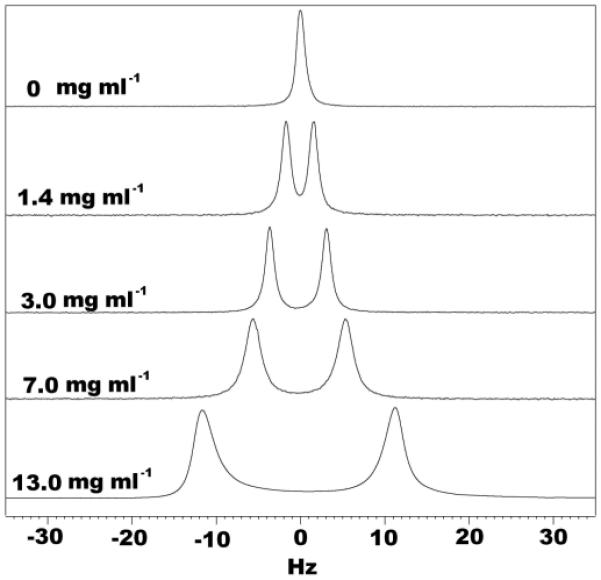
1D 2H spectra of collagen gel samples in 200 mM tris, 200 mM NaCl, pH 8.0, 90%/10% H2O/D2O with different initial collagen concentration. These spectra were recorded at 37°C with four scans and a sweep width of 300 Hz on a Bruker DMX 600 MHz spectrometer.
Figure 2A and B show 1H-15N IPAP-HSQC17 spectra for the uniformly 13C/15N labeled GlnBP in the absence and presence of 13 mg ml−1 collagen. Figure 2C and D show the correlation between Dmeas and Dcalc of GlnBP in 1.4 mg ml−1 and 13 mg ml−1 collagen gels, respectively. The magnitude of the RDCs observed in 1.4 mg ml−1 ranges from −1 to +1 Hz. The relatively large experimental error (0.4 Hz) compared to the magnitude of RDCs causes the dispersion between the measured and calculated RDCs (R=0.796). A 13 mg ml−1 collagen gel yielded about 10 times larger RDCs than 1.4 mg ml−1 collagen gel, and the dipolar couplings show good agreement with the X-ray structure (R=0.98).
Figure 2.
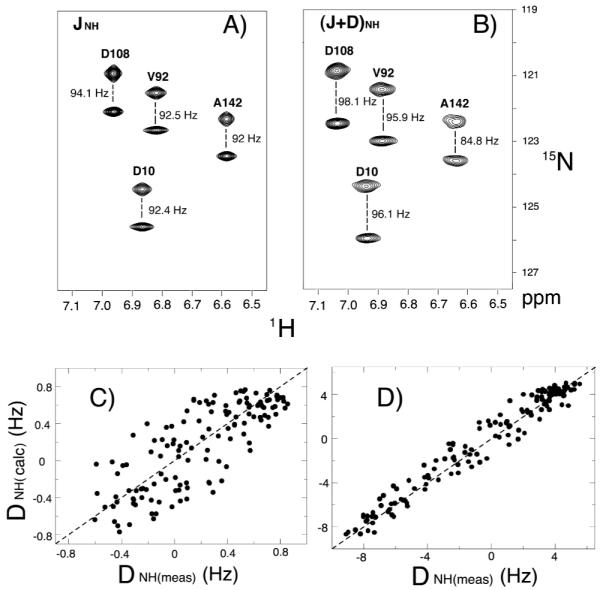
1H-15N IPAP-HSQC17 spectra for 150 μM uniformly 13C and 15N labeled GlnBP in 200 mM tris, 200 mM NaCl, pH 7.5 buffer condition. JNH splittings are shown for the isotropic sample (A). (J+D)NH splittings are obtained from the 13 mg ml−1 collagen gel (B). Spectra containing the upfield and downfield 15N-{1H} doublet components of the 1H-15N IPAP-HSQC spectra are superimposed. Measurements were conducted on a Bruker 600 MHz DMX NMR spectrometer at 37 °C. Correlation between measured 15N-1H RDCs of GlnBP in 1.4 mg ml−1 (C) and 13 mg ml−1 (D) collagen gel and couplings calculated on the basis of 1.94 Å X-ray crystal structure of GlnBP.13 Uncertainties in the measured dipolar couplings are (C) 0.4 Hz and (D) 0.6 Hz, the correlation r –factor between the measured and calculated is (C) 0.796 and (D) 0.981.
The collagen gels also showed good pH and detergent tolerance. The collagen gels (1.4 mg ml−1) were still stable after being soaked in low pH acetate buffer solution (pH=4) or in 100 mM DPC solution for several days. This was evaluated by the 2H splitting that could still be observed (Supporting Information).
The collagen molecule is a rod about 1.5 nm in diameter and 300 nm long, substantially smaller than Pf1 phage particles, which have a diameter of ~6.5 nm and 2000 nm in length.6c The smaller size and their organization lead to a smaller net susceptibility for the collagen monomer, thus necessitating a longer time to align in the magnetic field. However polymerization essentially locks that alignment so that the sample is permanently ordered after the initial preparation. Previous studies revealed the structure of collagen.18 It is made up of three left-handed polypeptide strands which are twisted together into a right-handed coiled coil. The numerous hydrogen bonds between proline and hydroxyproline will form a cooperative quaternary structure that highly stabilizes the collagen helix. This highly stable structure makes collagen gel quite suitable for biomolecular NMR measurements over a wide range of temperature or in various physical conditions, such as low pH value, high ionic strength or detergents. At the above concentrations, the collagen gels are very soft. This permits buffer exchange readily through diffusion. This also allows the addition or removal of protein after polymerization in the same manner.
The collagen gel provides a robust and cost effective alternative to align large biomolecules and membrane proteins in a magnetic field. It can be polymerized at various alignment directions relative to the field, thus affording another mechanism to modulate the size of the observed RDCs.7b, 19
Supplementary Material
Acknowledgment
This work was supported by the Intramural Research Program of the NIH, National Heart, Lung, and Blood Institute to NT and R01CA123363 from NCI and R01AR040618 from NIAMS to GIG. We thank Dr. Chien Ho for providing us with the GlnBP plasmid.
Footnotes
Supporting Information Available: Photographs of collagen samples before and after polymerization; 2H splitting of collagen samples as a function of temperature, at low pH value buffer condition and in DPC solution; 2H splitting of collagen as a function of the length of polymerization time.
References
- (1).Bax A, Kontaxis G, Tjandra N. Meth. Enzym. 2001;339:127–174. doi: 10.1016/s0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- (2).Clore GM, Gronenborn AM, Tjandra NJ. Magn. Reson. 1998;131:159–162. doi: 10.1006/jmre.1997.1345. [DOI] [PubMed] [Google Scholar]
- (3).Blackledge M. Prog. Nucl. Magn. Reson. Spectrosc. 2005;46:23–61. [Google Scholar]
- (4).(a) Veglia G, Opella SJ. J. Am. Chem. Soc. 2000;122:11733–11734. [Google Scholar]; (b) Peti W, Meiler J, Bruschweiler R, Griesinger C. J. Am. Chem. Soc. 2002;124:5822–5833. doi: 10.1021/ja011883c. [DOI] [PubMed] [Google Scholar]; (c) Briggman KB, Tolman JR. J. Am. Chem. Soc. 2003;125:10164–10165. doi: 10.1021/ja035904+. [DOI] [PubMed] [Google Scholar]; (d) Bouvignies G, Markwick P, Brüschweiler R, Blackledge M. J. Am. Chem. Soc. 2006;128:15100–15101. doi: 10.1021/ja066704b. [DOI] [PubMed] [Google Scholar]; (e) Lange OF, Lakomek NA, Farès C, Schröder GF, Walter KF, Becker S, Meiler J, Grubmüller H, Griesinger C, de Groot BL. Science. 2008;320:1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- (5).(a) Tolman JR, Flanagan JM, Kennedy MA, Prestegard JH. Proc. Natl. Acad. Sci. U.S.A. 1995;92:9279–9283. doi: 10.1073/pnas.92.20.9279. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tjandra N, Grzesiek S, Bax A. J. Am. Chem. Soc. 1996;118:6264–6272. [Google Scholar]
- (6).(a) Tjandra N, Bax A. Science. 1997;278:1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]; (b) Ottiger M, Bax AJ. J. Biomol. NMR. 1999;13:187–191. doi: 10.1023/a:1008395916985. [DOI] [PubMed] [Google Scholar]; (c) Hansen MR, Mueller L, Pardi A. Nat. Struct. Biol. 1998;5:1065–1074. doi: 10.1038/4176. [DOI] [PubMed] [Google Scholar]; (d) Rückert M, Otting G. J. Am. Chem. Soc. 2000;122:7793–7797. [Google Scholar]; (e) Prosser RS, Losonczi JA, Shiyanovskaya IV. J. Am. Chem. Soc. 1998;120:11010–11011. [Google Scholar]
- (7).(a) Tycko R, Blanco FJ, Ishii Y. J. Am. Chem. Soc. 2000;122:9340–9341. [Google Scholar]; (b) Sass HJ, Musco G, Stahl SJ, Wingfield PT, Grzesiek S. J. Biomol. NMR. 2000;18:303–309. doi: 10.1023/a:1026703605147. [DOI] [PubMed] [Google Scholar]; (c) Chou JJ, Gaemers S, Howder B, Louis JM, Bax A. J. Biomol. NMR. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- (8).Douglas SM, Chou JJ, Shih WM. Proc. Nat. Acad. Sci. U.S.A. 2007;104:6644–6648. doi: 10.1073/pnas.0700930104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lorieau J, Yao LS, Bax A. J. Am. Chem. Soc. 2008;130:7536–7537. doi: 10.1021/ja801729f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ottiger M, Bax A. J. Biomol. NMR. 1998;12:5822–5833. doi: 10.1023/a:1008366116644. [DOI] [PubMed] [Google Scholar]
- (11).Barrientos LG, Dolan C, Gronenborn AM. J. Biomol. NMR. 2000;16:329–337. doi: 10.1023/a:1008356618658. [DOI] [PubMed] [Google Scholar]
- (12).Zhang XZ, Yang YY, Chung TS, Ma KX. Langmuir. 2001;17:6094–6099. [Google Scholar]
- (13).(a) Hsiao CD, Sun YJ, Rose J, Wang BC. J. Mol. Biol. 1996;262:225–242. doi: 10.1006/jmbi.1996.0509. [DOI] [PubMed] [Google Scholar]; (b) Sun YJ, Rose J, Wang BC, Hsiao CD. J. Mol. Biol. 1998;278:219–229. doi: 10.1006/jmbi.1998.1675. [DOI] [PubMed] [Google Scholar]
- (14).Myllyharju J, Kivirikko KI. Ann. Med. 2001;33:7–21. doi: 10.3109/07853890109002055. [DOI] [PubMed] [Google Scholar]
- (15).(a) Kadler KE, Hojima Y, Prockop DJ. J. Biol. Chem. 1987;262:15696–15701. [PubMed] [Google Scholar]; (b) Velling T, Risteli J, Wennerberg K, Mosher DF, Johansson S. J. Biol. Chem. 2002;277:37377–37381. doi: 10.1074/jbc.M206286200. [DOI] [PubMed] [Google Scholar]
- (16).Saffarian S, Collier IE, Marmer BL, Elson EL, Goldberg G. Science. 2004;306:108–111. doi: 10.1126/science.1099179. [DOI] [PubMed] [Google Scholar]
- (17).Ottiger M, Delaglio F, Bax A. J. Magn. Reson. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- (18).Ramachandran GN, Kartha G. Nature. 1954;174:269–270. doi: 10.1038/174269c0. [DOI] [PubMed] [Google Scholar]
- (19).Ruan K, Tolman JR. J. Am. Chem. Soc. 2005;127:15032–15033. doi: 10.1021/ja055520e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.